
Abstract
Tissue regeneration is a complex process involving large changes in cell proliferation, fate determination, and differentiation. Mitochondrial dynamics and metabolism play a crucial role in development and wound repair, but their function in large-scale regeneration remains poorly understood. Planarians offer an excellent model to investigate this process due to their remarkable regenerative abilities. In this study, we examine mitochondrial dynamics during planarian regeneration. We find that knockdown of the mitochondrial fusion gene, opa1, impairs both tissue regeneration and stem cell pluripotency. Interestingly, the regeneration defects caused by opa1 knockdown are rescued by simultaneous knockdown of the mitochondrial fission gene, drp1, which partially restores mitochondrial dynamics. Furthermore, we discover that Mitolow stem cells exhibit an enrichment of pluripotency due to their fate choices at earlier stages. Transcriptomic analysis reveals the delicate mitonuclear balance in metabolism and mitochondrial proteins in regeneration, controlled by mitochondrial dynamics. These findings highlight the importance of maintaining mitochondrial dynamics in large-scale tissue regeneration and suggest the potential for manipulating these dynamics to enhance stem cell functionality and regenerative processes.
Similar content being viewed by others
Introduction
Functional regeneration is critical for maintaining organ health and facilitating a healthy lifespan. The process of regeneration, which requires the precise coordination of cell fate decisions with positional information, is highly conserved across different organisms1,2,3,4. While several signaling have been identified as evolutionally conserved to govern the regeneration, the metabolism programs remain largely unknown.
Mitochondria have been realized as crucial signaling centers in cell reprogramming and lineage commitment, and they may play a pivotal role in organ health, aging, and degenerative diseases5,6,7,8,9,10,11,12. Mitochondrial dynamics, including their fusion, fission, selective degradation, transport, and associated metabolism, are critical for stem cell fate, development, and cellular repair7,8,9,10,11,13,14,15,16,17,18,19,20,21. Specifically, in the case of Caenorhabditis elegans, changes in mitochondrial dynamics are key to epidermal cell membrane wound healing22. Similarly, in axolotls (Ambystoma mexicanum), a population of cells enriched with mitochondria was identified during limb regeneration23,24. These findings collectively point toward the role of mitochondria underlying tissue regeneration. However, the mechanistic contributions of mitochondrial dynamics to large-scale tissue regeneration, including body regeneration, remain largely unknown25.
Investigating highly regenerative animals provides valuable insights into the mechanisms of regeneration. The planarian flatworm is an ideal research organism for tissue regeneration studies, including at the whole-body scale26,27,28. The regenerative process is primarily credited to adult stem cells, known as neoblasts, in this flatworm29,30,31. These neoblasts exhibit regenerative and quasi-immortal attributes and are thus key reservoirs of body regeneration4,32,33. It remained unclear whether and how the metabolism mechanism controls the fitness of these adult stem cells to coordinate the cell fate plasticity and respond to unpredicted injuries. Previous studies have crucially noted a bimodal distribution of low and high mitochondrial membrane potential (MMP) within these neoblasts; the resulting cell groups were named Mitolow and Mitohigh cells34,35. This suggests a correlation between the regulation of cell fate and mitochondrial phenotypes in this flatworm. However, it remained unknown whether the influence of any mitochondrial dynamics can help study this connection between mitochondrial dynamics and body regeneration in planarians.
To address this gap, we used the planarian Schmidtea mediterranea (S. med.) as a model organism to examine the role of mitochondrial dynamics, particularly mitochondrial fusion and fission in body regeneration. We first showed the mitochondrial dynamics during planarian regeneration and in different cell types. Particularly, Mitolow cells that retain pluripotency contain tubular mitochondria, and Mitohigh cells have granular mitochondria. Regulation of the mitochondrial fusion–fission equilibrium by opa1 and drp1 plays a critical role in controlling planarian regeneration and cell pluripotency. This process is crucial for mitochondrial metabolism and mitonuclear balance, thereby regulating the expression of mitochondrial protein-encoding genes essential for planarian regeneration and the fitness of neoblasts in response to injuries. Considering the evolutionary significance of planarians as highly regenerative animals, this study highlights potential conserved mitochondrial functions retained early for stem cell pluripotency and tissue regeneration. Understanding this mechanism could potentially yield strategies to enhance regenerative capacity in various organisms, including humans.
Results
Mitochondria dynamics in planarian regeneration and various cell populations
Previous studies have used vital dye combination, including SiR-DNA for DNA content, CellTracker Green (CTG) for cell volume, and Mito Tracker/PK Mito for MMP, to enrich Mitolow cells from planarian neoblasts34,35,36. To comprehensively compare the mitochondrial dynamics in various cell populations, we employed the same strategy to distinguish five cell populations based on cell cycle and mitochondrial states from wild-type planarians. Based on DNA contents, 2N and 4N cells were grouped (Fig. 1A). 4N cells were divided into two groups based on their CTG staining intensity as 4N CTGlow and 4N CTGhigh (Fig. 1B)37. Within 4N CTGhigh cells, 4N CTGhighMitolow (Mitolow) and 4N CTGhighMitohigh (Mitohigh) cells were isolated based on the level of MMP (Fig. 1C). To facilitate reproducibility, we displayed a summary of sequential plots illustrating the gating strategy for Mitolow and Mitohigh cells (Fig. S1A). To validate the Mitolow and Mitohigh cells from planarian neoblasts, we performed FACS analysis by comparing the wild type with irradiated planarians (100 Gy). The results showed that Mitolow and Mitohigh cells from SiRNeoblasts are irradiation-sensitive (Fig. S1B). We also compared the cell cycle distribution of Mitolow and Mitohigh cells with that of the nucleated cells sorted after Hoechst 33342 staining. Mitolow, Mitohigh, and nucleated cells were immediately fixed after sorting and stained with DAPI for cell cycle analysis. Both Mitolow and Mitohigh cells displayed enrichment in the S/G2/M phase (Fig. S1C). We further analyzed the ratio of Mitolow and Mitohigh cells in both X1 cells and SiRNeoblasts and found that X1 and SiRNeoblasts have similar proportions of Mitolow and Mitohigh cells (Fig. S1D). These results suggest that Mitolow and Mitohigh cells are similarly proliferating cells at S/G2/M cell cycle stages.

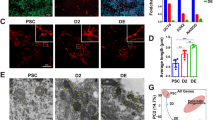
A Density plot of FACS gating based on DNA content to isolate SiR-DNA 4N (red circle) and SiR-DNA 2N (yellow circle) cells. B Pseudocolor dot plot shows the CTGhigh (red circle, SiRNeoblasts) and CTGlow (left of the blue dot line) cell populations among SiR-DNA 4N cells. C Pseudocolor dot plot of the gating strategy for two cell populations from CTGhigh with different PK Mito Red intensity, named Mitolow and Mitohigh. D Peaks in piwi-1 intensity for the enrichment of piwi-1high, piwi-1low, and piwi-1- in each indicated cell population as determined via FACS analysis. E Colorimetric WISH images show the piwi-1 probe staining in 60 Gy-irradiated hosts at 7 days post-transplantation with Mitolow or Mitohigh cells. The numbers indicate the ratio of hosts harboring piwi-1+ neoblasts. Scale bar, 500 μm. F Rescue efficiency of Mitolow or Mitohigh cells transplanted into lethally irradiated hosts. The upper right images show a rescued animal at the indicated time points. n = 10 hosts in each of three independent experiments. Scale bar, 500 μm. G Representative Hessian-SIM images of mitochondria in Mitolow (n = 33), Mitohigh (n = 32), CTGlow (n = 39), and 2N (n = 48) cells. Scale bars, 1 μm. H–K Dot plots of mitochondrial size (H), total length (I), branch length (J), and length/branch ratio (K). The quantification is shown as mean ± SEM. The unpaired two-tailed Student’s t-test was used to determine the significance of differences between two conditions. L Pseudocolor dot plots show Mitolow and Mitohigh cells with their percentages from regenerating planarians, including 0 hpa, 1 hpa, 6 hpa, 12 hpa, 1 dpa, 2 dpa, 3 dpa, 5 dpa, and 7 dpa. The quantification is shown as mean ± SEM. M Line plot shows the changes in the ratio of Mitolow and Mitohigh cells from wide-type regenerating planarians. The quantification is shown as mean ± SEM. n = 6 for 0 hpa and 4 for other time points. P-values were calculated by two-way ANOVA followed by Dunnett’s multiple comparisons.
To validate the enrichment of pluripotency in Mitolow cells in our study, we used the pan-neoblast marker piwi-1 to examine the piwi-1+ and piwi-1high cells within the five cell populations. A comparison among these populations showed that Mitolow cells contained the highest ratio of piwi-1+ cells (Fig. S1E, F) and enriched the highest proportion of piwi-1high neoblasts than the other cell populations as well as controls of Mitolow cells stained with piwi-1 sense probe or reagents without probes (Fig. 1D). We thereafter performed a transplantation assay to compare the pluripotency of Mitolow cells to Mitohigh cells. Following bulk-cell transplantation into neoblast-depleted planarian hosts, Mitolow cells proliferated more successfully than Mitohigh cells, forming colonies with an efficiency rate of 91% (n = 32). This rate surpassed that of the Mitohigh cells, which had a 4% efficiency rate (n = 25) (Fig. 1E). Additionally, only Mitolow cells rescued the viability of stem cell-depleted hosts that were irradiated with 60 Gy (Fig. 1F). Hence, these findings validated the pluripotency of Mitolow cells.
To explore the mitochondrial morphological dynamics in planarians, we examined whether mitochondria exhibit different morphologies in Mitolow, Mitohigh, CTGlow, and 2N cells. Leveraging the capacity of Hessian structured illumination microscopy (SIM), we directly observed the mitochondria in Mitolow, Mitohigh, CTGlow, and 2N cells. Our analysis revealed that Mitolow cells housed elongated, tubular mitochondria, whereas Mitohigh cells contained short, globular mitochondria (Fig. 1G). Quantitative analysis indicated a significant increase in mitochondrial size, total length, branch length, and ratio of length/branch of mitochondria from the Mitolow cells compared with the Mitohigh cells, consistent with the results of Hessian SIM (Fig. 1H–K). Mitochondrial morphology undergoes changes throughout the cell cycle. To further assess whether Mitolow and Mitohigh cells originate from similar cell cycles, we compared X1 cells from the S phase and G2/M phase with X2 and Xins cells. Our results revealed no obvious difference between cells in the S phase and G2/M phases, unlike those in X2 and Xins populations (Fig. S2A–E). This supports the aforementioned results of the cell cycle analysis, indicating that Mitolow and Mitohigh cells are in similar cell cycle stages. Additionally, we isolated Mitolow and Mitohigh cells from X1 to compare their mitochondrial morphology. We found similar mitochondrial morphology differences as we observed in the Mitolow and Mitohigh cells from SiRNeoblasts (Fig. S2F–J). By performing staining with MitoTracker Orange to measure the MMP, MitoTracker Green to measure the mitochondrial mass, and MitoSOX to measure reactive oxygen species (ROS) levels, we confirmed that Mitolow and Mitohigh cells exhibited lower and higher MMP (Fig. S2K), mitochondrial mass (Fig. S2L), and ROS levels (Fig. S2M), respectively.
We then examined the dynamics of the Mitolow and Mitohigh cell ratio during regeneration. Quantitative analysis of FACS data showed that the Mitolow cell ratio increased to its peak at 12 hours post-amputation (hpa), while the Mitohigh cell ratio decreased initially and then increased after 12 hpa, reaching its peak at 3 days post-amputation (dpa) (Fig. 1L, M). During planarian regeneration, neoblasts respond to injury by undergoing hyperproliferation, resulting in two peaks at 6 and 48 hpa, respectively38. The peak ratios of Mitolow and Mitohigh cells at 12 hpa and 3 dpa, respectively, may be the consequence of the substantial proliferation and differentiation of neoblasts. These observations prompted us to hypothesize that these variations in mitochondrial morphology might be related to changes in planarian cell fate status during body regeneration. Meanwhile, we acknowledge the limitation of this method for detecting the mitochondria in cells in vivo. Tomograph and other higher resolution in situ methods for mitochondria in planarians need to be developed to validate the dynamics in the future.
The mitochondrial fusion gene opa1 is required for planarian regeneration
To investigate the hypothesis suggested by our staining results, we next examined the relationship between mitochondria and regeneration, particularly the role of mitochondrial morphology changes during regeneration. The homologous gene candidates of six mitochondrial dynamics-related genes were identified in planarian S. med., including mitochondrial fusion genes Smed-opa1, Smed-mfn1, Smed-mfn2, mitochondrial fission genes Smed-drp1 and Smed-fis1, and mitochondrial transportation gene Smed-miro-1 (henceforth referred to as opa1, mfn1, mfn2, drp1, fis1, and miro-1) (Supplementary Data 1). Bacteria expressing dsRNA were used to feed planarians to knock down gene expression, and regeneration was tested after amputation. Through assessment for defects in regeneration but not homeostatic viability, we discovered that the mitochondrial fusion gene opa1 was essential for planarian regeneration postamputation, while knockdown of the mitochondrial fission gene drp1 resulted in only a minor regeneration delay for tail fragments (Fig. 2A, Supplementary Data 1). Quantitative real-time PCR (qPCR) showed a substantial reduction in transcripts after opa1 RNAi and drp1 RNAi (Fig. S3A, B). We then investigated organ integrity at 7 and 14 dpa via in situ hybridization and found that regeneration of the central nervous system (chat+), intestine (gata4/5/6+), and pharynx (laminin+) was blocked in planarians with opa1 knockdown compared with egfp RNAi controls (Fig. 2B, Fig. S3C).

A Representative images show regeneration outcomes in live animals of egfp RNAi, opa1 RNAi, and drp1 RNAi animals at 7 dpa. Dpa, days post-amputation. n = 10 for each RNAi treatment. Scale bar, 500 μm. B In situ probes for chat and gata4/5/6 were used to show regeneration defects of the central nervous system and intestine in opa1 RNAi animals compared with egfp RNAi and drp1 RNAi animals at 7 dpa. n = 4 for each condition. Scale bar, 500 μm. C TEM images of planarian neoblasts (upper row) display the morphology of mitochondria in egfp RNAi, opa1 RNAi, and drp1 RNAi animals. Cells with neoblast characteristics were selected for observation. The experiment was repeated three times. The lower-row images correspond to the boxed areas in the upper-row images. Scale bars, 1 μm. D Representative Hessian-SIM images of mitochondria in Mitolow cells from egfp RNAi, opa1 RNAi, and drp1 RNAi planarians. Scale bar, 1 μm. E–H Dot plots of mitochondrial size (E), total length (F), branch length (G), and length/branch ratio (H) of Mitolow cells from egfp RNAi (n = 24), opa1 RNAi (n = 38), and drp1 RNAi (n = 35) planarians. The quantification is shown as mean ± SEM. Unpaired two-tailed Student’s t test was used to determine the significance of differences between two conditions. I Pseudocolor dot plots show Mitolow and Mitohigh cells with their percentages from egfp RNAi and opa1 RNAi animals. Mitolow: p-value = 2.56 × 10-5 (72.9 ± 1.13% vs. 88.8 ± 0.80%); Mitohigh: p-value = 7.78 × 10-5 (20.6 ± 0.67% vs. 6.4 ± 0.74%) as opa1 RNAi versus egfp RNAi controls. The quantification is shown as mean ± SEM. Unpaired two-tailed Student’s t test was used to determine the significance of differences between two conditions.
To validate the effects of opa1 RNAi on mitochondrial morphology, we first showed that the ectopic expression of Myc-tagged planarian Opa1 could localize to the mitochondria in mammalian cells (Fig. S3D) due to the lack of an antibody against Opa1 and technical limitations of exogenous gene expression in planarian cells. Thereafter, transmission electronic microscopy (TEM) was used to compare the mitochondrial morphology in tissue sections from opa1 RNAi, drp1 RNAi, and egfp RNAi control animals. Previous studies have indicated that neoblasts could be identified with TEM through their lack of cytoplasmic differentiation and the presence of prominent decondensed chromatin30,39. Therefore, to compare among similar cell types, mitochondria were compared in neoblasts and were found that mitochondria within opa1 RNAi planarians were notably smaller and had lower cristae maturation than those within the egfp RNAi controls (Fig. 2C). In contrast, we observed increased mitochondrial interconnection and size in drp1 RNAi planarians compared with egfp RNAi controls (Fig. 2C). We also examined the consequence of mitochondrial morphology in mfn1;mfn2 RNAi, and fis1 RNAi animals. Despite significant reduction in their transcript levels (Fig. S3E, G), the mitochondrial morphology did not display any obvious changes in mfn1;mfn2 RNAi and fis1 RNAi compared to those in opa1 RNAi and drp1 RNAi animals (Fig. S3F, H). The lack of effect on mitochondrial morphology may explain the absence of defective regeneration in these RNAi conditions.
To confirm the changes in mitochondrial morphology elicited by opa1 RNAi and drp1 RNAi, we directly observed the mitochondria by Hessian SIM in isolated Mitolow cells from egfp RNAi, opa1 RNAi, and drp1 RNAi planarians. Our analysis revealed that egfp RNAi Mitolow cells housed elongated, tubular mitochondria, whereas opa1 RNAi Mitolow cells contained short, globular mitochondria (Fig. 2D) with a significant reduction in mitochondrial size (Fig. 2E), total length (Fig. 2F), branch length (Fig. 2G), and the ratio of length/branch of mitochondria (Fig. 2H). In contrast, drp1 RNAi Mitolow cells contained mitochondria with a significant increase in mitochondrial size, total length, and branch length compared with egfp RNAi Mitolow cells (Fig. 1D–H). These results confirmed the impact on the mitochondrial morphology caused by opa1 RNAi and drp1 RNAi. Concurrent with these alterations in mitochondrial dynamics, the application of opa1 RNAi significantly reduced the ratio of Mitolow cells compared to the egfp RNAi controls (Fig. 2I). These results suggest that defects in mitochondrial fusion by opa1 RNAi impact the pluripotency of Mitolow cells.
Reducing drp1 expression mitigates opa1 RNAi-induced regeneration impairment and mitochondrial morphological disruption
To examine whether mitochondrial dynamics plays a decisive role in cell fate and regeneration, we sought to ameliorate the deficiencies observed in animals subjected to opa1 RNAi. The drp1 RNAi resulted in an opposite morphology compared with that after opa1 RNAi (Fig. 2C–H). We, therefore, investigated whether drp1 RNAi ameliorated mitochondrial anomalies and regeneration defects in opa1 RNAi planarians. Subsequently, in addition to performing egfp;egfp RNAi, opa1;egfp RNAi, and drp1;egfp RNAi, we generated planarians with simultaneous opa1 and drp1 knockdown (referred to as opa1;drp1 RNAi animals). Unlike prior observations with opa1 RNAi animals, opa1;drp1 RNAi animals showed noticeable regeneration following amputation, mirroring the level of regeneration observed in the egfp;egfp RNAi and drp1;egfp RNAi animals (Fig. 3A). Probing for the intestinal and central nervous systems at 7 dpa via fluorescence in situ hybridization of mat and pc2 confirmed the rescue of the regenerative defects in opa1;drp1 RNAi animals (Fig. 3B). To validate the efficiency of gene suppression, we performed the qPCR analysis. The results showed that opa1;egfp RNAi and drp1;egfp RNAi significantly decreased the transcript levels of opa1 and drp1, respectively, compared to the egfp;egfp RNAi condition (Fig. S4A, B). Meanwhile, opa1;drp1 RNAi exhibited a comparable reduction in the transcript level of opa1 and drp1 to opa1;egfp RNAi and drp1;egfp RNAi conditions, respectively (Fig. S4A, B).

A Images of live egfp;egfp RNAi, drp1;egfp RNAi, opa1;egfp RNAi, and opa1;drp1 RNAi animals at 7 dpa. n = 30 for each treatment. Scale bar, 500 μm. B In situ probes for mat and pc2 show the regeneration outcomes of intestinal, and neuronal cells, respectively, in egfp;egfp RNAi, opa1;egfp RNAi, and opa1;drp1 RNAi animals at 7 dpa. n = 3 for each treatment. Scale bar, 500 μm. C TEM images of planarian neoblasts (upper row) display the morphology of mitochondria in egfp;egfp RNAi, drp1;egfp RNAi, opa1;egfp RNAi, and opa1;drp1 RNAi animals. The lower-row images correspond to the boxed areas in the upper-row images. The experiment was repeated three times. Scale bars, 1 μm (upper row) and 200 nm (lower row). D Representative Hessian-SIM images of mitochondria in egfp;egfp RNAi, drp1;egfp RNAi, opa1;egfp RNAi, and opa1;drp1 RNAi Mitolow cells. Scale bars, 1 μm. E–H Dot plots of mitochondrial size (E), total length (F), branch length (G), and length/branch ratio (H) egfp;egfp RNAi (n = 30), drp1;egfp RNAi (n = 28), opa1;egfp RNAi (n = 29), and opa1;drp1 RNAi (n = 35) Mitolow cells. The quantification is shown as mean ± SEM. Unpaired two-tailed Student’s t test was used to determine the significance of differences between two conditions.
We then investigated the phenotypic consequences of concurrent opa1 and drp1 knockdown on mitochondrial morphology via TEM. The results showed rescue of mitochondrial morphology in neoblasts of the opa1;drp1 RNAi animals similar to that in the egfp;egfp RNAi animals but in contrast to those in the opa1;egfp RNAi cohort (Fig. 3C). Furthermore, we directly observed mitochondria by Hessian SIM in isolated Mitolow cells from egfp;egfp RNAi, drp1;egfp RNAi, opa1;egfp RNAi, and opa1;drp1 RNAi planarians (Fig. 3D). Our analysis revealed that the mitochondrial morphology index values, which included the mitochondrial size (Fig. 3E), total length (Fig. 3F), branch length (Fig. 3G), and ratio of length/branch of mitochondria (Fig. 3H), were increased in the opa1;drp1 RNAi Mitolow cells to a similar level as in the egfp;egfp RNAi Mitolow cells, in contrast to those in the opa1;egfp RNAi Mitolow cells. A similar consequence was also observed in Mitohigh cells (Fig. S4C–G). The effect on the ratio of Mitolow and Mitohigh cells in different RNAi conditions was also assessed. Our analysis revealed that the ratio of Mitolow cells decreased concurrently with alterations in mitochondrial dynamics in both homeostasis (Fig. S4H, I) and at 1 dpa of opa1;egfp RNAi worms (Fig. S4J, K). This ratio was restored in opa1;drp1 RNAi worms. In contrast to the decrease in Mitolow cells, there was an increase in Mitohigh cells in opa1;egfp RNAi worms, which was also restored in opa1;drp1 RNAi Mitohigh cells.
Mitochondrial fusion–fission equilibrium is essential for neoblast proliferation and differentiation potency
To further evaluate the phenotypes of opa1 RNAi on regeneration, we examined its effects on cell proliferation and differentiation. In physiological homeostasis, no discernible decrease in neoblasts (Fig. 4A) or H3P+ proliferating neoblasts was detected upon opa1 RNAi (Fig. 4B). During regeneration in wild-type S. med., neoblasts are known to initiate two substantial proliferation bursts at intervals of 6 and 48 hpa38. We next examined the consequence of opa1 RNAi on neoblast proliferation during regeneration. In amputated opa1 RNAi worms, the ratios of H3P+ cells were significantly reduced at both 6 and 48 hpa compared to those in egfp RNAi controls (Fig. 4C–E), indicating a deficiency in the capacity of neoblasts to respond to tissue loss. In line with this restoration of mitochondrial morphology, cell proliferation rates during regeneration were also increased in opa1;drp1 RNAi animals compared with opa1;egfp RNAi animals according to the density of H3P+ cells at 6 and 48 hpa, respectively (Fig. 4D, E).

A Representative images of FISH staining for piwi-1 in egfp RNAi and opa1 RNAi planarians in homeostasis at 7 days post-feeding (dpf). Scale bar, 200 μm. B Images of egfp RNAi and opa1 RNAi planarians at 7 dpf stained with an anti-H3P antibody. Scale bar, 200 μm. C Changes in H3P+ cell density during regeneration of different RNAi planarian tails. n = 4 tails biological replicates from each RNAi condition at every time point. D, E Changes in H3P+ cell density at 6 hpa (D) and 48 hpa (E) for the indicated RNAi planarians. Besides drp1 RNAi animal tail samples at 48 hpa (n = 4), n = 5 tail samples were used from each of the other RNAi conditions at 6 hpa and 48 hpa. Each dot indicates an independent individual animal. F Representative images of piwi-1+ signals in different RNAi planarians at 7 and 14 dpi following 12.5 Gy of irradiation. Scale bars, 500 μm. G Quantification of piwi-1+ cell density at 14 dpi following 12.5 Gy of irradiation of the indicated RNAi animals. Besides opa1 RNAi animal samples (n = 9), n = 10 independent individual samples from each of the other RNAi conditions. H–J Quantification of co-stained cells positive for PIWI-1 and ston2 (H), hnf4 (I), or prog-1 (J) at 2 dpa in different RNAi planarians. Besides opa1 RNAi animal samples (n = 4), n = 3 samples from each of the other RNAi conditions (H), and n = 4 biological replicates from each RNAi condition (I–J), each dot indicates an independent data point. K Bar plot shows the ratio of X1 and X2 cells in different RNAi planarians. n = 4 biological replicates for each RNAi condition. L Bar plot shows the ratio of S and G2/M stages in X1 cells from different RNAi planarians. n = 4 biological replicates for each RNAi condition. The quantification in the analysis of all data in Fig. 4 (C–E) and (G–L) is shown as mean ± SEM, a two-tailed Student’s t-test was used to calculate the p-value versus egfp RNAi controls.
Consistently, we noticed significant suppression of neoblast repopulation following sublethal irradiation in organisms subjected to opa1 RNAi compared to egfp RNAi controls (Fig. 4F, G; Fig. S5A, B). Neoblast repopulation was increased to normal in opa1;drp1 RNAi animals compared with egfp;egfp RNAi and drp1;egfp RNAi controls (Fig. 4F, G). To determine whether the defects in neoblast proliferation are cell autonomous, we transplanted neoblasts isolated from opa1 RNAi planarians into neoblast-depleted wild-type hosts and vice versa and performed the same procedure. As predicted, the neoblasts of opa1 RNAi planarians failed to exhibit engraftment and clonogenic capacity in contrast to neoblasts of egfp RNAi control animals, as indicated by the significant difference in piwi-1+ cell number at 7 days post-transplantation (7 dpt) (Fig. S5C, D). In contrast, transplanted healthy, wild-type neoblasts engrafted and proliferated in irradiated opa1 RNAi worms at a level higher than that in irradiated egfp RNAi worms (Fig. S5E). These results suggest that Opa1-regulated mitochondrial fusion is cell-autonomously essential for maintaining neoblast proliferation in response to regeneration.
Interestingly, when we examined whether drp1 RNAi-induced mitochondrial interconnection affects neoblast proliferation during regeneration, we found significant reductions in H3P+ cells at both 6 and 48 hpa (Fig. 4D, E), but only slightly reduced repopulation after sublethal irradiation (Fig. 4F, G; Fig. S5A, B) and no difference after transplantation of drp1 RNAi neoblasts compared to egfp RNAi controls (Fig. S5C–E). Considering the successful regeneration of drp1 RNAi planarians, these results suggest that the reduced proliferation by drp1 RNAi is a non-cell-autonomous effect on neoblasts and that reduced proliferation alone does not explain the regeneration defects in opa1 RNAi planarians.
To further elucidate the function of mitochondria in cell differentiation during regeneration, we harvested regenerating trunks at 2 dpa and proceeded with whole-mount labeling of the neural progenitor marker ston2, intestinal progenitor marker hnf4, and early epidermal progenitor marker prog1. Immunofluorescence staining revealed significantly lower levels of ston2, hnf4, and prog-1 co-expression with PIWI-1 in the regenerating blastema region upon opa1 RNAi compared with egfp RNAi controls (Fig. 4H–J, Fig. S5F–H). The differentiation rates during regeneration were also increased in opa1;drp1 RNAi animals compared with opa1;egfp RNAi animals according to the density of progenitors of ston2+PIWI-1+ (Fig. 4H), hnf4+PIWI-1+ (Fig. 4I), and prog-1+PIWI-1+ (Fig. 4J) cells in the blastema at 2 dpa. Staining the patterning markers at 3 dpa for anterior sfrp-1 and notum and posterior wnt-1, wnt11-2, and wntP-2 revealed that the reestablishment of tissue patterning was disturbed in opa1 RNAi planarians compared to egfp RNAi controls (Fig. S5I). To assess whether the defects in cell differentiation could be attributed to an influence on the cell cycle, we performed cell cycle analysis on the proportion of X1 cells and the S/G2/M stages. The statistical results showed no significant reduction in neoblasts in opa1;egfp RNAi planarians on the proportion of X1 and X2 cells (Fig. 4K, Fig. S5J). Further analysis of the cell cycle of X1 cells revealed a decrease in the ratio of cells in the G2/M phase and an increase in the S phase (Fig. 4L, Fig. S5K), which potentially influenced the cell fate in opa1;egfp RNAi planarians. These above findings indicate that maintaining the equilibrium of mitochondrial dynamics is fundamental for determining stem cell fate when these cells must replace missing tissues and meet the demands of regeneration.
Mitolow cells are enriched at earlier stages of fate choices than Mitohigh Cells
Before delving into the role of opa1 in neoblast pluripotency, we first conducted experiments to elucidate the relationship between neoblast pluripotency and mitochondrial dynamics. After isolating cells from wild-type intact planarians using flow cytometry, we employed single-cell RNA sequencing (scRNA-seq) and subjected the cells to UMAP analysis, which resulted in 23 distinct clusters (Fig. 5A). Each cluster was assigned to a specific cell type based on the expression of neoblast subtype markers40,41,42,43,44,45 (Fig. 5A, Fig. S6A, Supplementary Data 2). Subsequently, we determined the lineage identity of each cell and compared the compositions of Mitolow and Mitohigh cells. Within the identified clusters, two clusters showed high expression of tgs-1, which we referred to as tgs-1+ neoblast c2 and c3 for further analysis. Analysis of individual scRNA-seq datasets revealed distinct cluster compositions between Mitolow and Mitohigh cells (Fig. 5B). While both Mitolow and Mitohigh cells comprised epidermal, muscular, cathepsin+/phagocytotic, parenchymal, protonephridia, and neural neoblasts, they differed in the composition of these clusters. Mitolow cells predominantly contained tgs-1+ neoblast c2 and c3, as well as intestinal neoblasts. In addition, Mitolow cells expressed higher levels of neoblast-enriched genes and lower levels of XBP1 and P4HB compared with Mitohigh cells (Fig. S6B), which matches the gene expression signatures of pluripotent neoblasts in the single-step fate model46. These results provide evidence to support the higher pluripotency status of Mitolow cells. To avoid the batch effect, we performed additional scRNA-seq analysis on Mitolow and Mitohigh cells isolated from tails and intact animals. The results showed a similar distinguishment in the enrichment of different cell clusters (Fig. S7A).

A UMAP plot of integrated single-cell data from Mitolow and Mitohigh cells. The cell types of each cluster in the same lineage are grouped with dashed shapes. B UMAP plots show clusters enriched in Mitolow and Mitohigh cells, respectively. The clusters grouped into the same cell lineages are shown in the same color. C UMAP plots show the subclustered epidermal neoblasts in Mitolow and Mitohigh cells, respectively. D Dot plot shows the expression of epidermal subtype markers across epidermal Mitolow and Mitohigh clusters. E Expression of subtype-specific markers in epidermal Mitolow and Mitohigh clusters. F UMAP plots show the subclustered muscular neoblasts in Mitolow and Mitohigh cells, respectively. G Dot plot shows the expression of muscular subtype markers across muscular Mitolow and Mitohigh clusters. H UMAP plots show the subclustered cathepsin+ neoblasts in Mitolow and Mitohigh cells, respectively. I Dot plot shows the expression of cathepsin+ subtype markers across cathepsin+ Mitolow and Mitohigh clusters. J UMAP plot shows the subclustered tgs-1+ neoblast cluster 2 (c2). K Expression of subtype-specific markers in the subclustered tgs-1+ neoblast c2. L UMAP plot shows the subclustered tgs-1+ neoblast c3. M Expression of subtype-specific markers in the subclustered tgs-1+ neoblast c3.
We next sought to elucidate the transcriptomic signatures associated with the pluripotency of Mitolow cells. We utilized the recently reported fate-specific transcription factors (FSTFs) to validate the subclusters within the epidermal, muscular, cathepsin+, and neural lineages33. By integrating and subclustering the epidermal cells from the Mitolow and Mitohigh cells, we identified 11 subclusters of epidermal neoblasts (Fig. 5C). The Mitolow epidermal neoblasts exhibited higher expression of canonical FSTFs such as zfp-1, co-expressing with soxP-3, and p53, while persisting the expression of soxP-3, and p53 in Mitohigh epidermal neoblasts (Fig. 5D, E). Conversely, the Mitohigh epidermal neoblasts predominantly expressed G0 progenitor FSTFs such as prog-2 and agat-3 (Fig. 5D, E). Notably, the dorsal and ventral epidermis, marked by the expression of PRDM-1 and kal1 respectively, were enriched in Mitohigh cells (Fig. 5D, E). These results suggest that the Mitolow epidermal neoblasts are more enriched at earlier stages of fate choices compared to Mitohigh cells. Subclustering muscular neoblasts revealed a total of 18 subclusters (Fig. 5F). Clusters 0, 1, 2, 3, 5, and 15 were enriched in Mitolow cells and displayed highly expressed HIST1H2AA and H2AFJ genes, which are core components of the nucleosome. In contrast, most subclusters in Mitohigh cells exhibited enriched expression of cell-type-specific FSTFs, including longitudinal muscle marker myoD, circular muscle marker nkx1-1, and intestinal muscle marker gata4/5/6-3 and PTPRD. Importantly, these TFs were already expressed during the Mitolow stage, suggesting the onset of muscular neoblast diversity within Mitolow cells (Fig. 5G, Fig. S7B). Our subclustering of cathepsin+ neoblasts revealed 5 clusters (Fig. 5H). Cluster 0, enriched in Mitolow cells, exhibited high expression of zfp-1 and TYMS genes. Conversely, the cathepsin+ cell marker CTSL was enriched in Mitohigh neoblasts, suggesting the enrichment of earlier phagocytic cell fates in Mitolow cells (Fig. 5I, Fig. S7C). When examining the subclusters of tgs-1+ neoblast c2 and tgs-1+ neoblast c3 within Mitolow cells, we identified 8 subclusters (Fig. 5J, L). Compared to tgs-1+ neoblast c3, tgs-1+ neoblast c2 had a higher enrichment of tgs-1+, gcm-1+, and ski-3+ cells. Of particular note, the neural FSTFs such as pou4l-1 and neuroD-1 were highly expressed in tgs-1+ neoblast c2 (Fig. 5K, M). Earlier stage FSTFs, such as the epidermal marker zfp-1, muscular marker myoD, and intestinal marker gata4/5/6-2, were enriched in the tgs-1+ neoblast c3 subcluster (Fig. 5K, M). Previous research proposed that tgs-1+ neoblasts might be neural-specialized neoblasts46. Our dataset, however, revealed previously undescribed cell clusters that co-express tgs-1 with multiple FSTFs. The lineage hierarchy of these cells warrants further detailed analysis in future studies. Altogether, our cell atlas of Mitolow and Mitohigh cells indicates that Mitolow cells show higher enrichment at earlier stages of fate choices compared to Mitohigh cells, as evidenced by the differential expression of FSTFs across various lineages.
Changes in mitochondrial dynamics regulate the gene expression in multiple lineages of neoblasts
To investigate gene expression changes in RNAi conditions, we performed bulk RNA-seq and scRNA-seq analyses (Fig. 6A). Bulk RNA-seq was conducted on egfp RNAi and opa1 RNAi tripartite fragments at 1 dpa as well as on tail fragments at various time points post-amputation (Fig. 6A). The results of bulk RNA-seq analysis were consistent with qPCR, indicating that the double knockdown of opa1 and drp1 effectively reduced the transcript levels of both genes to a similar extent as their respective single knockdown conditions (Fig. S8A, B). To assess the wound response and neoblast response in the early stages of regeneration, we analyzed the expression of wound-induced genes and neoblast-enriched genes. The bulk RNA-seq analysis showed that opa1 RNAi animals, which can normally activate the expression of early wound-induced genes, exhibited higher expression levels of pim-3, runt-1, and wnt-1 at 1 dpa. (Fig. 6B, Supplementary Data 3). The heatmap also showed a decrease in the expression levels of neoblast-enriched genes, particularly piwi-1 and histone2B, in opa1 RNAi animals compared to egfp RNAi animals at 1 dpa. (Fig. 6B, Supplementary Data 3). Additionally, piwi-1 and histone2B exhibited reduced transcript levels at 2 dpa, along with other neoblast-enriched transcripts including mcm4, vasa-1, and cyclinB-1 (Fig. S8C, Supplementary Data 3). These results are consistent with the findings from H3P antibody staining experiments conducted at both 6 and 48 hpa in opa1 RNAi animals compared to egfp RNAi controls (Fig. 4C–E). It further supports the result that there is a significant deficit in neoblasts’ capacity to respond to injury. Furthermore, the bulk RNA-seq data showed a decrease in the expression of anterior PCGs, including ndl-2, wnt2, ndl-5, fz5/8-4, sfrp-1, ndk, and fz4-4 in opa1 RNAi (Fig. S8C, Supplementary Data 3), consistent with the FISH results that opa1 RNAi animals were unable to establish the positional information during regeneration (Fig. S5I).

A Illustration of gene expression analysis for bulk RNA-seq of tripartite fragments at 1 dpa from egfp RNAi, opa1 RNAi, and drp1 RNAi, tail fragments at various time points post-amputation from egfp RNAi and opa1 RNAi, and Mitolow scRNA-seq at 1 dpa. B Heatmap shows the expression of signature genes after opa1 knockdown. The red asterisk (*) means DEGs compared opa1 RNAi with egfp RNAi control group. The up- (rose boxes) and down- (black boxes) regulated genes were identified with the threshold of Fold change > 1.2 and adjusted p-value < 0.05. C UMAP plot of integrated single-cell data from Mitolow cells of the indicated RNAi animals at 1 dpa. The clusters categorized into the same cell types are shown in the same color. D Differential gene expression analysis comparing opa1;egfp RNAi and egfp;egfp RNAi Mitolow cells shows up- and down-regulated genes across all cell clusters. Top: Bar graph showing the number of DEGs in each cluster. Middle: For each cluster, the change in expression of significantly changed genes (p adj <0.05, |log2FC | > 0.585) is shown. Bottom: The cell fate to which different clusters belong. The colors indicate the clusters. LogFC, log fold change. E Venn diagram presents the number of genes significantly downregulated in opa1;egfp RNAi compared with egfp;egfp RNAi, but significantly upregulated in opa1;drp1 RNAi compared with opa1;egfp RNAi Mitolow cells. F KEGG pathway enrichment and (G) Protein-protein interaction networks (PPI) display genes selected from the Venn diagram in (E). The Benjamini-Hochberg method was used to adjust the p values. The adjusted p-value < 0.05 was set to determine the significantly changed expression level. H Venn diagram presents the number of genes significantly upregulated in opa1;egfp RNAi compared with egfp;egfp RNAi, but significantly downregulated in opa1;drp1 RNAi compared with opa1;egfp RNAi Mitolow cells. I KEGG pathway enrichment and (J) Protein-protein interaction networks (PPI) display genes selected from the Venn diagram in (H). The Benjamini-Hochberg method was used to adjust the p values. The adjusted p-value < 0.05 was set to determine the significantly changed expression level.
To investigate the impact of opa1 knockdown on neoblast pluripotency and the proliferation reduction observed during regeneration, we compared the scRNA-seq data of Mitolow cells at 1 dpa from animals subjected to egfp;egfp RNAi, drp1;egfp RNAi, opa1;egfp RNAi, and opa1;drp1 RNAi. In total, 39,596 cells were collected from all four groups at 1 dpa, and egfp;egfp RNAi were performed twice as a technical replicate. Cells were categorized into 17 clusters by utilizing UMAP analysis on the expression of enriched genes (Fig. 6C). The same gene signatures that were employed to annotate the Mitolow cells from wild-type intact planarians were used to confirm the identity of each cluster (Fig. S9A, Supplementary Data 2). Consistency in cell clusters and cell ratios between the two egfp RNAi replicates indicated the robustness of the experimental results (Fig. S9B, C). Compared to Mitolow cells from wild-type intact planarians, the integrated cell atlas from the above RNAi animals at 1 dpa also comprised epidermal, protonephridia, intestinal, cathepsin+, muscular, parenchymal, and tgs-1+ neoblasts. Notably, the extended source of cells enabled the distinction of foxA+ and NKX2-3+ parenchymal neoblasts (Fig. 6C). We also identified a previously unreported cell cluster characterized by the expression of runt-1. Since runt-1 is a wound-induced gene in neoblasts47, this cell population might represent a specific neoblast population induced in regeneration. Furthermore, we also identified an unknown neoblast cluster exhibiting high expression of histone-encoding genes HIST1H2AA and H2AFJ. These newly identified cell clusters present intriguing targets for future studies into their functions in planarians.
To understand the impact of RNAi treatment, we compared the proportion of each cell cluster across different RNAi groups. No obvious difference was observed (Fig. S9B, C). We next analyzed the differential expressed genes (DEGs) in each cell cluster by comparing them with egfp;egfp RNAi controls (Supplementary Data 4). We identified 152 upregulated genes and 760 downregulated genes when comparing opa1;egfp RNAi with egfp;egfp RNAi (Fig. 6D). A noticeable fewer DEGs were observed in drp1;egfp RNAi (Fig. S9D), supporting the findings of minor influence on neoblast pluripotency by drp1 knockdown (Fig. 3D–G). To further explore the rescue of gene expression, we compared the gene expression in opa1;drp1 RNAi with opa1;egfp RNAi (Supplementary Data 4). 61 genes that were downregulated in opa1;egfp RNAi were significantly increased by opa1;drp1 RNAi (Fig. 6E), including genes associated with oxidative phosphorylation (OXPHOS), mitochondrial biogenesis, and PPAR signaling pathways. Importantly, the expression of 60 out of these 61 genes was rescued in opa1;drp1 RNAi by comparing with egfp;egfp RNAi treatment (Supplementary Data 4). The recovery of genes enriched in the ribosome and PPAR signaling pathway suggests that the cellular translation activity was restored (Fig. 6F, G). Intriguingly, egr-4, which is essential for planarian head regeneration48, was downregulated in opa1;egfp RNAi but rescued in opa1;drp1 RNAi. Moreover, fos-1 as one of the first wave genes in response to wound47, was downregulated in opa1;egfp RNAi and restored in opa1;drp1 RNAi planarians. This suggests that the mitochondrial dynamics, affected by opa1 and drp1, are also involved in the regulation of early response signals that are critical for planarian regeneration. 59 genes shown as upregulated in opa1;egfp RNAi were significantly decreased by opa1;drp1 RNAi (Fig. 6H). These genes, including RARS1, NARS1, DARS1, MARS1, and EPRS1 in the aminoacyl-tRNA biosynthesis and CARS in the transfer RNA biogenesis pathways, were enriched in these DEGs (Fig. 6I, J; Supplementary Data 4). A group of the upregulated DEGs (19/59) were rescued in cluster 11 muscular neoblasts, like RPS29, a 40S ribosome protein. This suggests that the opa1;egfp RNAi causes abnormal translation, leading to upregulated transcription of genes associated with the tRNA biosynthesis, which is restored in opa1;drp1 RNAi planarians.
To assess the impact of opa1 RNAi across all cell types besides stem cells, we compared the single-cell atlas of egfp;egfp RNAi, drp1;egfp RNAi, opa1;egfp RNAi, and opa1;drp1 RNAi animals. Given the potential of dissociation manipulation to result in the loss of fragile cells and induce stress responses, we employed single-nucleus RNA sequencing (snRNA-seq) to characterize the cellular components in RNAi planarians. This analysis yielded a total of 42,965 nuclei, which were subsequently categorized into 26 clusters through UMAP based on the expression of enriched genes in each cluster. The average number of genes per cell nucleus was 1,031, consistent with the typical range for snRNA-seq. Using marker genes, we identified 9 major cell types in the cell atlas (Fig. S10A, B, Supplementary Data 2). These clusters were then validated using classic planarian cell type markers (Fig. S10A), including neoblast, epidermal, muscular, intestinal, cathepsin+/phagocytic, protonephridia, pharyngeal, neural, and parenchymal cells. The diversity of cell types and gene heterogeneity were consistent with previous reports42,43.
We also compared the proportion of each cell cluster among different RNAi groups. No obvious difference was observed (Fig. S10C). We then examined the DEGs in each cell cluster by comparing them with egfp;egfp RNAi controls (Fig. S11A–C, Supplementary Data 5). We identified 10 downregulated genes in opa1;egfp RNAi that were rescued by opa1;drp1 RNAi (Fig. S11D, E). These included MT-ND1, MT-ND3, ND4, ATP6, MT-COX1, MT-COX2, and MT-COX3, which are related to OXPHOS and encoded by the mitochondrial genome (Supplementary Data 5). These genes were downregulated in epidermal, neoblast, and intestinal clusters, but their expression was restored in neoblasts following opa1;drp1 RNAi (Fig. S11A, D, E; Supplementary Data 5). Similarly, we observed these genes downregulated in 1 dpa mitolow scRNA-seq analysis after opa1 knockdown (Fig. 6E, Supplementary Data 4). Furthermore, we identified 12 upregulated genes that were rescued by opa1;drp1 RNAi (Fig. S11F, Supplementary Data 5). Genes encoding IARS1, QARS1, AARS1, and RARS1, which are involved in aminoacyl-tRNA biosynthesis, were upregulated in neoblasts, epidermal cells, and intestinal cells after opa1 knockdown. Their expression was restored in epidermal cells following opa1;drp1 RNAi. This indicates that opa1 knockdown stimulates these genes specifically in epidermal cells. Despite these findings, it is important to note that the number of genes differentially expressed following opa1 knockdown in other cell types was smaller than in neoblasts. This suggests that opa1-regulated mitochondrial dynamics primarily influence genes and functions involved in neoblasts during the early stages of regeneration, although we cannot exclude the plausibility of other cell types.
To complement the single-cell transcriptional analysis and identify gene expression changes specific to opa1 RNAi at 1 dpa, we further utilized bulk RNA-seq data from intact and 1 dpa time points (Fig. 6A). By comparing with egfp RNAi and drp1 RNAi, we identified 111 genes that were specifically downregulated, and 26 genes upregulated in opa1 RNAi planarians at 1 dpa (Fig. S12A–F, Supplementary Data 6). Pathway enrichment analysis found that UQCR10, UQCRH, and UQCRQ, which are associated with OXPHOS, were detected in bulk RNA-seq data (Fig. S12G, H). Histone proteins such as H4C1, H3-5, H2AZ1, and H3C1 showed lower expression in opa1 RNAi worms (Supplementary Data 6). In addition, genes involved in RNA splicing, including H4C1, YBX3, SNRPF, SNRPD1, SNRPD2, and SF3B5, were downregulated (Fig. S12H). The translation initiation factor EIF1, along with downregulated ribosomal protein genes RPLP2 and RPS25, suggested that opa1 knockdown decreases transcription and translation during regeneration (Fig. S12H). Furthermore, RARS1 associated with arginyl-tRNA was upregulated at 1 dpa (Fig. S12I), suggesting a feedback regulation to compensate for reduced translation following opa1 RNAi. These DEGs were enriched in similar signaling pathways compared to those enriched in the scRNA-seq analysis (Fig. 6F, G, I, J; Fig. S11E, F), offering potential insights into the regulatory pathways affected by mitochondrial fusion defects.
Mitochondrial Fusion–Fission Equilibrium is Critical for Mitonuclear Balance
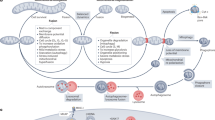
To facilitate the study of DEGs related to mitochondrial dynamic changes, we comprehensively categorized the annotated genes encoded by the nuclear genome and mitochondrial DNA (mtDNA). Previous studies have estimated that the mitochondrial proteome in yeast and humans contains at least 1000 ∼ 1500 different proteins49,50. In a study of the planarian mtDNA, it was found that the sexual and asexual biotypes of Schmidtea mediterranea have mtDNA with lengths of 27,133 bp and 17,176 bp, respectively. However, both biotypes have the same number of genes encoding for 12 proteins (ND1, ND2, ND3, ND4, ND4L, ND5, ND6, COX1, COX2, COX3, CYTB, and ATP6), 2 rRNA, and 22 tRNAs51. Although the planarian mitochondrial proteome has not been reported, gene homologs have been identified by searching the nuclear genome sequence for known mitochondrial-related genes (Supplementary Data 7)52. The identified proteins were then grouped according to their major functions, including mitochondrial detoxification, mitochondrial dynamics and surveillance, mitochondrial ribosome assembly, translation, OXPHOS, and mitochondrial metabolism. Consequently, we integrated and classified the genes that were transcriptionally up-regulated and down-regulated in association with opa1 RNAi, compared with egfp RNAi controls (Supplementary Data 7, Fig. 7A).

A Summary of the DEGs in multi-transcriptomic analysis. Genes are summarized with subcellular localization and annotated functions. Downregulated and upregulated genes are in blue and red, respectively. B Left: representative WB images of egfp;egfp RNAi, opa1;egfp RNAi, drp1;egfp RNAi, and opa1;drp1 RNAi planarians treated with puromycin (puro) or puromycin and cycloheximide (CHX). Ponceau staining was used to confirm equal protein loading. n = 3 biological replicates for each condition; each biological replicate was the mixture of proteins of six RNAi worms. Right: Quantitative representation of puromycin incorporation observed in left WB images. The data are shown as the mean ± SEM. P-value was calculated by unpaired two-tailed Student’s t-test versus egfp;egfp RNAi controls. C Quantification of the relative expression level of UPR signals in opa1 RNAi animals compared with egfp RNAi animals. n = 3 biological replicates for each RNAi condition. The quantification is shown as mean ± SEM. P-value was calculated by unpaired two-tailed Student’s t-test versus egfp RNAi controls. D Representative live images show the block of regeneration after mrpl2 RNAi and uqcr10 RNAi. Scale bar, 500 μm. E Quantification of H3P+ cell density at indicated regeneration time points of egfp RNAi, mrpl2 RNAi, and uqcr10 RNAi animals. Besides uqcr10 RNAi animal samples at 48 hpa and 72 hpa (n = 4 biological replicates for each condition), n = 3 samples from each of the other RNAi conditions at each time point. The data are shown as the mean ± SEM. Unpaired two-tailed Student’s t-test was used to determine the significance of differences at each time point versus egfp RNAi controls. F Representative FISH images of anterior and posterior patterning markers at 72 hpa. n = 3 worms of biological replicates for each RNAi treatment. Scale bar, 50 μm.
Through above transcriptome analysis, we found that the defects in opa1-regulated mitochondrial dynamics caused the downregulation of nuclear genome encoded H4C1, YBX3, SF3B5, SNRPD1, SNRPD2, and SNRPF, which are enriched in the U2-type prespliceosome assembly pathway, upregulation of nuclear genome encoded RARS1, EPRS1, IARS1, AARS1, and QARS1, which are enriched in the cytosolic tRNA aminoacylation signaling pathway, but also caused the downregulation of mitochondrial DNA encoded MT-ND1, MT-CO1, and ATP6 genes, which are involved in the electron transport chain, and MRPL2, MRPL17, and MRPL24 genes, which are involved in mitochondrial protein translation (Fig. 7A). Mitonuclear balance refers to the delicate equilibrium between the expression and function of genes encoded in the nuclear DNA and those encoded in the mitochondrial DNA (mtDNA)20,53,54. The above analyses suggest that there is an imbalance in the transcription of genes for mitochondrial proteins and cytosolic proteins (Fig. 7A). To assess the translation level, a puromycin incorporation experiment was performed. The translation inhibitor CHX was used as a negative control, and no significant decrease in puromycin incorporation was observed when comparing opa1;egfp RNAi with egfp;egfp RNAi control animals at 1 dpa (Fig. 7B). This suggests that there is no significant decrease in protein translation levels at the bulk-tissue level between opa1;egfp RNAi and egfp;egfp RNAi controls (Fig. 7B). However, considering the gene expression difference in neoblasts, we could not rule out the possibility that a reduction in protein translation occurred in neoblasts and other specific cell types.
Proper mitochondrial fusion and fission play pivotal roles in maintaining mitonuclear balance, facilitating coordinated gene expression, efficient energy production, and effective stress responses9,55. Previous studies have shown that loss-of-function of opa1 could result in the downregulation of mitochondrial genes encoding for components of the OXPHOS, mitochondrial ribosomal proteins, and other mitochondrial biogenesis-related genes, and upregulation of genes of heat shock proteins (HSPs), reactive oxygen species (ROS) scavengers, pro-apoptotic factors, and genes involved in UPR56,57,58,59,60,61. Knockout of either opa1 or drp1 would result in an enhanced UPR55,62. To validate this response, we attempted to identify homologous genes involved in mito-nuclear signaling pathways, such as atfs-1, atf4/5, and Nrf2, that are found in the roundworm C. elegans and mice. However, no obvious homologs were identified, likely due to the divergence of the DNA sequences with evolutionary distance. (Supplementary Data 7)63. Instead, 29 other homologous genes associated with UPRmt and UPRER were identified in planarians64,65. These genes included HSP10, HSP60, eIF2α, sirt1, sirt3, catalase, PERK, and among others. This divergence can obscure the identification of direct orthologs. Despite this challenge, we were able to quantify the transcription levels of genes associated with the UPRER under opa1 RNAi condition (Fig. 7C). This approach not only validates our response but also underscores the complexity and conservation of mito-nuclear signaling pathways across different organisms. The lack of many core conserved components of UPRmt in planarians suggests that UPRmt regulation in these organisms may involve other, yet undiscovered, transcription factors.
Given the altered expression of nuclear genome-encoded mitochondrial subunits, we hypothesize that the changes in mitochondrial dynamics may decrease the function of the electron transport chain and mitochondrial ribosomal subunits, thereby exerting functional effects. To further validate our hypothesis, we then performed an RNAi screen targeting the DEGs identified from opa1 RNAi animals. We focused on the nuclear genome-encoded genes with homologs in mammals and ultimately identified 62 genes that met these criteria (Supplementary Data 8). RNAi knockdown of two genes, mrpl2 and uqcr10, which encode proteins located in mitochondria, resulted in defective regeneration without noticeable homeostasis defects (Fig. 7D, Supplementary Data 8). Furthermore, neoblast hyperproliferation during regeneration was reduced in mrpl2 RNAi and uqcr10 RNAi animals compared with egfp RNAi controls (Fig. 7E). Additionally, the expression patterns of sfrp-1, notum, and wnt-1 were disorganized (Fig. 7F), similar to the observations in opa1 RNAi animals. These results indicate that the genes regulated by mitochondrial dynamics indeed contribute to the function of tissue regeneration. Moreover, upregulated genes such as IARS1, QARS1, and RARS1 are also essential for planarian normal physiology (Supplementary Data 8) and likely act as compensatory mechanisms in response to changes in mitochondrial dynamics by increasing their expression levels. Taken together, our findings suggest a mechanism in which the mitochondrial morphology dynamics are crucial for coordinating the mitonuclear balance, likely through the regulation of metabolism and translation in mitochondria.
Mitochondrial fusion–fission equilibrium is critical for metabolism in regeneration
Consistent with previous studies60,66,67,68,69,70,71, our results showed the downregulation of mitochondrial genes encoding for components of the OXPHOS in opa1;egfp RNAi animals. To examine the influence on mitochondria, Seahorse assays were employed to measure the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). OCR measurements suggested a delayed response but comparable OXPHOS in opa1;egfp RNAi animals, which was restored in opa1;drp1 RNAi animals (Fig. S12J). ECAR measurements suggested an increase in glycolysis in opa1;egfp RNAi and drp1;egfp RNAi, but this was recovered in opa1;drp1 RNAi animals (Fig. S12K). ATP levels were measured and found to be significantly reduced in opa1;egfp RNAi animals at 1 dpa (Fig. 8A), and in all drp1;egfp RNAi, opa1;egfp RNAi, and opa1;drp1 RNAi animals compared to egfp RNAi controls at 3 dpa (Fig. 8B). These observations suggest that compromised mitochondrial dynamics reduced the function of OXPHOS in planarians.

A, B The bar plot shows the relative level of ATP in different RNAi planarians at 1 dpa (A) and 3 dpa (B). n = 4 independent biological replicates for each RNAi condition. The data are shown as the mean ± SEM. P-value was calculated by unpaired two-tailed Student’s t-test versus egfp;egfp RNAi controls. C Oil Red O staining for lipid droplets on the cryosections of the indicated RNAi planarians. Scale bar, 50 μm. D Bar plot shows the density of the lipid droplets in size and the ratio among three ranges of size ( < 6, 6–20, and > 20 μm2) in egfp;egfp RNAi (n = 4580), opa1;egfp RNAi (n = 5134), drp1;egfp RNAi (n = 6706), and opa1;drp1 RNAi (n = 4667). the n value represents the number of lipid droplets. Each dot indicates a lipid droplet. Data are shown as mean ± SEM P values were calculated by one-way ANOVA followed by Tukey’s multiple comparisons. E Body length of egfp;egfp RNAi (n = 17), opa1;egfp RNAi (n = 15), drp1;egfp RNAi (n = 15), and opa1;drp1 RNAi planarians (n = 17) at 7 dpa. The quantification is shown as mean ± SEM. P-value was calculated by unpaired two-tailed Student’s t-test. F Heatmap shows the changes of metabolites in indicated RNAi animals at 1 dpa. Four replicates for each RNAi were grouped in the indicated colors. The metabolites with significant changes in abundance (z score) in opa1;egfp RNAi compared to egfp;egfp RNAi were shown. The Benjamini-Hochberg method was used to adjust the p values. The adjusted p-value < 0.05 and fold change > 1.2 was set to determine the significantly changed expression level. G Cartoon illustration for different morphologies of mitochondria and pluripotency in Mitolow cells and Mitohigh cells. H Illustration for the regeneration defects by opa1 RNAi and rescue by opa1;drp1 RNAi. I The balance of mitochondrial fusion and fission is regulated by opa1 and drp1. J opa1 RNAi causes the malfunction of mitochondria, elevated UPR, and mitochondrial imbalance.
A recent study suggested that inhibition of the mitochondrial translation and depletion of respiratory chain subunits would result in the accumulation of large lipid droplets in mouse intestine72. In planarian tissues, phagocytes in the intestine are the major cell type responsible for lipid storage73,74,75. Lipid staining on planarian cryosections also showed a significant increase in lipid droplets in the opa1;egfp RNAi and drp1;egfp RNAi animals compared with opa1;drp1 RNAi and egfp RNAi controls (Fig. 8C, D). To exclude the possibility that larger planarians contain bigger lipid droplets, we compared the worm size and found no significant increase in length after opa1;egfp RNAi and drp1;egfp RNAi compared with opa1;drp1 RNAi and egfp;egfp RNAi controls (Fig. 8E). We then quantified cellular metabolomics data from the four RNAi conditions at 1 dpa (Supplementary Data 9). The results showed that the levels of certain amino acids, including L-citrulline, citrulline, and guanine, decreased in both opa1;egfp RNAi and drp1;egfp RNAi planarians, with a slight recovery in opa1;drp1 RNAi, suggesting a reduction in anabolism when the mitochondrial dynamics was abolished (Fig. 8F). Similarly, metabolites involved in energy metabolism, such as ADP and adenosine 3’,5’-diphosphate disodium salt, increased in both opa1;egfp RNAi and drp1;egfp RNAi planarians and restored in opa1;drp1 RNAi (Fig. 8F), consistent with the reduction of ATP production (Fig. 8A, B). These findings suggest the impaired fatty acid oxidation, in line with the accumulation of lipid droplets in both opa1;egfp RNAi and drp1;egfp RNAi planarians. However, five metabolites, including L-leucine, L-norleucine, glutaryl-carnitine lithium salt, DL-2-aminoadipic acid, and L-2-aminoadipic acid, were significantly increased only in opa1;egfp RNAi and exhibited a recovery in opa1;drp1 RNAi (Fig. 8F). This suggested a unique change in the metabolism after opa1 knockdown. Although a direct target has not been identified thus far, our results suggest the possibility of unraveling the regulation of metabolism by the mitochondrial fusion-fission equilibrium during planarian regeneration.
In summary, we propose a working model that the mitochondrial dynamics can determine the pluripotency of planarian stem cells and is critical for planarian regeneration (Fig. 8G, H). Maintaining the mitochondrial fusion-fission equilibrium is required for the normal transcriptional program in the mitonuclear balance and metabolism (Fig. 8I, J).
Discussion
In this study, our findings revealed a critical role of mitochondrial dynamics in tissue regeneration. Specifically, we observed that Mitolow cells, which possess tubular mitochondria as opposed to the granular morphology seen in Mitohigh cells (Fig. 8G), contribute significantly to pluripotency enrichment. Previous studies reported variations in mitochondrial states associated with stemness34,35. However, the regulation of stemness by mitochondria and cell composition has not been investigated. Regarding the lineage development for neoblasts, it has been proposed that neoblasts exist in a hierarchy with a particular pluripotent class44 or a specialized neoblast can divide to produce cells with asymmetric outcomes, where one daughter becomes a neoblast capable of choosing a new fate in its subsequent cell division46,76. In the latter model, called the single-step fate model, pluripotency can be distributed across multiple neoblast classes with low expression of postmitotic markers such as P4HB and XBP146. Another two studies recently showed that neoblasts choose fate in highly intermingled neighborhoods lacking clear structure32 and generated an atlas of stem cell fates with associated transcription factor signatures33. Our previous studies showed the similarity between SiRNeoblasts and X1 cells36,45. In this study, we identified Mitolow and Mitohigh cells in both SiRNeoblasts and X1 cells. Our scRNA-seq analyses revealed that Mitolow cells are enriched with cycling neoblasts expressing low levels of P4HB and XBP1, in agreement with the gene expression signatures of pluripotent neoblasts in the single-step fate model46. We further showed evidence that Mitolow cells enriched transcription factor signatures at the earlier stages of fate choices. These results elucidate the mechanism by which Mitolow cells enrich the pluripotency by providing a cell atlas and suggest the utilization of Mitolow cells to enrich neoblasts at the earlier stages of fate commitment. Compared with Mitolow cells, Mitohigh cells exhibit relatively lower levels of piwi-1 but higher levels of P4HB and XBP1, markers previously associated with postmitotic cells. Although the exact mechanism remains unclear, the coexistence of Mitolow and Mitohigh cells in the S/G2/M phases of the cell cycle suggests a transition toward cell fate commitment during neoblast division. An alternative possibility is that the Mitohigh cells are indeed bona fide postmitotic progeny, with their apparently increased DNA content reflecting, for example, a change in chromatin state. Further experiments are needed to clarify the nature of the Mitohigh fraction and, more generally, when and how neoblast fate is regulated within the cell cycle.
Several studies have shown the role of mitochondrial dynamics, which regulates fission and fusion cycles, in cell culture and physiological development in animal models54,77, including stem cell pluripotency in culture78,79,80,81, neural stem cell and T-cell differentiation in development15,82,83,84, and competence of skeletal muscle stem cells85. Previous research in cell lines, such as HeLa, HEK293, NIH3T3, and MEFs, has observed the typically fragmented mitochondria during the G2/M phases and the interconnected network during the G1 phase of the cell cycle86. Observations in our study on mitochondrial morphologies in Mitolow and Mitohigh cells highlight the sophisticated functions of mitochondrial dynamics in coordinating both the cell cycle and cell fates. Different mitochondrial morphologies during the G2/M phases of neoblasts may be essential for meeting the diverse metabolic demands associated with cell fate commitment in vivo. Our study has also revealed the critical role of these genes in the cellular processes in planarian body regeneration. Specifically, deficiency in mitochondrial fusion induced by opa1 knockdown inhibited both stem cell pluripotency and the regeneration process. Conversely, the simultaneous knockdown of opa1 and drp1 mitigated these regenerative defects (Fig. 8G, H). The results indicated that the changes in mitochondrial dynamics do not merely arise as a consequence of cell differentiation, but also actively contribute to the regulation of cell fate determination. This aligns with previous studies, which have highlighted the regulatory function of mitochondrial dynamics in animal development across species15,17,19,82,83,84,87. These findings also noted a differential regulation in various tissues. During brain development in mice, mitochondrial fusion, mediated by opa1, is essential for neuronal stem cell differentiation82. On the other hand, drp1-mediated mitochondrial fission plays a significant role in the regeneration of mouse skeletal muscle cells85. The differential influence of opa1 and drp1 in various cell types underscores the specific roles of mitochondrial fusion and fission in cellular processes. Taken together, our findings not only validate the critical role of mitochondrial dynamics but also open new avenues for exploring their potential in regulating cell fate and tissue regeneration, particularly large-scale regeneration in the future.
Our findings provide valuable insights into the principle that the maintenance of a balance in mitochondrial dynamics is pivotal for maintaining mitonuclear balance, which in turn is essential for stem cell potency and tissue regeneration (Fig. 8I). Disruption of this equilibrium can lead to alterations in the cellular metabolic status and mitonuclear imbalance, ultimately impeding normal regeneration in opa1 RNAi animals. Despite the critical functions of mitochondria, their complex nature has posed significant challenges to extensive research, particularly in the context of macroscale damage. Currently, a significant body of research is focused on revealing the mechanisms through which mitochondria regulate nuclear transcription in development and cancer pathologies13,15,20,78,80,81,82,84. However, the applicability of these findings in the context of planarians and their relevance to tissue regeneration remains an open question. Our study contributes to this ongoing discourse by providing evidence that changes in mitochondrial dynamics can induce UPR and transcriptional changes. The necessity of the opa1-regulated mitochondrial respiratory chain complex III gene uqcr10 and mitochondrial ribosome gene mrpl2 for such regulatory activities during tissue regeneration further supports the existence of a mito-nuclear signaling pathway to regulate regeneration (Fig. 8J). However, a comprehensive understanding of this regulatory pathway necessitates further investigations to precisely identify the implicated components and mechanisms.
Our study found Mitolow and Mitohigh cells based on the MMP levels, which may correlate with the mitochondrial mass. One possibility is that the mitochondria in Mitohigh cells exhibit an increased surface area when they divide into a fragmented morphology. This suggests a greater requirement for mitochondrial membrane. Another possibility is that the effectiveness of staining mitochondria with dyes is a concern in characterizing mitochondrial mass dependent on mitochondrial membrane potential. When the mitochondria within the cell have similar membrane potential, the level of mitochondrial mass can be robustly compared. However, when the mitochondrial membrane potential is low, the dye may not fully penetrate the mitochondria and accurately reflect the level of mitochondrial mass. The quantitative mitochondrial proteomic analysis for Mitolow versus Mitohigh cells is useful to clarify this point. One of the challenges we face in our research is the lack of antibodies against mitochondrial membranes, which hampers our ability to specifically target and study these organelles in planarians. We are currently working on isolating mitochondria from these organisms. However, a significant obstacle is the inability to separate the desired cell populations to acquire sufficient sample volumes for proteomics analysis.
Mitochondrial biogenesis involves the synthesis and assembly of mitochondrial components, including proteins, lipids, and DNA, as well as the growth and division of existing mitochondria. Mitochondrial biogenesis and mitochondrial fusion/fission are two distinct but interconnected processes that regulate the number, distribution, and health of mitochondria within a cell49,88. During planarian regeneration, mitochondrial biogenesis assumes a vital function in augmenting the cellular capacity to meet high energy demands and generate new mitochondria required for cell proliferation. Mitochondria are involved in diverse signaling pathways. By increasing mitochondrial mass through biogenesis, cells may enhance their ability to signal and coordinate the regenerative response. It is apparent that mitochondrial biogenesis is intimately connected with mitochondrial fusion and fission to regulate important processes such as mitochondrial quality control, energy distribution, stress response, and metabolic shifts. Close coordination among these processes ensures the efficiency and adaptation of the regenerative process to the energetic and metabolic needs of the tissue. Further refinement of our system to dissect the intricate process is necessary for future studies.
Mitochondrial dynamics often decrease in aged tissues, a phenomenon that can contribute to diminished stem cell activity and impaired regenerative capacity. Our study highlights the crucial regulatory role of mitochondrial dynamics in maintaining cell pluripotency and regeneration ability, extending our understanding of these processes beyond cellular mechanisms to a physiological context of tissue regeneration. For a considerable time, mitonuclear imbalance has been associated with promoting longevity77,89. Our study suggests that while changes in mitochondrial dynamics lead to mitonuclear imbalance, they may concurrently attenuate tissue regeneration in planarians viewed as complete animals. This finding again underscores the complex role of the mitonuclear imbalance as a double-edged sword – while potentially contributing to longevity, it may also impede tissue regeneration. Indeed, our study brings to light the intricate interplay between mitochondrial dynamics, mitonuclear balance, and their collective influence on tissue regeneration in aged tissues. Deeper insights into these processes could pave the way for developing strategies to modulate mitochondrial dynamics, potentially enhancing regenerative capacity in aged tissues. This work stands as a testament to the profound complexity of life at both cellular and organismal levels, as well as the limitless possibilities for future research in this fascinating area.
Methods
Worm husbandry and radiation treatment
Asexual S. med. (strain CIW4) specimens were maintained and propagated in the dark at 20 °C in 1× Montjuïc salts as previously described90. All animals were randomly selected at 8 ∼ 10 mm for flow cytometry and 2 ∼ 3 mm for in situ hybridization and RNAi and then starved for 7-10 days before the experiments. Animals were exposed to 12.5 Gy for sublethal irradiation experiments using an RS2000 Pro X-ray irradiation apparatus (Rad Source Technologies). Animals exposed to 60 Gy of X-rays were used as transplant hosts as previously described31. After transplantation, hosts were maintained in Montjuïc water with 50 µg/mL gentamicin (GeminiBio, 400-100 P), and the Montjuïc water was changed every day.
Plasmid construction and cell transfection
The full-length cDNA sequence of smed-opa1 (SMED30010319) codon-optimized for human cell expression (Genewiz, Inc.) was cloned into the vector pcDNA4-3×Myc for transfection. To perform the transfection, HeLa cells were cultured in DMEM (Shanghai Basal Media Technologies Co., Ltd., L110KJ) supplemented with 10% fetal bovine serum (FBS) (Excell Bio, FSP500) at 37 °C under a 5% CO2 atmosphere. HeLa cells were seeded on coverslips in a 6-well plate and allowed to reach 70% confluence for transfection using the Lipofectamine 3000 reagent (ThermoFisher Scientific, L3000001).
Histology
Cryosection
Planarians were anesthetized with 66% MgCl2 (Sangon Biotech, A601336-0500) for 15 seconds to ensure complete extension of the worms. Subsequently, they were transferred to the embedding box. The animals were then embedded in Tissue-Tek® O.C.T. Compound (TYO, 4583) in the embedding box and rapidly frozen using dry ice. Cryosections with a thickness of 12 μm were obtained using a cryostat microtome (DAKEWE, CT520) and stored at −20 °C.
Oil Red O staining
Animals with similar sizes were used for comparison. Cryosections collected on glass slides were balanced to room temperature for 30 min and then rinsed with the appropriate amount of staining wash solution (Beyotime, C01558M-2) for 20 s. The slides were subsequently stained with Oil Red O staining solution (Beyotime, C01558M-1) for 20 min. After staining, the slides were washed with a staining wash solution for 30 s at room temperature. Following this, the slides were stained with filtered Hematoxylin staining solution (Beyotime, C0107) for 1 min, and rinsed in distilled water for 10 min.
TEM
TEM was performed as described previously39. Worms were fixed with a 2.5% glutaraldehyde (25% glutaraldehyde, Ted Pella Co., USA, 18426) and 2% paraformaldehyde (16% paraformaldehyde, Ted Pella Co., USA, 18505) mixture in 0.1 M cacodylate buffer at 4 °C overnight and then subjected to secondary fixation with 1% osmium tetroxide (4% OsO4, Ted Pella Co., USA, 18465) in 0.1 M cacodylate buffer for 90 min on ice in the dark. After dehydration, the worms were embedded in Spurr’s resin and sectioned with a microtome. Finally, the sections were imaged at 80 kV on an FEI Talos L120C transmission electron microscope.
SiRNeoblast-Mito Staining and Flow Cytometry
Cells from 8-10 mm planarian flatworms were treated as previously described with minor modifications31,45. Briefly, cells were macerated in calcium- and magnesium-free buffer with 1% bovine serum albumin, and debris was filtered through a 70 μm cell strainer. After centrifugation at 290 × g for 10 min, the cells were resuspended in isotonic planarian medium (IPM) with 10% fetal bovine serum (FBS, CellMax SA211.02) at the density of 4 × 106 cells/mL and incubated with SiR-DNA (1 µM, Cytoskeleton Inc., CY SC007) for 1 hour, CellTracker dyes (Thermo Fisher Technologies, C7025, C2110, C34551) for 10 min, and the following mitochondrial dyes: PK Mito dyes34 for 45 min, MitoTracker Orange for 45 min, (Thermo Fisher Technologies, M7510), CellROX Green (Thermo Fisher Technologies, C10492) for 15 min, MitoSOX Red (Thermo Fisher Technologies, M36008) for 10 min, and MitoTracker Green (Thermo Fisher Technologies, M7514) for 45 min. The sorting strategy for Mitolow and Mitohigh cells is based on cell size (FSC), internal complexity (SSC), and the PK Mito Red signal in 4N cells. Fig. S1A shows the gating strategy to enrich the target populations. The SSC-A/FSC-A plot displays all cells in the sample. The P1 gate enriches intact cells, excluding cell debris. The FSC-H/FSC-A plot shows all cells from the P1 gate. The P2 gate enriches single cells and excludes doublets. SSC-A/DAPI-A plot displays all cells from the P2 gate. The P3 gate enriches live cells based on the signal intensity of DAPI staining. SSC-A/APC-A (SiR-DNA) plot displays all live cells from the P3 gate. P4 gate enriches 4N cells based on higher DNA staining (SiR-DNA) intensity. The SSC-A/FITC (Cell Tracker) plot displays 4N cells from the P4 gate. P5 gate enriches SiRNeoblasts with higher Cell Tracker staining intensity. SSC-A/PE-A (PK Mito Red) plot shows SiRNeoblasts from the P5 gate. Based on PK Mito Red staining, the P6 gate (Mitolow cells) enriches cells with lower PK Mito intensity, and the P7 gate (Mitohigh cells) enriches cells with higher PK Mito intensity. Since 4N cells include both SiRNeoblasts and CTGlow cells, with the latter being differentiation-prone and exhibiting higher PK Mito signals, using 4N cells can distinguish between high and low levels of PK Mito signals. Low and high gates are then set based on the distinct cut-off of CTGlow cells. SiRNeoblasts with PK Mito signals lower and higher than this cut-off are defined as Mitolow and Mitohigh cell populations, respectively. These gates are applied to the SiRNeoblast population, which is used for analyzing and sorting Mitolow and Mitohigh cell populations. These Mitolow and Mitohigh cell populations constitute approximately 90% and 8% of the SiRNeoblasts, respectively. The dynamics of Mitolow and Mitohigh cell ratio during planarian regeneration was assessed using FACS at nine different time points after amputation: 0 hpa, 1 hpa, 6 hpa, 12 hpa, 1 dpa, 2 dpa, 3 dpa, 5 dpa and 7 dpa. 0 hpa refers to samples collected immediately after amputation. Cell composition analyses were performed using a Beckman CytoFLEX LX with the CytExpert v2.4.0.28 software. The cell populations were classified based on the example shown in Fig. S1 and a wild-type control was used as a quality control for each experiment. Targeted cells were sorted using a BD Fusion cell sorter with BD FACSDiva v9.0 software equipped with a 100 nm nozzle and purity sort mode. The data in FACS was analyzed with the software Flowjo v10.
In situ hybridization
Whole-mount in situ hybridization (WISH) was carried out as previously described in refs. 91,92. After 7 ∼ 10 days of starvation, the worms were subjected to the following series of steps. First, they were incubated in a 5% N-acetylcysteine solution for 5 min to remove mucus. The worms were then fixed in a 4% formaldehyde solution for 45 min. After two washes in PBSTx0.5%, the worms were dehydrated in 50% methanol for 5 min. The samples were finally stored in 100% methanol at −20 °C before use. Samples were bleached by exposure to a light source in a solution containing 5% formamide, 0.5× SSC, and 1.2% hydrogen peroxide for 2 hours. Following the bleaching process, the samples were subjected to overnight hybridization with DIG- and FITC-labeled probes, conducted at 56 °C. Samples were then incubated with anti-DIG-conjugated alkaline phosphatase (AP) antibody (Roche, 11093274910) at 4 °C overnight for colorimetric WISH experiments. The development of color was achieved using NBT (Roche, 11383213001) and BCIP (Roche, 11383221001). For the FISH experiment, the samples were incubated overnight at 4 °C with anti-DIG antibodies conjugated to peroxidase (POD) (Roche, 11207733910) or anti-FL antibodies conjugated to peroxidase (POD) (Roche, 11426346910). The fluorescence signals were generated by incubating the samples with tyramine-labeled with a fluorescent tag at room temperature for one hour. To enhance the optical clarity after signal development, a Sca/A2 solution containing 75% glycerol and 2 M urea was used for colorimetric WISH. A solution of 20% glycerol, 2.5% DABCO, and 4 M urea Sca/A2 was used for FISH.
In situ hybridization (ISH) of sorted cells was performed as described for WISH with minor alterations36. Cells adhered to poly-L-lysine (50 μg/mL, Sigma‒Aldrich, P1399)-coated culture plates were fixed with 4% formaldehyde (Sigma‒Aldrich, F8775) or 4% paraformaldehyde (Electron Microscopy Sciences, 15710) for 20 min. Fluorescent signals were developed by Cy5-tyramide (Sigma‒Aldrich, PA15101) signal amplification for 10 min at room temperature.
Antibody immunostaining
For H3P immunostaining, an anti-phospho-histone H3 (Ser10) (H3P) antibody (1:1,000, Abcam, ab32107) and an Alexa 555-conjugated goat anti-rabbit secondary antibody (1:1,000, Abcam, ab150086) were used to stain proliferating cells in the G2/M cell cycle phase. The anti-Smed-PIWI-1 antibody was a gift from Dr. Jochen Rink and was used at a 1:10000 dilution.
To immunofluorescently stain cultured cells, the HeLa cells were fixed with 4% paraformaldehyde after 24 hours of transfection. Primary antibodies used in this study included anti-Myc antibody (1:400 dilution; 9E10, Santa Cruz) and anti-TOMM20 antibody (1:500 dilution; ab186734, abcam). The fixed cells were incubated with the primary antibodies overnight and secondary antibodies conjugated with Alexa Fluor 555 (1:1000 dilution; goat anti-rabbit antibody, ab150086) or Alexa Fluor 488 (1:1000 dilution; goat anti-mouse antibody, ab150117, abcam) were incubated with the cells for two hours. To visualize the cell nuclei, the cells were stained with 4,6-diamidino-2-phenylindole (DAPI, 0.1%; ThermoFisher Scientific, D3571) for 15 min before being mounted in Prolong gold antifade reagent (ThermoFisher Scientific, P36934).
Cell transplantation
SiRNeoblasts were suspended in IPM with 10% FBS and diluted to a concentration of 1,000 cells/µL. Approximately 1 µL of the cell suspension (∼1000 cells/µL) was injected into either the postpharyngeal midline (of asexual CIW4 hosts) or the postgonopore midline (of sexual S2F1L3F2 hosts) at 0.75–1.0 psi (Eppendorf FemtoJet) using a borosilicate glass microcapillary (Sutter Instrument Co., B100-75-15) as described previously36.
Microscopy and image analysis
Colorimetric WISH samples and live worms were imaged using a Leica M205 FA stereomicroscope using Leica Application Suite X software. Fluorescence images were obtained with Nikon C2Si laser scanning confocal using NIS-Elements C Version 5.11.01 software, Zeiss 900 confocal using NIS Element AR 5.30.06 software, and Nikon CSU-W1 spinning-disk confocal microscopes using NIS-Elements AR 5.30.06 software. Objectives of 10× NA 0.45, 20× NA 0.75, 20× NA 0.8 and 60× NA 1.4 were used. Linear adjustments of brightness and contrast equally across controls and experimental images. Maximum intensity projections through areas of interest were performed using Fiji software (version 2.0.0)93.
The procedure of Hessian-SIM imaging of mitochondria was detailed in a previous study94. Mitochondrial images were acquired and reconstructed using a commercial High Intelligent and Sensitive Structured Illumination Microscope (HIS-SIM, Olympus) with a 100× NA 1.5 oil immersion objective. The cells were seeded in a confocal dish and maintained at room temperature for 3D-SIM imaging. To further improve the resolution and contrast in reconstructed images, sparse deconvolution was performed using Microscope X FINER software with level 2 sparse analysis95.
Analysis of mitochondrial morphological characteristics
To achieve the best identification of mitochondrial morphology, 2D images were projected from 3D images and converted into 8-bit form. The scale was set to microns to facilitate Fiji analysis. Then, the images were preprocessed by Mitochondria Analyzer with the plugins ‘Subtract Background’, ‘Sigma Filter Plus’, ‘Enhance Local Contrast’, and ‘Adjust Gamma’ to remove the background noise and smooth the signals. To correctly identify mitochondria in the images, we selected a block size set to an equivalent of 1.05 µm, and the optimal C value was empirically determined for each image with the ‘2D Threshold’. The resulting binary images were used as the input for the ‘2D Analysis’ command to measure the ‘area’, ‘perimeter’, ‘branch length’ and ‘aspect ratio’ of the mitochondria in every cell.
T4P cloning and probe synthesis
The cDNA sequences used for cloning were referenced from Planosphere, and the primers were designed with Primer396. All primer sequences are listed in Supplementary Data 10. PCR products were amplified from asexual CIW4 cDNA and ligated into the pPR-T4P vector as described previously97. The RNA probes were synthesized by labeling products of the reverse transcription reaction with T7 RNA polymerase (Promega, PAP 2077) with 10× RNA labeling (DIG RNA Labeling Mix: Roche 11277073910; Fluorescein RNA Labeling Mix: Roche 11685619910; DNP-11-UTP: PerkinElmer NEL555001EA).
RNAi feeding
RNAi experiments were carried out as previously described in ref. 98. RNAi feeding material was prepared by mixing 125 μL of liver paste (9:1 liver:water) with a bacterial pellet obtained from a 50-mL HT115 bacterial culture expressing dsRNA. EGFP, an Aequorea victoria gene showing no nucleotide sequence homology in planarians, was used for control RNAi experiments. The animals were fed once every three days, four times before sublethal irradiation treatment and six times for homeostasis and regeneration experiments. The animals were starved for seven days after the last feeding before irradiation exposure and amputation procedures.
qPCR
Total RNA was extracted from wild-type or RNAi worms using TRIzol according to the manufacturer’s protocol, and 500 ng of RNA was used to prepare cDNA. Biological replicate data obtained from qPCRsoft4.0 were averaged, and log2(fold change) values were determined by the DDCt method by calculating the differences from the average DCt value of the control RNAi replicate. The Ct values of the samples were normalized to the average Ct values of the housekeeping transcripts gapdh and β-tubulin. The primers used are listed below.
gapdh-forward: AGCTCCATTGGCGAAAGTTA
gapdh-reverse: CTTTTGCTGCACCAGTTGAA
β-tubulin-forward: TGGCTGCTTGTGATCCAAGA
β-tubulin-reverse: AAATTGCCGCAACAGTCAAATA
drp1-forward: CAAATTGCATCATCAGCGAG
drp1-reverse: TTTCACGAATCTCGCTGATG
opa1-forward: AGAGACACCATGAAACC
opa1-reverse: GCAGTGCCAGCAATG
mfn1-forward: TAATTCGATGCTAGGACAGA
mfn1-reverse: CCATTAGTGTCAGAAGTGCCC
mfn2-forward: CGGACGAACATCTAACGG
mfn2-reverse: CCCTGATTATCGTCTGTTCC
fis-1-forward: CGGAAATTGCTGTTTTACGA
fis-1-reverse: AGACAAGCTGGATCTTTTGA
artrogin1-forward: AATTTTAGTCAACGCGCCTG
artrogin1-reverse: GCCAAAATATAGATGCGCAC
musa1-forward: GAATTCGGTTTCGCTATTGG
musa1-reverse: TTTAAAGACGAGCTCCACAG
gabarap-forward: CAATTGCGTTCAGAAGATGC
gabarap-reverse: CTTTCATCACTGTACGCAAC
gabarapL2-forward: ATGTGGATTCTAAGGAAGCG
gabarapL2-reverse: CCGTCTTCATCCCTAAAGTC
trim9-forward: TGAGATGCTACGAGAAAGTG
trim9-reverse: CTATGGCTCATTTTTGGCTC
wwp1-forward: CGTCGATGATTGGGAAAAGC
wwp1-reverse: ACTTTTGGGGCCCATTGC
wwp2-1-forward: CATGAGAGTCAAACCTAGGG
wwp2-1-reverse: CGTGTGATAGCTGGAAAAAC
wwp2-2-forward: ATCGTCTGAGTCAGCATAAC
wwp2-2-reverse: ACTCGCTTATTTCTGGGATC
map1lc3c-forward: TCAAATCCGATCAGACGATC
map1lc3c -reverse: ACCAAACATTTCCTGGGAC
cathepsin L2-forward: GCTCGTCAAAAATAGTTGGG
cathepsin L2-reverse: TAGACCAAAGGATAGCTTGC
Single-cell and single nucleus sequencing and analysis
Cell samples for single-cell RNA sequencing were collected from two conditions, each containing tissues from the different regions: (1) Samples of sorted Mitolow and Mitohigh cells were from intact wild-type planarian and tails, respectively; (2) Samples of sorted Mitolow cells from RNAi worms included tissues from the mix of heads, trunks, and tails at 1 dpa. Single nucleus RNA sequencing samples were collected from RNAi worms, including tissues from the mix of heads, trunks, and tails at 1 dpa. An Illumina NovaSeq 6000 was used, and ∼310-320 million reads were sequenced per run. The reads were aligned to the SMED transcriptome by CellRanger v.3.1.0 with the default parameters99. rRNA genes (SMED30032663, SMED30027845, SMED30032887, SMED30015328, SMED30012309, SMED30027385, and SMED30023781) were deleted from the matrix. Cells with nFeature_RNA < 200 or nFeature_RNA > 6000 and genes expressed in no more than three cells, were removed from the gene expression matrix to build the Seurat object with Seurat package v.3.1.1100. The data were normalized and scaled, and the top 2000 variable features were selected to run PCA via RunPCA with the default parameters. Then, Harmony was used to remove batch effects from all the principal components (PCs), and a resolution of 0.6 was used for clustering101. The clusters were plotted by UMAP and calculated by ten Harmony dimensions. The function FindAllMarkers was used to calculate cluster markers. Differential gene analysis was conducted on single-cell RNA-seq data using Seurat package v.3.1.1. Genes were considered significantly downregulated if their avg_log2FC values were less than −0.585 and upregulated if the values were greater than 0.585 with an adjusted p value < 0.05. Results were visualized using ggplot. Heatmap was generated in R package pheatmap (1.0.12). Single-cell RNA sequencing libraries were generated on the 10× Genomics platform and are available at the Gene Expression Omnibus (GEO) with the Accession number GSE244070.
Bulk RNA sequencing
Collect worms from each RNAi group and three worms were used to constitute one sample for three biological replicates. After obtaining total RNA using 1.0 mL of TRIzol per tube according to the manufacturer’s protocol, mRNA was purified from total RNA using poly-(T)-attached magnetic beads. Then, the NEBNext Ultra RNA Library Prep Kit was used for library construction. Clustering of the index-coded samples was performed on a cBot cluster generation system using a TruSeq PE Cluster Kit v3-cBot-HS (Illumina). After cluster generation, the library preparations were sequenced on the NovaSeq 6000 system, and 150-bp paired-end reads were generated according to the procedure provided by Novogene Inc. Raw data were aligned to the transcriptome smed_20140614. The total number of reads per transcript was calculated. Differential expression transcripts were identified using DEseq2 in the R version 4.0.3. The results of RNA-seq analysis are processed via Sangerbox http://sangerbox.com/home.html.
Puromycin incorporation and western blotting
Amputated opa1;egfp RNAi, drp1;egfp RNAi, opa1;drp1 RNAi and egfp;egfp RNAi control animals were subjected to planarian water containing 0.5 mg/mL puromycin (1:20 dilution; beyotime, ST551)102, and collect the samples at 1 dpa. A cycloheximide (MCE, HY-12320) control was established using a concentration of 0.1 mg/mL47. The experiment was replicated three times independently to ensure data reliability.
For the Western blot analysis, every six RNAi animals, approximately 4-5 mm in size, were collected and transferred into 1.5 mL microcentrifuge tubes. They were then suspended in 40 µL of Medium RIPA Lysis Buffer (GenStar, 122-01) and homogenized for 20 minutes. Each RNAi condition contains three biological replicates. Following this, the samples were centrifuged at 15,000 rpm for 10 minutes. The supernatant was transferred to a new tube and the protein concentration was determined using the BCA assay (Genstar, E162-01). The protein samples were then boiled at 99 °C for 10 minutes, followed by a 4 °C centrifugation for 5 minutes. A total of 15 µg of protein was loaded onto SurePAGE™, BIS-tris 4-20% gels (GeneScript, M00675). Proteins were separated using standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to BioTrace™ NT nitrocellulose membranes (Pall Corporation, 55253088). The membranes were washed three times in TBST (0.5‰ Tween20 in TBS) and then blocked in 5% milk for 1 hour before incubation with an anti-puromycin antibody (1:10000 dilution; 12D10, Millipore) overnight at 4 °C. The next day, membranes were incubated with a secondary HRP-conjugated Goat Anti-mouse IgG(H + L) antibody (1:20000 dilution; ImmunityWay, RS0001) overnight. ECL Ultra (NCM, P10200) was used for visualizing the protein bands labeled with HRP. The nitrocellulose membranes were submerged in Ponceau S staining solution (Biosharp, BL519A) and gently agitated for 10 minutes. After staining, the membranes were rinsed with distilled water 2-3 times to reveal clear bands. The Ponceau S staining served as a loading control for the same total amount of protein loaded. Quantitative analysis of the protein bands obtained from GE AI680 was conducted using ImageJ 1.54k to measure protein grayscale values. Statistical graphs and analyses were generated using GraphPad Prism 9.0.
Metabolism and seahorse analyses
ATP detection
Amputated RNAi worms at 1 dpa and 3 dpa were collected and lysed in the ATP detection lysate (Beyotime, S0027). Typically, single worms of 4-5 mm in length require a 150 µL lysis buffer. Subsequently, the samples were combined with CellTiter-Glo® Luminescent Cell Viability Assay (CTG) reagent (Promega, G7570) in a 1:1 volume ratio and incubated at room temperature for 10 minutes. The signal was measured using a Thermo Varioskan LUX microplate reader (ThermoFisher Scientific, SIA-PR007). Protein concentrations were measured using the BCA kit (Genstar, E162-05). The final ATP content of each worm is presented as ATP/protein (μM/μg).
QTOF MS for untargeted metabolomics
Approximately 40 worms were placed in a 1.5 mL centrifuge tube for a single sample. Four biological replicates were set for each condition. Before the experiment, the samples were carefully weighed to ensure they had a similar weight of 30 mg. To initiate the extraction process, 400 μL of 80% (v/v) HPLC-grade methanol (pre-cooled to −80 °C) was added to the 1.5 mL centrifuge tube. The samples were then ground at 4 °C for approximately 1-2 min until dissolved in methanol. The samples were then left overnight at −80 °C. Following this, the samples underwent centrifugation at 14,000 g for 20 minutes, after which the supernatant was transferred to a new 1.5 mL centrifuge tube. Speedvac was used to dry the pellet at room temperature without heat. The pellets were dried using a Speedvac at room temperature without heating. Finally, the samples were stored at −80 °C. Each sample was dissolved in 100 μL of 80% methanol and loaded onto an amide chromatography column for QTOF MS analysis.
The detailed analysis was performed using an LC-MS system, which includes an Agilent 1290 Infinity II UHPLC system and an Agilent 6545 Q-TOF/MS (Agilent). The chromatography column was an ACQUITY UPLC BEH Amide (100 mm × 2.1 mm, 1.7 μm). The mobile phase consists of 15 mM ammonium acetate-0.3% NH3·H2O in water (A) and 15 mM ammonium acetate-0.3% NH3·H2O in 9:1 acetonitrile/water (B), with a flow rate of 0.3 mL/min. The column was eluted with 95% mobile phase B for 1 min, followed by a linear gradient to 50% mobile phase B over 8 min, then 50% mobile phase B for 3 min, and a linear gradient back to 95% mobile phase B over 0.5 min, followed by 95% mobile phase B for 1.5 min. The injection volume was 5 μL.
Mass spectrometry data were acquired in the mass range of 50-1250 m/z using electrospray ionization in both positive and negative ion modes. Other MS settings included: sheath gas temperature 350 °C, sheath gas flow 11 L/min, VCap 4000 V, nozzle voltage 1000 V, gas temperature 350 °C, nebulizer gas 30 psi, drying gas flow 8 L/min, fragmentor 110 V, and skimmer 65 V. Raw data were processed using Profinder 10.0 (Agilent) for peak detection, alignment, and integration. The raw data and processed data are shown in Supplementary Data 9.
Seahorse assays
The Seahorse XFe96 Analyzer (Agilent-Sea horse Bioscience) and Seahorse XFe96/XF Pro FluxPak Mini (Agilent, 103793-100) were used to measure the mitochondrial oxygen consumption rate (OCR) and Extracellular Acidification Rate (ECAR) for worms with RNAi treatment. The instrument temperature was set to 25 °C one day in advance. To prepare the culture plate, it was coated with 25 μL of 25 μg/mL polyethyleneimine (PEI) overnight at 4 °C. The tails of RNAi worms were then transferred into the culture plate one day after amputation in 1× Montjuïc salt solution. The culture plate was incubated in an incubator for 30 min at 25 °C. ECAR was measured both in basal conditions and after the injection of 200 mM D-glucose, 5 μM oligomycin, and 100 mM 2-deoxyglucose (2-DG). The OCR was measured under basal conditions and after the step-wise addition of oligomycin (5 μM), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (1 μM), and Sodium azide (NaN3) (50 mM). The collected data were analyzed using the Seahorse Wave controller software (Agilent Technologies, Seahorse Bioscience).
Statistical analyses
Microsoft Excel Professional Plus 2021 and GraphPad Prism 9.0 were used for statistical analysis. The data in all graphs are shown as the mean ± SEM. An unpaired two-tailed Student’s t test was used to determine the significance of differences between two conditions. One-way ANOVA followed by Tukey’s multiple comparisons were used to determine the significance of the difference in the analysis of lipid droplets. Two-way ANOVA followed by Sidak multiple comparisons were used to determine the significance of differences in the Seahorse assay.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw sequencing data generated in this study are available at the GEO with the Accession number GSE244070. Data supporting the findings of this study are provided in Supplementary data files and Source data files. Source Data are provided with this paper and in the Zenodo database publicly (https://zenodo.org/records/14058603)103. Source data are provided with this paper.
Code availability
All codes and a README file with detailed explanations of the code structure and functionality are available at: https://github.com/leilabteam/mitoproject. The codes can also be accessed via Zenodo (https://zenodo.org/records/14058603)103.
References
Tanaka, E. M. & Reddien, P. W. The cellular basis for animal regeneration. Developmental Cell 21, 172–185 (2011).
Reddien, P. W. The Cellular and Molecular Basis for Planarian Regeneration. Cell 175, 327–345 (2018).
Bideau, L., Kerner, P., Hui, J., Vervoort, M. & Gazave, E. Animal regeneration in the era of transcriptomics. Cell Mol. Life Sci. 78, 3941–3956 (2021).
Reddien, P. W. Positional Information and Stem Cells Combine to Result in Planarian Regeneration. Cold Spring Harb. Perspect. Biol. 14, a040717 (2022).
Panopoulos, A. D. et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 22, 168–177 (2012).
Gu, W. et al. Glycolytic Metabolism Plays a Functional Role in Regulating Human Pluripotent Stem Cell State. Cell Stem Cell 19, 476–490 (2016).
Ito, K. & Ito, K. Metabolism and the Control of Cell Fate Decisions and Stem Cell Renewal. Annu Rev. Cell Dev. Biol. 32, 399–409 (2016).
Zhang, H., Menzies, K. J. & Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 145, dev143420 (2018).
Chan, D. C. Mitochondrial Dynamics and Its Involvement in Disease. Annu Rev. Pathol. 15, 235–259 (2020).
Tan, J. X. & Finkel, T. Mitochondria as intracellular signaling platforms in health and disease. J. Cell Biol. 219, e202002179 (2020).
Chakrabarty, R. P. & Chandel, N. S. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 28, 394–408 (2021).
Shen, K. et al. Mitochondria as Cellular and Organismal Signaling Hubs. Annu Rev. Cell Dev. Biol. 38, 179–218 (2022).
Kasahara, A. et al. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science 342, 734–737 (2013).
Varanita, T. et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 21, 834–844 (2015).
Buck, M. D. et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 166, 63–76 (2016).
Chen, H. & Chan, D. C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 26, 39–48 (2017).
Sênos Demarco, R., Uyemura, B. S., D’Alterio, C. & Jones, D. L. Mitochondrial fusion regulates lipid homeostasis and stem cell maintenance in the Drosophila testis. Nat. Cell Biol. 21, 710–720 (2019).
Sênos Demarco, R. & Jones, D. L. Mitochondrial fission regulates germ cell differentiation by suppressing ROS-mediated activation of Epidermal Growth Factor Signaling in the Drosophila larval testis. Sci. Rep. 9, 19695 (2019).
Corrado, M. et al. Deletion of the mitochondria-shaping protein Opa1 during early thymocyte maturation impacts mature memory T cell metabolism. Cell Death Differ. 28, 2194–2206 (2021).
Zhu, D., Li, X. & Tian, Y. Mitochondrial-to-nuclear communication in aging: an epigenetic perspective. Trends Biochem Sci. 47, 645–659 (2022).
Han, S. et al. Mitochondrial integrated stress response controls lung epithelial cell fate. Nature 620, 890–897 (2023).
Fu, H. et al. Wounding triggers MIRO-1 dependent mitochondrial fragmentation that accelerates epidermal wound closure through oxidative signaling. Nat. Commun. 11, 1050 (2020).
Gerber, T. et al. Single-cell analysis uncovers convergence of cell identities during axolotl limb regeneration. Science 362, eaaq0681 (2018).
Qin, T. et al. Single-cell RNA-seq reveals novel mitochondria-related musculoskeletal cell populations during adult axolotl limb regeneration process. Cell Death Differ. 28, 1110–1125 (2021).
Zhao, Y., Gao, C., Pan, X. & Lei, K. Emerging roles of mitochondria in animal regeneration. Cell Regen. 12, 14 (2023).
Elliott, S. A. & Sánchez Alvarado, A. The history and enduring contributions of planarians to the study of animal regeneration. Wiley Interdiscip. Rev. Dev. Biol. 2, 301–326 (2013).
Rink, J. C. Stem cell systems and regeneration in planaria. Dev. Genes Evol. 223, 67–84 (2013).
Liberali, P. & Schier, A. F. The evolution of developmental biology through conceptual and technological revolutions. Cell 187, 3461–3495 (2024).
Baguñà, J., Saló, E. & Auladell, C. Regeneration and pattern formation in planarians. III. Evidence that neoblasts are totipotent stem cells and the source of blastema cells. Development 107, 77–86 (1989).
Reddien, P. W. & Alvarado, S. ánchez A. Fundamentals of planarian regeneration. Annu Rev. Cell Dev. Biol. 20, 725–757 (2004).
Wagner, D. E., Wang, I. E. & Reddien, P. W. Clonogenic neoblasts are pluripotent adult stem cells that underlie planarian regeneration. Science 332, 811–816 (2011).
Park, C., Owusu-Boaitey, K. E., Valdes, G. M. & Reddien, P. W. Fate specification is spatially intermingled across planarian stem cells. Nat. Commun. 14, 7422 (2023).
King, H. O., Owusu-Boaitey, K. E., Fincher, C. T. & Reddien, P. W. A transcription factor atlas of stem cell fate in planarians. Cell Rep. 43, 113843 (2024).
Yang, Z. T. et al. Cyclooctatetraene-conjugated cyanine mitochondrial probes minimize phototoxicity in fluorescence and nanoscopic imaging. Chem. Sci. 11, 8506–8516 (2020).
Mohamed Haroon, M. et al. Mitochondrial state determines functionally divergent stem cell population in planaria. Stem Cell Rep. 16, 1302–1316 (2021).
Lei, K. et al. Pluripotency retention and exogenous mRNA introduction in planarian stem cells in culture. iScience 26, 106001 (2023).
Zhang, W., Li, X., Zhao, Y. & Lei, K. Protocol for culturing and functionally manipulating planarian neoblasts using SiRDNA-based flow cytometry. STAR Protoc. 5, 102877 (2024).
Wenemoser, D. & Reddien, P. W. Planarian regeneration involves distinct stem cell responses to wounds and tissue absence. Dev. Biol. 344, 979–991 (2010).
Brubacher, J. L., Vieira, A. P. & Newmark, P. A. Preparation of the planarian Schmidtea mediterranea for high-resolution histology and transmission electron microscopy. Nat. Protoc. 9, 661–673 (2014).
van Wolfswinkel, J. C., Wagner, D. E. & Reddien, P. W. Single-cell analysis reveals functionally distinct classes within the planarian stem cell compartment. Cell Stem Cell 15, 326–339 (2014).
Molinaro, A. M. & Pearson, B. J. In silico lineage tracing through single cell transcriptomics identifies a neural stem cell population in planarians. Genome Biol. 17, 87 (2016).
Fincher, C. T., Wurtzel, O., de Hoog, T., Kravarik, K. M. & Reddien, P. W. Cell type transcriptome atlas for the planarian Schmidtea mediterranea. Science 360, eaaq1736 (2018).
Plass, M. et al. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science 360, eaaq1723 (2018).
Zeng, A. et al. Prospectively Isolated Tetraspanin(+) Neoblasts Are Adult Pluripotent Stem Cells Underlying Planaria Regeneration. Cell 173, 1593–1608.e1520 (2018).
Niu, K. et al. Canonical and early lineage-specific stem cell types identified in planarian SirNeoblasts. Cell Regen. 10, 15 (2021).
Raz, A. A., Wurtzel, O. & Reddien, P. W. Planarian stem cells specify fate yet retain potency during the cell cycle. Cell Stem Cell 28, 1307–1322.e5 (2021).
Wenemoser, D., Lapan, S. W., Wilkinson, A. W., Bell, G. W. & Reddien, P. W. A molecular wound response program associated with regeneration initiation in planarians. Genes Dev. 26, 988–1002 (2012).
Fraguas, S., Barberan, S., Iglesias, M., Rodriguez-Esteban, G. & Cebrià, F. egr-4, a target of EGFR signaling, is required for the formation of the brain primordia and head regeneration in planarians. Development 141, 1835–1847 (2014).
Pfanner, N., Warscheid, B. & Wiedemann, N. Mitochondrial proteins: from biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 20, 267–284 (2019).
Song, J., Herrmann, J. M. & Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 22, 54–70 (2021).
Ross, E., Blair, D., Guerrero-Hernandez, C. & Alvarado, S. A. Comparative and Transcriptome Analyses Uncover Key Aspects of Coding- and Long Noncoding RNAs in Flatworm Mitochondrial Genomes. G3 (Bethesda) 6, 1191–1200 (2016).
Rath, S. et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547 (2021).
Isaac, R. S., McShane, E. & Churchman, L. S. The Multiple Levels of Mitonuclear Coregulation. Annu Rev. Genet 52, 511–533 (2018).
Sutandy, F. X. R., Tascher, GossnerI. & Munch, G. C. A cytosolic surveillance mechanism activates the mitochondrial UPR. Nature 618, 849–854 (2023).
Chen, H. et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289 (2010).
Amati-Bonneau, P. et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 131, 338–351 (2008).
Givvimani, S. et al. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PLoS One 7, e32388 (2012).
Bertholet, A. M. et al. OPA1 loss of function affects in vitro neuronal maturation. Brain 136, 1518–1533 (2013).
Cogliati, S. et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171 (2013).
Tezze, C. et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 25, 1374–1389.e1376 (2017).
Pereira, R. O. et al. OPA1 Regulates Lipid Metabolism and Cold-Induced Browning of White Adipose Tissue in Mice. Diabetes 71, 2572–2583 (2022).
Romanello, V. et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 29, 1774–1785 (2010).
Melber, A. & Haynes, C. M. UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res 28, 281–295 (2018).
Lima, T., Li, T. Y., Mottis, A. & Auwerx, J. Pleiotropic effects of mitochondria in aging. Nat. Aging 2, 199–213 (2022).
Cilleros-Holgado, P. et al. Mitochondrial Quality Control via Mitochondrial Unfolded Protein Response (mtUPR) in Ageing and Neurodegenerative Diseases. Biomolecules 13, 1789 (2023).
Hu, C. et al. OPA1 and MICOS Regulate mitochondrial crista dynamics and formation. Cell Death Dis. 11, 940 (2020).
Jang, S. & Javadov, S. OPA1 regulates respiratory supercomplexes assembly: The role of mitochondrial swelling. Mitochondrion 51, 30–39 (2020).
Romanello, V. & Sandri, M. The connection between the dynamic remodeling of the mitochondrial network and the regulation of muscle mass. Cell Mol. Life Sci. 78, 1305–1328 (2021).
Noone, J., O’Gorman, D. J. & Kenny, H. C. OPA1 regulation of mitochondrial dynamics in skeletal and cardiac muscle. Trends Endocrinol. Metab. 33, 710–721 (2022).
Nyenhuis, S. B. et al. OPA1 helical structures give perspective to mitochondrial dysfunction. Nature 620, 1109–1116 (2023).
Zanfardino, P. et al. OPA1 mutation affects autophagy and triggers senescence in autosomal dominant optic atrophy plus fibroblasts. Hum. Mol. Genet 33, 768–786 (2024).
Moschandrea, C. et al. Mitochondrial dysfunction abrogates dietary lipid processing in enterocytes. Nature 625, 385–392 (2024).
Thommen, A. et al. Body size-dependent energy storage causes Kleiber’s law scaling of the metabolic rate in planarians. Elife 8, e52613 (2019).
Forsthoefel, D. J., Cejda, N. I., Khan, U. W. & Newmark, P. A. Cell-type diversity and regionalized gene expression in the planarian intestine. Elife 9, e52613 (2020).
Wong, L. L. et al. Intestine-enriched apolipoprotein b orthologs are required for stem cell progeny differentiation and regeneration in planarians. Nat. Commun. 13, 3803 (2022).
Lei, K. et al. Egf Signaling Directs Neoblast Repopulation by Regulating Asymmetric Cell Division in Planarians. Dev. Cell 38, 413–429 (2016).
Xin, N. et al. The UPRmt preserves mitochondrial import to extend lifespan. J. Cell Biol. 221, e202201071 (2022).
Luchsinger, L. L., de Almeida, M. J., Corrigan, D. J., Mumau, M. & Snoeck, H. W. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 529, 528–531 (2016).
Prieto, J. et al. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 7, 11124 (2016).
Pei, S. et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 23, 86–100.e106 (2018).
Zhong, X. et al. Mitochondrial Dynamics Is Critical for the Full Pluripotency and Embryonic Developmental Potential of Pluripotent Stem Cells. Cell Metab. 29, 979–992.e974 (2019).
Khacho, M. et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 19, 232–247 (2016).
Iwata, R., Casimir, P. & Vanderhaeghen, P. Mitochondrial dynamics in postmitotic cells regulate neurogenesis. Science 369, 858–862 (2020).
Baixauli, F. et al. An LKB1-mitochondria axis controls T(H)17 effector function. Nature 610, 555–561 (2022).
Hong, X. et al. Mitochondrial dynamics maintain muscle stem cell regenerative competence throughout adult life by regulating metabolism and mitophagy. Cell Stem Cell 29, 1298–1314.e1210 (2022).
Horbay, R. & Bilyy, R. Mitochondrial dynamics during cell cycling. Apoptosis 21, 1327–1335 (2016).
Bean, C. et al. The mitochondrial protein Opa1 promotes adipocyte browning that is dependent on urea cycle metabolites. Nat. Metab. 3, 1633–1647 (2021).
Dorn, G. W. 2nd. Evolving Concepts of Mitochondrial Dynamics. Annu Rev. Physiol. 81, 1–17 (2019).
Houtkooper, R. H. et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457 (2013).
Newmark, P. A. & Sánchez Alvarado, A. Bromodeoxyuridine specifically labels the regenerative stem cells of planarians. Dev. Biol. 220, 142–153 (2000).
Pearson, B. J. et al. Formaldehyde-based whole-mount in situ hybridization method for planarians. Developmental Dyn.: Off. Publ. Am. Assoc. Anatomists 238, 443–450 (2009).
King, R. S. & Newmark, P. A. In situ hybridization protocol for enhanced detection of gene expression in the planarian Schmidtea mediterranea. BMC Dev. Biol. 13, 8 (2013).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Huang, X. et al. Fast, long-term, super-resolution imaging with Hessian structured illumination microscopy. Nat. Biotechnol. 36, 451–459 (2018).
Zhao, W. et al. Sparse deconvolution improves the resolution of live-cell super-resolution fluorescence microscopy. Nat. Biotechnol. 40, 606–617 (2022).
Davies, E. L. et al. Embryonic origin of adult stem cells required for tissue homeostasis and regeneration. Elife 6, e21052 (2017).
Adler, C. E., Seidel, C. W., McKinney, S. A. & Sánchez Alvarado, A. Selective amputation of the pharynx identifies a FoxA-dependent regeneration program in planaria. Elife 3, e02238 (2014).
Adler, C. E. & Alvarado, S. ánchez A. Systemic RNA Interference in Planarians by Feeding of dsRNA Containing Bacteria. Methods Mol. Biol. 1774, 445–454 (2018).
Robb, S. M., Gotting, K., Ross, E. & Sánchez Alvarado, A. SmedGD 2.0: The Schmidtea mediterranea genome database. Genesis 53, 535–546 (2015).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Tiku, V. et al. Nucleolar fibrillarin is an evolutionarily conserved regulator of bacterial pathogen resistance. Nat. Commun. 9, 3607 (2018).
Pan, X. et al. mitoproject: Mitochondrial dynamics govern whole-body regeneration through stem cell pluripotency and mitonuclear balance. Zenodo, https://doi.org/10.5281/zenodo.14058603 (2024).
Acknowledgements
We thank Min Jiang, Xian-Jie Yang, and Xiang-Dong Fu for reading the manuscript and for the insightful discussion. We thank Xiaoshuai Huang for his assistance with mitochondrial imaging. We acknowledge the High-Performance Computing Center and the Core facilities of Flow Cytometry, Molecular Biology, Genomics, Microscopy, Mass Spectrometry & Metabolomics, and General Equipment & Autoclave Service of Westlake University for technical support. We acknowledge Yinping Gao, Fang Xiao, Ping Deng, Haiyan Zhang, Guicun Fang, Jinhui Li, Yanan Tang, Xilai Ding, Shan Feng, and Yalin Wang for their assistance with equipment. We thank Guangzhou CSR Biotech Co. Ltd for live-cell imaging by using their commercial super-resolution microscope (HIS-SIM), data acquisition, SR image reconstruction, analysis and discussion. K.L. is supported by the National Natural Science Foundation of China (32122032, 31970750, and 32470878), the “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2024SSYS0030), Zhejiang Provincial Key Laboratory Construction Project, and the start-up fund from the Westlake Education Foundation. C.G. is supported by the National Natural Science Foundation of China (32000498).
Author information
Authors and Affiliations
Contributions
K.L., X.P., Y.Z., and Y. L. conceived the project, designed experiments, analyzed data, and wrote the manuscript. K.L., X.P., J.C., W.Z., L.Y., Y.Z.X., Y.Y., H.X., Y.Z., Y.S., N.L., and C.G. performed all experiments and data acquisition and analyzed data. Y.L. performed the scRNA-seq analyses. L.C. designed and provided the HIS-SIM. Z.C. designed and provided the PK Mito dyes.
Corresponding authors
Ethics declarations
Competing interests
Z.C. holds the patent of PK Mito dyes (CN111454193). The remaining authors declare no other competing interest.
Ethical approval
We support inclusive, diverse, and equitable conduct of research.
Peer review
Peer review information
Nature Communications thanks Dasaradhi Palakodeti and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pan, X., Zhao, Y., Li, Y. et al. Mitochondrial dynamics govern whole-body regeneration through stem cell pluripotency and mitonuclear balance. Nat Commun 15, 10681 (2024). https://doi.org/10.1038/s41467-024-54720-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54720-1
