
Abstract
The structural basis for the allosteric interactions within G protein-coupled receptors (GPCRs) heterodimers remains largely unknown. The metabotropic glutamate (mGlu) receptors are complex dimeric GPCRs important for the fine tuning of many synapses. Heterodimeric mGlu receptors with specific allosteric properties have been identified in the brain. Here we report four cryo-electron microscopy structures of mGlu2-4 heterodimer in different states: an inactive state bound to antagonists, two intermediate states bound to either mGlu2 or mGlu4 agonist only and an active state bound to both glutamate and a mGlu4 positive allosteric modulator (PAM) in complex with Gi protein. In addition to revealing a unique PAM binding pocket among mGlu receptors, our data bring important information for the asymmetric activation of mGlu heterodimers. First, we show that agonist binding to a single subunit in the extracellular domain is not sufficient to stabilize an active dimer conformation. Single-molecule FRET data show that the monoliganded mGlu2-4 can be found in both intermediate states and an active one. Second, we provide a detailed view of the asymmetric interface in seven-transmembrane (7TM) domains and identified key residues within the mGlu2 7TM that limits its activation leaving mGlu4 as the only subunit activating G proteins.
Similar content being viewed by others


Introduction
G protein-coupled receptor (GPCR) heterodimers are allosteric machines allowing the control of one subunit by the other1,2. Among the large GPCR family, metabotropic glutamate (mGlu) receptors form constitutive dimers, activated by the excitatory neurotransmitter glutamate and play important roles in modulating synaptic transmission and neuronal excitability3,4. These receptors are categorized into three groups, namely group I (mGlu1 and mGlu5), group II (mGlu2 and mGlu3) and group III (mGlu4, mGlu6, mGlu7 and mGlu8)3,4. They form either homodimers or intra-group heterodimers such as mGlu1-5, as well as inter-group heterodimers between group II and III such as mGlu2-4 and mGlu2-75. Interestingly, mGlu4 predominantly forms heterodimers in the forebrain, revealing novel opportunities for controlling brain functions6. G proteins are activated by one subunit only in both mGlu homo- and heterodimers7,8,9,10,11. Although mGlu2 homodimers perfectly activate G proteins, only the mGlu4 and mGlu7 subunits interact with and activate G protein in the mGlu2-4 and mGlu2-7 heterodimers7,8,9,10,11,12, respectively. Surprisingly, the G protein coupling efficacy of mGlu48 and mGlu79,13 is much higher when associated with mGlu2 than with themselves. How such exclusive asymmetry is structurally controlled remains unknown.
The mGlu subunits consists of a bilobate Venus flytrap (VFT) domain that contains the orthosteric binding site interconnected through a cysteine-rich domain (CRD) to a seven-transmembrane domain (7TM)4. Agonists bind to the upper and lower lobes of VFTs, stabilizing their closed (c) state14. The closure of both VFTs results in a reorientation of ~70° of the VFTs leading to the active closed-closed (Acc) state of the VFT dimer14,15. Such movement brings the CRDs into close proximity, leading to a relative rotation of the 7TM domains, reaching a TM6-TM6 interface and a specific contact between the CRDs and the extracellular loop 2 (ECL2)9,10,11,16,17. This state allows a slight conformational change in one 7TM domain only, enabling G protein activation10,11. Although such an asymmetric active conformation is well identified in mGlu homodimers10,11, how the oriented asymmetry is controlled in heterodimeric receptors remains unknown.
Here, we show the structural basis for the oriented asymmetric activation of the mGlu2-4 heterodimer. We present the cryo-electron microscopy (cryo-EM) structures of this heterodimer in four different conformational states, including the active G protein bound complex. In association with the functional analysis of several mutants and single-molecule FRET data, our study helps elucidate the allosteric interactions between the various domains of this mGlu heterodimer leading to its oriented asymmetric activation. In addition to the description of a unique positive allosteric modulator (PAM) site in the 7TM domain of mGlu4, our data explain why agonist occupancy of a single binding domain partially activates this heterodimer. We also provide an explanation why the mGlu4 7TM is preferentially activated upon the formation of an asymmetric dimer. Our study will help design ways to specifically modulate such heterodimer to better identify their function.
Results
Structure determination of mGlu2-4 heterodimer in different states
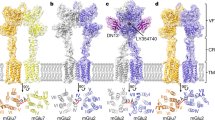
To facilitate structural investigations of the mGlu2-4 heterodimer, we integrated a LgBiT tag and a HiBiT tag at the C-termini of mGlu2 and mGlu4 receptors, respectively, with the purpose of enhancing the stability of their heterodimeric formation. This pair of fusion tags forms a stable complex and has already been used to stabilize GPCR-G protein complexes18. The human mGlu2-HiBiT and mGlu4-LgBiT were co-expressed and purified in full-length. Here, we determined four structures of the mGlu2-4 heterodimer (Fig. 1, Supplementary Table 1) by single-particle cryo-EM. The structure of fully inactive state was determined using two antagonists, LY34149519 and CPPG20 (Fig. 1a, Supplementary Fig. 1a–c). In an attempt to stabilize one specific VFT of mGlu2-4 in a closed state, we employed the selective agonist LY37926821 for mGlu2 or the selective agonist L-AP422 for mGlu4, respectively (Supplementary Fig. 1d–i). Cryo-EM data analysis revealed two substantially different conformations of monoliganded mGlu2-4 heterodimer, with the population distribution of 25.2% (intermediate state I) and 74.8% (intermediate state III) in the mGlu2 agonist bound state, or 55.4% (intermediate state II), 34.2% (intermediate state IV) and 10.4% other state in the mGlu4 agonist bound state (Fig. 1b, c, Supplementary Fig. 2). Among these four intermediate states, intermediate state I with bound mGlu2 agonist (Fig. 1b), and intermediate state II with bound mGlu4 agonist (Fig. 1c) could be solved at a good resolution (5.95 Å and 4.46 Å, respectively). This enabled unambiguous domain-wise docking of the main chains of mGlu2-4 and ligands for these two intermediate states. We further assembled the mGlu2-4 and Gi protein complex in presence of the endogenous ligand glutamate (Glu) occupying both VFTs and the mGlu4 PAM VU015504123 (Fig. 1d, Supplementary Fig. 1j–l). By performing local refinement separately on the ECD, 7TM and Gi protein, we got 3D reconstructions at resolutions of 3.5 Å, 3.75 Å and 3.3 Å, for these three domains respectively (Supplementary Fig. 3). High-quality density map for mGlu2-4G complex, where the G protein is coupled to mGlu4 7TM, allows the unambiguous placement of agonist, PAM, most side-chain amino acids of the receptor including all the ECLs and intracellular loops (ICLs) except ICL2 of mGlu2, and the heterotrimeric Gi protein (Fig. 1d, Supplementary Fig. 4).

a Structural model for the mGlu2-4 heterodimer in inactive state with two potent competitive antagonists of the group-II and -III mGlu receptors, LY341495 and CPPG. Structural models for mGlu2-4 heterodimer in two intermediate states with mGlu2 agonist LY379268 bound to mGlu2 subunit (b) or with mGlu4 agonist L-AP4 bound to mGlu4 subunit (c). d Structural model for mGlu2-4 heterodimer in the active state in complex with heterotrimeric Gi3 protein and in the presence of Glu and the mGlu4 PAM VU0155041, where two molecules of Glu stabilize the close state of both mGlu2 and mGlu4 VFTs.
A unique PAM binding site in the mGlu4 7TM
VU0155041 acts as a PAM for both mGlu4-4 homodimer and mGlu2-4 heterodimer23,24. Inside the allosteric pocket, VU0155041 is clearly observed in the structure in contact with residues from TM3, TM5, TM6, TM7 and ECL2 (Fig. 2a, b). The conformation of VU0155041 was also confirmed by molecular dynamics simulations (Supplementary Figs. 5-7), which supports the orientation of VU0155041 shown in our structure. The binding pocket of VU0155041 exhibits spatial proximity to the ECL2, distinguishing it from all PAMs and NAMs of other class C GPCRs, only the lower part of this site overlapping those of FITM and cinacalcet in mGlu1 and CaSR, respectively (Fig. 2a, Supplementary Fig. 8a–h). The chemical structure of VU0155041 is composed of the dichlorophenyl side and the carbamoyl group23, that interact with the protein along two parts (Fig. 2b, Supplementary Fig. 8i). In Part I, the dichlorophenyl side towards TM3 and TM5 is involved in hydrophobic interactions with M6432.56, R6553.32, R6563.33, D7505.37 and L7535.40 (superscript numbers refer to the GPCRdb numbering scheme). In Part II, the carbamoyl group directly adjacent to ECL2 establishes direct hydrophobic interactions with I748ECL2, F8056.57, T8197.29 and L8227.32, thereby modulating the structural arrangement and stabilization of the TMD-ECL2 region. In addition, there is a potential salt bridge between the Cyclohexane-1-carboxylic acid and K745ECL2. This binding mode is supported by the mutagenesis and functional analyses. Indeed, the R6553.32A, R6563.33A, C746ECL2A, I748ECL2A, D7505.37A, L7535.40A, K745ECL2E, and F8056.57A, substitutions abolished the potentiation effect of VU0155041 on Glu potency in mGlu2-4 measured by G protein trimer rearrangement in BRET assay (Fig. 2c, Supplementary Fig. 9a–j) and significantly decreased the VU0155041 potency (Supplementary Fig. 9k).

a Comparison of the PAM binding pocket in mGlu4 and mGlu2 subunit. The mGlu4 PAM VU0155041 is shown as magenta sticks. The mGlu2 PAM JNJ-40411813 is shown as yellow sticks. b Detailed binding mode of VU0155041 in mGlu4 subunit. c Potency (pEC50) of Glu with or without VU0155041 on mGlu2-4 chimeric heterodimer (composed of mGlu2C1 and mGlu4C2, referred as Ctr) and the indicated mutants. Data points represent mean ± s.e.m. from independent experiments (n = 3, 5, 4, 3, 3, 4, 4, 4, 4) and analyzed using unpaired student t test (two-tailed) to determine significance between pEC50 of Glu without and with VU0155041 treatment for each receptor. ***P < 0.001, not significant (ns) >0.05. P values for (c): 0.0042, 0.5357, 0.3131, 0.1191, 0.6036, 0.6994, 0.0884, 0.3710, 0.2541 (from left to right). Source data are provided as a Source Data file.
Inter-subunit rearrangement of 7TMs in mGlu2-4 heterodimer
Structural analysis suggests that the transmembrane domains of the mGlu2-4 dimer rearranged from TM5-TM5 interaction in inactive state to TM6-TM6 or TM6-TM7 interaction in active state (Fig. 3a, b). To further explore these conformational rearrangements associated with mGlu2-4, activation, we used cysteine crosslinking between selected mutants, at the surface of live cells. We used a quality control system to allow cell surface targeting of the mGlu2-4 heterodimer only8,25, by introducing complementary coiled-coil regions (C1 and C2) derived from the GABAB receptor and intracellular retention signals in engineered chimeric mGlu2 and mGlu4 subunits (mGlu2C1 and mGlu4C2) (Supplementary Fig. 10a). To examine any possible Cys cross-linking between the subunits, the cysteines at the top of VFT of mGlu2C1 and mGlu4C2 (mGlu2C1C121A and mGlu4C2C136A) were substituted to prevent this covalent natural link between the subunits (Fig. 3c). One endogenous cysteine in mGlu4 (C7154.48) was also mutated to alanine to avoid possible unspecific cross-linking. To detect the crosslinked heterodimer in blots, SNAP-tagged mGlu2C1 and Halo-tagged mGlu4C2 were used to label specifically the cell surface mGlu2-4 heterodimers using non cell-permeate fluorescent substrates26,27,28. We showed that mGlu2C1C121A and mGlu4C2C136A-C715A mutants with cysteine substitution in TM4, TM5 and TM6 could be symmetrically cross-linked in reducing conditions with CuP treatment under non-stimulated conditions, further indicating that the dimerization interface involves TM5 and TM6, and also TM4 from both subunits (Supplementary Fig. 10b–e). Occupancy of both binding sites with glutamate clearly decreased TM4-TM4 and TM5-TM5 cross-linking, while TM6-TM6 cross-linking was enhanced (Supplementary Fig. 10d) indicating that the interface of mGlu2-4 likely shifts from TM4-TM5 to TM6 upon activation. As the TM4-TM4 interface is shown in our two intermediate mGlu2-4 structures with bound LY379268 or L-AP4 (intermediate states I and II, Fig. 1b, c), it suggests that these structures do not correspond to an active state. An additional cross-linking between TM7 of mGlu2 and TM6 of mGlu4 (with mGlu2-V7897.27C/mGlu4-P8036.55C and mGlu2-T7937.31C/mGlu4-P8036.55C) can also be detected after glutamate application (Supplementary Fig. 10d), consistent with our structure of the mGlu2-4G complex. In agreement with this asymmetric interaction being associated with the active state, this mGlu2-TM7 and mGlu4-TM6 cross-linked heterodimer clearly displays a high constitutive activity, although lower than the agonist-induced one (Fig. 3d). This partial constitutive activity lower than the agonist-induced one resulting from the crosslinking might be due to a fraction of the dimers not being crosslinked.

The interface of the 7TM domains of the mGlu2-4 heterodimer in inactive (a) and active (b) state. c Schematic indication of the constructs used in (d). Ctr, mGlu2C1-mGlu4C2 heterodimer. Ctr-CA, Ctr with C121A mutation in mGlu2 and the two C136A and C715A mutations in mGlu4. Ctr-CA+T7937.31C+P8036.55C, Ctr-CA with one additional Cys mutation at residue 793 of mGlu2 and one at residue 803 of mGlu4. d IP1 production in Mock cells, cells transfected with Ctr-CA, or Ctr-CA+T7937.31C+P8036.55C together with Gqi9 and treated with CuP, and without or with Glu treatment. Data points represent mean ± s.e.m. from four independent biological replicates each performed in triplicates and analyzed using one-way ANOVA with Dunnett’s multiple comparison test to determine significance. **P < 0.01, *P < 0.05. P values for (d): 0.0037, 0.0286 (from left to right). Source data are provided as a Source Data file.
To further determine the mechanism of the asymmetric activation of G protein through the mGlu4 subunit in the mGlu2-4 heterodimer, we compared the mGlu4 subunit in the mGlu2-4G complex with the mGlu4 subunit without G protein bound (Gfree) in the mGlu4-4 homodimer (PDB: 8JD6)10 (Supplementary Fig. 11a). The upper end of TM6 of mGlu4 in mGlu2-4G further moved away from mGlu2 compared with Gfree mGlu4, to avoid TM6-TM6 clash in mGlu2-4G (Supplementary Fig. 11a). Such a movement may favor the TM6-TM6 interaction allowing activation of the mGlu4 7TM domain. When we use the mGlu2 subunit with G protein bound (Gbound mGlu2) from the mGlu2G-4 complex (PDB: 8JD5)12 and superimpose it with the mGlu2 subunit in the mGlu2-4G complex, the two TM6 are very close which generates potential steric hindrance preventing a correct TM6-TM6 interface leading to G protein activation (Supplementary Fig. 11b). Such observation explains how the interface of the mGlu2-4 heterodimer allows the allosteric control of one subunit by the other, favoring the activation of mGlu4 7TM in the mGlu2-4 heterodimer8.
Structural basis of the partial activation of mGlu2-4 heterodimer
How can a symmetric closure of the dimeric VFT lead to a fully asymmetric coupling of G proteins by the 7TM dimer? Indeed, a very precise association of the CRDs is required to bring both 7TMs in a correct position for their activation as well illustrated by several structures of the activated states of mGlus such as mGlu2-2 and mGlu4-4 homodimers in complex with G proteins10. However, it remains unclear whether and how the asymmetrical closure of one VFT within a dimer leads to partial receptor activation as observed in either mGlu homodimers or heterodimers25,27,29. In the LY379268-bound mGlu2-4 structure, the mGlu2 VFT is in a closed state, but that of mGlu4 remains in an open state, and the dimer resides in a resting orientation (Rc2o4 state) (Fig. 4a). Similarly, and in a symmetric fashion, in the L-AP4-bound mGlu2-4 structure, the mGlu4 VFT is closed, while that of mGlu2 is open and the dimer is still in the resting state (Ro2c4 state) (Fig. 4b). Nevertheless, despite these differences in the VFT dimer, in both structures, the CRD of mGlu2 underwent bending and moved closer to mGlu4, while the CRD of mGlu4 remains unchanged, when compared to the inactivate state. Meanwhile, a TM4-TM4 interface was observed and validated (Supplementary Fig. 12a, b, Supplementary Fig. 10d) in these two solved states, a conformation unlikely to generate a signal, as all class C receptor-G protein complexes revealed a TM6-TM6 interface30. These data suggest that an allosteric modulation leading to a partial bending of mGlu2 CRD is not sufficient to rearrange the TMDs into the active TM6-TM6 interface. Note that for each complex we also observed another state with no bending of the mGlu2 CRD (intermediate states III and IV, Fig. 1b, c), but where a symmetrical TM4/5 interface is formed between mGlu2 and mGlu4 (Supplementary Fig. 12a, b).

Side views of the superimposed structures of mGlu2-4 heterodimer in inactive state with two intermediate states, either mGlu2 agonist-bound intermediate state I (a) or mGlu4 agonist-bound intermediate state II (b). Single molecule FRET histograms of mGlu2-4 reorientation sensor in the absence of ligand (Apo) or in the presence of 10 μM L-AP4, 1 μM LY379268 or 10 mM L-Glu without G protein (c) or with G protein (d). FRET histograms show the apparent FRET efficiency (EPR) as the mean ± s.e.m of three independent biological replicates. Histograms display the fitting with four gaussians (white (1) = very low FRET, orange = low FRET, grey = high FRET, white (2) = very high FRET) together with the global fit (black, dotted). The mean high and low FRET efficiencies of the Apo and L-Glu conditions are indicated by dashed lines for comparison. e Fraction of molecules in the active low FRET state without or with G protein. Shown are the means ± SD of at least three independent biological replicates obtained from the number of molecules found in the low FRET population (orange) over the sum of all molecules found in the low FRET (orange) and high FRET (grey) populations. Data were analyzed using one-way ANOVA with a Tukey’s post-hoc multiple comparison test to determine significance. ***P < 0.001, *P < 0.05, not significant (ns) >0.05. P values for (e): Apo no Gi vs Apo + Gi: 0.2319, Apo no Gi vs L-AP4 no Gi: 0.0005, L-AP4 no Gi vs LY379268 no Gi: 0.0346, L-AP4 + Gi vs LY379268 + Gi: 0.1843. Source data are provided as a Source Data file.
Although these different states are unlikely able to activate G proteins, even partially, the mGlu2-4 heterodimer does partially signal when activated by either an mGlu2 or an mGlu4 selective agonists29. Such partial activity is not inhibited by locking the associated agonist-free subunit in the open state with an antagonist, demonstrating that the partial activity clearly results from a closed-open state of the VFT dimer. This is observed either with the mGlu2 agonist LY379268 and the mGlu4 antagonist MSOP (Supplementary Fig. 12c), or with the mGlu4 agonist L-AP4 and the mGlu2 antagonist LY341495 (Supplementary Fig. 12d). Such a conclusion is further supported by an mGlu2-4 heterodimer carrying two mutations (SATA)25 in either the mGlu2 or the mGlu4 binding site, largely decreasing glutamate affinity. Whether the mutation is in mGlu2 or in mGlu4, glutamate generates only a partial response compared to the wild-type dimer, while the receptor activity is abolished in the case of the double mutation (Supplementary Fig. 12e–i).
In order to better understand the partial activation of the mGlu2-4 receptor occupied by a single agonist, we performed single molecule FRET (smFRET) experiments monitoring the reorientation of the VFT dimer, from the R to the A state, as previously reported14,31,32. Receptors were produced and solubilized as previously described31,32, and smFRET acquisitions were performed in detergent micelles with 0.0025% LMNG (w/v), 0.00025% CHS tris (w/v), and 0.0025% GDN (w/v). Under these conditions, the receptor is fully functional and sensitive to allosteric modulators31,32. Receptors were labeled with d2 on a SNAP-Tag29 on the N-terminus of the mGlu4 subunit, and with Cy3B-tetrazine on a trans-Cyclooct-2-en-L-Lysine (TCO*A) incorporated at position 358 of mGlu233. This FRET sensors reports on the reorientation of the VFT dimer from the resting to the active state as a decrease in FRET, similar to our previously used SNAP/SNAP and ncAA/ncAA sensors for mGlu2 homodimers29,31,32,34.
The smFRET histograms show a multimodal distribution (Fig. 4c), indicating the co-existence of several conformational states. We fitted all distributions with four Gaussians (corresponding to the very low FRET (VLF), the low FRET (LF), the high FRET (HF) and the very high FRET (VHF) populations). The major changes in response to ligands were found to result from the depopulation of the HF state accompanied by an increase of the LF state, reporting on the reorientation of the VFT dimer toward the active state. To gain a quantitative view of this reorientation, we calculated the fraction of active molecules, defined as the fractional amplitude of the molecules found in the LF state relative to the HF + LF states. As no notable changes were observed in the two minor populations at VLF, E ~ 0.02 and VHF, E ~ 0.88, (Supplementary Table 2, Supplementary Fig. 13a, b) that are only observed with SNAP tagged subunits32, these were not included in the analysis. We nevertheless verified that calculating the fraction of the active molecules as VLF + LF relative to all molecules led to similar results. In the absence of ligand, a main high FRET population is centered at 0.56 and shifts to 0.3 in the presence of Glu (Fig. 4c, Supplementary Table 3). LY379268 or L-AP4 used at a concentration that selectively activate the mGlu2 or mGlu4 subunit, respectively (1 µM and 10 µM, respectively) (Supplementary Fig. 13c–e) applied alone are also able to increase the population of this low FRET active state, but not as efficiently as glutamate (Fig. 4c), consistent with the partial activation of the mGlu2-4 heterodimer by these selective agonists (Supplementary Fig. 14). This observation is similar to what we obtained on mGlu2 homodimers bound by partial agonists32, and indicates a partial activation of mGlu2-4. In the presence of the Gi protein heterotrimer, the fraction of receptors in the active state (LF state) in the presence of either LY379268 or L-AP4 largely increases (Fig. 4d, e, Supplementary Table 3), consistent with the G protein stabilizing the active conformation of the monoliganded mGlu2-4 heterodimer.
These data together suggest that the closure of only one VFT can shift the VFT dimer conformation equilibrium towards an active orientation at least for a proportion of mGlu2-4 dimers, then producing partial activity. These active forms of the heterodimer were not observed under the experimental conditions used for cryo-EM.
CRD-ECL conformational change of mGlu2-4 heterodimer upon activation
It has been proposed that the ECL2-CRD interaction of class C GPCRs plays an important role in signal transmission upon activation9,10,16,17. The interaction between ECL2 and CRD in mGlu4 is mediated by an hydrogen bond formed by D563CRD and Q730ECL2 and an ionic bond formed by E568CRD and R738ECL2 (Fig. 5a). In contrast, only the E555CRD and Q558CRD form hydrogen bonds with K707ECL2 in mGlu2. Functional data show that substitutions of these residues in either mGlu4 or mGlu2 to alanine substantially reduced by 26–40% of the maximal Gi activation (Emax) induced by glutamate in the heterodimer (Supplementary Fig. 15a–h, Supplementary Table 4). Signaling of the heterodimer induced by the mGlu4 agonist is also decreased in heterodimers carrying the CRD mutation in either mGlu2 (Fig. 5b, Supplementary Fig. 15i, j, Supplementary Table 4) or mGlu4 (Fig. 5c, Supplementary Fig. 15k, l, Supplementary Table 4) (18–60%). However, although the signal induced by the mGlu2 agonist LY379268 is also decreased in the mGlu2-4 mutated in the mGlu2 CRD by 40–45% (Fig. 5b), it is increased by 37% if the mGlu4 carries the CRD mutation D563A-Q730A (Fig. 5c). This suggests that the mGlu2 subunit activates mGlu4 more easily when its CRD-ECL2 contact is weakened. These data reveal the critical role of the CRD-ELC2 contact in controlling mGlu2-4 heterodimer activation.

a Critical residues involved in the interaction between CRD and ECL2 in mGlu2 subunit (left) and mGlu4 subunit (right) in the mGlu2-4G complex. L-AP4 (left) and LY379268 (right) induced G protein activation in mGlu2C1-mGlu4C2 heterodimer (Ctr) without or with the indicated mutations in the CRD and ECL2 of mGlu2 subunit (b) or mGlu4 (c) subunit. Data are normalized by using the maximum response (Emax) of mGlu2-4 Ctr and present as mean ± s.e.m. from at least three independent biological replicates, each performed in triplicate. The indicated number of independent experiments (n): (b), Ctr, E555A-K707A, Q558A-K707A, left: n = 3, 3, 3; right: n = 5, 5, 4; (c), Ctr, E568A-R738A, D563A-Q730A, left: n = 7, 5, 5; right: n = 8, 4, 5. The inserted graphs correspond to representative Emax and analyzed using one-way ANOVA with a Dunnett’s post-hoc multiple comparison test compared with Ctr to determine significance. ****P < 0.0001, ***P < 0.001, **P < 0.01, not significant (ns) >0.05. P values for (b): 0.0015, 0.0030, 0.0003, 0.0004; for (c), 0.0050, <0.0001, 0.0039, 0.8085 (from left to right). Source data are provided as a Source Data file.
Intra-subunit conformational change of mGlu2-4 heterodimer upon activation
When superimposing mGlu4 subunits from mGlu2-4G (this study) and mGlu2G-4 (PDB: 8JD5)12, intra-subunit conformational changes at the intracellular ends of TM3 and TM5 of mGlu4 can be observed (Fig. 6a). A conserved amino acid, Y7926.44 in TM6 of mGlu4, undergoes considerable rotation upon binding to the G protein, transitioning from interaction with I7725.59 in the Gfree state to participating in stabilizing the interface in the Gbound state, thereby facilitating the release of TM5. As a result, the bottom of TM5 in mGlu4 deflects by 3.6 Å towards TM3, while Y6783.55 on TM3 rotates by 12° away from TM5 to avoid steric hindrance (Fig. 6b). The conformational changes observed in mGlu2 subunits before and after coupling to the G protein are similar to mGlu4 (Fig. 6b), but the corresponding key residues 3.55 and 5.59 are not conserved between different mGlu groups, contrary to Y6.44 (Supplementary Fig. 16a). In the TM3-5-6 network of the mGlu2 subunit, Y7676.44 and F7475.59 contribute to a robust contact between TM5 and TM6, while A6583.55 on TM3 causes minimal steric hindrance, resulting in a 2.5 Å shift at the bottom of TM5 and an angle of only 10° relative to TM5 (Fig. 6b). Moreover, when comparing heterodimers and homodimers, we observe that both mGlu2 and mGlu4 subunits undergo identical conformational changes when coupled to the G protein (Supplementary Fig. 16b–d). Conserved intra-subunit conformational changes have also been observed in GABAB heterodimers35,36 (Supplementary Fig. 16f), suggesting the preservation of the TM activation mechanism in class C GPCRs.

a Comparison of the conformational changes within mGlu4 7TM between mGlu4 subunit in mGlu2-4G complex and mGlu4 subunit in mGlu2G-4 complex (PDB: 8JD5). Left: Side view. Right: magnified view. b Comparison of the conformational changes within mGlu2 7TM between mGlu4 subunit in mGlu2-4G complex and mGlu4 subunit in mGlu2G-4 complex (PDB: 8JD5). Left: Side view. Right: magnified view. c, d Glu-induced G protein activation in mGlu2C1-mGlu4C2 heterodimer (Ctr), mGlu2-2 (WT) or mGlu4-4 homodimer (WT), without or with the indicated mutations in TM3, TM5 and TM6. Data are normalized by using the maximum response (Emax) of Ctr or indicated WT and present as mean ± s.e.m. from at least three independent biological replicates, each performed in triplicate. The indicated number of independent experiments (n): (c), left: Ctr, Y6783.56A, I7725.59A, Y7926.44A, n = 11, 5, 3, 4; right: WT, Y6783.56A, I7725.59A, Y7926.44A, n = 4, 4, 3, 4; (d), left: Ctr, A6583.55Y, F7475.59A, Y7676.44A, n = 7, 4, 5, 4; right: WT, A6583.55Y, F7475.59A, Y7676.44A, n = 9, 7, 4, 6. The inserted graphs correspond to representative Emax and analyzed using one-way ANOVA with a Dunnett’s post-hoc multiple comparison test compared with Ctr or WT to determine significance. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, not significant (ns) >0.05. P values for (c): 0.0080, 0.5383, 0.0304, <0.0001, 0.9794, 0.0001; for (d), 0.9997, 0.2513, 0.9758, <0.0001, 0.0048, 0.0083 (from left to right). Source data are provided as a Source Data file.
To further illustrate the influence of these pockets formed by residues in 3.55, 5.59, 6.44, we generated mutations in both mGlu2C1 and mGlu4C2 constructs, replacing these residues with an alanine (or with a tyrosine if already an alanine). The mutations Y6763.55A and Y7926.44A in mGlu4 decreased glutamate-induced mGlu2-4 activation, while the mutation in I7725.59A had no effect (Fig. 6c, Supplementary Fig. 17a, Supplementary Table 4). Similar, but stronger effects can be observed in mGlu4-4 homodimer relative to the heterodimer (Fig. 6c, Supplementary Fig. 17b, Supplementary Table 4) (3.55: 71.9% vs 60.3%, 6.44: 68.4% vs 23.4%). Despite the fact that mutations of the equivalent residues in mGlu2 (A6583.55Y, F7475.59A and Y7676.44A) strongly increased mGlu2-2 homodimer activation to around 1.5–2 folds, almost no effect was observed in the mGlu2-4 heterodimer (Fig. 6d, Supplementary Fig. 17c, d, Supplementary Table 4), consistent with the mGlu4 subunit being responsible for signaling in this heterodimer.
We then examined which of the subunit is indeed responsible for the G protein activation by the heterodimer. For this we took advantage of a mutation of a conserved residue (F781 in mGlu4) in the ICL3 known to prevent G protein activation in mGluRs8,10,37. In agreement with mGlu4 being responsible for Gi and Go activation in the heterodimer, the mGlu2-4 heterodimer carrying the F781S mutation in mGlu4 only is not functional (Supplementary Fig. 17e, f). When we co-express the mGlu2C1-A6583.55Y and mGlu4C2-FS, the activity of mGlu2-4 heterodimer was recovered, which can be further enhanced with the mGlu2 PAM LY487379 (Supplementary Fig. 17g–i), demonstrating a switch from mGlu4 to mGlu2 for G protein activation by this heterodimer. This suggests that the bulky tyrosine in TM3 may create clashes with TM5 in mGlu2, which then favors the active state of this domain.
Coupling of Gi through ICLs in mGlu2-4 heterodimer
The G protein binding pocket is formed by the intracellular ends of TM3 and three ICLs of the mGlu4 subunit (Supplementary Fig. 18a). The structure of the mGlu4 Gbound complex in the mGlu4-4G homodimer10 and in the mGlu2-4G heterodimer are very similar. Indeed, the only minor difference is a downward deflection of the αN helix of the G protein in the heterodimer (Fig. 7a). In our mGlu2-4G complex, the ICL2 and TM3 interact with the α5 and αN helices, squeezing in between the helices to reach the β2-β3 linker of the Gαi protein (Fig. 7b). The ICL1 and ICL3 of the mGlu4 subunit and the C-terminal ‘hook-like’ region of the Gi protein form a hydrophobic interaction, involving K619ICL1, A620ICL1 and F781ICL3 of mGlu4, and I343 and L353 of the Gαi protein (Fig. 7c). These interactions are supported by our functional data, as the mutation of these residues in either the receptor or the G protein, resulted in decreased potency or Emax of Glu (Fig. 6d, e, Supplementary Fig. 18b–h, Supplementary Table 4).

a Comparison of the Gi binding pocket between mGlu2-4G complex and the mGlu4-4G (PDB: 8JD6). Detailed interactions in mGlu2-4G structure of mGlu4 ICL2 and the intracellular end of TM3 with Gαi3 (b), mGlu4 ICL1 and ICL3 with Gαi3 (c), respectively. d Potency (pEC50) of Glu in mGlu2-4 Ctr and indicated mutants measured by IP1 assay. e Change of IP1 production (referred as Span) in mGlu2-4 Ctr and indicated mutants. Data in (d) and (e) are present as mean ± s.e.m. from at least three independent biological replicates, each performed in triplicate. The indicated number of independent experiments (n): (d), n = 10, 4, 4, 4, 3, 3, 4, 3, 3, 4, 4, 3, 3, 3; (e), n = 10, 4, 5, 5, 3, 4, 4, 3, 3, 5, 4, 3, 5, 3. Data are analyzed using one-way ANOVA with a Dunnett’s post-hoc multiple comparison test compared with Ctr to determine significance. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, not significant (ns) >0.05. nd, not determined (data for which a robust concentration response curve or plateau could not be established within the concentration range tested). P values for (d): 0.9266, 0.1049; for (e): 0.0138, 0.0006, 0.0004, 0.8300, 0.1592, 0.0134, 0.0039, 0.0284, <0.0001, 0.0008, 0.0076, 0.0001, 0.0187 (from left to right). Source data are provided as a Source Data file.
The ICLs of mGlu play a critical role in G protein binding and activation, particularly ICL2, which contributes significantly to this interaction10,12. While the overall conformation of the receptor and G protein remains similar to the recently resolved mGlu2G-4 structure12, there are changes in the specific residues involved and their surface area (Fig. 8a, Supplementary Fig. 18i, j). The apex of ICL2 protrudes more prominently in the latter case, and these structures are in closer proximity to Gαi, resulting in additional interactions with the α5-αN pocket (Fig. 8a). When we mutated G667 or A668 at the top of ICL2 of mGlu2 subunit into tryptophan with bulkier side chains, the mGlu2 subunit becomes able to activate G protein in the mGlu2C1 and mGlu4C2-FS heterodimer (Fig. 8b, c, Supplementary Fig. 18k, Supplementary Table 4). This effect was even more pronounced for a double mutation (Fig. 8b, c). Sequence alignment shows that the top of ICL2 in mGlu group II including the mGlu2 subunit are glycine and alanine, while in group III mGluRs including mGlu4 these are serine and valine (Fig. 8d). Increased potency was also observed in the ICL2 mutant (G667A, A668W) in mGlu2-2 homodimer (Fig. 8e, f, Supplementary Fig. 18l, Supplementary Table 4). These findings suggest that the spatial relationship between ICL2 and α5-αN participates in the G protein preference of the mGlu4 subunit within the mGlu2-4 heterodimer.

a Comparison of the conformation of ICL2 in mGlu4 subunit of the mGlu2-4G complex and in mGlu2 subunit in the mGlu2G-4 complex (PDB: 8JD5), which interacts with α5 and αN of Gαi3. b Glu-induced Ca2+ release in mGlu2C1-mGlu4C2 heterodimer with F781S (FS) mutation in mGlu4 subunit (mGlu4C2-FS + mGlu2C1, Ctr), without or with the indicated single mutation (G667W, A668W) or double mutations (G667W-A668W) in mGlu2 ICL2. Data are normalized by using the maximum response (Emax) of Glu in cells transfected with mGlu4C2-FS + mGlu2C1-G667W-A668W and present as mean ± s.e.m. from at least three independent biological replicates, each performed in triplicate. The indicated number of independent experiments (n): Ctr, G667W, A668W, G667W-A668W, n = 6, 6, 5, 7. c Representative Emax of Ca2+ release in (b). d Sequence alignment of the four residues in the top of ICL2 from mGlu 1-8. e Glu-induced Ca2+ release in mGlu2-2 homodimer WT or the indicated mutants. Data are normalized by using the maximum response (Emax) of Glu in cells transfected with mGlu2-2 WT and present as mean ± s.e.m. from at least three independent biological replicates, each performed in triplicate. The indicated number of independent experiments (n): Ctr, G667W, A668W, n = 8, 6, 6. f pEC50 of Glu in mGlu2-2 WT or indicated mutants in (e). Data in (c) and (f) are analyzed using one-way ANOVA with a Dunnett’s post-hoc multiple comparison test compared with Ctr or WT to determine significance. ****P < 0.0001, ***P < 0.001. P values for (c): 0.0004, <0.0001, <0.0001; for (f): < 0.0001, <0.0001 (from left to right). Source data are provided as a Source Data file.
Discussion
In this work, we report structural information essential to understand the oriented asymmetry of the mGlu2-4 heterodimer. These include four structures describing an inactive state, two intermediate states and an active state in complex with the G protein. In addition to the description of a unique mode of interaction of an mGlu4 PAM, our structural, functional and smFRET data of wild-type and mutant receptor dimers illustrate the inter- and intra-subunit conformational changes in mGlu2 and mGlu4, leading to the peculiar asymmetric activation of this heterodimer (Fig. 9).

Cartoon illustrates activation-related transitions including inter-subunit and intra-subunit from the inactive, through one agonist-bound intermediate state to the agonist, PAM and G protein-bound full active state.
The structure of the inactive state stabilized with both mGlu2 and mGlu4 antagonists confirms that the VFT dimer is in a resting orientation with both VFTs open, leading to the 7TM domains facing each other via TM5, similar to what was observed for some other inactive class C GPCRs9,17,26,28,35,38.
Several studies reported that binding of a single agonist in one or the other subunit generated partial activity in both mGlu homo25,27 and heterodimers27,29. Our cryo-EM structures revealed that the selective mGlu2 and mGlu4 agonists stabilize only one VFT in a closed state, while the other stays open. However, the VFT dimer remains in its resting orientation (Rco), therefore unable to push the 7TMs in their TM6 interface required for activity. When examined in an optimized solubilization condition31,32, at the single molecule level, we observed that only a fraction of the dimers occupied by a single agonists reaches the active orientation. This explains the partial activity measured in functional assays, likely due to a conformational equilibrium between agonist-bound intermediate and active states. This active state very likely corresponds to an active closed-open (Aco) conformation as competitive antagonists acting at the subunit not occupied by the agonist show no effect. This indicates that the unbound VFT is in the open state. These data explain the lower stability of the active VFT orientation, providing an explanation for the partial agonist activity of the selective mGlu2 and mGlu4 agonists at this heterodimer.
One key feature of the mGlu2-4 heterodimer is its oriented asymmetric activation, best illustrated by the G protein activation mediated by the mGlu4 subunit only8. Our data provide a number of possible explanations for this complex allosteric interaction between the subunits. First, our cryo-EM structure of the active heterodimer-G protein complex revealed an asymmetric 7TM dimer interface involving TM6 of mGlu4, and both TM6 and TM7 of mGlu2. Of interest, a more symmetric TM6-TM6 interface is observed in the forced active mGlu2G-4 heterodimer12. However, this structure is stabilized by an mGlu2 PAM12, making it compatible with the activation of the mGlu2 subunit, rather than the mGlu4 7TM. Secondly, we observed a different flexibility of the VFT-CRD connection in mGlu2 compared to mGlu4, illustrated by the peculiar angle of the mGlu2 CRD in the intermediate states occupied by a single agonist, independently of the agonist-occupied subunit (mGlu2 vs. mGlu4). Thirdly, although the CRD-ECL2 interaction is essential for the signal transmission to the 7TMs, both in the homo and heterodimer, the activity resulting from the mGlu2 subunit activation is favored in a mutant that disrupts this interaction in the mGlu4 subunit. This illustrates a role of the mGlu4 CRD-ECL2 interaction in the asymmetric allosteric interaction between the subunits. Fourthly, within the mGlu2 and mGlu4 7TMs, we identified residues important for their activity, including three residues that limit mGlu2 receptor homodimer activity, as their mutations increased coupling efficacy in the homodimer. However, these mutations have almost no effect when introduced in the mGlu2 subunit within the mGlu2-4 heterodimer. These data show that these mutations potentiating mGlu2 activity are not able to influence the asymmetric activation of the mGlu4 7TM. Of interest, these mGlu2 mutants also illustrate that the mGlu2 sequence is not optimal for an efficient G protein activation by the homodimer, suggesting that the evolution is not only directed by the function of mGlu2 homodimers, consistent with the functional relevance and existence of mGlu heterodimers containing an mGlu2 subunit. Eventually, we also show key features at the 7TM-G-protein interaction that orient the G protein towards the mGlu4 subunit. Indeed, the mGlu4-G protein interaction shows more contacts than the mGlu2-G protein one, and favoring the later with point mutations reorients the G protein coupling of the heterodimer towards mGlu2. These data strengthen our view that the oriented asymmetry of the active form of mGlu2-4 is likely responsible for the G protein activation by mGlu4, rather than a better G protein recruitment of the G protein by mGlu4 leading to the observed asymmetry. Indeed, mGlu4 homodimer is less efficient in activating G proteins than mGlu2 homodimers, but the mGlu4 subunit becomes as efficient to activate G protein in the heterodimer, as mGlu2 in the homodimer8,29.
In conclusion, our structural and functional analysis of the mGlu2-4 heterodimer illustrate how the symmetric Acc state of the VFT dimer leads to the activation of the G protein by the mGlu4 7TM only, through multiple allosteric switches. Such information may help elucidate the asymmetric activation of other mGlu dimers and will help develop tools to specifically target these receptors abundant in the brain for which the function still remains elusive.
Methods
Constructs
For protein production, human full-length mGlu2 (UniProt: Q14416) and mGlu4 (UniProt: Q14833) cDNAs were cloned into the pEGBacMam vector. To facilitate purification, the original mGlu2 and mGlu4 signal peptide was substituted by haemagglutinin (HA) signal sequence. For mGlu2, a Flag tag and a T4L tag were inserted just after this signal peptide before the sequence coding for the VFT domain. For mGlu4, 2*MBP tag and an 8× His tag was added at the C-terminal end of the receptor. To assist formation of mGlu2-4 heterodimer, two fusion tags (LgBiT and HiBiT)18 were added to the C-terminus of mGlu2 and mGlu4 respectively, to increase the stability of the dimer.
For signaling assay, the pRK5 plasmids encoding the HA-tagged wild-type mGlu2 and Flag-tagged mGlu4 from human were used. The site-directed mutations of mGlu2 or mGlu4 were generated using QuikChange mutagenesis protocol (Agilent Technologies, Santa Clara, CA, USA). We first generate no-disulfide bond-mGlu2-4 mutants to removal the cysteine at the top of VFT of both mGlu2 and mGlu4 (mGlu2C121A and mGlu4C136A-C715A). A quality control system containing complementary coiled-coil regions (C1 and C2) derived from the GABAB receptor and intracellular retention signals (KKXX) was used in engineered chimeric mGlu2 and mGlu4 subunits (mGlu2C1 and mGlu4C2) to allow cell surface targeting of the heterodimer8. Additional SNAP-tag was added in the N-terminus of mGlu2 for the cross-linking experiments.
For smFRET experiments, the pcDNA3 plasmids encoding human mGlu2 or human mGlu4 with N-terminal Flag (Sigma-Aldrich) and SNAP-tags (New England Biolabs) were a gift from Revvity (Codolet, France). N-terminal Flag and SNAP-tags were removed using the In-Fusion® HD cloning kit (Takara Bio Europe) according to the manufacturer’s protocol. A premature TAA was introduced by mutation of Arg358 of the human mGlu2 subunit using the QuikChange site-directed mutagenesis kit (Agilent Technologies). The plasmid used for TCO*A incorporation in response to the TAA stop codon (PylRSAFM15TAA) was a kind gift from Irene Coin.
Expression and purification of heterotrimeric Gi3
The dominant-negative Gαi3 was introduced four mutations (S47N, G203A, E245A and A326S) and human β1γ2 subunits (β1–8×His tag) were used as previously described39. The Trichuplusia ni Hi5 insect cells (Expression Systems) were co-infected with both Gαi3 and Gβ1γ2 virus in a 3:1 ratio at a density of 2.2 × 106 cells/mL. Cells were harvested after 48 h post infection. Cells were thawed in hypotonic buffer (10 mM HEPES pH 7.5, 100 μM MgCl2, 100 μM TCEP, 50 μM GDP and 1× protease inhibitor cocktail) and using dounce homogenization to homogenize. The membrane fraction was collected and further solubilized with a buffer A (20 mM HEPES pH 7.5, 100 mM NaCl, 2 mM MgCl2, 100 μM TCEP and 50 μM GDP) with addition 1×Protease Inhibitor Cocktail, 1.0% (w/v) sodium cholate hydrate (Sigma-Aldrich), 0.05% (w/v) n-dodecyl-β-D-maltopyranoside (DDM, Anatrace) and protease inhibitors for 1 h at 4 °C. The supernatant was collected and bound to Ni-NTA beads. Then the beads were washed with buffer A containing 0.1% (w/v) lauryl maltose neopentyl glycol (LMNG, Anatrace) and 30 mM imidazole and eluted with buffer A containing 0.01% LMNG and 300 mM imidazole. The purified heterotrimeric DN-Gi3 protein was incubated with scFv16 and further purified by Superdex 200 column, then concentrated and stored at −80 °C for later use.
Expression and purification of scFv16
The scFv16 protein was expressed and purified as previously described40. In brief, the Hi5 insect cells were infected by 6×His-tagged scFv16 virus and the scFv16 was produced in a secreted form for 48 h. The expressed scFv16 was purified using a Ni-NTA resin. The C-terminal 6×His tag of the Ni-NTA eluent was cleaved by 3C protease and further purified by gel filtration chromatography using a Superdex 200 column. Finally, the purified scFv16 protein was concentrated and stored at −80 °C for later use.
Protein expression and purification of mGlu2-4 heterodimer
HEK293F cells were co-transfected with Flag-tagged mGlu2 (full length)-TEV-HiBiT and HA-mGlu4 (full length)-TEV-LgBiT-MBP-OMBP-8×His at a cell density of 2.6–2.8 × 106/mL at 37 °C. After 16 h, 10 mM sodium butyrate was added and expression at 30 °C for 72 h. Cells were collected and washed with phosphate-buffered saline buffer. Finally, the cell pellets were rapidly frozen in liquid nitrogen and stored at 80 °C for later use.
For the preparation of the mGlu2-4-Gi3 complex, cells were thawed and suspended in lysis buffer (50 mM HEPES, 150 mM NaCl, 2 mM MgCl2, 10% glycerol, DNase and 1×protease inhibitor cocktail) dounced homogenization, then solubilized with 0.5% (w/v) LMNG (w/v) and 0.002% CHS (w/v) and eluted with buffer B containing 3 mM CaCl2 and 10 mM maltose. The eluted proteins were bound to M1 anti-Flag affinity resin. After washed with buffer B with addition 3 mM CaCl2 and eluted with buffer B containing 5 mM EGTA and 0.1 mg/mL Flag peptide. TEV was added to remove the NanoBiT moities and the MBP-OMBP-8×His tags. Then, the mGlu2-4 heterodimer protein was mixed with a 1.2 molar excess of DNGi3-scFv16 at 25 °C for 1 h and was followed by the addition of 0.2 μ/ml apyrase for an additional 1 h at 24 °C.
For inactive state and intermediate state mGlu2-4 heterodimer, cells were thawed and suspended in lysis buffer dounced homogenization, then solubilized with 1% (w/v) DDM, 0.2% (w/v) CHS and 0.025% (w/v) digitonin (Biosynth). The supernatant was collected and incubated with MBP beads. Protein-bound gel was washed with buffer C1 (50 mM HEPES, 150 mM NaCl, 2 mM MgCl2, 0.05% DDM (w/v), 0.025% digitonin (w/v) and 0.01% CHS (w/v)) and eluted with buffer C1 containing 3 mM CaCl2 and 10 mM maltose. The eluted proteins were bound to M1 anti-Flag affinity resin. After washed with buffer C1 containing 3 mM CaCl2, then washed with buffer C2 (50 mM HEPES, 150 mM NaCl, and 2 mM MgCl2) containing 3 mM CaCl2, 0.02% DDM (w/v), 0.004% CHS (w/v) and 0.05% digitonin (w/v), then washed with buffer C2 containing 3 mM CaCl2 and 0.05% digitonin (w/v), the proteins were eluted with buffer C2 containing 0.05% (w/v) digitonin, 5 mM EGTA and 0.1 mg/mL Flag peptide. TEV (1/100 w/w) was used to remove NanoBiT and MBP. Finally, the proteins were purified by size-exclusion chromatography on a Superose 6 increase 10/300GL column (GE Healthcare). Throughout purification, buffers were supplemented with either 10 mM Glu and 50 μM VU0155041 for mGlu2-4-Gi3 complex, 15 μM LY341495 and 5 μM CPPG for inactive state, 100 μM LY379268 for intermediate state-mGlu2 agonist, or 100 μM L-AP4 for intermediate state-mGlu4 agonist. Purification procedure of all samples was accomplished in 24 h. The sample was pre-screened for sample quality by negative-stain transmission electron microscopy and then immediately prepared on cryo-EM grids for structural determination. For mGlu2-4-Gi3 complex, protein was collected and concentrated to 25 mg/mL for electron microscopy studies. For inactive state and intermediate state mGlu2-4 heterodimer, protein was collected and concentrated to 20 mg/mL for electron microscopy studies.
Cryo-EM grid preparation and data collection
For inactive state sample, 3 μL of sample was applied at a concentration of 20 mg/mL with 0.03% (w/v) β-octyl glucoside detergent to glow-discharged holey carbon grids (Quantifoil Au300-R1.2/1.3, and R0.6/1). For intermediate state sample, 3 μL of sample was applied at a concentration of 20 mg/mL to glow-discharged holey carbon grids (Quantifoil Au300-R0.6/1). For mGlu2-4-Gi3 complex, 3 μL of sample was applied at a concentration of 25 mg/mL to glow-discharged holey carbon grids (Quantifoil Au300-R1.2/1.3). The grids were blotted for 3.5 s using an FEI Vitrobot Mark IV (Thermo Fisher Scientific) at 4 °C and 100% humidity and plunge-frozen into liquid ethane. Micrographs for mGlu2-4-Gi3 complex were recorded in the Center of Cryo-Electron Microscopy (Zhejiang University). Micrographs for other heterodimers were recorded in the Center of Cryo-Electron Microscopy (Liangzhu Laboratory). All of image stacks were acquired at a dose rate of about 7.0 electrons per Å2 per second with a defocus ranging from –1.0 to –2.5 μm on Falcon 4 direct electron detector with a pixel size of 0.93 Å using EPU. The total exposure time was 6 s. A total of 7647, 17,122, 35,658, and 16,112 movies were collected for the inactive state, two intermediate states and mGlu2-4G complex, respectively.
Imaging processing and 3D reconstruction
Image stacks for the various state mGlu2-4 heterodimers were subjected to beam-induced motion correction using MotionCor239. Contrast transfer function parameters for non-dose-weighted micrographs were determined by Gctf41. Particles were auto-picked using the reference-based and Laplacian of Gaussian picking algorithm in Relion42. Auto-picked particle projections were transferred to CryoSPARC (v3.3.2 and v4.0.3)43 to eliminate damaged particles and contaminants, multiple rounds of two-dimensional reference-free alignment were performed. The initial models for both maps were obtained using a stochastic gradient descent algorithm in CryoSPARC. The particles in clearly defined 2D classes were then subjected to several rounds of heterogeneous refinement to isolate unique conformational states and eliminate particles in poorly defined classes. Final global refinement of the heterodimer maps were performed with the CryoSPARC non-uniform refinement and local refinement with masks including only the VFTs with CRDs, 7TMs, or G protein. Composite maps were generated with Chimera (v.1.15)44. Map sharpening was performed with CryoSPARC and deepEMhancer45. Validation of cryo-EM maps and models was performed with Phenix (v.1.20.1-4487). UCSF Chimera (v.1.15) and Chimera X (v.1.5 and v1.6.1)46 were used for map/model visualizations and figure preparation. Transfer of particles from CryoSPARC to Relion was performed by UCSF pyem (v.0.5).
Model building and refinement
The initial models of inactive state and partial active state mGlu2-4 heterodimer were generated structure of a single subunit based on AlphaFold47 prediction. The initial model for the mGlu2-4-Gi3 complex was generated based on the agonist-PAM-bound mGlu4-Gi3 structure (Protein Data Bank (PDB) code 7E9H)10. All of the models were docked into the electron microscopy density maps in UCSF Chimera46. The docked model was subjected to flexible fitting using Rosetta48 and was further rebuilt in COOT49, followed by real-space-refinement in Phenix50. Ligand (antagonist, agonist and PAM) coordinates and geometry restraints were generated using a phenix.elbow50. The final refinement statistics were validated using the module “comprehensive validation (cryo-EM)” in Phenix50. The goodness-of-fit of the model to the map was determined using a global model-versus-map Fourier shell correlation. Structure figures were created using UCSF Chimera and the UCSF Chimera X package.
Molecular dynamics simulations
From the structure of the mGlu2-4 heterodimer complex (PDB ID: 8WGB), the CRD regions were directly removed, and the retained mGlu2-4 heterodimer complex with two possible poses of VU0155041 (recognized as PAM), pose-1 with inward-aromatic ring and pose-2 with outward-aromatic ring, was used as the initial configurations for all-atom molecular dynamics (MD) simulations, respectively (Supplementary Fig. 5a–c). The mGlu2-4 heterodimer complex was embedded into a lipid bilayer (232 POPC molecules), which was built by the Membrane Builder module in the CHARMM-GUI server51. The protonation state of the complex was determined by H++52. Subsequently, the system was solvated in a box (10.0 × 10.0 × 15.6 nm3) with TIP3P waters and 0.15 M Na+/Cl– ions (Supplementary Table 5). The CHARMM36m forcefield was used to describe the system53, and all MD simulations were performed using GROMACS-2019.454. After 5000 steps of energy minimization performed by the steepest descent algorithm, then a 250 ps NVT equilibration simulation was performed at 310 K. Subsequently, a cumulatively 1.65 ns NPT equilibration to 1 atm was performed by using the Berendsen barostat. During the pre-equilibrium MD simulation, a harmonic restraint potential of 10 kCal/mol ∙ Å–2 was adopted and gradually reduced to zero. Long-range electrostatic interactions were treated by the particle-mesh Ewald method. The short-range electrostatic and van der Waals interactions both used a cutoff of 10 Å. All bonds were constrained by the LINCS algorithm. Finally, to mitigate the uncertainties associated with sampling results, three replicates of the production run with different initial velocities were performed. The overall stability of the system was evaluated by calculating the RMSD of the heavy atoms of the mGlu2-4 heterodimer complex (except the loops in the extracellular and intracellular regions), and the Cα atoms of the transmembrane helices were used for structural alignment (Supplementary Fig. 6). GROMACS’s rms function was used to calculate RMSD. For the stability analysis of ligand pose of pose-1 system, the first 50 ns was ignored and only the last 450 ns MD simulation data was used (Supplementary Fig. 6). The stability was evaluated by overall structural alignment of the representative conformations (saved every 100 ns for each trajectory) for the three replicas and the distance distribution of important contacts (Supplementary Fig. 7). The distances between the chloride atoms (ClA and ClB) of ligand and the Cα atoms of R655/R656 were applied to monitor part-1 stability of ligand, and the distance between the C14 atom of ligand and the Cα atom of T819 was applied to monitor part-2 stability of ligand. The analyses results confirmed the stability of ligand pose in pose-1 system (Supplementary Fig. 7). All the analysis of the structures from the calculation results were displayed by using ChimeraX46 and PyMol.
Bioluminescence resonance energy transfer (BRET) assay
For the detection of the G protein activation by the homodimers mGlu2 and mGlu4 and the heterodimer mGlu2-4 wild-type or mutants, HEK293 cells (ATCC, CRL-1573, lot: 3449904) were transfected with wild-type or mutant mGlu subunits, Gαi3-Nluc, Gβ1, Venus-Gγ9 and EAAC1. For the detection of the activation of Gi3 wild-type or mutants, HEK293 cells were transfected with the constructs mGlu2C1, mGlu4C2, wild-type or mutant Gαi3-Nluc, Gβ1, Venus-Gγ9 and EAAC1. Lipofectamine 2000 was used for the cDNA transfection into HEK293 cells. After 36 h transfection, cells were washed and starved in PBS at 37 °C for 1 h. BRET1 measurements were performed using the Mithras LB 940 (Berthold Technologies, German)51 or PHERAstar FS (BMG Labtech, USA)52. The signals emitted by the donor (460–500 nm band-pass filter, Em 480) and the acceptor entity (510–550 nm band-pass filter, Em 530) were recorded after the addition of 10 μM furimazine. The BRET signal was determined by calculating the ratio between the emission of acceptor and donor (Em 530/Em 480). The basal BRET ratio (BRETbasal) of cells was recorded before the stimulation with drugs or buffer. The change in BRET ratio (net BRET) was obtained by subtracting the BRET ratio after agonist treatment from the basal BRET (BRETbasal-BRETagonist).
IP1 production measurements
IP1 production was measured using the IP-One HTRF kit (Revvity, Codolet, France). HEK293 Cells were co-transfected with the plasmids of receptor, glutamate transporter EAAC1 and a chimeric G protein Gαqi9, which allows efficient signaling through the phospholipase C pathway, at a ratio of 2:1:1. Transfected HEK293 cells were seeded in a 96-well plate, and 36 h after transfection, cells were treated with glutamate diluted in stimulation buffer of the kit for 30 min at 37 °C. Then, cryptate-labeled anti-IP1 monoclonal antibody and d2-labeled IP1 in lysis buffer were added to the wells. After 1 h of incubation at 25 °C, the plates were read in PHERAstar FS with excitation at 337 nm and emission at 620 and 665 nm. The accumulation of IP1 concentration (nM) was calculated according to a standard dose response curve.
Intracellular calcium release measurements
After 36 h transfection with plasmids encoding the indicated mGlu subunits and a chimeric protein Gqi9, HEK-293 cells were loaded with 1 μM Ca2+-sensitive fluorescent dye Fluo-4 AM (Molecular Probes, Eugene, OR, USA) for 1 h at 37 °C. After a wash, cells were incubated with buffer and glutamate solution at various concentrations was added after 20 s of recording. Fluorescence signals (excitation 485 nm, emission 525 nm) were measured by using the fluorescence microplate reader Flexstation (Molecular Devices, Sunnyvale, CA, USA) at sampling intervals of 1.5 s for 60 s. The response of Ca2+ release was generated by the peak value of fluorescence signals after drug injection subtracting the mean value of fluorescence signals before drug injection. All the values of drug treatment were subtracted the value in buffer treatment as the net Ca2+ release. Data were analyzed with the program Soft Max Pro (Molecular Devices, Sunnyvale, CA, USA).
Cross-linking and fluorescent-labeled blot experiments
HEK293 cells were transfected with HA-SNAP-mGlu2C1 and Flag-HALO-mGlu4C2 or indicated mutants by lipofectamine 2000 and plated in 12-well plates for 36 h. Then cells were labeled with 100 nM SNAP-649 non cell permeant in culture medium at 37 °C for 2 h. Cells were incubated with glutamate (1 mM) or PBS at 37 °C for 30 min. Afterwards, cross-link buffer (1.5 mM Cu(II)-(o-phenanthroline), 1 mM CaCl2, 5 mM Mg2+, 16.7 mM Tris HCl, pH 8.0 and 100 mM NaCl) was added at 37 °C for 20 min. After incubation with 10 mM N-ethylmaleimide at 4 °C for 15 min to stop the cross-linking reaction, cells were lysed with lysis buffer (containing 50 mM Tris pH 7.4, 150 mM NaCl, 1% NP-40 and 0.5% sodium deoxycholate) at 4 °C for 1.5 h. After centrifugation at 12,000 × g for 30 min at 4 °C, supernatants were mixed with loading buffer at 37 °C for 10 min. In reducing conditions, samples were treated without DTT in loading buffer for 10 min before loading the samples. Equal amounts of proteins were resolved by 59:1 acrylamide:bisacrylamide and 6% SDS-PAGE. Proteins were transferred to nitrocellulose membranes (Millipore). Membrane was imaged on an Odyssey CLx imager (LI-COR Bioscience, Lincoln, NE, USA) at 700 nm and 800 nm.
Cell surface quantification by ELISA
ELISA was performed to detection of the HA- and Flag-tagged constructs at the cell surface. 36 h after transfection, the HEK293 cells were fixed with 4% paraformaldehyde, blocked with 10% FBS. HA tagged constructs were detected with a monoclonal rat anti-HA antibody (with horseradish peroxidase) 3F10 (Roche) at 0.5 μg/mL. Flag-tagged constructs were detected with the mouse monoclonal anti-Flag antibody (with horseradish peroxidase) M2 (Sigma, St. Louis, MO) at 1.0 μg/mL. Bound antibodies coupled to horseradish peroxidase were detected by chemoluminescence using SuperSignal substrate (Pierce) and the EnVision™ Multilabel Plate Reader (Cat.2103-0010, Perkin Elmer, Waltham, MA, USA).
smFRET experiments
Adherent HEK293T cells (American Type Culture Collection CRL-3216, LGC Standards S.a.r.l., France) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich) at 37 °C with 5% CO2. HEK293T cells were transfected using JetPrime transfection reagent (Polyplus-transfection SA, Illkirch-Graffenstaden, France) in standard flat 6-well TC-plates (Sarstedt). After treatment with a 28.5 μg/µl solution of poly-l-ornithine-hydrobromide in DPBS w/o Ca2+ and Mg2+ (Thermo Fischer Scientific, France) at 37 °C for 15 min, wells were rinsed once with DPBS and a total of 1.2 × 106 cells were seeded in 2 ml of DMEM per well 24 h before transfection. Transfections were performed using 2 μg of total DNA per well with a 2:1:3 ratio of vectors coding for mGluR2:mGluR4:PylRSAFM15TAA in 200 μl of JetPrime buffer (Polyplus). 4 μl of JetPrime Reagent per well were added and incubated for 25 min at RT. Last, the transfection mixture was added to the cells and incubated at 37 °C for 6 h. Then, for incorporation of the unnatural amino acid, the medium was replaced with 2 ml fresh DMEM supplemented with 0.2 mM trans-Cyclooct-2-en-L-Lysine (TCO*A; Sirius Fine Chemicals Sichem GMBH). The medium was further exchanged with fresh TCO*A-containing medium after 24 and 48 h.
SNAP-tag labeling of the human mGlu4 subunit was performed on adherent cells at 37 °C and 5% CO2 for 2 h using a final concentration of 300 nM SNAP-d2 in Gibco DMEM GlutaMax without phenol red, supplemented with GlutaMAX and pyruvate (Thermo Fisher Scientific, France). Following labeling, excess dye was removed by three cycles of washing with DPBS without Ca2+ and Mg2+ (Thermo Fischer Scientific, France) at ambient temperature.
Labeling of the TCO*A-bearing hmGlu2 subunit was performed right after SNAP-tag labeling using a final concentration of 2 μM Tetrazine-Cy3b (AAT Bioquest) in acquisition buffer (20 mM Tris-HCl pH 7.4, 118 mM NaCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 4.7 mM KCl and 1.8 mM CaCl2) incubated for 15 min at 37 °C. Excess dye was removed by three cycles of washing with DPBS without Ca2+ and Mg2+ (Thermo Fischer Scientific, France) at 25 °C.
For crude membrane fractions preparation, adherent cells were detached mechanically using a cell scraper in DPBS w/o Ca2+ and Mg2+ (Thermo Fisher Scientific, France) and collected at 500 × g and 22 °C for 5 min. Subsequently, cells were re-suspended in cold hypotonic lysis buffer (10 mM HEPES pH 7.4 and cOmplete™ protease inhibitor), frozen, and stored at −80 °C. After thawing, cells were passed through a 200 µl pipette tip 50 times on ice. After two rounds of centrifugation at 500 × g and 4 °C for 5 min, the supernatant was centrifuged at 21,000 × g and 4 °C for 30 min to collect crude membranes. The pellets were washed once with acquisition buffer, flash-frozen in liquid N2, and stored at −80 °C until solubilization.
Receptors were solubilized using 10 μl of acquisition buffer (see below) containing 1% LMNG (w/v) and 0.1% CHS Tris (w/v) per membrane fraction (corresponding to cells cultured in one well of a six-well plate) for 15 min on ice. Subsequently, the solubilization mixture was centrifuged for 10 min at 4 °C and 21,000 × g, and the supernatant was mixed with 90 μl of acquisition buffer containing 0.11% GDN (w/v). The diluted sample was then passed through a Zeba Spin desalting column (0.5 mM, 7 kDa cutoff; Thermo Fisher Scientific, France) equilibrated in acquisition buffer containing 0.005% LMNG (w/v), 0.0005% CHS Tris (w/v) and 0.005% GDN (w/v). The eluate was then diluted 10 times in acquisition buffer and stored on ice in the dark.
smFRET experiments with pulsed interleaved excitation (PIE)–multiparameter fluorescence detection (MFD) were performed on a homebuilt confocal microscope using the SPCM 9.85 software (B&H) as described previously53. Modifications are described in the following. A combination of 530/20 (530AF20, Omega Optical, Brattleboro, VT, USA) and 530/10 nm (FLH532-10, Thorlabs, Maisons-Laffitte, France) bandpass filters was used for Cy3B excitation. A 635/10 (FLH635-10, Thorlabs, Maisons-Laffitte, France) bandpass filter was used for D2 excitation. The excitation power was 25 μW (prompt at 535 nm) and 12.5 μW (delayed at 635 nm) at the entrance into the microscope. Inside the microscope, the light was reflected by dichroic mirrors that match the excitation/emission wavelengths of the respective fluorophore combinations (CFF545/650- Di01, Semrock, Rochester, NY, USA) and coupled into a 100×, numerical aperture 1.4 objective (Nikon, France). The following emission filters were used: Cy3B parallel and perpendicular ET BP 585/65 (Chroma, Bellows Falls, VT, USA); red/d2 parallel and perpendicular FF01-698/70-25 (Semrock, Rochester, NY, USA). Dual color emission was separated using FF649LP long pass filters (parallel and perpendicular, Semrock, Rochester, NY, USA).
smFRET measurements were performed on SensoPlate 384-well plates (nontreated, Greiner Bio-One, France) passivated with bovine serum albumin (1 mg/ml) in acquisition buffer with 0.0025% LMNG (w/v), 0.00025% CHS Tris (w/v) and 0.0025% GDN (w/v) for at least 1 h before sampling application. Samples containing 30 to 100 pM labeled receptors were measured in acquisition buffer (20 mM Tris-HCl pH 7.4, 118 mM NaCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 4.7 mM KCl and 1.8 mM CaCl2) with 0.0025% LMNG (w/v), 0.00025% CHS Tris (w/v) and 0.0025% GDN (w/v). Measurements at saturating ligand concentrations were performed at 10 mM Glu, 10 μM LY379268 and 100 μM L-AP4.
smFRET data analysis was performed using the PAM 1.3 software package54. A single-molecule event was defined as a burst containing at least 30 photons with a maximum allowed interphoton time of 0.16 ms and a Lee-filter of 30. Apparent FRET efficiencies (EPR) and stoichiometry (S) were calculated as described previously following the recommendations made in previous reports32.
To display FRET (EPR) histograms, an ALEX 2CDE55 filter was applied with a maximum value set at 35 and a minimum value at 0. Doubly labeled molecules with an S = 0.3 to 0.7 were selected and normalized to the total number of molecules for individual biological replicates. In this way, average histograms with the mean apparent FRET efficiency ± s.e.m were obtained. Fitting was performed using a homemade python multi gaussian fitting script for average FRET histograms and for each individual dataset separately to derive data shown in scatterplots. Scatter plots were displayed using GraphPad Prism 7.05 and statistical analysis was done using a one-way ANOVA with Tukey’s multiple comparisons test.
Statistical analysis
Statistical analyses were performed on at least three individual data sets and analyzed in GraphPad Prism software version 8. Data are present as mean ± s.e.m or mean ± s.d. from at least three independent experiments performed in technical triplicate, with the number of independent experiments indicated in the figure legend. For dose-response curves, data are analyzed using nonlinear curve fitting for the log (agonist) vs. Response (three parameters) curves. Bars represent the agonist potency (pEC50), maximum agonist response (Emax) of expression of receptors. Data are analyzed using the one-way ANOVA with Dunnett’s multiple comparison test or unpaired student t test to determine significance. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, not significant (ns) >0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated in this study are included in the main text or the Supplementary Information. Cryo-EM maps have been deposited in the Electron Microscopy Data Bank under accession codes: EMD-37509 (mGlu2-4 bound with LY341495 and CPPG), EMD-37508 (mGlu2-4 bound with LY379268), EMD-37506 (mGlu2-4 bound with L-AP4), and EMD-37507 (mGlu2-4-Gi3 complex). The atomic coordinates have been deposited in the Protein Data Bank under accession codes: 8WGD (mGlu2-4 bound with LY341495 and CPPG), 8WGC (mGlu2-4 bound with LY379268), 8WG9 (mGlu2-4 bound with L-AP4), and 8WGB (mGlu2-4-Gi3 complex). The MD simulation data (cleaned trajectories, start structure, simulation parameters) generated in this study have been deposited in the github (https://github.com/Yanzhang-ZJU/mGluDimer.git) and Zenodo (https://doi.org/10.5281/zenodo.13293875). Source data are provided with this paper.
References
Kenakin, T. G protein coupled receptors as allosteric proteins and the role of allosteric modulators. J. Recept. Signal Transduct. Res. 30, 313–321 (2010).
Kenakin, T. & Miller, L. J. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharm. Rev. 62, 265–304 (2010).
Pin, J. P. & Bettler, B. Organization and functions of mGlu and GABA(B) receptor complexes. Nature 540, 60–68 (2016).
Gregory, K. J. & Goudet, C. International Union of Basic and Clinical Pharmacology. CXI. Pharmacology, signaling, and physiology of metabotropic glutamate receptors. Pharm. Rev. 73, 521–569 (2021).
Doumazane, E. et al. A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J. 25, 66–77 (2011).
Meng, J. et al. Nanobody-based sensors reveal a high proportion of mGlu heterodimers in the brain. Nat. Chem. Biol. 18, 894–903 (2022).
Hlavackova, V. et al. Evidence for a single heptahelical domain being turned on upon activation of a dimeric GPCR. EMBO J. 24, 499–509 (2005).
Liu, J. et al. Allosteric control of an asymmetric transduction in a G protein-coupled receptor heterodimer. eLife 6, e26985 (2017).
Du, J. et al. Structures of human mGlu2 and mGlu7 homo- and heterodimers. Nature 594, 589–593 (2021).
Lin, S. et al. Structures of G(i)-bound metabotropic glutamate receptors mGlu2 and mGlu4. Nature 594, 583–588 (2021).
Seven, A. B. et al. G-protein activation by a metabotropic glutamate receptor. Nature 595, 450–454 (2021).
Wang, X. et al. Structural insights into dimerization and activation of the mGlu2-mGlu3 and mGlu2-mGlu4 heterodimers. Cell Res. 33, 762–774 (2023).
Habrian, C. H. et al. Conformational pathway provides unique sensitivity to a synaptic mGluR. Nat Commun 10, 5572 (2019).
Olofsson, L. et al. Fine tuning of sub-millisecond conformational dynamics controls metabotropic glutamate receptors agonist efficacy. Nat. Commun. 5, 5206 (2014).
Rondard, P. & Pin, J. P. Dynamics and modulation of metabotropic glutamate receptors. Curr. Opin. Pharm. 20, 95–101 (2015).
Koehl, A. et al. Structural insights into the activation of metabotropic glutamate receptors. Nature 566, 79–84 (2019).
Fang, W. et al. Structural basis of the activation of metabotropic glutamate receptor 3. Cell Res. 32, 695–698 (2022).
Duan, J. et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 11, 4121 (2020).
Kingston, A. E. et al. LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology 37, 1–12 (1998).
Ebrahimi-Ghiri, M., Khakpai, F. & Zarrindast, M. R. Combined treatment of scopolamine and group III mGluR antagonist, CPPG, exerts antidepressant activity without affecting anxiety-related behaviors. Physiol. Behav. 224, 113034 (2020).
Imre, G. The preclinical properties of a novel group II metabotropic glutamate receptor agonist LY379268. CNS Drug Rev. 13, 444–464 (2007).
Fedosiewicz-Wasiluk, M., Hoły, Z. Z. & Wiśniewski, K. L-AP4, a potent agonist of group III metabotropic glutamate receptor, decreases central action of angiotensin II. Pol. J. Pharm. 54, 415–422 (2002).
Williams, R. et al. Synthesis and SAR of a novel positive allosteric modulator (PAM) of the metabotropic glutamate receptor 4 (mGluR4). Bioorg. Med. Chem. Lett. 19, 4967–4970 (2009).
Yin, S. et al. Selective actions of novel allosteric modulators reveal functional heteromers of metabotropic glutamate receptors in the CNS. J. Neurosci. 34, 79–94 (2014).
Kniazeff, J. et al. Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat. Struct. Mol. Biol. 11, 706–713 (2004).
Xue, L. et al. Major ligand-induced rearrangement of the heptahelical domain interface in a GPCR dimer. Nat. Chem. Biol. 11, 134–140 (2015).
Scholler, P. et al. HTS-compatible FRET-based conformational sensors clarify membrane receptor activation. Nat. Chem. Biol. 13, 372–380 (2017).
Xue, L. et al. Rearrangement of the transmembrane domain interfaces associated with the activation of a GPCR hetero-oligomer. Nat. Commun. 10, 2765 (2019).
Moreno Delgado, D. et al. Pharmacological evidence for a metabotropic glutamate receptor heterodimer in neuronal cells. eLife 6, e25233 (2017).
Liu, L. et al. Asymmetric activation of dimeric GABAB and metabotropic glutamate receptors. Am. J. Physiol. Cell Physiol. 325, C79–C89 (2023).
Cao, A. M. et al. Allosteric modulators enhance agonist efficacy by increasing the residence time of a GPCR in the active state. Nat. Commun. 12, 5426 (2021).
Lecat-Guillet, N. et al. Concerted conformational changes control metabotropic glutamate receptor activity. Sci. Adv. 9, eadf1378 (2023).
Serfling, R. et al. Designer tRNAs for efficient incorporation of non-canonical amino acids by the pyrrolysine system in mammalian cells. Nucleic Acids Res. 46, 1–10 (2018).
Doumazane, E. et al. Illuminating the activation mechanisms and allosteric properties of metabotropic glutamate receptors. Proc. Natl Acad. Sci. USA 110, E1416–E1425 (2013).
Mao, C. et al. Cryo-EM structures of inactive and active GABA(B) receptor. Cell Res. 30, 564–573 (2020).
Shen, C. et al. Structural basis of GABA(B) receptor-G(i) protein coupling. Nature 594, 594–598 (2021).
Francesconi, A. & Duvoisin, R. M. Role of the second and third intracellular loops of metabotropic glutamate receptors in mediating dual signal transduction activation. J. Biol. Chem. 273, 5615–5624 (1998).
Ling, S. et al. Structural mechanism of cooperative activation of the human calcium-sensing receptor by Ca(2+) ions and L-tryptophan. Cell Res. 31, 383–394 (2021).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Koehl, A. et al. Structure of the micro-opioid receptor-G(i) protein complex. Nature 558, 547–552 (2018).
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Scheres, S. H. Processing of structurally heterogeneous cryo-EM data in RELION. Methods Enzymol. 579, 125–157 (2016).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Wang, R. Y. et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife 5, e17219 (2016).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Gales, C. et al. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol. 13, 778–786 (2006).
Hauser, A. S. et al. Pharmacogenomics of GPCR drug targets. Cell 172, 41–54.e19 (2018).
Hellenkamp, B. et al. Precision and accuracy of single-molecule FRET measurements-a multi-laboratory benchmark study. Nat. Methods 15, 669–676 (2018).
Schrimpf, W., Barth, A., Hendrix, J. & Lamb, D. C. PAM: a framework for integrated analysis of imaging, single-molecule, and ensemble fluorescence data. Biophys. J. 114, 1518–1528 (2018).
Tomov, T. E. et al. Disentangling subpopulations in single-molecule FRET and ALEX experiments with photon distribution analysis. Biophys. J. 102, 1163–1173 (2012).
Acknowledgements
We thank the Research Core Facilities of Life Science (HUST, Wuhan) for their assistance in functional measurements. We thank the Cryo-Electron Microscopy Center of Liangzhu laboratory for the cryo-EM data collection. Cryo-EM specimens were screened at the Center of Cryo-Electron Microscopy, Zhejiang University. Sample preparation for cryo-EM studies was supported by the Protein Facility, Zhejiang University School of Medicine. We thank I. Coin, and R. Serfling (Universität Leipzig, Germany) for providing the plasmid for ncAA incorporation. This project was supported by National Key R&D Program of China (2022YFE0116600 and 2021ZD0203302 to J.L.), the National Natural Science Foundation of China Grant (82320108021 and 32330049 to J.L.), the Key R&D Projects of Zhejiang Province (2021C03039 to Y.Z.), the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang 569 (2020R01006 to Y.Z.). The Fondation pour la Recherche Médicale (EQU202303016470 to P.R.) and the Agence Nationale pour la Recherche (ANR 18-CE11-0004-01 to J.P.P. and E.M.), the Major Project of Guangzhou National Laboratory (GZNL2023A03007 to J.L.), the Key Technologies R&D Program of Guangdong Province (2010A080813001 to J.L.) and the interdisciplinary Research Programme of HUST (2023JCYJ006 to C.X. and 5003170102 to J.L). The Postdoctoral Innovative Talent Support Program (grant number BX20220122 to C.S.). The Centre de Biologie Structurale is a member of France-BioImaging national infrastructure (Grant ANR-10-INBS-04, “Investments for the future).
Author information
Authors and Affiliations
Contributions
J.L. and Y.Z. conceived and supervised the whole project with important inputs from J.P.P; C.S., C.X., and W.H. designed the experiments for functional assays; W.H. generated the constructs and performed the functional experiments; N.J. designed the constructs, expressed, and purified the mGlu2-4 complex; K.X. did the MD analysis. J.G. prepared the cryo-EM grids, performed cryo-EM map calculation, model building; D-D.S. evaluated all the protein samples by negative-stain EM; J.G. and J.Q. collected the cryo-EM data; L.B., R.Q., E.M., designed and performed the smFRET experiments; Y.S. helped W.H. for the plasmid preparation and BRET experiments. W.H., N.J., C.X., J.G., Y.L., and C.S. prepared the figures; C.X. and C.S. participated in manuscript writing advised by J.L.; Y.G. advised the cryo-EM data processing; P.R and J.P.P participated in the interpretation of the data and the preparation of the manuscript; J.L., J.P.P., and Y.Z. wrote the manuscript with inputs from all the authors.
Corresponding authors
Ethics declarations
Competing interests
P.R. and J.P.P. are involved in a collaborative team between the CNRS and Cisbio Revvity, developing htrf-based tools. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Antonella Di Pizio who co-reviewed with Alessandro Nicoli; Karen Gregory, Xin-Yun Huang and David Millar for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, W., Jin, N., Guo, J. et al. Structural basis of orientated asymmetry in a mGlu heterodimer. Nat Commun 15, 10345 (2024). https://doi.org/10.1038/s41467-024-54744-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54744-7
