
Abstract
The CRISPR-associated endonuclease Cas9 derived from prokaryotes is used as a genome editing, which targets specific genomic loci by single guide RNAs (sgRNAs). The eukaryotes, the target of genome editing, store their genome DNA in chromatin, in which the nucleosome is a basic unit. Despite previous structural analyses focusing on Cas9 cleaving free DNA, structural insights into Cas9 targeting of DNA within nucleosomes are limited, leading to uncertainties in understanding how Cas9 operates in the eukaryotic genome. In the present study, we perform native-polyacrylamide gel electrophoresis (PAGE) analyses and find that Cas9 targets the linker DNA and the entry-exit DNA region of the nucleosome but not the DNA tightly wrapped around the histone octamer. We further determine cryo-electron microscopy (cryo-EM) structure of the Cas9-sgRNA-nucleosome ternary complex that targets linker DNA in nucleosomes. The structure suggests interactions between Cas9 and nucleosomes at multiple sites. Mutants that reduce the interaction between nucleosomal DNA and Cas9 improve nucleosomal DNA cleavage activity in vitro, although inhibition by the interaction between Cas9 and nucleosomes is limited in vivo. These findings will contribute to the development of novel genome editing tools in chromatin.
Similar content being viewed by others


Introduction
CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats and CRISPR associated) systems in bacteria provide adaptive immunity against foreign nucleic acids1. Cas9 associates with either dual RNA guides (CRISPR RNA [crRNA] and a trans-activating crRNA [tracrRNA]) or a combined single guide RNA (sgRNA). These RNA modules direct Cas9 to target specific DNA sequences, facilitating the cleavage of double-stranded DNA. The target DNA is complementary to an approximately 20 nt guide segment in the guide RNA and is flanked by a protospacer adjacent motif (PAM). Cas9 from Streptococcus pyogenes especially exhibits robust nuclease activities in eukaryotic cells, and is widely used as versatile genome-engineering tool2.
CRISPR-Cas9 systems are derived from bacteria with genomes typically composed of supercoiled circular DNA with nucleoid-associated proteins3. In contrast, eukaryotes, the targets of genome editing, store genome DNA in chromatin, in which the primary structure is the nucleosome4,5. The nucleosome contains four histone proteins, H2A, H2B, H3, and H4, which form a histone octamer that is wrapped by 145 ~ 147 bp DNA6. In the nucleosome, the DNA directly interacting with histones (core DNA) extends linker DNAs, which are histone-free DNA regions next to both sides of the nucleosomal DNA ends.
Various structural analyses have sought to clarify the DNA cleavage mechanism of Cas9, but all have focused on how Cas9 cleaves free DNA7,8,9,10,11,12. However, due to a lack of structural insights into Cas9 targeting DNA in nucleosomes, the principles governing Cas9 targeting the eukaryotic genome remain unclear. In addition, nucleosomes reportedly suppress Cas9 DNA cleavage both in vitro and in vivo13,14,15, indicating that many genomic DNA sites escape Cas9 targeting. This problem has largely limited the applications of Cas9 for eukaryotic genome editing, which has not been resolved due to the lack of structural information about Cas9 and nucleosome interactions.
In the present study, we found from native-polyacrylamide gel electrophoresis (PAGE) analyses that Cas9 targets the linker DNA and the nucleosome DNA end region and differences in the efficiency of Cas9-mediated cleavage of nucleosomal DNA between the DNA entry and exit sites. We also determined cryo-electron microscopy (cryo-EM) structure of the Cas9-sgRNA-nucleosome ternary complex that targets linker DNA in nucleosomes. The structure suggested interactions between Cas9 and nucleosomes at multiple sites. Mutants that reduce the interaction between nucleosomal DNA and Cas9 improved nucleosomal DNA cleavage activity in vitro, although inhibition by the interaction between Cas9 and nucleosomes was limited in vivo.
Results
Cas9 targets nucleosomal DNA end regions
To elucidate where Cas9 binds and cleaves the DNA in the nucleosome, we performed a native-PAGE analysis (Fig. 1a).

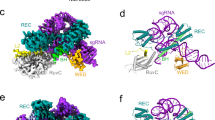
a Schematic of the experimental setup for in vitro binding and cleavage assays. The sgRNA that recognizes each target site was incubated with Cy5-labeled SpCas9-NG mutant that recognizes the NG PAM sequence. Mononucleosomes contained Cy3-labeled histone H4 and 5’ -end-labeled DNA with 6 FAM were then added to Cas9-sgRNA ribonucleoproteins. The 5’ -end-labeled DNA is colored light purple, and the non-labeled DNA is colored dark purple. Histones H2A, H2B, H3, and H4 are colored yellow, light red, light blue, and light green, respectively. The figure was created by UCSF Chimera X referred Apo-SpCas9 (PDB ID: 4CMP), SpCas9-sgRNA binary complex (PDB ID: 4ZT0), Nucleosome (3LZ0). b Schematic of the nucleosome DNA containing the Widom 601 sequence. The location of the histone core is indicated by the light gray oval. The location of PAMs within the double-stranded sequence are indicated with arrows, pointing in the 5’ to 3’ orientation of the NG sequence. The dark red and the dark blue arrows show the NG sequences on the solvent and histone sides, respectively. 6 FAM is attached to the 5’ end of the left side of the DNA. c and d In vitro nucleosome DNA cleavage activities of the SpCas9-NG for each PAM sequence. The reaction products were resolved, visualized with an Amersham Imager 680 (Cytiva). Data are mean ± s.d. (n = 3). The experiments were repeated three times with similar results. Source data is provided as a Source data file. e and f Native-PAGE gels showing the results of a binding assay targeting the indicated PAMs. The experiments were repeated three times with similar results. Data for representative gels are shown in this figure. The native-PAGE gels were imaged in merged three-color channels: 6-FAM on the nucleosome DNA end (blue), Cy3 on histone H4 (green) and Cy5 on Cas9 (red). The results of PAM1 to PAM15 and PAM16 to PAM28 are shown in e and f, respectively.
The reconstituted nucleosome contained Cy3-labeled histone H4 and 6 FAM end-labeled DNA consisting of a Widom 601 positioning sequence16 for core nucleosomal DNA (histone-DNA contacting region) with 25 base pairs of linker DNA (histone-free DNA region) on both sides (Fig. 1a, b). The wild-type (WT) Cas9 recognizes NGG sequences to cleave DNA, limiting the number of targetable DNA regions on the nucleosome DNA sequence. To overcome this limitation, we utilized the SpCas9-NG that recognizes the NG PAM sequence17, enabling the targeting of 28 sites (PAM1–PAM28) in each superhelical locations (SHLs) in the nucleosome core particle and linker DNA regions (Fig. 1b and Supplementary Fig. 1a).
The native-PAGE analysis showed that Cas9 cleaved the linker DNA (at PAM1 and PAM28) (Fig. 1c, d), consistent with the previous studies18,19,20. In addition, DNA cleavage by Cas9 at SHL + 6 (PAM27) was observed in the core DNA region (Fig. 1d). By contrast, DNA binding and cleavage by Cas9 were not observed at SHL0–SHL ± 5 (PAM2–PAM14 and PAM19–PAM26) (Fig. 1c, d, e, f). A previous study reported that the DNA region around SHL ± 6 to SHL ± 7 transiently unwrapped from the histone surface (DNA unwrapping)21, while the histone-DNA contact is stable at SHL0–SHL ± 522. Therefore, DNA unwrapping may enable Cas9 to bind and cleave the region from SHL + 6 to the linker DNA, whereas stronger histone and DNA interactions may prevent Cas9 from even binding at SHL0–SHL ± 5.
Despite the nucleosome’s symmetrical structure with SHL0 as the central axis, the native-PAGE analysis also showed that the efficiency of nucleosome DNA cleavage by Cas9 significantly differs between the DNA entry/exit sites (Fig. 1c, d). Specifically, the DNA cleavage activity was lower for PAM1 (at DNA entry side) compared to PAM28 (at DNA exit side), and the DNA cleavage observed at SHL + 6 (PAM27) was not observed at SHL-6 (PAM18) (Fig. 1c, d and Supplementary Fig. 1a). Previous studies reported that the Widom601 sequence has more flexibility on the DNA exit side across the dyad than on the DNA entry side, depending on the internal base sequence of the core DNA23,24,25. Thus, flexibility differences at the DNA entry/exit sites, probably caused by the base sequences of the core DNA, may impact Cas9’s DNA cleavage activity.
There was no cleavage at PAM14 and PAM17 although they are in SHL7, where nucleosome unwrapping occurs. Based on the superimposed model of Cas9 and nucleosome (Supplementary Fig. 1b, c), the position of Cas9 may make the capability of nucleosome DNA cleavage. In Supplementary Fig. 1b, Cas9 seemed to physically collide with the histone core, occluding access to PAM14 and PAM17. Additionally, SHL5 is the region where the interaction between histone and DNA is strong, causing Cas9 not to pull away for DNA cleavage.
Cryo-EM analysis for Cas9-sgRNA-nucleosome complex targeting linker DNA
Unexpectedly, we observed ternary complex formation of Cas9, sgRNA, and nucleosome in PAM1 (Fig. 1e). The 6-FAM fluorescence on the 5’ end of the DNA was absent, indicating that the ternary complex probably represented a post-DNA cleavage state (Supplementary Fig. 3). In PAM28, the formation of the ternary complex formation was not observed, which was presumably due to Cas9 departing from the nucleosome after cleaving the nucleosome DNA (Supplementary Fig. 3). Therefore, the formation of the ternary complex in PAM1 is anticipated to be due to the presence of an unknown interaction between the nucleosome and Cas9.
To investigate how Cas9 targets nucleosomes to form the complex, we performed a cryo-EM analysis of the Cas9-sgRNA-nucleosome complex targeting the PAM1 site. We used wild-type Cas9, instead of SpCas9-NG, for this cryo-EM analysis because of the higher reconstitution efficiency of the complex. During the data processing, we observed fluctuations in the proximity of Cas9 to the nucleosome (Supplementary Fig. 4 and Supplementary Table 1). We determined the structure of the Cas9-sgRNA-nucleosome complex in a state where Cas9 contacts the core DNA, which we refer to as the “DNA attached state” (Fig. 2).

a The domain organization of S. pyogenes Cas9. Residues 178 to 297 and 766 to 924 were not included in the final model. b Cryo-EM density maps of the Cas9-sgRNA-nucleosome complex in DNA attached state. c Overall structure of the Cas9-sgRNA-nucleosome complex in DNA attached state.
In the present structure, the HNH and REC2 domains were mostly disordered, resembling the post-DNA cleavage state (Fig. 2a, c and Supplementary Fig. 5a, b), as previously reported10,26. Furthermore, no densities of the non-target DNA strand and the phosphate backbone in the target DNA strand at the cleavage site near the bridge helix were observed, indicating the double-strand cleavage of nucleosomal DNA by Cas9 (Supplementary Fig. 5c, d). Therefore, these findings indicated that the complex was in a post-DNA cleavage state, consistent with the results of biochemical experiments (Supplementary Fig. 3).
Within the complex, approximately 15 base pairs of the nucleosome DNA were peeled off from the histone core exposing the N-terminal α-helix in H3 to the solvent (Supplementary Fig. 6a, b). The length of the detached DNA corresponded to the length of DNA that dissociates from the histone core during nucleosome DNA unwrapping. Therefore, Cas9 may bind to the nucleosome upon DNA unwrapping, and maintain the DNA detachment from histones, preventing the DNA from rewrapping around histones. Alternatively, Cas9 binding may induce the nucleosome DNA unwrapping around the entry-exit site. This binding mode, which maintains the DNA dissociation histones, bears similarity to the binding mode observed in previously reported complex structures of endogenous nucleosome-binding proteins, such as RNA polymerase, and nucleosomes27 (Supplementary Fig. 6c).
To our knowledge, there have been no previous reports of bacterial proteins binding to nucleosomes in such a manner that partially alters the nucleosome structure. This observation leads us to speculate that this binding mode may be one of the reasons why Cas9 is functional in eukaryotic cells.
Interactions between PI domain in Cas9 and nucleosome
The Cas9-sgRNA-nucleosome structure suggested several interactions between the PI domain in Cas9 and nucleosome (Fig. 3).

a Close-up view of the suggested Cas9-histone tail interacting regions. The cryo-EM density enclosed by a dashed oval indicates the EM density of the histone tail. b In vitro binding activities of the wild-type Cas9 for canonical nucleosomes and tail-less nucleosomes. The nucleosome was incubated with the Cas9-sgRNA complex at 37 °C for 10 min. The Cy5 fluorescence of Cas9 in reaction products were visualized by Native-PAGE analysis with an Amersham Imager 680 (Cytiva), and quantified by ImageJ. Data are mean ± s.d. (n = 3). The experiments were repeated three times with similar results. c In vitro nucleosome DNA cleavage activities of the wild-type Cas9 for canonical nucleosomes and tail-less nucleosomes. The nucleosome was incubated with the Cas9-sgRNA complex at 37 °C for 10 min. The EtBr stained reaction products were visualized by Native-PAGE analysis with an Amersham Imager 680 (Cytiva), and quantified by ImageJ. Data are mean ± s.d. (n = 3). The experiments were repeated three times with similar results. (d) Close-up view of the suggested interacting regions of the PI edge and nucleosomal DNA. The lysine (K) residues that potentially contact the DNA are highlighted. e In vitro binding activities of the wild-type Cas9 and Cas9 mutants to reduce interaction between PI edge and nucleosome DNA entry. The Cy3 fluorescence of nucleosomes in reaction products were visualized by Native-PAGE analysis with an Amersham Imager 680 (Cytiva), and quantified by ImageJ. Data are mean ± s.d. (n = 3). The experiments were repeated three times with similar results. f In vitro nucleosome DNA cleavage activities of the wild-type Cas9 and Cas9 mutants to reduce interaction between PI edge and nucleosome DNA entry. The 6-FAM fluorescence of nucleosomes in reaction products were visualized by Native-PAGE analysis with an Amersham Imager 680 (Cytiva), and quantified by ImageJ. Data are mean ± s.d. (n = 3). The experiments were repeated three times with similar results.
A weak EM density was observed in the space between Cas9 and the nucleosome, which appears to be an H2A histone-tail (Fig. 3a). Although, we could not identify which residues of the histone tail interact with Cas9 due to the poor density, the histone tail is positively charged, while the Cas9 PI region likely interacting with the histone tail exposes acidic residues (Supplementary Fig. 7), suggesting that the histone tail and Cas9 electrostatically interact. To examine the effects of the histone tail on the binding and DNA cleavage efficiency of Cas9, we performed native-PAGE analyses using tail-less nucleosomes, in which the histone tails were removed. Unexpectedly, there were no significant changes in the binding affinity or DNA cleavage activity of WT Cas9 against tail-less nucleosomes compared to canonical nucleosomes (Fig. 3b, c). These results suggested that the interaction between the histone tail and the PI domain occurs after complex formation and does not play a role in Cas9’s recognition of the target sequence within the nucleosome.
The loop between the two β-sheets (β18-β19) of the PI domain, referred to as the PI edge in this study, were suggested to interact with the DNA near the DNA entry site (Fig. 3d). The PI edge contains numerous lysine residues, which were implemented to recognize the phosphate backbone of DNA (Fig. 3d). To investigate the impact of the PI edge on the stability of the complex and Cas9’s DNA cleavage activity, mutations were introduced at K1151, K1153, K1155, and K1156. Native-PAGE results showed a reduction in complex formation for the mutants (Fig. 3e), suggesting that these lysine residues are crucial for the stability of the Cas9-sgRNA-nucleosome complex obtained in this study. In contrast, there were no significant differences in DNA cleavage efficiencies between the WT and mutants (Fig. 3f). After DNA cleavage, Cas9 reportedly moves away from the DNA, including the PAM28. Therefore, while Cas9 would generally dissociate after cleavage, Cas9 entanglement in the nucleosome through the PI edge may lead to continuous complex formation even after DNA cleavage.
Nonspecific interactions between Cas9 and core DNA affects DNA cleavage efficiency in vitro and in vivo
Two loops, bridging helices 49 and 50 and helices 51 and 52 in the PI domain, were suggested to associate with the phosphate backbone of the core DNA (Fig. 4a). Mutations were introduced into five basic residues (H1264, K1296, H1297, R1298, and K1300) in these loops. We then explored their effects on the binding affinity and DNA cleavage activity towards the nucleosome at PAM1 using native-PAGE analyses (Fig. 4b, c). Interestingly, mutants incorporating H1264, R1298, and K1300 exhibited increases in nucleosome binding and DNA cleavage activities (Fig. 4b, c). Given that Cas9-sgRNA-nucleosome complex formed in PAM1 was in post-DNA cleaved state, it is plausible that binding and cleavage activities increase simultaneously. These results suggested that the cleavage of nucleosomal DNA may be hindered by non-specific interactions between Cas9 and core DNA in vitro, even in the regions where Cas9 can cleave DNA.

a Close-up view of the suggested interactions between the PI domain and nucleosomal core DNA. The lysine (K), arginine (R) and histidine (H) residues that potentially contact the DNA are highlighted. b In vitro binding activities of wild-type Cas9 and Cas9 mutants reducing interaction between Cas9 and core DNA for nucleosomes. The nucleosome was incubated with the Cas9-sgRNA complex at 37 °C for 10 min. The Cy3 fluorescence of nucleosomes in the reaction product was visualized by Native-PAGE analysis, and quantified by ImageJ. Data are mean ± s.d. (n = 3 biologically independent samples). The experiments were repeated three times with similar results. c In vitro nucleosome DNA cleavage activities of wild-type Cas9 and mutants reducing interaction between Cas9 and core DNA. The nucleosome was incubated with the Cas9-sgRNA complex at 37 °C for 30 min. The 6-FAM fluorescence of nucleosomes in the reaction product was visualized by Native-PAGE analysis, and quantified by ImageJ. Data are mean ± s.d. (n = 3 biologically independent samples). The experiments were repeated three times with similar results. t tests, * P < 0.05, ** P < 0.01. d Schematic of the experimental setup for in vitro binding and cleavage assays. e In vivo mutation efficiencies at three target sites (SPL7, SPL17 and GAPDH) in transgenic rice calli of the wild-type Cas9 and Cas9 mutants reducing interaction between Cas9 and core DNA for nucleosomes. Dots indicate the mutation frequency for each independent calli. Bars indicate the average mutation frequency. Error bars indicate the standard error (SE). (The number of calli used for each WT, H1264D/R1298Q, H1264E/R1298Q and H1264A/R1298Q was SPL17; n = 14, 17, 17, 17: SPL17; n = 15, 17, 17, 17: GAPDH; n = 15, 17, 17, 17).
In the proximal CRISPR (proxy-CRISPR) method, the binding of catalytically inactive Cas9 to proximal regions reportedly induces changes in the chromatin structure of the target locus, thus enhancing the genome editing efficiency of another nuclease-active Cas929. Therefore, we conducted mutagenesis in the rice callus by targeting regions where the proxy-CRISPR method enhanced mutation efficacy, as they are presumed to bear chromatin and nucleosome structures (Fig. 4d). Some Cas9 mutants that reduced interaction between the PI domain and core DNA induced mutations at efficiencies similar to those of WT Cas9 (Fig. 4e, Supplementary Fig. 8). In Supplementary Fig. 8, mutation induction efficiency was little higher in Cas9 mutants than that of WT Cas9. It was surprising that the mutants designed to target a specific position within linker DNA exhibited slight but detectable activity even in vivo. Factors contributing to the limited improvement in genome editing activity in vivo compared to in vitro results may include the non-static nature of nucleosome positioning due to as the cell cycle, as well as the potential limitation of the designed mutant in a more dynamic in vivo environment, which contrasts with the controlled conditions in vitro. Therefore, although the Cas9 mutants have a constrained efficacy in the more dynamic conditions present in vivo, these results indicated that the interaction between the PI domain and core DNA may negatively influence genome editing efficiency in vivo.
Discussion
In this study, our native-PAGE analysis showed that Cas9 can target nucleosome DNA end regions, linker DNA and SHL + 6 regions. The native-PAGE analysis also revealed significant differences in the efficiency of Cas9-mediated cleavage of nucleosomal DNA between the DNA entry and exit sites. It is hypothesized that these differences are attributable to the varying flexibility of the nucleosomal DNA between the entry and exit sites23,24,25, which in turn may affect Cas9 accessibility. Thus, flexibility differences at the DNA entry/exit sites, probably caused by the base sequences of the core DNA, may impact Cas9’s DNA cleavage activity. Additionally, since the flexibility of the DNA near the ends of the nucleosomal DNA affected the cleavage efficiency of linker DNA, it is possible that even if target sites are set to avoid nucleosome positions, the sequence and flexibility of the DNA within nearby nucleosomes could control Cas9 access and thereby influence genome editing activity. Future research is expected to elucidate these factors.
We further determined the structure of the Cas9-sgRNA-nucleosome complex in a post-cleaved state. The structure revealed several non-specific interactions between Cas9 and the nucleosome. The native-PAGE analysis for Cas9 mutants suggested that the interaction between the PI domain and core DNA may especially hinder Cas9 DNA cleavage in vitro. We also found a modest increase in genome editing efficiency in vivo, though the efficiency was limited compared to our in vitro data.
Based on these observations, we here propose the following mechanism for the nucleosome-mediated inhibition of DNA cleavage by Cas9 occurred locally (Fig. 5). The inhibition occurs in two stages. First, Cas9 is restricted from accessing the target sequence in nucleosomes by the low flexibility of the DNA end regions. In living organisms, the flexibility of the nucleosome DNA end, which is regulated by the internal sequence of the nucleosomal DNA and post-translational modifications (PTMs), controls the interactions of nucleosome-binding factors30,31. Thus, the DNA accessibility of Cas9 may also be regulated by the nucleosome DNA end flexibility. Second, even if Cas9 overcomes the restriction and binds to the target sequence, non-specific interactions with nucleosomal DNA reduce the motions of Cas9 needed for DNA cleavage, thus decreasing the DNA cleavage efficiency. The specific manner by which the non-specific interactions hinder Cas9 and the inhibited entity are presently unclear. However, given that the PI domain is involved in PAM sequence recognition, it is possible that the interaction of the PI domain may influence PAM recognition.

In the first step, Cas9 is restricted from accessing the target sequence by the flexibility of the DNA end regions. Subsequently, even if Cas9 overcomes the restriction and binds to the target sequence, non-specific interactions with the core DNA reduces helicase activity of Cas9 leading to the decrease of DNA cleavage efficiency. The figure was created by UCSF Chimera X referred SpCas9-sgRNA binary complex (PDB ID: 4ZT0) and structure obtained in this study.
Considering the in vivo results, we speculate that the proposed model above has a limited impact on the in living organism. This study focused on the one-to-one interactions between Cas9 and nucleosomes, but the actual chromatin structure in vivo is far more complex. Nevertheless, we anticipate that further research into these mechanisms and the insights gained will lead to the development of novel tools.
Diverse types of Cas effectors, each exhibiting significant variations in their structures and properties, have been discovered32. The mechanisms of nucleosome access and DNA cleavage activities are also anticipated to diverge among these Cas effectors. The experimental series conducted in this study, ranging from the exploration of binding and DNA cleavage sites on nucleosomes to structural analysis, can be applied to other Cas effectors. This approach may clarify the interplay between various Cas effectors and nucleosomes, leading to the development of novel genome editing tools. This study offers a new perspective on the development of genome editing tools by highlighting the interaction between Cas proteins and chromatin structures.
Methods
Expression and purification of proteins
The genes encoding full-length S. pyogenes Cas9 (residues 1–1368) and S. pyogenes Cas9 mutant R1335V/G1288R/T1337R/L1111R/A1322R/E1219F/D1135V (NG SpCas9)17 were separately cloned into a pET28 vector with an N-terminal hexa-histidine (His6) tag. The WT SpCas9 and SpCas9 mutant were expressed at 20 °C in Escherichia coli Rosetta 2 (DE3) (Novagen), and purified by chromatography on Ni-NTA Superflow resin (QIAGEN). The eluted protein was incubated overnight at 4 °C with TEV protease to remove the His6–tag, and further purified by chromatography on Ni-NTA, Hitrap SP HP (GE Healthcare) and HiLoad Superdex 200 16/60 (GE Healthcare) columns in buffer consisting of 20 mM Tris-HCl (pH 8.0) with 150 mM NaCl.
Recombinant histone proteins were prepared as recombinant proteins27. For the preparation of fluorescent labeled nucleosomes, E63 in H4 was substituted with Cysteine. Histone proteins H2A, H2B, H3, and H4 were expressed as His6-tagged proteins in E. coli cells, and then purified by Ni-NTA column chromatography (Qiagen) under denaturing condition. After His6 tag cleavage by thrombin protease (Wako), the resulting histone proteins were further purified by MonoS cation exchange column chromatography (GE Healthcare). The purified histone proteins were lyophilized and stored at 4 °C.
Preparation of fluorescent DNA and proteins
Cas9 was mixed with a three-fold molar excess of Cy5-NHS-ester (Lumiprobe), and further incubated at 4 °C for 2 h. Subsequently, Cy5-labeled Cas9 was purified by HiLoad Superdex 200 16/60 (GE Healthcare) columns in buffer (20 mM Tris-HCl (pH 8.0), 150 mM NaCl) (Supplementary Fig. 2).
For the preparation of an histone octamer containing fluorescent H4, E63C in H4 was labeled with Cy3-maleimide (Lumiprobe). After labeling, H2A, H2B, H3, and H4 were mixed under denaturing conditions. The mixture was dialyzed against high-salt buffer, and the octamer was reconstituted and purified by Superdex 200 gel filtration chromatography.
For the preparation of DNA fragment containing 6-FAM, a 193 bp DNA containing the Widom 601 sequence was PCR-amplified using a primer labeled with 6-FAM at the 5’ end synthesized by FASMAC. The product was then phenol-chloroform extracted, ethanol precipitated, and purified by non-denaturing PAGE.
Nucleosome reconstitution
The 193 bp Widom 601 DNA16 without 6-FAM was prepared by amplification of the plasmid containing the 193 bp DNA fragment in E. coli cells and extracted by EcoRV33. The nucleosome was reconstituted by the salt dialysis method, and purified by non-denaturing electrophoresis using a Prep Cell apparatus (Bio-Rad) (all other experiments)27. The purified nucleosomes were dialyzed in 20 mM Tris-Cl buffer (pH7.5) containing 1 mM DTT and 5 % glycerol. The resulting nucleosomes were flash-frozen in liquid nitrogen and stored at −80 °C.
Single-guide RNA (sgRNA) preparation
With respect to the location of PAM sequences in the nucleosomal DNA, we designed sgRNA sequences to target PAM sequences present on at least one histone side and one solvent side at each super helix location (SHL) in the nucleosome (Fig. 1b and Supplementary Fig. 1a). The 100-nt sgRNA was transcribed in vitro with T7 polymerase using a DNA template purchased from Eurofins Genomics, and was purified by 10% denaturing polyacrylamide gel electrophoresis. The sequences of sgRNAs used in this study are listed in Supplementary Table. 1.
Native-PAGE analyses
Nucleosome, sgRNA, and Cas9 (WT/NG/Mutants) binding assays for the gel shift assays in this study were all conducted in the same manner. First, the complete Cas9-sgRNA ribonucleoprotein complex was formed by incubating a two-fold molar excess of the chosen sgRNA with Cas9 at room temperature for 5 min. Next, 0.5 µM DNA substrate (either naked DNA or nucleosome) was mixed with Cas9 and sgRNA (molar ratio, 1:3:6) in buffer containing 10 mM Tris-HCl pH 8.0, 15 mM NaCl, 5 mM MgCl2, and 1 mM DTT. For the detection of Cas9-mediated DNA cleavage, the Cas9-sgRNA ribonucleoprotein (RNP) incubated with nucleosomes was deproteinized by Proteinase K treatment at 37 °C for 30 min. Products were separated on a native 5% polyacrylamide gel with 0.5x TBE as the running buffer. Cy3, Cy5 and 6 FAM fluorescence signals were scanned by using the Amersham Typhoon scanner (Cytiva) before gels were stained with ethidium bromide.
Preparation of the Cas9-sgRNA-nucleosome complex for cryo-EM
We utilized WT SpCas9 instead of the SpCas9-NG mutant for cryo-EM analysis because of the higher complex reconstruction efficiency. For the preparation of SpCas9-sgRNA-nucleosome complexes crosslinked for single particle analysis, the SpCas9-sgRNA-nucleosome binding reaction was performed by mixing nucleosome (0.5 μM), SpCas9 (2 μM), and sgRNA (2 μM) in 2 mL of reaction solution (18 mM Tris-Cl (pH 7.5), 30 mM NaCl, 1.2 mM DTT, 5 mM MgCl2 and 1% glycerol) at 37 °C for 30 min. To stabilize and purify the Cas9-sgRNA-nucleosome complex, the reaction mixture was fractionated by the GraFix method34. Gradient solutions were prepared using low sucrose concentration solution (10 mM HEPES-NaOH (pH 7.5), 20 mM NaCl, 1 mM DTT, and 5% sucrose) and high sucrose concentration solution (10 mM HEPES-NaOH (pH 7.5), 20 mM NaCl, 1 mM DTT, 20 % sucrose, and 1 % glutaraldehyde [diluted from glutaraldehyde solution, Electron Microscopy Sciences, 16220]). Half of the reaction mixture was loaded on the top of the gradient solution. The sample was centrifuged at 125,000 x g at 4 °C for 16 h using a Beckman SW41 rotor. After centrifugation, 750 μL fractions were taken from the top of the gradient, and the fractions were analyzed by Native-PAGE. The Cas9-sgRNA-nucleosome complex sample was dialyzed in a buffer containing 20 mM HEPES-NaOH (pH 7.5) and 1 mM DTT. The resulting samples were concentrated using Amicon Ultra 30 K filters (Merck Millipore). The DNA concentrations of the Cas9-sgRNA-nucleosome complexes plunge frozen for cryo-EM was 367.7 μg/mL.
Electron microscopy and data collection
The purified complex solution was applied to freshly glow-discharged Cu/Rh 200 mesh R1.2/1.3 grids (Quantifoil), in a Vitrobot Mark IV (FEI) at 16 °C with a blotting time of 4 s under 100% humidity conditions.
The cryo-EM data were collected using a Titan Krios G3i microscope (Thermo Fisher Scientific), running at 300 kV and equipped with a Gatan Quantum-LS Energy Filter (GIF) and a Gatan K3 Summit direct electron detector in the electron counting mode. Each movie was recorded at a nominal magnification of 81,000 x, corresponding to a calibrated pixel size of 1.05 Å (The University of Tokyo, Japan) at the electron exposure of 11.59 e/pix/sec for 5.656 s, resulting in an accumulated exposure of 54.5 e/ Å2. The data were automatically acquired by the image shift method using the SerialEM software35, with a defocus range of 1.0 to 2.5 mm, and 7875 movies were acquired. The dose-fractionated movies were subjected to beam-induced motion correction and dose-weighting, using the MotionCor236 algorithm implemented in RELION-3.137, and the contrast transfer function (CTF) parameters were estimated using CTFFIND438.
Electron microscopy data processing
All cryo-EM data processing steps were conducted using the cryoSPARC v3.2.0. software package39 (Supplementary Fig. 5). From the motion-corrected and dose-weighted micrographs, 4,459,962 particles were automatically picked using Blob Picker in cryoSPARC. The particles were subjected to several rounds of reference-free 2D classifications, followed by cryoSPARC Ab-Initio Reconstruction to curate particle sets. The 1,087,299 particles were further curated by cryoSPARC Heterogenous Refinement (N = 3), using the map derived from the cryoSPARC Ab-Initio Reconstruction as the template. The selected 305,595 particles were further subjected to 3D variability analysis40. The resulting maps with different conformations were refined using non-uniform refinement41, yielding maps at resolutions of 4.56 Å, according to the FSC criterion of 0.14342. The local resolution was estimated with BlocRes in cryoSPARC. Cryo-EM data collection, refinement, and validation statistics are available in Supplementary Table 3.
Model building
Rigid docking was performed in COOT43 using the crystal structures of the Cas9-sgRNA-target DNA complex (PDB code 4UN3) and the nucleosome (PDB code 3LZ0).
Figure preparation
Figures were prepared using UCSF ChimeraX44, CueMol (http://www.cuemol.org). To quantify the binding ratio and DNA cleavage efficiency, we used ImageJ and GraphPad Prism. Both binding ratio and DNA cleavage efficiency were estimated. To quantify the binding ratio of Cas9 to nucleosomes and the efficiency of nucleosomal DNA cleavage, we quantified the bands in Native-PAGE using ImageJ. We quantified the fluorescence of Cy3 in H4 in the gel for the binding ratio, and 6-FAM at the 5’-end of nucleosomal DNA for the DNA cleavage efficiency. However, since it was difficult to label tail-less nucleosomes with fluorescence, we quantified the fluorescence of Cy5 in Cas9 for the binding rate and the band of EtBr-stained DNA for the DNA cleavage efficiency. The quantified data was imaged by GraphPad Prism. The binding ratio was calculated as the ratio of the intensity of the band indicating the complex to the sum of the intensity of the bands indicating the complex and the nucleosome. For calculating DNA cleavage efficiency, the difference between the intensity of the band nucleosomes were loaded and the band Cas9 added was divided by the intensity of the band only nucleosomes loaded.
Transformation of CRISPR/Cas9 vector into rice callus
Binary vectors containing three sgRNAs, rice codon-optimized SpCas9-WT or -H1264A/R1298Q, H1264D/R1298Q, H1264E/R1298Q, and hygromycin phosphotransfease (HPT) expression cassettes (Fig. 4d and Supplementary Table 2) were transformed into scutellum-derived calli or rice (Oryza sativa L. cv. Nipponbare) by Agrobacterium45. Briefly, 11 to 14-day-old cultured rice primary calli were infected with Agrobacterium [strain EHA10546]. After 3 days of co-cultivation, the calli were transferred to callus induction medium containing 50 mg/l hygromycin B (Wako Pure Chemicals) and 25 mg/l meropenem (Wako Pure Chemicals). All the hygromycin resistant secondary calli that emerged from the single primary callus were pooled together and extracted DNA for sequencing analysis47.
Amplicon sequence analysis
The first PCR was conducted with the primer sets listed in Supplementary Table 2. PCR products were purified using Agencourt AMPure XP beads (Beckman Coulter, CA, USA) and used as templates for a 2nd round of PCR to attach sequence adapters (IDT for Illumina DNA/RNA UD indexes, Illumina, CA, USA) for amplicon-sequencing libraries. The second round PCR products were purified using Agencourt AMPure XP beads. The fragment sizes were checked by agarose gel electrophoresis, and the concentration were measured using a Qubit 2.0 Fluorometer and a Qubit dsDNA HS Assay Kits (ThermoFisher Scientific). The sequencing libraries were subjected to paired-end sequencing on a Miseq sequencer (Illumina). Amplicon sequence results were analyzed using CRISPResso2 (http://crispresso2.pinellolab.org)48.
Statistical analysis
Data are presented as means ± s.d. The alpha level was 0.05 or 0.01. Statistical analyses were performed in GraphPad Prism, using t tests as indicated.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic models have been deposited in the PDB under the accession codes 8YNY. The cryo-EM density map has been deposited in the Electron Microscopy Data Bank under the accession codes EMD-39431. Source data are provided with this paper.
References
Hille, F. et al. The Biology of CRISPR-Cas: Backward and Forward. Cell 172, 1239–1259 (2018).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Travers, A. & Muskhelishvili, G. Bacterial chromatin. Curr. Opin. Genet. Dev. 15, 507–514 (2005).
Woodcock, C. L., Safer, J. P. & Stanchfield, J. E. Structural repeating units in chromatin. I. Evidence for their general occurrence. Exp. Cell Res. 97, 101–110 (1976).
Luger, K., Mäder, A. W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260 (1997).
Zhou, K., Gaullier, G. & Luger, K. Nucleosome structure and dynamics are coming of age. Nat. Struct. Mol. Biol. 26, 3–13 (2019).
Anders, C., Niewoehner, O., Duerst, A. & Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573 (2014).
Nishimasu, H. et al. Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA. Cell 156, 935–949 (2014).
Jiang, F. et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871 (2016).
Zhu, X. et al. Cryo-EM structures reveal coordinated domain motions that govern DNA cleavage by Cas9. Nat. Struct. Mol. Biol. 26, 679–685 (2019).
Cofsky, J. C., Soczek, K. M., Knott, G. J., Nogales, E. & Doudna, J. A. CRISPR-Cas9 bends and twists DNA to read its sequence. Nat. Struct. Mol. Biol. 29, 395–402 (2022).
Pacesa, M. et al. R-loop formation and conformational activation mechanisms of Cas9. Nature https://doi.org/10.1038/s41586-022-05114-0 (2022) .
Yarrington, R. M., Verma, S., Schwartz, S., Trautman, J. K. & Carroll, D. Nucleosomes inhibit target cleavage by CRISPR-Cas9 in vivo. Proc. Natl Acad. Sci. Usa. 115, 9351–9358 (2018).
Horlbeck, M. A. et al. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife 5, (2016).
Hinz, J. M., Laughery, M. F. & Wyrick, J. J. Nucleosomes Inhibit Cas9 Endonuclease Activity in Vitro. Biochemistry 54, 7063–7066 (2015).
Lowary, P. T. & Widom, J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276, 19–42 (1998).
Nishimasu, H. et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259–1262 (2018).
Hinz, J. M., Laughery, M. F. & Wyrick, J. J. Nucleosomes Selectively Inhibit Cas9 Off-target Activity at a Site Located at the Nucleosome Edge. J. Biol. Chem. 291, 24851–24856 (2016).
Makasheva, K. et al. Multiplexed Single-Molecule Experiments Reveal Nucleosome Invasion Dynamics of the Cas9 Genome Editor. J. Am. Chem. Soc. 143, 16313–16319 (2021).
Handelmann, C. R., Tsompana, M., Samudrala, R. & Buck, M. J. The impact of nucleosome structure on CRISPR/Cas9 fidelity. Nucleic Acids Res. https://doi.org/10.1093/nar/gkad021 (2023).
Li, G. & Widom, J. Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol. 11, 763–769 (2004).
Bilokapic, S., Strauss, M. & Halic, M. Histone octamer rearranges to adapt to DNA unwrapping. Nat. Struct. Mol. Biol. 25, 101–108 (2018).
Li, M. & Wang, M. D. Chapter Two - Unzipping Single DNA Molecules to Study Nucleosome Structure and Dynamics. in Methods in Enzymology (eds. Wu, C. & Allis, C. D.) vol. 513 29–58 (Academic Press, 2012).
Ngo, T. T. M., Zhang, Q., Zhou, R., Yodh, J. G. & Ha, T. Asymmetric unwrapping of nucleosomes under tension directed by DNA local flexibility. Cell 160, 1135–1144 (2015).
Mauney, A. W., Tokuda, J. M., Gloss, L. M., Gonzalez, O. & Pollack, L. Local DNA Sequence Controls Asymmetry of DNA Unwrapping from Nucleosome Core Particles. Biophys. J. 115, 773–781 (2018).
Zuo, Z. & Liu, J. Allosteric regulation of CRISPR-Cas9 for DNA-targeting and cleavage. Curr. Opin. Struct. Biol. 62, 166–174 (2020).
Kujirai, T. et al. Structural basis of the nucleosome transition during RNA polymerase II passage. Science 362, 595–598 (2018).
Shibata, M. et al. Real-space and real-time dynamics of CRISPR-Cas9 visualized by high-speed atomic force microscopy. Nat. Commun. 8, 1430 (2017).
Chen, F. et al. Targeted activation of diverse CRISPR-Cas systems for mammalian genome editing via proximal CRISPR targeting. Nat. Commun. 8, 14958 (2017).
North, J. A. et al. Regulation of the nucleosome unwrapping rate controls DNA accessibility. Nucleic Acids Res. 40, 10215–10227 (2012).
Tóth, K. et al. Histone- and DNA sequence-dependent stability of nucleosomes studied by single-pair FRET. Cytom. A 83, 839–846 (2013).
Bhatia, S., Pooja & Yadav, S. K. CRISPR-Cas for genome editing: Classification, mechanism, designing and applications. Int. J. Biol. Macromol. 238, 124054 (2023).
Arimura, Y., Tachiwana, H., Oda, T., Sato, M. & Kurumizaka, H. Structural analysis of the hexasome, lacking one histone H2A/H2B dimer from the conventional nucleosome. Biochemistry 51, 3302–3309 (2012).
Kastner, B. et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat. Methods 5, 53–55 (2008).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zivanov, J., Nakane, T. & Scheres, S. H. W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A. & Fleet, D. J. 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 213, 107702 (2021).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Goddard, T. D. et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Toki, S. Rapid and efficientAgrobacterium-mediated transformation in rice. Plant Mol. Biol. Rep. 15, 16–21 (1997).
Hood, E. E., Gelvin, S. B., Melchers, L. S. & Hoekema, A. NewAgrobacterium helper plasmids for gene transfer to plants. Transgenic Res. 2, 208–218 (1993).
Edwards, K., Johnstone, C. & Thompson, C. A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res. 19, 1349 (1991).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Acknowledgements
We thank Y. Matsuzaki and M.Nishimura for assistance with Native-PAGE analyses, Y. Kise for assistance with electron microscopy, Y. Itoh for assistance with model building and PDB entry, and the members of the O.N. laboratory for comments and discussions. We thank R.Nakagawa for crucial comments and thoughtful advice. Electron microscopy data were collected at the cryo-EM facility at the University of Tokyo. M.E., S.T. and H.S. was supported by Cross-ministerial Strategic Innovation Promotion Program (SIP) “Building a Resilient and Nourishing Food Supply Chain Management for a Sustainable Future” from Bio-oriented Technology Research Advancement Institution under Grant Number JPJ012287. H.K. was supported by JSPS KAKENHI Grant Number JP23H05475 and JP24H02328, Research Support Project for Life Science and Drug Discovery (BINDS) from AMED under Grant Number JP23ama121009 and JP24ama121009, JST CREST Grant Number JPMJCR24T3 and JST ERATO Grant Number JPMJER1901. T.Kujirai was supported by JSPS KAKENHI Grant Number JP20H03201, JP20H05690, JP22K15033, JP23K17392, JP24H00062. O.N. was supported by AMED grant numbers JP23fa627001 and JP19am0401005; the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under grant numbers JP23ama121002 (support number 3272) and JP23ama121012; and the Cabinet Office, Government of Japan, Public/Private R&D Investment Strategic Expansion Program (PRISM), grant number JPJ008000.
Author information
Authors and Affiliations
Contributions
R.N. and Y.S. prepared SpCas9 and SpCas9 mutants samples. J.K. and T.Kujirai prepared nucleosome samples. R.N. performed biochemical analyses with assistance from J.K. and T.Kujirai. R.N. performed structural analyses with assistance from T.Kujirai and T.Kusakizako. R.N performed model building and structural refinement with assistance from T.Kujirai, H.H. and T.Kusakizako. M.E. and H.S. performed mutagenesis assays in rice calli with assistance from S.T.. R.N. wrote the manuscript with help from all authors. O.N. and H.K. supervised the research.
Corresponding authors
Ethics declarations
Competing interests
O.N. is a co-founder, board member and scientific advisor for Curreio. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Martin Depken and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nagamura, R., Kujirai, T., Kato, J. et al. Structural insights into how Cas9 targets nucleosomes. Nat Commun 15, 10744 (2024). https://doi.org/10.1038/s41467-024-54768-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54768-z
