
Abstract
Bacteria withstand antibiotic treatment through three alternative mechanisms: resistance, persistence or tolerance. While resistance and persistence have been described, whether drug-induced tolerance exists in cancer cells remains largely unknown. Here, we show that human cancer cells elicit a tolerant response when exposed to commonly used chemotherapy regimens, propelled by the pervasive activation of autophagy, leading to the comprehensive activation of DNA damage repair pathways. After prolonged drug exposure, such tolerant responses morph into persistence, whereby the increased DNA damage repair is entirely reversed. The central regulator of mitophagy PINK1 drives this reduction in DNA repair via the cytoplasmic relocalization of the cell identity master HNF4A, thus hampering HNF4A transcriptional activation of DNA repair genes. We conclude that exposing cancer cells to relevant standard-of-care antitumour therapies induces a pervasive drug-induced tolerant response that might be broadly exploited to increase the impact of first-line, adjuvant treatments and debulking in advanced cancers.
Similar content being viewed by others
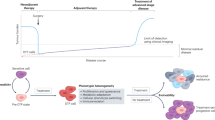

Introduction
Antibiotic treatment failures could emerge from the acquisition of resistance mutations but may also arise from two alternative mechanisms including tolerance and persistence, described as early as 19441. Resistance is an inherited trait acquired by bacteria strains through gene transfer or mutations in the resistome2. It is assessed by the minimum inhibitory concentration (MIC), whereby resistant strains are able to grow at higher MIC3. In tolerance, the bacteria population withstands transient exposure to high antibiotic concentrations otherwise lethal, with no changes in MIC and weakly dependency on the drug concentration administered. This property of tolerant cells is reflected in the time-kill curve, which shows a monophasic delay in reducing the fraction of survivors when compared with susceptible bacteria. Importantly, the time-kill curves reach a plateau, even with very high drug concentrations, implying that the fraction of surviving cells only weakly depends on the drug concentration, in clear contrast to resistance. As such, tolerance is measured with time, that is, the minimum duration of treatment that kills a certain percentage of the bacterial population (MDK), and not by drug concentration (MIC).
Similarly to tolerance, persistence is not measured by concentration but by time. However, in this case, the majority of the population is eliminated to a faster rate than the minority of persister cells upon treatment. Hence, in persistence, the time-kill curves are biphasic3,4,5.
Cellular persistence has been described in mammalian cancer cells6,7,8, where upon several weeks of chemotherapy or targeted therapy treatment, a small sub-population (usually accounting for <1% of the population) survives. In most documented cases of bacterial tolerance or persistence, the cells are inherently tolerant before exposure to the drug, for example by having a decreased metabolism. In that case, finding specific pathways to target for decreasing survival is very difficult due to the pleiotropy of the phenotype. However, a particular form of tolerance called drug-induced tolerance has been shown to depend on the activation of stress-response pathways upon exposure to the drug. Specifically, drug-induced tolerance in bacteria has been shown to be due to the activation of the DNA repair response, and blocking these pathways enables to decrease drug-induced tolerance9. Therefore, identifying the type of tolerance may have important therapeutic consequences.
Whether drug-induced tolerance exists in cancer cells, and its potential effects during cancer treatment, largely remains unknown. Here we show that drug-induced tolerance exists in human cancer cells and it is propelled by autophagy that mediates the DNA damage repair response to chemotherapy. Persistence instead is featured by a complete reversal of this phenotype through the central regulator of mitophagy, PINK1 that determines the re-localization of the cell identity master HNF4A.
Results
Cancer cells exhibit tolerance to cancer therapy
To determine whether drug-induced tolerance exists in cancer, we exposed five patient derived organoids (PDOs) obtained from metastatic colorectal cancer (mCRC) to the liver to established therapeutic regimens based on FOLFOX (5-FU + oxaliplatin) and FOLFIRI (5-FU + SN38) and assessed cell survival at early time points (from 16 up to 120 hours), in line with the experiments conducted in bacteria to differentiate tolerance from persistence, where the emergence of tolerance ranges from minutes to several hours, depending on the antibiotic used10 (Fig. 1a and Supplementary Fig. 1a). These anticancer drugs were administered at increasing concentrations, starting at a half maximal inhibitory concentration (IC50) and scaled up to 50-fold higher. In most PDOs, the killing rate (or survival fraction at a given time point) did not scale with the concentration of the drug but rather approached saturation, leading to a reduced concentration dependance at high dosage, a pattern consistent with tolerance (Fig. 1a). Notably, the tolerant response was present in the entire cell population rather than in a subpopulation of cells as in persistence, suggesting that at early time points all the cells showed a tolerant response3,4,11. To discern more rigorously these different patterns of response, again in line with the framework applied in bacteria, we applied to our data the same mathematical tools utilized in the bacterial field. The modelling revealed that most, albeit not all, cancer models (including several patient-derived samples) showed a robust, single exponential decay response to treatments, thus defining a drug-induced tolerant response. As in bacteria, this initial pattern morphed with time towards a biphasic behaviour, typical of a persistent response (Supplementary Fig. 1a). Intriguingly, the time of the transition was different between PDOs. For example, PDO17 showed a shift from tolerance to persistence at 48 hours, while others did not complete this shift within the experiment time frame (120 hours). Also, some PDOs displayed a tolerant phenotype to certain drugs, while being resistant to others, as it is the case of the PDO61 and PDO68, that were resistant to FOLFIRI, and tolerant to FOLFOX (Supplementary Fig. 1b). We extended our analysis to additional established and primary patient-derived cancer cells obtained from various tissues, that were exposed to the treatment regimens used in patients in the respective conditions, even at concentrations far exceeding the ones used in the clinical practice (Fig. 1b–e; Supplementary Fig. 1a). Two CRC cell lines, HCT116 and HT29, were hence treated with FOLFOX or FOLFIRI (Fig. 1b, c; Supplementary Fig. 1a and c); two breast cancer (BC) models, a cell culture obtained from a metastatic BC patient-derived xenograft (MBC26)12 and the MDA-MB-231 cell line were treated with cisplatin (Fig. 1d and Supplementary Fig. 1a, c); and two primary cell cultures derived from ovarian cancer (OC; OC_AS05 and OC_AS17) patients13 were treated with carboplatin and paclitaxel (CPT + PTX) (Fig. 1e and Supplementary Fig. 1a, c). Again, in all cellular models the early time-kill profiles were typical of a tolerant response but evolved with time towards persistence. As above, the timing of this shift depended on the drug used and was variable, with some cancer cells turning towards persistence within 48 hours of treatment (i.e. HT29 FOLFOX-treated, OC_AS05, OC_AS17), or even 24 hours as in MDA-MB-231 breast cells. Again, in line with the results obtained in bacteria, the transient exposure to the drugs did not select a resistant subpopulation, as after five to seven weeks of suspension of the treatment (DH), the sensitivity of tolerant cells to the compounds was entirely restored (Supplementary Fig. 1d).

a Time-kill curves of three PDOs from mCRC patients treated with FOLFOX or FOLFIRI (n=mean of three experimental replicates ± SD). Time-kill curves of HCT116 (b) and HT29 (c) CRC cells in response to FOLFOX or FOLFIRI (n=mean of three experimental replicates ± SD). Time-kill curves of MDA-MB-231 and MBC26 patient-derived xenograft breast cancer cells (d) treated with cisplatin and two models of primary ovarian cancer cells (OC_AS05 and OC_AS17) treated with carboplatin and paclitaxel (CPT + PTX) (e). The drugs concentrations ranged from the half maximal inhibitory concentration (IC50) to 50- to 100-fold higher. Cell viability was measured at 0, 16, 24, 48, 72 and 120 hours (hrs). Fraction of surviving cells (expressed in Log10 scale) was calculated for each concentration at each time point with respect to the time zero (n=mean of three experimental replicates ± SD). f Dose response curve of NCI-H23 lung cancer cells to cisplatin 5 μg/ml (n = 3 experimental replicates; mean ± SD). Source data are provided as a Source Data file.
Notably, a model of lung adenocarcinoma, the NCI-H23 cell line, among those tested did not show resistant, tolerant or persistence responses, even at the lowest dose of the drug, which was able to entirely eliminate cells (Fig. 1f), suggesting that not all tumour cells activate a tolerant or persistence response during chemotherapy.
Overall, these results suggest that most human cancer cells present a drug-induced tolerant response when exposed to compounds commonly used in the clinic, even at exceedingly high concentrations. The results that we observed in cancer cells are similar to established findings in bacteria, where drug exposure to DNA-damaging agents induces an early, tolerant response4.
Autophagy elicits DNA damage repair
To shed light on the mechanisms underlying drug-induced tolerance in cancer, we reasoned that the early tolerant response seen in human cells could stem from a modified response to the DNA insult. As anticipated, treatment with these agents elicited widespread DNA damage. We hence assessed the extent of DNA damage at the single-cell level, as measured by phosphorylation of the histone H2AX (γH2AX), at early time points (8 and 16 hours) following exposure to drugs (dose IC50x10). In the subset of cells undergoing apoptosis there was an increase in the γH2AX signal, as anticipated (Supplementary Fig. 2a–c). However, in the non-apoptotic cell subgroup, γH2AX levels surprisingly decreased at 16 hours compared with 8 hours, despite the continuous presence of agents at high concentration (Fig. 2a). To corroborate these observations, we evaluated γH2AX expression by flow cytometry and confirmed a strong reduction in DNA damage at 16 hours, compared to the signal detected at 8 hours (Fig. 2b, Supplementary Fig. 2d). To ascertain the engagement of the downstream effectors of the DNA damage-sensing kinases ATR and ATM, we then assessed the phosphorylation status of the serine/threonine-specific kinases CHK1 and CHK2, respectively. Notably, phosphorylation of both proteins was also reduced at 16 hours (Fig. 2c) compared with the signal at 8 hours. In all, these results suggest that cancer cells mount a strong drug-induced tolerant response which leads to an overall early reduction in DNA damage during the early hours after exposure to high levels of DNA-damaging agents.

a Imagestream analysis of γH2AX in HCT116 cells treated for 8 h and 16 h with FOLFOX or FOLFIRI. γH2AX expression in the annexin V negative (-) population is expressed as mean intensity in Log10 scale (distribution of two independent experiments were merged considering CTRL n = 12473; FOLFOX 8 h n = 24742; FOLFOX 16 h n = 18282; FOLFIRI 8 h n = 22867; FOLFIRI 16 h n = 19223). ANOVA followed by Tukey post-test was applied (****p< 0.0001)(median; first and third quartile are shown). b Flow cytometry analysis of γH2AX positivity in the annexin V(-) population of HCT116 cells treated as mentioned above (n = 5 experimental replicates; median; whiskers: min to max; 25th and 75th percentiles are shown). Significant differences were calculated by an ANOVA followed by Bonferroni post-test for multiple comparisons. c Representative western blot analysis for phosphorylated (p-) CHK1 and CHK2 performed on HCT116 cells. Corresponding proteins level quantification calculated as ratio to GAPDH, used for normalization, is reported. d RNA-seq analysis was performed in HCT116 cells treated for 8 h and 16 h with FOLFOX or FOLFIRI. Gene ontology (GO) analysis was performed on differentially expressed genes common to the two treatments. Significant ontologies (P ≤ 0.05) clustered according to their semantic similarity are ranked by -log10(p-value). e GSEA of the three most significant functions for the interaction of ULK1 inhibitor (SBI-0206965) groups and chemotherapies. f. Expression of genes quantified by RT-qPCR relative to the non-treated group (n = 3; mean ± SD). RPLP0 was used to normalize expression. Significant differences among groups were calculated by a two-sided Student’s t-test. g. DNA repair proficiency of the HCT116 cells treated for 16 h with FOLFOX or FOLFIRI alone or in combination with a ULK1 inhibitor (ULK1i). Reporter fluorescence is expressed relatively to the non-treated sample (mean of n = 2 experimental replicates). Source data are provided as a Source Data file.
We surmised that this reduced DNA damage may emerge from an increased ability of the tolerant cells to repair DNA. Indeed, transcriptomic analyses revealed, alongside DNA damage response, cell cycle arrest and apoptosis, a pervasive increase in the expression levels of the entire set of DNA damage repair (DDR) pathways, including homologous recombination (HR), base excision (BER) and nucleotide excision repair (NER), which started at 8 hours, but was especially intense at 16 hours after treatment (Fig. 2d and Supplementary Data 1).
To uncover the mechanisms underlying this widespread transcriptional upregulation of DNA repair after exposure to chemotherapy, we explored additional deregulated pathways. We found there was a specific, robust upregulation of the genes involved in autophagy (Fig. 2d and Supplementary Data 1). Specifically, as confirmed by RT-qPCR, several key autophagic players including ULK1 and ATG2A were upregulated upon treatment (Supplementary Fig. 2e).
We then tested whether autophagy itself could regulate the expression of DDR genes in the tolerance window to chemotherapies. We thus targeted cells with a compound (SBI-0206965) that inhibits the Unc-51-like autophagy activating kinase 1 (ULK1)14, which initiates autophagy in mammalian cells. RNA-seq, followed by a gene set enrichment analysis, revealed that the ULK1 inhibitor (ULK1i), besides the p53 response and TNFα–NF-κB inflammatory pathways, blunted the increase in the expression of DNA repair genes (Fig. 2e, f, Supplementary Fig. 3a, Supplementary Data 2). We confirmed these results with a genetic approach. To this end, we silenced the main genes governing different steps of autophagy15, namely ATG2A, ATG5, and ATG12, with specific shRNAs (Supplementary Fig. 3b). The knock-down of each gene blunted or reduced the DNA repair response of cancer cells to the treatments (Supplementary Fig. 3c).
To determine if the changes in the expression of DNA repair genes correspond to the actual modulation of cellular DNA repair, we exploited a fluorescence multiplex host cell reactivation (FM-HCR) assay16. This assay measures the ability of human cells to repair plasmid reporters, each bearing a different kind of DNA damage. DNA repair mediated by the various pathways was prevented by ULK1 inhibition, confirming that autophagy during tolerance mediates the activation of DNA repair (Fig. 2g and Supplementary Fig. 3d).
Altogether, these data demonstrate that after exposure to DNA damage, mammalian cells mount a tolerant response that is driven by autophagy and leads to empowerment of DNA repair.
Inhibiting autophagy improves the cellular response to cancer therapy
We next explored if the inhibition of autophagy could reduce the survival promoted by the tolerant response upon treatments. To this end, we probed the impact of the genetic silencing of ATG2A, ATG5 and ATG12 on cell viability at 16 h of treatment. We confirmed that the suppression of autophagy significantly reduced cell survival, by hampering the DNA damage response (Supplementary Fig. 4a). To further support the protective and central role of autophagy in drug-induced tolerance, we stimulated autophagy with two different mTOR inhibitors (Temsirolimus and Rapamycin), previously shown to increase the autophagic response17. Indeed, these treatments increased the number of viable cells upon chemotherapy (Supplementary Fig. 4b, c).
We expanded our analysis with a clonogenic assay in HCT116 cells, which revealed that the combination of FOLFOX and FOLFIRI with two autophagy inhibitors with different mechanisms of action, ULK1i and hydroxychloroquine (HCQ; which inhibits the fusion of lysosome and autophagosome18), profoundly reduced the number of colonies when compared with FOLFOX and FOLFIRI alone (Supplementary Fig. 4d), although the effect of HCQ was not as pronounced as that of ULK1i. Colony formation was significantly abrogated also by the genetic silencing of ATG2A, ATG5 and ATG12, confirming the impact of autophagy on cell survival during chemotherapy (Supplementary Fig. 4e).
To confirm the role of autophagy in driving the survival of tolerant cells, we determined the effect of ULK1i combined with either FOLFOX or FOLFIRI in the mCRC PDOs models. Once more, ULK1 inhibition greatly increased the antitumour effects of FOLFOX and FOLFIRI (Fig. 3a and Supplementary Fig. 4f). We next tested the efficacy of the autophagy inhibition and chemotherapy combination in vivo (Fig. 3b). HCT116 CRC cells were subcutaneously transplanted in immunocompromised mice (n = 8 per group) and animals treated with these combinations for two weeks. Autophagy inhibition strongly enhanced the impact of chemotherapy in reducing tumour volumes (Fig. 3b). To formally demonstrate the potential synergistic effect of autophagy inhibition and chemotherapy, we generated a population genetics model19 that describes tumour growth (see Supplementary Methods). We fit this model to data from experiment shown in Fig. 3b, using advanced Bayesian inference methodology to generate probability distributions over drug-associated death rates, the kinetic parameters we use as proxy to measure autophagy inhibition. Our approach confirmed the growth inhibition of drug-exposed populations relative to control, across all classes of drugs and combinations (monotherapy and doublet therapy) (Fig. 3c). The most striking evidence was however a non-additive but synergistic effect for combination treatments, supported by 36% and 65% decreased growth rate when ULK1i was combined with FOLFOX and FOLFIRI, which was far stronger than the expected additive effect model (Fig. 3d).

a Viability of five PDOs from mCRC patients treated with FOLFOX, FOLFIRI, and ULK1i (SBI-0206965). Each dot represents the measurement of a single organoid after one week of treatment. Significant differences among groups calculated by applying a two-sided Student’s t-test (mean ± SD) (exact P values are indicated in figure; ****P < 0.0001). b Tumour growth curves of HCT116 cells subcutaneously transplanted in immunocompromised mice (n = 8 mice per group). Mice were treated twice a week for two weeks with FOLFOX or FOLFIRI alone or in combination with ULK1i. Tumour volume was measured twice a week and significant differences among groups calculated by applying a two-sided Student’s t-test (mean ± SEM). A population genetics model describing tumour growth during treatment. A decrease in growth rate occurred with respect to the CTRL sample in mice treated with ULK1i, FOLFOX, FOLFOX+ULK1i, FOLFIRI and FOLFIRI+ULK1i (c) (n = 8 per group; median; whiskers: min to max; first and third quartile are shown). Growth curves of synergistic and additive inhibition for the combinatorial treatments (d). e, f Quantification of immunohistochemistry analysis for Ki67 and γH2AX performed on tumours after two weeks of treatment. Three different areas of each tumour slide were considered and significant differences among groups were calculated by an ANOVA followed by Bonferroni post-test (n = 21; median; whiskers: min to max; 25th and 75th percentiles are shown; ****P < 0.0001). g Quantification of viability of MDA-MB-231 and MBC26 breast cancer cells treated with cisplatin and ULK1i after one week (MDA-MB-231 - n = 7; mean ± SD) or three days (MBC26 - n = 3; mean ± SD). Differences among groups were calculated by ANOVA followed by Tukey post-test (exact P values are indicated in figure; ****P < 0.0001). h Cell viability of two primary cancer models of ovarian cancer treated with carboplatin and paclitaxel and ULK1i (n = 3 to 4 experimental replicates; mean ± SD). Differences among groups were calculated by a two-sided Student’s t-test (exact P values are indicated in figure; ****P < 0.0001). Source data are provided as a Source Data file.
To corroborate the synergistic effect exerted by the combinatorial treatments on tumours, we performed an immunohistochemistry analysis which confirmed a reduction in the abundance of the proliferative marker Ki67 (Fig. 3e). Of note, we found an increase in γH2AX (Fig. 3f) in all combinatorial groups, confirming the crucial role of autophagy in DNA repair during chemotherapy administration.
We extended the analysis to additional cancer models. The microsatellite-stable HT29 CRC cell line displayed a similar tolerant-like response in terms of DDR and autophagy activation upon high concentrations of chemotherapy (Supplementary Fig. 4g, h) and was responsive to autophagy inhibition and its combination with chemotherapy (Supplementary Fig. 4i). We then explored whether the tolerant response seen in breast and ovarian cancer was also linked to increased DDR driven by autophagy. Indeed, both DDR and autophagy genes were increased in breast (MDA-MB-231) and ovarian primary cancer cells (Supplementary Fig. 4j, k). The effects of the cisplatin and ULK1 inhibition combination were strongly synergistic in MDA-MB-231 cells and in a BC PDX derived cells model (Fig. 3g), as were carboplatin and paclitaxel in two primary OC models (Fig. 3h).
Notably, we confirmed that the role of autophagy in tolerance is linked to the induction of DNA damage, as the combination of autophagy inhibition with other drugs with a different mechanism of action (i.e. inducing block of proliferation) did not impact on cell survival (Supplementary Fig. 4l).
These data suggest that autophagy inhibition may be a successful strategy to improve the response to therapy in many cancer types.
DNA repair is abrogated in persister cells arising from drug-induced tolerance
Bacteria exposed to antibiotics transition from a tolerant state shared by the entire population to persistence, in which only a subpopulation can withstand and survive treatment3,4. Our data support the notion that cancer cells as well transition from drug-induced tolerance into persistence. We next sought to define whether persister cells displayed an increased DDR, as tolerant cells. We thus exposed cells to chemotherapy for two additional weeks beyond the initial 16 hours. Drugs were then withdrawn from the cell culture for five weeks (drug holiday, DH) (Supplementary Fig. 5a). Persister cells emerged, as confirmed also by scRNA-seq experiments which showed a population of slow-cycling cells (Supplementary Fig. 5b). As expected, persister cells regained sensitivity to chemotherapy after the drug holiday (Supplementary Fig. 5c). Moreover, persister cells were clearly distinguishable from cells that had been exposed to drugs for 8 and 16 hours, and from DH cells (Fig. 4a).

a UMAP embeddings of single-cell RNA sequencing in HCT116 cells treated for 8 h, 16 h, 2 weeks (PCs), and after five weeks of drug holiday (DH). Expression clustering at different time points and the specific DNA damage repair machineries of two experimental replicas are reported. b Density plots showing the relation between genomic instability (x-axis) and chromatin accessibility (y-axis) at different time points. c Distribution of the expression level at different time points of the top 200 genes mostly correlated with the overall transcription rate from the CytoTrace model. d Barplots reporting the cumulative frequency of the barcodes in tolerant cells (TCs; 8 h), persister cells (PCs) and DH cells for different treatments (CTRL, FOLFOX or FOLFIRI) and time points (TCs, PCs and DH). Each horizontal grey rectangle in the bars corresponds to the relative frequency of each barcode detected in that sample (number copies of that barcode over the total number of copies of all barcodes). Each bar is annotated with the corresponding entropy value (H). e HCT116 cells clustered for their phenotypic/transcriptomic similarities and diverse subpopulations (each represented by a different colour) quantified during treatment. UMAP embedding of single-cell RNA and single-cell GET sequencing showing cells coloured by multimodal clusters (f) and by time treatment (g), obtained by multilevel Nested Stochastic Block Models. Heatmaps report the fraction of cells at each time point found in the group highlighted in the inset. Source data are provided as a Source Data file.
Based on the robust activation of the DDR pathway in tolerant cells, we expected that during chemotherapy exposure persister cells would maintain the transcriptional activation of the DDR pathway seen in the tolerance phase. To our surprise, we found that in these cells, all DDR pathways, including HR, NHEJ, MMR, BER, and NER, were consistently downregulated (Fig. 4a). Intrigued by these results, we reasoned that the concomitant reduction of DDR pathways could be associated with a reduced ability to accurately repair DNA in persisters, leading to an enhanced accumulation of genomic instability. To measure genomic instability, we assessed the excess of structural variants at a given sequencing coverage (see Supplementary Methods) using scGET-seq20, a single-cell technology that we recently developed, able to probe single-cell epigenetic and genetic landscapes. Persister cells, but not the control cells, did indeed show increased genomic instability (Fig. 4b and Supplementary Fig. 5d), when compared with cells after 8- and 16-hour treatments. Notably, this increase in instability was associated with a sizeable compaction of chromatin (Fig. 4b and Supplementary Fig. 5d) and remarkably, with an overall decrease in the transcriptional output (Fig. 4c). These findings suggest that persister cells enter frozen epigenetic and transcriptional states.
Since a critical link between genomic instability and clonal selection during drug treatment has been proposed21,22, we then wanted to determine whether genetic clonal evolution phenomena were present, and if any specific genetic subclone became dominant in this population. To this end, we barcoded cells and compared the variation of the Shannon entropy in time and the distribution of the barcode relative frequencies via a permutation test with resampling. Control cells and treated samples showed a similar gradual reduction of the entropy in time, due to the underlying stochastic dynamics (Fig. 4d), as anticipated. Additionally, the distribution of barcode frequencies of control cells and both treated samples showed no statistically significant differences at all time points (Supplementary Fig. 5e). Both these results support the hypothesis that genetic clonal evolution does not occur during chemotherapy, in line with previous findings8,23,24,25.
To explore the possibility that alternative evolutionary strategies are deployed by cancer cells during chemotherapy, we analysed transcriptomic data. scRNA-seq revealed the emergence, in independent experiments, of a dominant but not unique persister subpopulation. This subpopulation featured a homogeneous transcriptional landscape, suggesting that cancer cells exposed to chemotherapy transition from a tolerant to a persistent state governed by phenotypic selection (Fig. 4e). As anticipated, this dominant clone (DC) was not genetically homogeneous but included genetic clones featuring various barcodes (Fig. 4d). Notably, while the transcriptomic features of DC were present only in persister cells, the epigenetic landscape defining this population was already present at 8 and 16 hours after treatment (Fig. 4f, g), suggesting that chromatin remodelling occurs before any gene expression changes, a pattern reminiscent of pioneering factors activity during fate decision in development26.
There were other smaller subpopulations within the persister population that did not share common transcriptomic features with other subpopulations or the DC (Fig. 4e). We then assessed the fitness potential of this dominant population and compared and contrasted it with those of the less-represented cell populations during DH. DC cells survived despite the presence of the drugs, but once the drug was removed (that is, the DH population) the transcriptomic as well as the epigenetic landscapes of the DC disappeared (Fig. 4e). Notably, the ability to repair DNA (Supplementary Fig. 5f), as well as the genomic stability state (Supplementary Fig. 5g), was restored in the remaining cell populations. In all, these results suggest that after drug-induced tolerance, cancer cells enter into persistence, where the predominant phenotypic state is preceded by pervasive chromatin compaction associated with blunted transcription, reduced DNA repair and increased genomic instability. Notwithstanding, no genetic fixation predisposing to resistance takes place, at least within the time frame of our experiments. This predominant population promptly disappears once the phenotypic selection pressure is withheld, leaving room for more diverse, and evolutionary-favoured, subpopulations.
Persistent cells rely on enhanced mitophagy for survival
Despite an overall reduction in transcription, there were a few loci and genes that showed strong expression in persister cells (Fig. 4c). Surprisingly, autophagy remained among the most upregulated and activated pathways in persister cells, as attested by increased autophagic vacuoles (Supplementary Fig. 6a, b), functional activation of autophagic flux attested by the reduction in pHluorin intensity27 (Supplementary Fig. 6c) and by the transcriptional activation of genes encoding autophagy effectors (Supplementary Fig. 6d). Since autophagy drives an increase in DDR response during the tolerant phase, we wondered which genes and pathways could antagonize the activation of DDR induced by autophagy in the persister population. Several pathways related to mitochondria biogenesis, and most prominently mitochondrial demise, were powerfully activated in persister cells, specifically the mitophagy pathway (Fig. 5a and Supplementary Data 1). Additionally, several other networks related to mitochondrial functions were upregulated, including genes regulating reactive oxygen species (ROS) (Fig. 5a and Supplementary. Data 1). Single-cell RNA-seq analysis confirmed this prominent increase in mitophagy in persister cells (Fig. 5b). We hence sought to determine the engagement of mitochondria in these cells, via flow cytometry analysis and electron microscopy. We found that the mitochondrial mass was vastly expanded in persister cells (Fig. 5c and Supplementary Fig. 6e). TEM analysis revealed a robust increase in the number of mitochondria (Fig. 5d). Also, mitochondrial morphology was profoundly affected, since mitochondria were significantly more elongated, as shown by reduced circularity and increased fusion/fission and replicative functions in comparison with control cells (Fig. 5e, f; Supplementary Fig. 6f). In addition, bafilomycin treatment, which blocks autophagosome-lysosome fusion, confirmed the activation of mitophagy (Fig. 5g). To demonstrate that the increased number of mitochondria was associated with functional sequelae, we exploited the MitoTimer fluorescent reporter protein, which monitors both mitochondria maturation and mitophagic flux28. We detected a shift in the abundance of newly expressed (green) versus mature (red) MitoTimer, thus confirming the increase in mitophagy during persistence (Supplementary Fig. 6g, h).

a Gene ontology analysis performed in persisters on differentially expressed genes common among the two treatments. The pathways related to mitochondria functions are reported, ranked by -LOG10(p-value). b UMAP embedding derived from scRNA-seq analysis highlighting the increased expression of a mitophagy signature in the persister population. c Mitochondrial mass measured in persisters by performing a mitochondrial green fluorescent assay. The Median Fluorescent Intensity (MFI) is reported for each treatment of one representative experiment. d Mitochondrial number, normalized to the cytosolic fraction occupied by mitochondria (n = 25 fields; median; whiskers: min to max; 25th and 75th percentiles are shown). Significant differences among groups were measured by an ANOVA followed by Dunnett’s post-test (****P < 0.0001). Representative images from Transmitted Electron Microscopy (TEM) (scale bar 1 µm) (e) to calculate mitochondria circularity (f). Significant differences among groups were measured by an ANOVA followed by Dunnett’s post-test (n = 25 fields; median; whiskers: min to max; 25th and 75th percentiles are shown) (****P < 0.0001). g Representative images of mitophagy from TEM in persisters after bafilomycin treatment. Asterisks indicate mitophagy events. Scale bar: 500 nm. h. Mitophagic flux measured as relative fluorescence intensity (RFI) by mt-mKeima reporter in HCT116 cells infected to silence PINK1 or a SCR control during FOLFOX or FOLFIRI treatment. Significant differences among groups were calculated by applying one-way ANOVA followed by Sidak’s post-test for multiple comparisons (n = 3; mean ± SEM) (****P < 0.0001; **P = 0.0049). Source data are provided as a Source Data file.
We hence sought to determine the molecular wiring responsible for the increased mitophagy. PINK1 was the top-ranked up-regulated gene involved in mitophagy regulation in persister cells (Supplementary Fig. 6f and i). PINK1 activates the clearance of damaged mitochondria29 and protects cells against oxidative stress-induced death30. We confirmed this role of PINK1 also in our system, leveraging on the mitophagy inducer CCCP (Supplementary Fig. 6j). We then tested whether the genetic silencing of PINK1 (shPINK1) reduced mitophagy in the presence of chemotherapy administration. We confirmed that the mitophagic flux was strongly enhanced by FOLFOX or FOLFIRI treatments (Fig. 5h) and that such increase was instead hampered by PINK1 knock-down during chemotherapy exposure (Fig. 5h). We next sought to determine whether chemotherapy generates mitochondrial ROS, as a result of increased mitochondrial generation, and if knock-down of PINK1 might blunt this effect. CCCP, as expected, increased the levels of mitochondrial ROS, an effect that was prevented by PINK1 knock-down (Supplementary Fig. 6k). Accordingly, the treatment of cancer cells with either FOLFOX or FOLFIRI strongly enhanced mitochondrial ROS levels, an increase that was abrogated by PINK1 downregulation (Supplementary Fig. 6l). In all, these data confirm a central role for PINK1 in driving mitophagy and maintaining mitochondrial turnover in persister cells.
PINK1 represses the DNA damage response through the inhibition of the cell-identity regulator HNF4A
We next asked whether PINK1 exerted any role in modulating the expression of DNA repair genes. The analysis of TCGA data sets of CRC, BC and OC patients demonstrated an inverse correlation between PINK1 expression and genes involved in error-free DNA repair (Supplementary Fig. 7a). RNA-seq analysis revealed that inhibition of PINK1 was strongly implicated with the transcriptional regulation of DDR, whereby PINK1 knock-down robustly increased the expression of genes involved in error-free DDR (i.e. HR, MMR and BER) (Fig. 6a; Supplementary Fig. 7b and Supplementary Data 3), beyond its canonical role in regulating genes involved in mitophagy and lysosomal regulation (Supplementary Data 3). We also determined whether the expression of DDR genes following chemotherapy exposure was affected by PINK1 silencing. Indeed, PINK1 knock-down partially reverted the inhibition of DDR elicited by chemotherapy (Supplementary Fig. 7b). The significant increase in the DNA repair proficiency of shPINK1 cells was further confirmed by the observed reduction in γH2AX levels in PINK1-silenced cells in the presence of chemotherapy (Supplementary Fig. 7c). Overall, these data showed that PINK1 has a central role in controlling DDR by modulating the activity of the DNA repair machinery.

a Gene ontology analysis of up-regulated genes in HCT116 cells genetically silenced for PINK1 expression (n = 3 experimental replicates; mean ± SD). The most pertinent functions were ranked by -Log10(p-value). b Ingenuity pathway analysis for upstream regulators performed on differentially expressed genes in persisters and transcription factors ranked for their significance (-Log10 p-value). The Venn diagram represents overlap of HNF4A targets and deregulated genes upon PINK1 silencing. c. Proximity Ligation Assay (PLA) foci quantified as PLA puncta/nuclei in HT29 SCR and shPINK1 conditions (Scale bar: 10 μm). Significant differences among groups were quantified by applying two-sided Student’s t-test (n: SCR = 92; shPINK1 = 61; P = 0.0003). d Confocal acquisition of immunofluorescence staining in HT29 cells for PINK1 and HNF4A. Nuclei were counterstained with DAPI. Scale Bar: 7 µm. e Quantification of HNF4A nuclei fluorescence performed by using Cell Profiler 4.2.1. Significant differences among groups were calculated by applying ANOVA followed by Tukey post-test (***P = 0.0002; **P = 0.0062). f Western blot and protein level quantification in HT29 silenced for PINK1 and an SCR control. g MCRC PDO17 infected to silence PINK1 or a SCR control and then treated with FOLFOX or FOLFIRI for one week. The dotted line represents a drug holiday. Significant differences among conditions were calculated by two-sided Student’s t-test (n = 3 experimental repeats; mean ± SEM). h Kaplan-Meier curves representing Disease Free Survival (DFS) in CRC patients, divided at the median expression of a mitophagy signature. Significant differences among low and high-risk population were calculated by Log-Rank test (HR: Hazard Ratio). Source data are provided as a Source Data file.
Since PINK1 is a serine/threonine protein kinase, we surmised that it could exert its repressive activity on DDR genes by regulating one or more DNA-interacting protein. The analysis of the upstream regulators of the most deregulated genes in persister cells upon PINK1 inactivation identified the cell-identity regulator hepatocyte nuclear factor 4-alpha (HNF4A) as the top-ranked transcription factor regulating this gene subset. Indeed, HNF4A target genes largely overlapped with those deregulated by PINK1 inactivation (Fig. 6b and Supplementary Data 3).
We hypothesized that HNF4A activated DDR in turn. Over-expression of HNF4A in HCT116 cells (Supplementary Fig. 7d) confirmed this observation: DDR genes were significantly up-regulated upon HNF4A expression both in basal conditions (Supplementary Fig. 7e), and upon chemotherapy administration (Supplementary Fig. 7f), mimicking the PINK1-silenced phenotype.
We next asked whether PINK1 may regulate the DNA damage response through HNF4A. We thus silenced HNF4A (siHNF4A) in a background of genetically silenced PINK1 cells. siHNF4A prevented the increase in RAD51, FEN1 and PCNA DDR gene expression elicited by PINK1 knock-down (Supplementary Fig. 7g). This result suggests that the phenotype associated with PINK1 silencing is at least in part the result of modulation in HNF4A activity. We highlighted a similar dynamic in an additional CRC model, namely HT29 (Supplementary Fig. 7h, i).
We then wondered how a cytoplasmatic factor (PINK1) may affect the activity of a transcription factor as HNF4A. Immune precipitation assay (IP) revealed that a potential, direct interaction was present between the two proteins (Supplementary Fig. 7j). We confirmed these results with a proximity ligation assay (PLA) that documented an in situ interaction between the two proteins (Fig. 6c). As expected, PINK1 silencing significantly reduced this interplay. Then, confocal analysis of the immunofluorescence revealed that PINK1 and HNF4A co-localized in the cytoplasm, hence outside of the nucleus (Fig. 6d, e), both in basal (SCR) and in mitophagy-induced conditions (+CCCP). Interestingly, when mitophagy was induced, HNF4A was shuffled from the nucleus to the cytoplasm. This effect was significantly abrogated in the presence of PINK1 silencing (Fig. 6d, e). These data suggested a direct interaction between PINK1 and HNF4A; we then explored whether this association might have any functional relevance. Several kinase cascades (PKC, AMPK and ERK 1/2) inhibit the trans-activation of HNF4A through phosphorylation31. We focused on the S313 phosphorylation site, one of the leading residues inhibiting the HNF4A transcriptional activity31. PINK1 silencing reduced the level of S313 phosphorylation (Fig. 6f), strengthening the hypothesis of the repressive activity of PINK1 on HNF4A. We then analysed the binding of HNF4A to DNA in SCR and shPINK1 conditions. Surprisingly, we found several HNF4A peaks that were impacted by PINK1 knockdown (Supplementary Fig. 7k; complete list of functions is reported in Supplementary Data 4), suggesting a prominent role of HNF4A in modulating the activity of PINK1. Among the 532 peaks showing an increased binding of HNF4A upon PINK1 knock-down, several were related to DNA repair, suggesting indeed a model where PINK1 represses DDR through inhibition of the transcriptional activity of HNF4A on DDR genes.
We next sought to determine whether PINK1 exerts any role in preserving persister cells. We found that indeed in a mCRC PDO, PINK1 inactivation reduced the number of persister cells during chemotherapy, affecting, most importantly, the ability of persister cells to recover during drug holiday (Fig. 6g).
Is there any association between mitophagy activation and survival in patients? To address this question, we used a mitophagy signature composed of 18 genes derived from our RNA-seq analysis (see Supplementary Data 4), to explore the association between the activation of mitophagy genes and patients’ outcome. Activated mitophagy was an independent factor predicting worse disease-free survival (DFS) (Fig. 6h) and progression free survival (PFS) (Supplementary Fig. 7l). These data suggested that mitophagy genes, including PINK1, identified a subpopulation of CRC patients with a high risk of relapse and death. Overall, these observations confirmed that mitophagy may have prognostic relevance in CRC patients.
Discussion
We provide evidence that cancer cells present a drug-tolerant response, that allow them to withstand even exceedingly high drug concentrations. The results that we observe in cancer cells are similar to established findings in bacteria, where the SOS DNA damage response underlies drug-induced tolerance32, suggesting that this atavistic, survival mechanism is conserved from unicellular to multicellular organisms21,33,34.
While a connection between autophagy and DNA repair has been previously reported35,36, our data suggest that autophagy is the central machinery engaged during tolerance, immediately after exposure to chemotherapy, temporarily enhancing the ability of cancer cells to repair DNA. As such, the inhibition of autophagy might be employed concomitantly to standard-of-care treatments, in many cancer types and for indications much broader than the currently targeted RAS/RAF-mutated tumours37,38. Autophagy inhibition could possibly be achieved by using inhibitors of upstream autophagy effectors, such as ULK1 inhibitors (Supplementary Fig. 8), which have proven more effective and less toxic than chloroquine and hydroxychloroquine in clinical trials37,38. Notably, even short-term treatments interfering with autophagy might be sufficient to hamper the tolerant response in the proper time window, without exposing patients to unwarranted toxicity. However, further evaluations are warranted to properly target tolerance into the clinical setting.
If drug exposure is prolonged, depending on the cancer model such tolerance response evolves towards persistence, as reported in bacteria. This switch to a second phenotype entails prominent chromatin compaction and abrupt transcriptional silencing. Nevertheless, a large body of literature has reported that pervasive chromatin relaxation occurs as a result of DNA damage39,40. However, most studies exploring the response of chromatin to DNA damage were based on short term treatments (i.e., a few hours). During prolonged DNA damage, as it is the case of persistence, it appears that chromatin relaxation occurs41,42. We propose that persister cells silence DNA repair mechanisms through chromatin compaction (which disrupts the accessibility of repair pathways to DNA), alongside the transcriptional and functional reduction of the entire DNA repair machinery itself, as it has been recently shown for targeted therapies43,44. It is tempting to speculate that this orchestrated response is an evolutionary conserved strategy to increase genomic instability, thus broadening the evolutionary pool that cancer cells can tap into to increase their survival chances under extreme stress conditions.
The involvement of the identity regulator HNF4A suggests that this response potentially touches and affects several key cellular nodes, temporarily reshaping cell identity, in line with the main activity of HNF4A45. PINK1 and mitophagy might represent the driving forces propelling this phenotypic transition, counteracting the activity of HNF4A and ultimately leading to chromatin compaction and a reduced ability to repair DNA (Supplementary Fig. 8). A role for PINK1 in chemo-resistance46 and in the maintenance of mitochondrial homoeostasis during persistence47,48 has been proposed. HNF4A coordinates cell identity of the endodermal organ49,50 by regulating multiple transcription factors51,52. The connection between PINK1 and HNF4A suggests that PINK1 and mitophagy might have a much wider, exploitable role in the management of drug resistance and the associated transient changes in cell identity than previously thought. Additionally, our results suggest that HNF4A acts as a gateway for the activity of PINK1, in turn repressing or activating key genes involved in mitochondrial activity and DDR pathways, among others. As for the mechanism, the repositioning of HNF4A outside the nucleus is an ingenious subterfuge exploited by PINK1 to remove a hindering HNF4A from the nucleus and inactivating it through the phosphorylation at serine 313.
DNA damaging agents remains the mainstay of cancer treatment53. Preventing the emergence of tolerance in cancer cells could increase the effectiveness of current standard-of-care chemotherapy regimens, akin to the use of radio-sensitizers in radiotherapy54. The reduction in cancer mortality achieved in the past decades is mainly the result of novel adjuvant therapies55. Therefore, blocking the tolerant response in conjunction with currently used anticancer treatments could improve the patient response in first-line treatments, as well as to debulk the tumour mass in more advanced diseases.
Methods
Cell lines and patient derived organoids
Experiments were performed in HCT116, HT29, MDA-MB-231 cell lines (from ATCC), maintained in their media as recommended by supplier. Cell line authentication was performed by Thermo Fisher, AmpFlSTR® Identifiler® Plus PCR Amplification Kit. All cell lines were tested for mycoplasma and resulted negative.
Samples from liver metastatic gastrointestinal cancers were obtained from San Raffaele Hospital (Milan, Italy). Patient Derived Organoids (PDO) cultures were established as previously reported20. In order to perform experimental assays, dissociated PDOs were suspended in growth factor-reduced Matrigel (Corning 356231, Fisher Scientific), seeded in single domes flat-bottom cell culture plate (Corning) and, after dome solidification, covered with complete human organoid medium. Primary cells from Patient-Derived Xenograft of Breast cancer patients were obtained from the European Institute of Oncology (Milan, Italy)12. Primary ovarian cancer (OC) cells were obtained upon informed consent from patients diagnosed with high-grade serous ovarian cancer undergoing surgery at the Gynaecology Division of the European Institute of Oncology (Milan, Italy)13.
CRC cell lines and PDOs are derived from patients of both sexes. Female patients were recruited for breast and ovarian cancer sample collection. The sex of participants was based on self-report. Sex was not considered in the study design and sex-based analysis was not performed.
In vitro treatments
Dose response curves to chemotherapy regimens
For dose-response curves, about 5000 cells from mCRC PDOs were tested in 96 well; about 10,000 HCT116, HT29 and NCI-H23 or 5000 MDA-MB-231 cells were plated on 48 wells in respective media; 2500 cells from primary OC samples and 3500 metastatic breast cancer PDX cells were plated on 96 wells. After 24 h, cells were treated with respective chemotherapy regimens and then fixed with crystal violet and analysed at each time points (0 h–16 h–24 h–48 h–72 h–120 h) by ImageJ. Fraction of cell survival was calculated for each concentration at each time, considering viable cells with respect to time 0 and expressed in Log10 scale.
To assess tolerance, FOLFOX or FOLFIRI were added at the following concentration: 5-FU 100 µM, oxaliplatin 100 µM, SN38 100 nM. Cisplatin was used at 50 µg/ml; carboplatin at 100 µM with paclitaxel 50 nM. ULK1 inhibitor (ULK1i: SBI-0206965) was added at 100 µM dose. After 8 h or 16 h, cells were harvested for experimental purposes. To induce autophagy, Temsirolimus and Rapamycin were added at 10 µM concentration for 8 h and 16 h.
To obtain persister cells, 1.2 x 106 HCT116 cells were plated on a 6 cm plate and continuously exposed to each treatment for two weeks at the following concentration: 5-FU 10 µM, oxaliplatin 1 µM, SN38 10 nM. Medium and drugs were refreshed every four days. During drug holiday, a complete medium was added to cells until the end of the experiment. Chemonaive cells were used as control group.
PDOs from mCRC patients were treated with FOLFOX: 10 µM 5-FU and 10 µM of oxaliplatin or FOLFIRI: 10 µM 5-FU and 10 nM SN38 in triplicate, and in combination with ULK1 inhibitor SBI-0206965 (10 µM). For proliferation assay, MDA-MB-231 and BC PDX cells were treated with cisplatin at 5 µg/ml. The ULK1 inhibitor was added at 10 µM concentration. After one week or three days of culture, respectively, cells were fixed in 10% methanol and stained with 0.5% Crystal Violet. OC cells were exposed to carboplatin at 10 µM with paclitaxel 5 nM. Cell viability was measured in quadruplicate after three or five days of treatment. Cell viability was quantified by ImageJ analysis and expressed relatively to the control group.
Clonogenic potential (1000 cells plated on a six-well plate) was measured in triplicate. 24 h after plating cells were treated with drugs. Doses of chemotherapies were adjusted taking into consideration the number of plated cells to avoid over-toxicity: 5-FU 1 µM, oxaliplatin 100 nM, SN38 1 nM; ULK1i 10 µM and HCQ 25 µM. After 7 days of culture, colonies were fixed in 10% methanol and stained with 0.5% Crystal Violet. Colonies growth was quantified by ImageJ analysis and expressed relatively to the control group.
DNA damage characterization
Imagestream acquisition and analysis
HCT116 cells were treated for 8 h and 16 h with FOLFOX or FOLFIRI combinations as described above. Then, cells were harvested and stained with Annexin V-FITC and fixed for further staining. Cells were incubated with a primary antibody against γH2AX (Ser139 - clone 2F3; Biolegend) and then with a secondary antibody Alexa Fluor 633 and counterstained with DAPI for nuclei labelling. Cells were acquired at ImagestreamX MarkII System (Amnis, LuMinex Corporation) with the 60X_0.9NA objective and the following laser settings: 405 nm (8 mW), 488 nm (200 mW) and 642 nm (150 mW). Single-stained samples were acquired with the identical laser settings of the samples and used for compensation. Annexin V positive and negative cells were quantified and expressed as percentage of total population acquired. Morphology mask in Ch01 was used to identify the nuclear region and cells with good nuclear staining were gated using Area DAPI versus Intensity DAPI features. H2AX phosphorylation was expressed as mean intensity in the nuclear region in Annexin V positive or negative population, respectively. Experiments were conducted in duplicate and at least 20,000 events for each condition were acquired.
Flow cytometry
HCT116 cells were treated for 8 h and 16 h with FOLFOX or FOLFIRI combinations as above. Then, cells were harvested and stained with ANNEXIN V-FITC and fixed for further staining. Cells were incubated with a primary antibody against γH2AX (Ser139 - clone 2F3; Biolegend) and then with a secondary antibody Alexa Fluor 633. To track autophagy in persister cells, HCT116 were transfected with a pEX-PK-hLC3 modified fluorescent reporter (pEX-PK-hLC3 was a gift from Isei Tanida - Addgene plasmid #61458; http://n2t.net/addgene:61458)27, in which mKate2 was used as an internal control. 72 h from transfection, cells were harvested and acquired to compare pHluorin and mKate2 fluorescence and estimate the autophagic flux. To track Mitophagy during persistence, we used pTRE-Tight-MitoTimer fluorescent reporter protein (pTRE-Tight-MitoTimer was a gift from Roberta Gottlieb - Addgene plasmid #50547; http://n2t.net/addgene:)28, under the control of a tetracycline response element. After transfection, synchronized expression of MitoTimer was achieved by inducing expression with the addition of doxycycline (Dox) pulse for 2 h, followed by removal of any residual Dox. The protein was monitored as it matures from green to red over 48 h and MitoTimer expression was used to detect mitophagy as previously reported56. Cells were acquired at CytoFLEX LX (Beckman Coulter) and analysed by FlowJo (Becton Dickinson v10.8.1). pHAGE-mt-mKeima was a gift from Richard Youle (Addgene plasmid # 131626; http://n2t.net/addgene:131626)57. After infection to silence PINK1 or an SCR control, cells were plated on 96 well for Incucyte Live Cell Analysis and fluorescence was quantified in the presence or absence of FOLFOX or FOLFIRI treatements.
DNA damage reporter assays
A Fluorescence multiplex host cell reactivation (FM-HCR)58 assay was applied to measure the DNA repair capacity of the HCT116 cells in response to chemotherapy. Briefly, 60,000 cells were plated on 12 well plates and after 48 h transfected with undamaged or damaged reporters to detect MMR (mOrange_G:G and mOrange_T:G), GFP_HR and BFP_NHEJ, respectively, by using Lipofectamine 3000 (Thermofisher). Immediately after transfection cells were treated with FOLFOX (5-FU 100 µM, oxaliplatin 100 µM) or FOLFIRI (5-FU 100 µM, SN38 100 nM) for 16 h and with ULK1 inhibitor (100 µM), then harvested, acquired by flow cytometry at CytoFLEX LX (Beckman Coulter) and analysed by FlowJo (Becton Dickinson v10.8.1). Reporter expression was assessed relatively to the non-treated sample and normalized to a transfection control both in damaged and undamaged groups.
Western blot
Cells were lysed in SDS buffer and processed, as previously described59. Membranes were probed with following antibodies: PARP (Cell signalling), pCHK2 (Cell signalling), pCHK1 (Cell signalling), GAPDH (GT239; GeneTex), p21 (12D1; Cell signalling), γH2AX (Cell signalling), PINK1 (NovusBio), HNF4A (C11F12; Cell Signalling), p-HNF4A Ser313 (Invitrogen), RAD51 (D4B10), FEN1 (E5X2T; Cell Signalling), EXO1 (Abcam), PCNA (D3H8P; Cell Signalling). For p-HNF4A Ser313 evaluation, cells were starved for 16 h and then harvested and processed as mentioned before. Images were cropped at specific protein bands of interest to improve the clarity of data presentation.
Mitochondria characterization
HCT116 cells were treated for two weeks with FOLFOX and FOLFIRI as previously specified. Then, cells were incubated with Mitotracker Green (Invitrogen M7514; 100 nM) for 20 min at 37 °C in the dark to detect mitochondrial mass or with MitoSOX Red (Invitrogen M36008; 5 μM) for 10 min for mitochondrial ROS detection. Mean Fluorescence intensity (MFI) of 20,000 acquired events was detected at CytoFLEX LX (Beckman Coulter) and analysed by FlowJo (Becton Dickinson v10.8.1) (Mitotracker at Ex/Em: 490/516 nm; Mitosox at Ex/Em: 510/580 nm). After genetic silencing of PINK1 or a SCR control, cells were treated with 50 µM CCCP for 2 h (as mitophagy induction control) or with FOLFOX or FOLFIRI for three days and then MitoSox levels evaluated as above.
Transmission electron microscopy (TEM) assay
For TEM analysis, cells treated for two weeks with FOLFOX or FOLFIRI were fixed for 1 h at room temperature in 2,5% glutaraldehyde in 100 mM NaCacodylate buffer pH 7.2, and then post-fixed in 1% osmium tetroxide, 1,5% potassium ferricyanide in 100 mM NaCacodylate buffer overnight at 4 °C. After block staining in 0,5% uranyl acetate ON at 4° C, samples were dehydrated with a crescent concentration of ethanol, infiltrated, and embedded in epoxy resin. Then, after being cured for 48 hours, resin blocks were cut into ultrathin sections (70–90 nm) by using an ultramicrotome (UC7, Leica microsystem, Vienna, Austria), collected, stained with uranyl acetate and Sato’s lead solutions, and observed in a Transmission Electron Microscope Talos L120C (FEI, Thermo Fisher Scientific) operating at 120 kV. Images were acquired with a Ceta CCD camera (FEI, Thermo Fisher Scientific). For morphometrical analysis, images of the random selected cellular profiles were analysed using ImageJ software. In particular, mitochondria were counted and their area was expressed as the ratio of mitochondria area and cytosol (mitochondrial fraction). Circularity was used to define mitochondria elongation. For the evaluation of mitophagy events, after two weeks of treatments, cells were treated with 5 nM Bafilomycin for 16 h and then processed as above.
Animals
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from Charles River. Female mice 6–12 weeks old (15–20 g weight) were used for experimental procedures. Housing conditions provided 12 hours of dark/light cycle, 22° C+/− 2° C temperature and 55% +/− 5% humidity.
Drug sensitivity in vivo
Mice were transplanted subcutaneously with 2.5 x 106 HCT116 cells in 200 µl of PBS/Matrigel (1:1). Animals were randomized and subjected to drug tests when estimated tumour volume reached ≈ 50 to 150 mm3 (calculated as L*l2/2, where L is the major length and l the minor length). Tumours were measured twice a week during treatment. Mice were distributed into the control and treatment groups (n = 8 per group). The treatment protocols were designed to reflect clinical regimens. In particular, for FOLFOX-like treatment, mice were injected intraperitoneally with 5-fluorouracil (30 mg/kg), oxaliplatin (5 mg/kg) and levofolinate calcium (10 mg/kg) twice a week for two weeks. For FOLFIRI-like treatment, mice were injected (intraperitoneally) with 5-fluorouracil (30 mg/kg), irinotecan (40 mg/kg) and levofolinate calcium (10 mg/kg) twice a week for two weeks. The ULK1 inhibitor SBI-0206965 was administered intraperitoneally at 30 mg/kg dose twice a week. The relative growth for each group was obtained by considering the ratio of tumour volume at the first day of treatment and at the end of the experiment. Tumour growth inhibition (TGI) was calculated according to the following formula:
TGI = relative tumour volume(treated) / relative tumour volume(control) x 100.
Differences among groups were calculated by applying an unpaired Student’s t-test.
Immunofluorescence analysis
HT29 cells were infected to silence PINK1 and an SCR control, plated on slides overnight and treated by carbonyl cyanide 3-chlorophenylhydrazone (CCCP; 25 µM) for 24 h. The next day, cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.5% Triton-X, and blocked for 1 h with 5% bovine serum albumin. The primary antibodies against HNF4A (K9218; Invitrogen) and PINK1 (NovusBio) were incubated for 1 h and 30’ at room temperature and then for 1 h with the secondary antibody. Slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for nuclei labelling and mounted on glass slides with ProLong Gold antifade reagent. Images were collected by TCS SP5 Laser Scanning Confocal Microscope (Leica) × 63 magnification.
For each cell, the number of HNF4A nuclear foci was evaluated using Cell Profiler 4.2.160. In order to count HNF4A foci, the Otsu thresholding method was applied. The pipeline detected cell nuclei in the DAPI channel as objects with a diameter of 65–600 pixels. HNF4A foci smaller than 10 pixels in the nucleus were then identified and mapped to the segmented parental nuclei. Cells with less than five foci per cell were excluded. Differences among groups were calculated by applying ANOVA followed by Tukey post-test.
Proximity ligation assay (PLA)
100’000 HT29 infected to silence PINK1 and a SCR control were plated on slides. The next day, cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.5% Triton-X and then proximity ligation assay was performed following customer instructions (PLA Duolink Merck). Briefly, cells were incubated with the primary antibodies against HNF4A (K9218; Invitrogen) and PINK1 (NovusBio) for 1 h and 30’ at room temperature. Then, incubation with the probes was performed. After ligation and amplification, slides were mounted by using in situ mounting media with DAPI and acquired at 100x magnification by Axio Imager.A2 fluorescence microscope (Zeiss). ImageJ was used to process each image. The PLA puncta were reliably isolated from the background and then counted. Quantification of nuclei proceeds similarly. The results were expressed as PLA puncta/nuclei. Significant differences among groups were calculated by applying a two-sided Student t-test.
Immunoprecipitation
HT29 CRC cells were lysed in a non-denaturating buffer (20 mM Tris HCl pH 8, 137 mM NaCl, 1% Nonidet P-40 (NP-40), 2 mM EDTA) supplemented with a protease and phosphatase inhibitor cocktail. Cell lysates (∼1.2 mg) were incubated with anti-PINK1 Antibody at 4°C overnight, followed by 3 h incubation with protein A beads in rotation. As a negative control, cell lysates were immunoprecipitated with anti-IgG antibody. Immunocomplexes were washed with lysis buffer, boiled in SDS/PAGE sample buffer and analysed by immunoblot analysis as described above.
Histology and immunohistochemistry
Tumour fragments from in vivo experiments were formalin-fixed and paraffin-embedded. After deparaffinization, sections were treated with citrate buffer for 20 min at 95 °C, followed by incubation with 3% hydrogen peroxide in distilled water for 5 min at RT. Sections were stained with monoclonal anti-Ki67 (CST#9027; 1:200) and γH2AX (Ser139; CST#9718; 1:400). Three non-overlapping regions of interest for each tumour section were randomly selected. Then, the total number of positive-staining tumour cells in each image/field and the total number of tumour cells in each image were counted to calculate the Ki67 index and the γH2AX positivity. Significant differences among groups were calculated by applying an ANOVA followed by Bonferroni post-test.
Quantitative RT-PCR
Total RNA was extracted as indicated below and reverse transcribed using ImProm II™ Reverse Transcription System (Promega A3800). Quantitative RT-PCR analyses were done on Viia7 Real-Time PCR System (Applied Biosystems) with the SYBR™ Green PCR Master Mix (Applied Biosystems™ 4309155) as instructed by the manufacturer (Applied Biosystems). The transcription level of the RPLP0 housekeeper gene was used as a normalizer. Complete primer sequences are reported in Supplementary Table 1.
Barcoded library, plasmids, infections and esiRNA
Human non-targeting library (a gift from Prof. Luca Magnani, Imperial College London) was engineered into the pScribe4-EFS-TagRFP-2A-PuropRSI-U6-RFP-2A-Puro lentiviral vector containing the puromycin-resistance and the RFP fluorescent marker. Each barcode cassette (BC) was composed of 148 degenerated, non-overlapping nucleotides. Library infection was performed using a Multiplicity of Infection of ~0.2 TU/cell to obtain about 5'000 barcoded cells. Then, cells were plated to perform chemotherapy drug treatments as indicated above. Barcodes amplification strategy and sequencing are indicated in Supplementary Methods and Supplementary Table 2.
For genetic silencing, HCT116 cells were infected with scrambled shRNA or specific shRNA targeting PINK1 (TRCN0000007101 and TRCN0000199193), ATG2A (TRCN0000172407), ATG5 (TRCN0000151474) and ATG12 (TRCN0000007392) cloned into pLKO vector. Lentiviral particles were added to HCT116 cells, together with 4 μg/mL polybrene (Sigma) for 16 hours. After 48 hours medium was replaced and 2 μg/mL of puromycin was added and maintained along chemotherapy treatment for 5 days. EsiRNA was obtained from Merck for silencing of LUC (EHURLUC), as control, and HNF4A (EHU066741). HNF4A over-expression was obtained by using a barcoded lentiviral vector to express HNF4A under control of EF1a promoter (Addgene plasmid #120450).
DNA amplification strategy for barcode sequencing
DNA for barcode analysis was obtained for the sc-RNAseq experiments performed on the Chromium platform. After ligation step, 1/4 of the total cDNA produced from 10X scRNA library was used as input for specific amplification of Cellecta barcodes, with KAPA HiFi HotStart ReadyMix (Roche), by using the following primers: 5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCCGACCACCGAACGCAACGCACGCA-3’ and ACACTCTTTCCCTACACGACGCTCTTCCGATCT, at the following PCR conditions: 95 °C for 3 minutes, 98°C for 20 seconds, 65°C for 15 seconds, 72 °C for 45 seconds and back to step 2, 13x followed by a 2 minutes hold at 72°C. After a cleanup-SPRI beads selection, the eluted samples were again amplified by using P5 primer 5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGA-3’ and multiplexed with ten different primers indicated in Supplementary Information and obtained from Illumina Inc. using the same PCR conditions and 6x rounds. The product was again purified by cleanup-SPRI beads selection, and the DNA quality checked by Bioanalyzer High Sensitivity analysis (Agilent). After sequencing a Chromium platform (10x Genomics), samples were demultiplexed and barcode abundances analyzed.
Barcodes processing and analysis
FASTQ files for each sample were analyzed using a custom script. First, we checked if the sequence of FBP1 primer (CCGACCACCGAACGCAACGCACGCA) is found at the 5’ allowing for an edit distance equal to 1. Second, we checked if the spacer sequence is found at position 14:18. Lastly, subsequences at position 25:39 and 43:73 are assigned to reference sequences in the CloneTracker barcode catalogue, allowing for an edit distance equal to 1. Raw counts of the final barcode sequences are used for subsequent analysis.
In order to assess whether the tested drugs may be selected for genetically distinct populations, we considered the distribution of the barcode frequencies in every batch, using the timepoints in the control experiment as a baseline.
The rationale is that any significant deviation from the barcode dynamics observed in the control is most likely due to the selection of genetically distinct subpopulations (notice that, since this is not a longitudinal experiment, the barcode pools are different across batches).
Thus, to analyze the distribution of barcodes across the different experiments, we first calculated the Shannon entropy in every batch, with the goal of quantifying the variability of barcode composition: lower values of the entropy indicate that there is a lower number of barcodes with higher frequency, while increasing values indicate a higher number of distinct barcodes characterized by lower frequencies.
The experimental setting was made of 9 batches -- 3 timepoints t, with t = {8 hours, Long Term, Drug Holiday} and 3 therapeutic strategies s, with s = {no drug for the control, FOLFOX and FOLFIRI}, that were used to calculate entropy for each condition.
Next, we computed the normalized frequency of each barcode in each batch, i.e., the number of copies of a given barcode over the total number of copies of all barcodes and, for every time point. A permutation test was performed to compare the distribution of normalized barcode frequencies in the control against the distribution in corresponding time point for either the FOLFOX or the FOLFIRI experiment. The permutation test assesses whether the provided data are sampled from the same distribution via resampling. In detail, we employed the function stats.permutation_test() from the scipy package in Python, setting the mean as the statistic to be computed on the data. Non-significant p-values (p > 0.05) for every test indicate that the barcode frequency distributions in both the FOLFOX and the FOLFIRI experiments at each timepoint are not different from the corresponding in the control.
CUT&Tag analysis
In order to evaluate HNF4A binding to the genome upon PINK1 silencing, we performed a Cleavage Under Targets and Tagmentation (CUT&Tag) assay61. The experiment was performed following the protocol: CUT&Tag-direct with CUTAC V.3 available at https://www.protocols.io62. Briefly, from SCR and shPINK1 HT29 samples and from HepG2 cell line as a positive control, we extracted and slightly fixed nuclei according to the protocol-appendix. A total amount of 100’000 nuclei for each reaction was prepared as starting material. The primary antibody (HNF4α, C11F12, Rabbit mAb #3113, Cell Signalling technology®, 1 µg/mL) incubation was performed overnight and one-hour incubation with the secondary antibody (Guinea Pig anti-Rabbit IgG, ABIN101961, antibodies-online.com) followed. Both immunoreactions were performed in a 100 µL volume. To ensure Tn5 binding, samples were incubated with the CUTANATM pAG-Tn5 (15-1017, Epicypher®, 1:20 dilution) for 2 hours at room temperature in a 50 µL volume. Tagmentation was performed according to the manufacturer guidelines. Sequencing tags were pseudoaligned to hg38 reference genome using chromap63 (parameters: --low-mem --trim-adapters -l 2000 --Tn5-shift --remove-pcr-duplicates -t 8). Peaks were identified from resulting BED files using MACS264. The union of peaks identified in all samples was used to create a fragment count matrix which was then analyzed using edgeR65. We considered peaks having |logFC|≥ 0.65 and p < 0.05 for further analysis.
RNA extraction and sequencing
Total RNA was extracted from cells by using Promega Blood and tissue kit. mRNA purification and NGS libraries were obtained following Illumina instruction (TruSeq RNA Sample Preparation) and sequenced on Illumina Platform (Paired End/2x100; 45 million reads for each sample). Bioinformatic analysis is fully described below.
scRNA-seq and GET-seq
scRNA-seq and scGET-seq were performed as previously described20. Briefly, scRNA-seq was performed on a Chromium platform (10x Genomics) using ‘Chromium Single Cell 3ʹ Reagent Kits v3’ kit manual version CG000183 Rev C (10x Genomics). Final libraries were loaded on a Novaseq6000 platform (Illumina) to obtain 50,000 reads per cell for scRNA-seq and 100,000 reads per nucleus for scGET-seq.
Analysis of bulk RNA-seq data
Read tags were processed using kallisto66 v0.48.0 using gencode v3967 as the gene model. Transcripts were summarized at gene level using txImport68. Differential expression was evaluated by means of linear models implemented in edgeR65. Preranked Gene Set Enrichment Analysis was performed using GSEApy69 using coefficients of linear models as ranking metric.
Functional annotation was performed using DAVID70 on genes selected at BH-corrected p-value lower than 0.01. The resulting terms were filtered for p-value lower than 0.05. To reduce redundancy, we calculated term similarity using GOSemSim71 using Wang similarity metric. Term-to-term similarity was clustered into seven groups, representing major processes at coarse level.
Processing of scRNA-seq data
Cell barcode quantification was performed using UMI-tools72, setting the expected number of cells to 5000. Read tags from 10x Chromium libraries were aligned to hg38 reference genome using STARsolo73 v2.7. Count matrices were imported into scanpy objects74. Doublets were identified with scrublet75 using default parameters. After concatenation of all data samples, predicted doublets, cells with more than 20% counts assigned to mitochondrial genes and cells with less than 2000 genes by counts were removed from the analysis. Counts were normalized and log1p transformed before cell cycle phase assignment. After that, we calculated cell cycle progression value (CC_diff) as G2M_score – S_score. We regressed out CC_diff and %MT before scaling.
We calculated cell kNNgraph using BBKNN76 setting the number of neighbours to half the square root of the number of cells. We applied diffusion maps on the resulting graph before reevaluating the neighbours. Finally, cell clusters were identified using schist77 with default parameters.
Quantification of multiple gene signatures was performed using ‘score_genes’ function in scanpy. DNA damage-related and mitophagy pathways were retrieved from Reactome_2016 gene set available from enrichr78.
CytoTrace score79 was calculated using cellrank80, using the geometric mean for aggregation. Additionally, we evaluated the distribution at all time points of the top 200 genes in the CytoTrace model that have the highest correlation with the overall level of expression.
To compute the cell-to-cell transition matrix we applied the Waddington OT kernel (wk) implemented in cellrank using the experimental time to set the progression. We also applied a connectivity kernel (ck) to stabilize the Schur decomposition. Finally, the transition matrix was calculated using a weighted kernel (0.9wk + 0.1ck). GPCCA method was then used to identify two major cell macrostates.
Processing of scGET-seq data
Raw reads were preprocessed as previously described81 using the scGET workflow82 with default parameters. Each sample consists of two matrices representing read counts for the hg38 genome binned at 5 kbp, one for Tn5 and one for TnH. Once count matrices have been generated, we fitted a zero-inflated Poisson distribution on Tn5 and TnH counts per each cell and generated the empirical distribution of the difference between the two transposases. Using such per-cell distribution, we computed the p-value of the difference Tn5-TnH for each genomic bin in each cell. p-values for each tail were log-transformed as previously detailed83, so that accessible regions are assigned a positive value and compact regions are assigned a negative value.
We removed regions that were not profiled in at least 90% of the cells, we removed cells with a total coverage lower than 2,000. Reduction of dimensionality was achieved by tensor train decomposition (TTD) using tensorly python library84. The first 20 components were used as a basis to compute the cell kNN graph, using BBKNN76 with cosine distance (‘angular’) and a number of neighbours equal to half the square root of the number of cells.
Chromatin Accessibility Score (CAS) was obtained as a fraction of regions with enriched Tn5 over TnH, as result of the signed log(p-value) previously calculated. Values were computed at cell level and evaluated only on the ~50k regions that were retained after filtering in the preprocessing stage. We defined the threshold for compaction as the median value of CAS in untreated cells at early time points (8 h and 16 h).
Analysis of genomic instability
We applied delly85 on aligned TnH reads to retrieve a list of structural variants (SV). As Delly returns the name of reads supporting each SV, we matched read names to single cells to build a count matrix of SV. We applied a linear model fitting the log10-coverage (as the number of TnH reads per cell) with the log(nSV + 1) (as the number of SV per cell). We then calculated the index of genome instability as the square root of the studentized residuals of the fit. Positive values indicate an excess of SV at a given coverage (hence high instability).
Combined analysis of scGET-seq and scRNA-seq data
Single-cell epigenetic and transcriptomic profiles have been collected from the same cell populations, therefore a correspondence between multiple modalities should exist. To integrate the two modalities we implemented a deep learning-based strategy (MultiOmic Wasserstein Generative Adversarial Network, MOWGAN, https://github.com/vgiansanti/MOWGAN86). At the core of our strategy, there is a Generative Adversarial Network (WGAN-GP) that uses Wasserstein loss function and gradient penalty to achieve Lipschitz continuity87.
The WGAN-GP training is performed in mini-batches, where cells are sampled from the whole dataset (i.e., from each molecular layer). Input data for WGAN-GP are the single-cell embeddings (PCA for scRNA and TTD for scGET), clipped to the same number of dimensions. To maximize the conservation of local structure in data during batch training, we sort cells in each modality according to the first component of the Laplacian Eigenmap. The sampled data are combined in a tensor of shape (\(N\), \(M\), \(C\)), where \(N\) is the number of cells in the mini-batch (256), \(M\) the number of modalities evaluated (2, in the case of the scRNA and scGET layers), and \(C\) is the number of components in each embedding used for the analysis. Mini-batches are created sampling from the same sample batches to avoid spurious associations. To couple mini-batches across modalities we select N cells from the scRNA layer, then we select n = 50 random mini-batches from scGET layer and run Bayesian Ridge regression to fit the best coupling. This step is repeated for every mini-batch that is used during training.
The network generator is designed with three convolutional 1D layers (Conv1D) and two batch normalization layers (BN). The network critic is designed with two Conv1D layers and a Dense layer with 1 unit. All Conv1D layers are characterized by a kernel size of \(M\), stride 1 and the ReLU activation function. Finally, different optimizers are used for each component: Adam optimizer for the generator with learning rate = 0.001, beta_1 = 0.5, beta_2 = 0.9, epsilon = 1\(e\) − 07 and the AMSgrad option; RMSprop optimizer with learning rate = 0.0005 for the critic. Once training has been performed, the MOWGAN can generate embeddings for synthetic cells that are artificially paired.
Using MOWGAN we generated 1500 cells for each sample batch. For each modality (scGET and scRNA) we created a single scanpy object for including the synthetic and the original data and applied Harmony88 to perform the integration. The new embeddings produced by Harmony were used to generate the kNN graphs for each modality. We then applied the multilevel Nested Stochastic Block Model77, for which a single kNN is created with labelled edges corresponding to each modality. Cells generated by MOWGAN are represented only once in the integrated graph and serve as anchors for graph analysis.
Population dynamic in vivo
In order to estimate the posterior distributions of the parameters describing the growth pattern of the HCT116 tumours treated with different drugs, a Bayesian model called biPOD was employed.
The model is based on the assumption that the population of cells is either undergoing an unconstrained linear birth-death process, which leads to exponential growth, or a growth pattern in which the environment imposes some constraint on the carrying capacity (i.e. the maximum size of the population). Six measurements of the tumour volume were used as a proxy of the population size, being the volume proportional to the number of cells. All the samples were untreated and evolved naturally up to the second time point, after which the different treatments began. Hence, we modelled the growth pattern as a piecewise evolutionary process, in which the growth parameters change when the treatment starts. The six different samples are merged into a single sample using a mean operation and the biPOD model is applied. The method performs model selection, discerning whether the most probable growth pattern is either exponential or logistic, and then produces the posterior distributions of the parameters of interest via a Markov Chain Monte Carlo sampling procedure. The growth rates ⍴ of the populations under different drugs can be estimated and compared against the control sample.
For all the samples the exponential growth was deemed as the best growth pattern. Hence, we also tested whether the effect of the two drugs combined was either statistically smaller, larger or equivalent to the sum of the effects of the two drugs when used singularly.
Co-expression analysis
Co-expression analysis of PINK1 and RAD51, EXO1, FEN1 and PCNA genes was evaluated in colorectal cancer patients (n = 592), breast invasive carcinoma patients (n = 1082) and in ovarian serous cystadenocarcinoma (n = 300) from TCGA data in cBioPortal dataset (https://www.cbioportal.org/). mRNA expression profiles were quantified considering the z-scores relative to diploid samples with a z-score threshold ± 2. X and Y axis are expressed in logarithmic scale. P values and Spearman correlation (r) are reported for each comparison.
Survival analysis
CRC metadata for survival analysis were retrieved from cBioPortal (coadread_tcga_pan_can_atlas_2018), together with gene expression values as Z-scores compared to normal adjacent tissue. Survival analysis was performed using lifelines python package89. Briefly, we performed Principal Component Analysis on expression values of selected genes upregulated (FDR ≤ 0.01) in HCT116 and annotated for Mitophagy term in Gene Ontology (gene list in Supplementary. Data 4). For each sample, we considered its loading on the first component as signature value. We set a threshold of the median value of the signature as threshold for Kaplan-Meier curves.
Statistical significance test for tolerance vs persistence response
To quantify the possibility that the observed data resembles a tolerant or a persistent response, we analysed the kill curves of the different cancer models under treatments. We assumed that a persistence response would give rise to a biphasic exponential curve due to the sub-population of persister cells that are affected differently by the treatment.
In some cases, the late response cannot be predicted by a single exponential (the null model). To quantify this, the early response of the killing phase was fitted to a 2-parameter linear fit on a semi-log scale. We simulated 1000 of these fits by generating sets of data points according to the mean (\({\mu }_{d}\left(t\right)\)) and standard deviation (\({\sigma }_{d}\left(t\right)\)) measured in the experiment. The mean (\({\mu }_{n}\)) and standard deviation (\({\sigma }_{n}\)) of the late response under the null model were linearly extrapolated from the simulated data and the z-score of the observed data can be calculated:
From here, we can calculate the p-value regarding the probability that the observed data can be predicted by the null model.
Where \({{{\rm{erf}}}}\left(z\right)\) is the standard error function.
Statistical analysis
Data are represented as mean ± SD (if not diversely indicated). Comparisons between two groups were assessed by using two-tailed Student’s t-test, or between more groups by using the one-way ANOVA followed by Dunnett post-test, Bonferroni post-test, Tukey post-test and Sidak’s post-test for multiple comparisons, as indicated in figure legend. P ≤ 0.05 and lower were considered significant. For RNA-seq analysis, statistical parameters are indicated above.
Data availability
The datasets generated in this study have been deposited in the ArrayExpress: for bulk RNA-seq studies E-MTAB-12946, E-MTAB-12948; E-MTAB-12947; E-MTAB-12952; E-MTAB-12953; for scRNA-seq data and barcode tracking E-MTAB-12949; E-MTAB-12950; E-MTAB-12951; for CUT&Tag data E-MTAB-13597. Source data are provided in this paper. All remaining data can be found in the Article, Supplementary and Source Data files. Source data are provided with this paper. Correspondence and requests for materials should be addressed to Giovanni Tonon and Simona Punzi.
Code availability
Code necessary to preprocess scGET-seq data is available at https://github.com/leomorelli/scGET82, https://github.com/dawe/scatACC90 and https://doi.org/10.5281/zenodo.13856260. Code for MOWGAN (v0.1) is available at https://github.com/vgiansanti/MOWGAN91.
References
Bigger, J. W Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 244, 497–500 (1944).
Wright, G. D. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 5, 175–186 (2007).
Brauner, A., Fridman, O., Gefen, O. & Balaban, N. Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 14, 320–330 (2016).
Balaban, N. Q. et al. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 17, 441–448 (2019).
Pu, Y. et al. Drug-tolerant persister cells in cancer: the cutting edges and future directions. Nat. Rev. Clin. Oncol. 20, 799–813 (2023).
Oren, Y. et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 596, 576–582 (2021).
Sharma, S. V. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 (2010).
Shaffer, S. M. et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435 (2017).
Dörr, T., Vulić, M. & Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 8, e1000317 (2010).
Zheng, E. J. et al. Modulating the evolutionary trajectory of tolerance using antibiotics with different metabolic dependencies. Nat. Commun. 13, 2525 (2022).
Sleeboom, J. J. F. et al. The extracellular matrix as hallmark of cancer and metastasis: From biomechanics to therapeutic targets. Sci. Transl. Med. 16, eadg3840 (2024).
Punzi, S. et al. Development of personalized therapeutic strategies by targeting actionable vulnerabilities in metastatic and chemotherapy-resistant breast cancer PDXs. Cells 8, 605 (2019).
Francavilla, C. et al. Phosphoproteomics of primary cells reveals druggable kinase signatures in ovarian cancer. Cell Rep. 18, 3242–3256 (2017).
Egan, D. F. et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell 59, 285–297 (2015).
Li, X., He, S. & Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 19, 12 (2020).
Nagel, Z. D. et al. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc. Natl. Acad. Sci. USA 111, E1823-32 (2014).
Kim, Y. C. & Guan, K.-L. mTOR: a pharmacologic target for autophagy regulation. J. Clin. Invest. 125, 25–32 (2015).
Mauthe, M. et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14, 1435–1455 (2018).
Caravagna, G. et al. Subclonal reconstruction of tumors by using machine learning and population genetics. Nat. Genet. 52, 898–907 (2020).
Tedesco, M. et al. Chromatin velocity reveals epigenetic dynamics by single-cell profiling of heterochromatin and euchromatin. Nat. Biotechnol. 40, 235–244 (2022).
Greaves, M. Evolutionary determinants of cancer. Cancer Discov. 5, 806–821 (2015).
Funnell, T. et al. Single-cell genomic variation induced by mutational processes in cancer. Nature 612, 106–115 (2022).
Rambow, F. et al. Toward minimal residual disease-directed therapy in melanoma. Cell 174, 843–855.e19 (2018).
Marine, J.-C., Dawson, S.-J. & Dawson, M. A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 20, 743–756 (2020).
Marsolier, J. et al. H3K27me3 conditions chemotolerance in triple-negative breast cancer. Nat. Genet 54, 459–468 (2022).
Balsalobre, A. & Drouin, J. Pioneer factors as master regulators of the epigenome and cell fate. Nat. Rev. Mol. Cell Biol. 23, 449–464 (2022).
Tanida, I., Ueno, T. & Uchiyama, Y. A Super-Ecliptic, pHluorin-mKate2, Tandem fluorescent protein-tagged human LC3 for the monitoring of mammalian autophagy. PLoS ONE 9, e110600 (2014).
Hernandez, G. et al. MitoTimer: A novel tool for monitoring mitochondrial turnover. Autophagy 9, 1852–1861 (2013).
Onishi, M., Yamano, K., Sato, M., Matsuda, N. & Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 40, e104705 (2021).
Deas, E., Plun‐Favreau, H. & Wood, N. W. PINK1 function in health and disease. EMBO Mol. Med 1, 152–165 (2009).
Vető, B. et al. The transcriptional activity of hepatocyte nuclear factor 4 alpha is inhibited via phosphorylation by ERK1/2. PLoS ONE 12, e0172020 (2017).
Dörr, T., Lewis, K. & Vulić, M. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 5, e1000760 (2009).
Israel, L. Tumour progression: Random mutations or an integrated survival response to cellular stress conserved from unicellular organisms? J. Theor. Biol. 178, 375–380 (1996).
Lambert, G. et al. An analogy between the evolution of drug resistance in bacterial communities and malignant tissues. Nat. Rev. Cancer 11, 375–382 (2011).
Eliopoulos, A. G., Havaki, S. & Gorgoulis, V. G. DNA damage response and autophagy: a meaningful partnership. Front. Genet. 7, 204 (2016).
Hewitt, G. & Korolchuk, V. I. Repair, reuse, recycle: the expanding role of autophagy in genome maintenance. Trends Cell Biol. 27, 340–351 (2017).
Bryant, K. L. et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med 25, 628–640 (2019).
Tolcher, A. W. et al. A phase 1/2 study of DCC-3116 as a single agent and in combination with trametinib in patients with advanced or metastatic solid tumors with RAS or RAF mutations. JCO 40, TPS3178–TPS3178 (2022).
Hauer, M. H. & Gasser, S. M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 31, 2204–2221 (2017).
Burgess, R. C., Burman, B., Kruhlak, M. J. & Misteli, T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 9, 1703–1717 (2014).
Guler, G. D. et al. Repression of stress-induced LINE-1 expression protects cancer cell subpopulations from lethal drug exposure. Cancer Cell 32, 221–237.e13 (2017).
Kurppa, K. J. et al. Treatment-induced tumor dormancy through YAP-mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell 37, 104–122.e12 (2020).
Knoechel, B. et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat. Genet 46, 364–370 (2014).
Russo, M. et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 366, 1–14 (2019).
Dubois, V., Staels, B., Lefebvre, P., Verzi, M. & Eeckhoute, J. Control of cell identity by the nuclear receptor HNF4 in organ pathophysiology. Cells 9, 2185 (2020).
MacKeigan, J. P., Murphy, L. O. & Blenis, J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat. Cell Biol. 7, 591–600 (2005).
Li, Y. et al. PINK1-mediated mitophagy promotes oxidative phosphorylation and redox homeostasis to induce drug-tolerant persister cancer cells. Cancer Res. 83, 398–413 (2023).
Li, Y. et al. Mitophagy is a novel protective mechanism for drug-tolerant persister (DTP) cancer cells. Autophagy 19, 2618–2619 (2023).
Chen, L. et al. A reinforcing HNF4–SMAD4 feed-forward module stabilizes enterocyte identity. Nat. Genet 51, 777–785 (2019).
Xia, B. et al. Machine learning uncovers cell identity regulator by histone code. Nat. Commun. 11, 2696 (2020).
Stefflova, K. et al. Cooperativity and rapid evolution of cobound transcription factors in closely related mammals. Cell 154, 530–540 (2013).
Ramaker, R. C. et al. Dissecting the regulatory activity and sequence content of loci with exceptional numbers of transcription factor associations. Genome Res. 30, 939–950 (2020).
Puigvert, J. C., Sanjiv, K. & Helleday, T. Targeting DNA repair, DNA metabolism and replication stress as anti-cancer strategies. FEBS J. 283, 232–245 (2016).
Petroni, G., Cantley, L. C., Santambrogio, L., Formenti, S. C. & Galluzzi, L. Radiotherapy as a tool to elicit clinically actionable signalling pathways in cancer. Nat. Rev. Clin. Oncol. 19, 114–131 (2022).
Esposito, M., Ganesan, S. & Kang, Y. Emerging strategies for treating metastasis. Nat. Cancer 2, 258–270 (2021).
Williams, J. A., Zhao, K., Jin, S. & Ding, W.-X. New methods for monitoring mitochondrial biogenesis and mitophagy in vitro and in vivo. Exp. Biol. Med. 242, 781–787 (2017).
Sun, N. et al. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat. Protoc. 12, 1576–1587 (2017).
Piett, C. G., Pecen, T. J., Laverty, D. J. & Nagel, Z. D. Large-scale preparation of fluorescence multiplex host cell reactivation (FM-HCR) reporters. Nat. Protoc. 16, 4265–4298 (2021).
Invrea, F. et al. Synthetic lethality screening highlights colorectal cancer vulnerability to concomitant blockade of NEDD8 and EGFR pathways. Cancers 13, 3805 (2021).
Stirling, D. R. et al. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinforma. 22, 433 (2021).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Henikoff, S., Henikoff, J. & Ahmad, K. CUT&Tag-Direct with CUTAC V3. https://www.protocols.io/view/cut-amp-tag-direct-with-cutac-bqwvmxe6, https://doi.org/10.17504/protocols.io.bqwvmxe6 (2020).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nat. Commun. 12, 6566 (2021).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Harrow, J. et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521 (2016).
Fang, Z., Liu, X. & Peltz, G. GSEApy: a comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 39, btac757 (2023).
Dennis, G. et al. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 4, R60 (2003).
Yu, G. et al. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 26, 976–978 (2010).
Smith, T., Heger, A. & Sudbery, I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017).
Ash Blibaum, Werner, J. & Dobin, A. STARsolo: single-cell RNA-seq analyses beyond gene expression. https://doi.org/10.7490/F1000RESEARCH.1117634.1 (2019).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: Computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019).
Polański, K. et al. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36, 964–965 (2020).
Morelli, L., Giansanti, V. & Cittaro, D. Nested Stochastic Block Models applied to the analysis of single cell data. BMC Bioinforma. 22, 576 (2021).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinforma. 14, 128 (2013).
Gulati, G. S. et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411 (2020).
Lange, M. et al. CellRank for directed single-cell fate mapping. Nat. Methods 19, 159–170 (2022).
Cittaro, D., Lazarević, D., Tonon, G. & Giannese, F. Analyzing genomic and epigenetic profiles in single cells by hybrid transposase (scGET-seq). STAR Protoc. 4, 102176 (2023).
Leomorell. & Cittaro, D. leomorelli/scGET: pre-release of scGET repository. Zenodo https://doi.org/10.5281/ZENODO.5095040 (2021).
de Pretis, S. & Cittaro, D. Dimensionality Reduction and Statistical Modeling of scGET-Seq Data. http://biorxiv.org/lookup/doi/10.1101/2022.06.29.498092, https://doi.org/10.1101/2022.06.29.498092 (2022).
Kossaifi J., Panagakis, Y., Anandkumar, A. & Pantic, M. TensorLy: Tensor Learning in Python. J. Mach. Learn. Res. 20, 1–6 (2019).
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012).
Giansanti, V. et al. Scalable integration of multiomic single-cell data using generative adversarial networks. Bioinformatics 40, btae300 (2024).
Arjovsky, M., Chintala, S. & Bottou, L. Wasserstein Generative Adversarial Networks. In Proceedings of the 34th International Conference on Machine Learning 214–223 (PMLR, 2017).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Davidson-Pilon, C. lifelines: survival analysis in Python. JOSS 4, 1317 (2019).
Cittaro, D. dawe/scatACC: Version 0.1. Zenodo https://doi.org/10.5281/ZENODO.5095157 (2021).
Giansanti, V. et al. Scalable Integration of Multiomic Single Cell Data Using Generative Adversarial Networks. http://biorxiv.org/lookup/doi/10.1101/2023.06.26.546547, https://doi.org/10.1101/2023.06.26.546547 (2023).
Acknowledgements
We thank all the members of the Tonon laboratory for the discussions, support and for critical reading of the manuscript. We are grateful to The Advanced Light and Electron Microscopy BioImaging Centre (ALEMBIC) and The Experimental Imaging Centre (CIS) at Ospedale San Raffaele IRCCS. We also thank Simone Cenci, Tommaso Perini, Luca Magnani, Gioacchino Natoli, Andrea Viale, Davide Gabellini, and Rosa Bernardi for helpful discussions and for reviewing the manuscript. We thank Luca Magnani for scientific discussion and providing reagents. Supplementary Fig. 5a and 8 were created with BioRender.com. This work was supported by a CRUK/AIRC Accelerator grant and AIRC investigator grant (to GT). GC acknowledges funding from the Italian Foundation for Cancer Research (AIRC) under MFAG 2020, ID 24913 project, PI Giulio Caravagna. GG was supported by an AIRC three-years fellowship Guglielmina Lucatello e Gino Mazzega Fellowship for Italy, ID 26728-2021.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
S.P. and G.C. performed in vitro treatments on CRC and BC cells; S.P. and C.B. performed in vitro experiments on OVC; S.P. and O.B. performed experiments on CRC organoids; S.P. performed in vivo experiments; S.P. prepared cells for RNAseq, scRNAseq and GETseq profiling; A.A.C. performed CUT&Tag experiments; I.V. and M.P. assisted with in vitro experiments; F.G. and C.O. prepared libraries and performed sequencing; D.C., V.G. and S.P. analysed data; S.P., G.G. and G.C. flow cytometry and imaging experiments; G.S. and G.C. performed analysis on population dynamic; L.P. and A.G. performed analysis on barcodes; A. Gu. developed modelling for tolerance; M.A. participated to bioinformatic analysis and revised the manuscript; L.L. and U.C. provided BC and OVC cells and revised the manuscript; D.L., Z.N. provided plasmids and reporters; N.B., G.D. and T.A.Y. assisted with the experimental design and reviewed the manuscript; S.P. and G.T. conceived the project and designed the experiments. S.P. and G.T. wrote the manuscript with input from all authors. G.T. supervised all aspects of the work.
Corresponding authors
Ethics declarations
Competing interests
G.T., D.C., and F.G. have submitted a patent application, pending, covering TnH and GET-seq20. The remaining authors declare no competing interests.
Ethic
Investigations have been conducted in accordance with ethical standards and according to National and International guidelines. In vivo studies were performed after approval from our fully authorized animal facility, notification of the experiments to the Ministry of Health (as required by the Italian Law) (IACUC N°1254), and in accordance to EU directive 2010/63. The maximum tumour volume allowed (1500 mm3) was not exceeded. Human tissue biopsies were collected from patients whose informed consent was obtained in writing according to the policies of the Ethics Committee of the San Raffaele Hospital and the regulations of the Italian Ministry of Health. The studies were conducted in full compliance with the Declaration of Helsinki.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Punzi, S., Cittaro, D., Gatti, G. et al. Early tolerance and late persistence as alternative drug responses in cancer. Nat Commun 16, 1291 (2025). https://doi.org/10.1038/s41467-024-54728-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54728-7
This article is cited by
-
Overcoming resistance mechanisms in cancer immunotherapy—novel approaches and combinations
Naunyn-Schmiedeberg's Archives of Pharmacology (2025)
