
Abstract
Protein translocation across cellular membranes is an essential and nano-scale dynamic process. In the bacterial cytoplasmic membrane, the core proteins in this process are a membrane protein complex, SecYEG, corresponding to the eukaryotic Sec61 complex, and a cytoplasmic protein, SecA ATPase. Despite more than three decades of extensive research on Sec proteins, from genetic experiments to cutting-edge single-molecule analyses, no study has visually demonstrated protein translocation. Here, we visualize the translocation, via one unit of a SecYEG-SecA-embedded nanodisc, of an unfolded substrate protein by high-speed atomic force microscopy (HS-AFM). Additionally, the uniform unidirectional distribution of nanodiscs on a mica substrate enables the HS-AFM image data analysis, revealing dynamic structural changes in the polypeptide-crosslinking domain of SecA between wide-open and closed states depending on nucleotides. The nanodisc-AFM approach will allow us to execute detailed analyses of Sec proteins as well as visualize nano-scale events of other membrane proteins.
Similar content being viewed by others

Introduction
Many nascent polypeptide chains with signal peptides synthesized by ribosomes in the cytoplasm are transported across the membrane through the Sec channel. The Sec pathway is conserved in all organisms1. In the model bacterium Escherichia coli, many periplasmic or outer membrane proteins are first synthesized in the cytoplasm, then directed to the Sec channel, the SecYEG complex, in the inner membrane based on the information of their signal peptides, and finally passed through the membrane, while remaining in unfolded states (Supplementary Fig. 1a)2,3. SecYEG is composed of three membrane proteins, SecY, SecE, and SecG, and provides a pathway specialized for unfolded nascent chains4. SecYEG does not function as it is: the protein transport is driven by the motor protein SecA ATPase5 and the membrane protein SecDF6, and proton motive force also contributes to protein transport7. In the case of nascent chains for inner membrane proteins, SecYEG mediates their integration into the cytoplasmic membrane in cooperation with the membrane chaperone/insertase YidC8,9. When nascent chains are for periplasm-localized proteins, their signal sequences are cleaved by signal peptidase10, and the membrane-translocated region folds into mature structures in the periplasm. This process is facilitated by chaperones such as the PpiD/YfgM complex and SurA11,12,13. When nascent chains are for outer membrane proteins, the regions translocated across the inner membrane are inserted into the outer membrane from the periplasm via the BAM complex14,15,16. If protein integration into the outer membrane stalls, the protein being transported is degraded by protease BepA and removed from this transport pathway17,18,19,20,21. In the Sec-mediated protein transport processes, several proteins are thus involved, but SecA, membrane-embedded SecYEG, and substrate proteins alone are sufficient to demonstrate minimal ATP-dependent membrane translocation activity across the membrane in vitro as the initial step reaction22.
In 2008, the crystal structure of the SecA-SecY channel complex was reported under a translocating peptide-free condition23. By improving the conditions, two detailed structures of the SecA-SecY complex disulfide-linked with translocating peptides were later reported by X-ray crystallography and cryo-electron microscopy24,25 (Supplementary Fig. 1b). All of these structures show monomeric SecA bound to one SecY channel, although some regions of the translocating peptide were partially disordered, and suggested that the functional unit consists of one SecYEG and one SecA. In a functional analysis based on structural information, the two-helix-finger (THF) domain of SecA plays a crucial role26, and a power stroke-type push-and-slide model in which SecA repeatedly pushes the translocating protein toward the Sec translocon was proposed27. The model is also based on the fact that SecA anchors the polypeptide in the ATP-bound state and releases it in the ADP-bound state28. In SecA, the clamp region between the polypeptide-crosslinking domain (PPXD) and the nucleotide-binding domain (NBD) is also an important substrate-binding site29. In addition, it was demonstrated that SecA produces mechanical force and actively drives the protein translocation30. Recently, detailed structures of the SecA-SecY channel complex with a translocating peptide without any artificially introduced disulfide bonds were reported in an ADP-BeF3− state as well as in an ADP-bound state31. An important finding in this report was that the interaction between SecA and a translocating peptide was partially but clearly confirmed. Comparing these structures yielded a hand-switching model in which a substrate-binding area of SecA, not the helical wing domain (HWD), changes its interaction with the translocating peptide depending on nucleotide-binding states. While detailed and concrete molecular mechanisms of SecYEG-SecA models have been proposed, SecA has also been reported to impale in the membrane and reach the periplasm32. In the SecA crystal structures, a flexible PPXD shows different orientations (Supplementary Fig. 1b and c). In this paper, we define the structure of SecA in the SecYEG-SecA complex as its closed form and the SecA structures reported by Osborne et al.33 and Hunt et al.34 as its open and wide-open forms, respectively. In fact, SecA undergoes conformational changes between these forms in solution35,36,37. Furthermore, conformational changes of the PPXD coupled with nucleotide-binding states facilitate the translocation of nascent chains38. In contrast, a Brownian ratchet model was inspired by molecular dynamics simulations based on the SecY-SecA structures39,40,41,42, in which Arg residues of the translocating protein play an important role in determining the direction of protein transport43. Other models have been proposed, such as a substrate catch-and-release model derived from SecA conformational changes of the substrate interaction region coupled with its nucleotide-binding states44, but no unified view has yet emerged.
SecA characteristically changes its oligomeric state between monomer and dimer45,46. This oligomeric change of SecA is reported to be important for its motor function; for example, when dimeric SecA in the cytoplasm is recruited to the membrane and then dissociates, SecA functions as a monomer on the membrane47,48. According to a recent super-resolution microscopy analysis, in vivo, most SecA molecules are localized as dimers on the inner membrane49. These are only a few examples; the oligomeric state of SecA is still controversial. Many studies could not control the state of SecA in the equilibrium of its monomer and dimer, nor the association/disassociation of SecA with SecYEG, which hindered precise analysis and led to frequent incorrect interpretations, making them prominent issues in SecA research. In an AFM observation of SecA alone, conformational changes were mainly caused by the PPXD, although the information is limited because SecA was not in an activated state for protein translocation50. AFM observations of membrane-reconstituted SecYEG and SecA have shown changes in the binding of SecA to SecYEG51 and changes in the SecA oligomeric state depending on nucleotides and substrates52. However, these observations of SecYEG-SecA complexes have not yet been able to detect the details of structural changes of domains in real-time. As mentioned above, due to the difficulty in controlling SecA oligomers and the resolution limit, definitive real-time images of the single-unit have not been obtained. In view of the spatiotemporal scale of the protein translocation reaction, high-speed atomic force microscopy (HS-AFM) is a suitable observation device, but optimizations of the experimental system are crucial for further detailed analysis. Consequently, it is necessary to (1) strictly adjust SecA-SecYEG stoichiometry, (2) provide the membrane environment necessary for protein translocation, and (3) isolate one unit completely.
Single-unit particles of membrane proteins embedded in nanodiscs, consisting of lipids and membrane scaffold proteins, can be isolated for analyses. Nanodiscs containing the bacterial SecY channel were used for single-particle analysis by cryo-electron microscopy24,53 and binding assays with SecA54,55,56. Taufik et al. showed that SecYEG-embedded nanodiscs could transport a preprotein (substrate), proOmpA, through the channel57, but they could not control the oligomeric state of SecA. Our group have been analyzing purified Thermus thermophilus Sec proteins because of their high thermal stability6,58. A fusion protein of T. thermophilus SecY and SecA enabled us to purify the SecYEG-SecA complex (SecYAEG), resulting in the stoichiometry of SecYEG and SecA definitely being 1:1. SecYAEG-reconstituted liposomes retained protein translocation activity59, although we could not rule out the possibility that the SecYAEG complex forms a further oligomeric state, such as (SecYAEG)2, on the liposomes during protein translocation. The SecYAEG complex was next reconstituted into nanodiscs for the construction of purified, homogeneous one-unit particles. The resulting particles (SecYAEG-NDs), at least possessing the ability to bind preprotein59, were observed by HS-AFM, showing that they are uniform60 (Fig. 1a upper). In that report, images of uniform particles consisting of two ellipses were observed, and larger ellipses were verified as the region of SecYEG embedded in the nanodisc by streptavidin labeling of SecY. That is, SecYAEG-embedded nanodiscs are immobilized with a side-on orientation on the mica surface, and we could distinguish the regions of SecYEG and SecA.

a Typical HS-AFM images of SecYAEG-ND (upper panels) and SecYA(ΔPPXD)EG-ND (lower panels) on a mica surface. The AFM image height (nm) is shown by the color bar. Representative data from more than 10 independent experiments with similar results are shown. b Cross-sectional height profiles for SecA regions along the red lines shown in (a). c Schematic representation of the structural models of SecYAEG-ND (upper) and SecYA(ΔPPXD)EG-ND (lower) at 80° in (f), accompanied by conical AFM probes utilized in the generation of simulated AFM images. PPXD is highlighted in yellow, the rest of SecA in blue, and SecYEG in magenta. d Simulated AFM images of SecYAEG-ND (upper) in wide-open form and SecYA(ΔPPXD)EG-ND (lower) in (c). The simulated AFM image height (nm) is shown by the color bar. e Cross-sectional height profiles for SecA regions along the red lines shown in (d). f Averaged image correlation scores as a function of rotation angles between the actual HS-AFM and the simulated AFM images. Black arrowhead indicates the angle with the highest correlation score. Source data of the graphs are provided as a Source Data file.
In this study, we initiate the protein translocation reaction in one unit of SecYAEG-ND in the presence of ATP and preproteins, and visualize it by HS-AFM in real time. Furthermore, structural changes caused by PPXD and depending on the type of nucleotides are confirmed. This is a critical first step toward clearly visualizing the protein transport, which is one of the nano-scale as well as essential events in the cell.
Results and discussion
Orientation of SecYAEG-ND on mica surface
For AFM observation of proteins in aqueous environments, freshly prepared cleaved mica is commonly employed as a support substrate because of the inherent cleanliness and atomic-level smoothness of the mica surface61. Since protein immobilization on the negatively charged mica surface occurs primarily through electrostatic interactions62, the orientations of protein molecules upon the mica are influenced by their three-dimensional conformation and surface charge distribution. Therefore, certain proteins can be placed on the mica surface in a homogeneous orientation.
Our previous HS-AFM observations have shown that SecYAEG-NDs presented homogeneous particles, consisting of a pair of elliptical particles, immobilized onto the mica surface in a side-on orientation. The larger and smaller elliptical particles, which were identified as the SecYEG-ND and SecA regions, respectively60, are shown in Fig. 1a, upper. Although these similar molecular images of SecYAEG-ND captured by HS-AFM suggested that SecYAEG-NDs were immobilized on the mica surface with a homogeneous orientation, the definitive orientation of SecYAEG-ND during HS-AFM imaging remains indeterminate. To observe structural changes of exposed SecA, the correlation between its 3D structure and its HS-AFM image must initially be clarified. SecA possesses the independent distinguishable PPXD domain (Supplementary Fig. 1c), which undergoes conformational transitions during protein translocation. Here, we attempted to determine the orientation plane of observed SecYAEG-ND images using a PPXD-deleted mutant complex, SecYA(ΔPPXD)EG.
First, compared to SecYAEG-ND images, SecYA(ΔPPXD)EG-ND images showed that a portion of SecA, probably corresponding to the PPXD domain, was missing, leading to a height reduction of 3.5 nm (Fig. 1a and b). Next, taking advantage of this distinct variation in height distribution in the absence of the PPXD, we ascertained the adsorption orientation of the ΔPPXD mutant on the mica surface by comparing the observed HS-AFM image with simulated AFM images generated from a predicted structure model (Fig. 1c lower and 1d lower)63. The predicted model was a stable conformation of SecYA(ΔPPXD)EG-ND obtained from molecular dynamics (MD) simulation using AlphaFold-predicted SecYA(ΔPPXD)EG complex (as detailed in Materials and Methods, Supplementary Fig. 2). Finally, we calculated and plotted two-dimensional image correlation average scores between the HS-AFM image of the ΔPPXD mutant (Fig. 1a lower) and simulated AFM images as a function of the rotational angles (Fig. 1f). The simulated AFM images were generated by systematically rotating the predicted structural model in 5-degree increments, covering the range 0 to 360 degrees around the nanodisc’s central axis (Fig. 1f right). The highest image score was observed at 80°, a reasonable angle considering the electrostatic interactions with the mica surface: the surface of SecYA(ΔPPXD)EG-ND in contact with the mica surface at this angle is predominantly positively charged (Supplementary Fig. 3a). Given the surface charge, SecYEG-ND, possessing the PPXD domain, is almost certain to be positioned on the mica substrate at the same angle (Supplementary Fig. 3b). Moreover, the structural changes in the flexible region (PPXD) of SecA, discussed later, consistently occur at the same location on each particle, further supporting this idea. The height variations deduced from cross-sectional analyses of the HS-AFM image and the simulated AFM image of the ΔPPXD mutant were 3.5 nm and 3.4 nm, respectively, demonstrating a close congruence (Fig. 1b and e). In addition, we note a maximum height discrepancy between the HS-AFM and simulated AFM images of SecYAEG-ND (Fig. 1b and e). The height of the HS-AFM images is approximately 4 nm lower than that of the simulated AFM images. This height difference may be due to nanodisc deformation from circular to elliptical shape, interactions with the mica substrate, mechanical effects from the AFM probe, and/or shifting of the SecYAEG complex’s position in the nanodisc. Indeed, an elliptical formation of nanodiscs containing SecY has been shown in cryo-EM analysis24. To demonstrate that these factors could contribute to the height difference, we generated a deformed nanodisc structure by scaling and created a simulated AFM image. Comparison of the simulated image with the actual HS-AFM image revealed that the height and size of the nanodisc corresponded closely (Supplementary Fig. 4). These results suggest that the observed height difference is attributable to substrate adsorption or mechanical effects induced by the AFM probe. While the overall height of SecYAEG-ND was not congruent, it is noted that the observed 3.5 nm height change of the SecA region arising from ΔPPXD is consistent with that in the simulated AFM images (3.4 nm), with similar shapes (Fig. 1b and e).
The simulated AFM image of SecYAEG-ND (Fig. 1d upper and Supplementary Fig. 5 right) was created for its wide-open form. In contrast, when simulated AFM images representing the open and closed forms at an 80° rotation angle (Supplementary Fig. 5 left and center) were compared with the HS-AFM images (Fig. 1a upper), the simulated AFM image of the wide-open structure showed the best correspondence. Consequently, we conclude that the resting state of SecYAEG-ND is the wide-open structure.
HS-AFM visualization of protein translocation by SecYAEG-ND
We embarked on the following experimental phase to directly observe the protein translocation mediated by SecYAEG-ND. As a model substrate protein for translocation, we employed proOmpA-sfGFP, which consists of three parts: an N-terminal signal sequence, unfolded OmpA region, and sfGFP at the C-terminus (Fig. 2a upper)59. Because the sfGFP tag readily folds in solution, we were able to observe these tags, which serve as C-terminal markers, as distinct particles by HS-AFM; this modification facilitated the visualization of the extended proOmpA in HS-AFM images. Initially, proOmpA-sfGFP molecules were imaged to confirm the anticipated characteristic string-like structures of unfolded peptides, which exhibit a flexible structure that alternates between loosely entangling and unfolding (Fig. 2a and Supplementary Movie 1). Many string-like particles showed some tangled regions. The histogram of the end-to-end distances between the center of the sfGFP tag and the opposite end, corresponding to the signal sequence, exhibits a single Gaussian distribution with a mean (±s.d.) value (X0) of 22 ± 9 nm. Next, using a mixture of SecYAEG-ND and proOmpA-sfGFP, we visualized string-like structures with a distinct sfGFP blob at one end, protruding from the SecYAEG-NDs (Fig. 2b and Supplementary Movie 2). In contrast, no string-like structures were observed in the absence of proOmpA-sfGFP60 (Fig. 1a). When observed with a wide scan range, some SecYAEG-ND particles were complexed with proOmpA-sfGFP, while others did not form substrate complexes (Supplementary Fig. 6). This HS-AFM observation is consistent with the partial formation of substrate-SecYAEG-ND complexes previously detected in a native PAGE experiment59. Approximately 16% of 262 observed molecules were accompanied by a string-like structure, existing as SecYAED-ND/proOmpA-sfGFP complexes. The fact that not all SecYAEG-NDs formed complexes with the substrate is also consistent with the native PAGE results59. Furthermore, when we used a signal sequence mutation of proOmpA, known as 3Q, which disrupts the interaction between proOpmpA and SecY25,59, string-like structures did not bind to SecYAEG-NDs and were separately observed (Supplementary Fig. 7). The X0 for the histogram of the end-to-end distances of the string-like structures extending from SecYAEG-ND was determined to be 20 ± 10 nm, which is 2 nm shorter than the observed distance for proOmpA-sfGFP alone. The observed distance difference is likely attributable to the signal sequence being bound to SecA. From these distance measurements, we conclude that recognition of the signal sequence leads to the formation of the SecYAEG-ND/substrate complex. In addition, E. coli precursor proteins preMglB and prePhoA can serve as substrates as well as proOmpA because their translocation by SecYAEG was detected (Supplementary Fig. 8). Using preMglB-sfGFP and prePhoA-sfGFP instead of proOmpA-sfGFP, we observed interactions between these substrates and SecYAEG-ND by HS-AFM imaging (Supplementary Fig. 9).

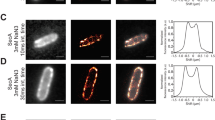
a Schematic model and HS-AFM image of proOmpA-sfGFP on a mica surface (upper) and histogram of the end-to-end distance, extracted from snapshots of the HS-AFM movies, between sfGFP and signal sequence (lower). The solid line represents a fitting curve following a Gaussian distribution and X0 is the center value of the distribution with standard deviation (n = 55). The AFM image height (nm) is shown by the color bar. A representative image from more than 20 independent experiments with similar results are shown. b Schematic model and HS-AFM image of proOmpA-sfGFP bound to SecYAEG-ND (upper) and histogram of the end-to-end distance, extracted from snapshots of the HS-AFM movies, between sfGFP and SecYAEG-ND (lower). The solid line represents a fitting curve following a Gaussian distribution and X0 is the center value of the distribution with standard deviation (n = 89). The AFM image height (nm) is shown by the color bar. A representative image from more than 10 independent experiments with similar results is shown. c Clipped HS-AFM images of proOmpA bound to SecYAEG-ND in the presence of 5 mM ATP. Time 0 s in the initial image indicates an arbitrarily chosen time point of the HS-AFM observation. The AFM image height (nm) is shown by the color bars. The proOmpA and sfGFP regions are given a reddish and greenish tint, respectively. The red arrowhead in the 10.2 s panel indicates the substrate that has inserted into the SecA side. In the 15.4 s image, the red arrowhead highlights the translocated proOmpA extending from the SecYEG-ND side. These images represent snapshots from the highest-resolution movie obtained across numerous experiments. Source data of the graphs are provided as a Source Data file.
Subsequently, during the observation of the SecYAEG-ND/substrate complexes, ATP was added to the observation solution in order to visualize the translocation of proOmpA. Initially, a string-like structure with sfGFP extends from the SecA side, indicating that translocation had just begun, with the substrate slightly visible from the SecYEG-ND side (see also Supplementary Movie 3). Notably, over time, the sfGFP approached SecA, and a string-like structure emerged distinctly from the SecYEG-ND side, namely from the side opposite to SecA (Fig. 2c, Supplementary Fig. 10 and Supplementary Movie 4). This experiment marks the visualization of real-time translocation to the best of our knowledge for the first time, achieved by observing the SecYAEG-ND/substrate complexes from a lateral perspective.
By quantifying the time-dependent elongation of the substrate polypeptide by SecYAEG-ND, we estimated an average translocation rate of approximately 0.9 nm/s for five particles, equivalent to an average rate of 2.2 amino acids per second (aa/s). A single-molecule FRET analysis reported that the protein translocation rate for E. coli SecYEG and SecA was ~40 aa/s64. Our study used Sec proteins from a thermophilic bacterium, T. thermophilus, which functions optimally at high temperatures. Compared to E. coli Sec proteins, our estimated protein translocation speed is slower but reasonable. Additionally, the N-terminus of SecA functions as an anchor to the lipid bilayer27, and the SecA membrane-binding affinity related to its N-terminus changed during the ATP hydrolysis cycle65. Therefore, the fusion protein used in this study, which links the C-terminus of SecY and the N-terminus of SecA, may restrict the conformational changes of SecA’s N-terminus, potentially causing the slower protein translocation rate.
We counted the number of SecYAEG-ND particles with proOmpA protruding from the SecYEG-ND side in HS-AFM images at 30 min after the addition of ATP. Among the 43 SecYAEG-ND particles that interacted with proOmpA-sfGFP, 23 retained substrate proteins protruding from the SecYEG-ND side, implying that the 23 particles successfully achieved protein translocation through the SecYAEG-NDs. The fact that not all of the molecules demonstrated translocation suggests that conformational changes were impeded by steric hindrance caused by adsorption on the mica substrate, potentially resulting in a substantial reduction or complete loss of translocation activity. Nonetheless, our single-molecule observations have demonstrated that one SecYEG in combination with one SecA is sufficient for protein translocation.
Nucleotide-dependent structural transition of SecA in SecYAEG-ND/substrate complex
An in-depth examination of the HS-AFM movies of the SecYAEG-ND/substrate complex in the protein translocation condition also uncovered conformational transitions in the SecA region (Fig. 3a and Supplementary Movie 5). Fig. 3a shows prominent high-low conformational transitions in SecA at the position indicated by the arrows. We note that these conformational transitions were specific to the SecYAEG-ND/substrate complex (Fig. 3a and Supplementary Movie 5) and were not evident in the SecYAEG-NDs in the absence of the substrate (Fig. 1a upper)60. Cross-sectional profiles for the two distinct conformational states showed a 2.0 nm step in the Low state but no step in the High state (Fig. 3b and c). In addition, the High state profile is essentially identical to that of SecYAEG-ND in the absence of substrate, which we defined above as the wide-open structure (Fig. 1a upper, 1c upper, 1d upper, 3b, 3d). A comparison of the cross-sectional profile of the observed Low state (Fig. 3c) with those of the open and closed images calculated by the predicted structural models (Fig. 3e, Supplementary Fig. 5 left, center and right) showed that the height gap of the Low state (2.0 nm) was closer to the open state (1.9 nm) and closed state (1.7 nm). The height gap between these two structures differs by only 0.2 nm, making it difficult to distinguish between them based solely on the simulated HS-AFM images. Nevertheless, the crystal structure of SecA in the open state (PDB ID 1TF5) was determined in the absence of its substrate and SecY, while the closed state was determined with its substrate and SecY. In this experiment, conducted under conditions where both the substrate and SecYEG were present, the Low state of the observed HS-AFM images is considered to represent the closed state. Furthermore, the shape of the height profile also showed similarities between the simulated image of the closed state and the observed HS-AFM images. In conclusion, our observations imply that the High and Low states captured by HS-AFM correspond to the wide-open and closed forms, respectively. The simulated AFM and HS-AFM images of SecYA(ΔPPXD)EG-ND (Fig. 1) show that the height gaps in the SecA region are 3.5 nm (observed) and 3.4 nm (predicted), due to the lack of the flexible PPXD (Fig. 3f, g, and Supplementary Fig. 1c), and are larger than those of the predicted open, closed (Fig. 3e, and Supplementary Fig. 5), and observed Low states (Fig. 3c). These findings suggest that the High and Low states are derived from structural dynamics of the PPXD, which is intimately associated with protein translocation.

a Real-time HS-AFM images of SecYAEG-ND complexed with proOmpA-sfGFP. Arrows indicate regions of height variation upon exposure to 5 mM ATP. Height profiles of the SecA region in the High (b) and Low (c) states, traced along the red lines. Height profiles of the SecA region in the wide-open (d) and closed (e) forms of the simulated AFM images generated from the predicted structures at an 80° angle, which is defined in Fig. 1f, traced along the red lines. Representations of the SecYAEG-ND in wide-open (f) and closed (g) forms predicted by MD simulation in two different orientations. PPXD is highlighted in yellow, the rest of SecA in blue, and SecYEG in magenta. The color bars show the AFM or simulated AFM image height (nm). Representative data from more than 5 independent experiments with similar results are shown. Source data of the graphs are provided as a Source Data file.
SecA functions as an ATPase motor, with ATP hydrolysis serving as a pivotal mechanism in mediating protein translocation. Conformational changes of PPXD have been proposed to occur in conjunction with the ATP hydrolysis cycle, as indicated by FRET analysis38. To investigate the correlation between the High and Low forms of SecA and the stages of the ATP hydrolysis cycle, we monitored structural transitions of the PPXD domain within the SecYAEG-ND/proOmpA in several buffer conditions. In detail, SecYAEG-ND and proOmpA were pre-incubated and then immobilized on a mica substrate. Either nucleotides or nucleotide analogs were then introduced into the buffer, and HS-AFM imaging was conducted at an imaging rate of 0.5 s/frame. In the captured snapshot images, the High and Low states of the SecA region were counted (Fig. 4a). As depicted in Fig. 1, the wide-open state appears highly stable in this HS-AFM observation, possibly due to sample adsorption on the mica. Despite this potential background effect, we were able to detect distinctive structural changes in each buffer condition. Under the nucleotide-free condition (Fig. 4a Apo), 55% of the SecYAEG-ND/proOmpA complexes were discerned in the High form. This proportion increased in the presence of 5 mM ATP, where 77% of the SecA region exhibited the High form (Fig. 4a ATP). In the presence of 1 mM AMP-PNP (Fig. 4a AMP-PNP), a nonhydrolyzable nucleotide analog assumed to mimic the ATP-bound state, 81% of the complexes became the High form, indicating that the SecA region tends to favor the High conformation when ATP is bound. When 5 mM ADP was present (Fig. 4a, ADP), 87% of the complexes exhibited the High form, indicating that the High form is the predominant state not only in ATP-bound but also in ADP-bound conditions. In contrast, upon the addition of ADP-AlF3, which mimics the ADP-Pi transition state (Fig. 4a ADP-AlF3), the occurrence of the High form decreased to 68%, indicating a shift towards a higher occurrence of the Low form. Furthermore, in the observation solution containing 5 mM ATP and 50 mM NaH2PO4, in which the ADP-Pi state is considered to predominate (Fig. 4a ATP-NaH2PO4), the complexes were 59% in the closed form and 41% in the High form. We thus propose the following structural changes of the SecA region during the ATP hydrolysis cycle for protein translocation (Fig. 4b): the transition to the ADP-Pi state during the ATP hydrolysis cycle favors a conformational shift towards the closed form. ATP binding prompts the SecA region to assume the High form. Subsequent ATP hydrolysis to the ADP-Pi state, corresponding to ADP-AlF3 and ATP-NaH2PO4 in Fig. 4a, induces a transition to the Low form, which then reverts to the High form upon phosphate release, resulting in the ADP-bound state. The Apo state observations suggest that SecA is in equilibrium between the High or Low forms upon ADP dissociation. These nucleotide-dependent changes in SecA observed in our study, while exhibiting differences in intensity compared to previous FRET results38, remain consistent with the conclusion. Therefore, we successfully visualized the structural transitions of the PPXD domains in protein translocation.

a Distribution of the High and Low forms in different nucleotide conditions, as counted from HS-AFM snapshot images. SecA can transition to the Low form upon interacting with its substrate, as opposed to being in the High state in the resting condition. The number of particle images for each state is as follows: Apo (n = 100), 5 mM ATP (n = 284), 1 mM AMP-PNP (n = 140), 5 mM ADP-AlF3 (n = 174), 5 mM ADP (n = 134), 5 mM ATP, and 50 mM NaH₂PO₄ (n = 134). b Schematic representation of SecA conformational dynamics during the ATP hydrolysis cycle. The diagram illustrates SecA conformational transitions between High (wide-open) and Low (closed) conformations in response to nucleotide state changes. Upon ATP binding, SecA adopts the High conformation, which transitions to the Low conformation after ATP hydrolysis. Following phosphate release, SecA returns to the High conformation.
As HS-AFM observation can monitor real-time structural changes, we now discuss the turnover of the PPXD domain. SecA in the closed form can hold the substrate tightly, based on previous structural studies. The critical role of the PPXD domain in the closed form is to bring the substrate close to the SecYEG channel and to hold it there31. After releasing the substrate, the PPXD domain interacts with the distal region of the substrate, towards its C-terminus, mediating protein translocation. Although other regions of SecA, including HWD, are important for protein translocation, we could not detect their structural changes in this study. During high-resolution real-time imaging of the SecYAEG-ND/proOmpA complex, the SecA region displayed repetitive conformational changes between the High and Low states, in both the presence and absence of ATP, but the transition rate was different. In the nucleotide-free (Apo) condition, which is a SecA inactive state, the mean cycle time Tcycle, indicative of the average duration for the PPXD domain to transition from the High to the Low state and back, was determined to be 13 ± 11 s. The similar magnitude of both the mean and standard deviation of Tcycle implies a wide distribution of cycle times, suggesting that the conformational transitions of PPXD in the Apo condition are primarily stochastic in nature, likely influenced by thermal fluctuations. On the other hand, the Tcycle in the presence of 5 mM ATP was estimated to be 2 ± 1 s, approximately 5 times faster than in the Apo condition (Supplementary Fig. 11). Hence, the Tcycle in the presence of ATP directly reflects the hydrolysis duration, equivalent to a rate of 0.5 ATP/s. If the structural transitions in the PPXD domain are linked to ATP hydrolysis, those in the Apo state might rarely occur, and the ATP hydrolysis cycle could be prioritized. In a previous study, the ATP hydrolysis rate of E. coli SecA during protein translocation was estimated at approximately 2 ATP/s38, which is faster than that of T. thermophilus SecA in this study. The rate difference is possibly due to their respective optimal temperatures.
This study, using the HS-AFM technique, has achieved visualization of protein translocation mediated by the Sec translocon. The crucial initial step was the successful integration of the stable SecYEG-SecA complex into nanodiscs, ensuring single-molecule observation. The constrained environment of the nanodiscs facilitated the simultaneous monitoring of dynamic events occurring on both sides of the lipid membrane. By fully utilizing image correlation analysis between simulated AFM and observed AFM images, aided by molecular dynamics simulations and imaging of domain-depleted mutants, we successfully identified the orientation of the particles on the surface of observation. This task was notably challenging due to the resolution limitations of HS-AFM, but our approach enabled precise determination of the observation plane, which is critical for accurate interpretation of the dynamic processes of target proteins. This foundational setup allowed us to observe the binding and translocation processes of SecYAEG-ND with unfolded substrate proteins, along with the capability to estimate the substrate translocation rate. Furthermore, the study notably captured the conformational transitions between the High and Low states of SecA depending on the PPXD transitions. We discerned a definitive correlation between the ATP hydrolysis cycle and PPXD conformational states and quantified the rate of these conformational changes. These insights shed light on the dynamic conformational changes in SecA during ATP hydrolysis, emphasizing their essential roles in substrate translocation. Consequently, this enhances our understanding of protein transport mechanisms across membranes, finally illuminating these complex processes at a molecular level. The research advances our comprehension of the dynamic conformational changes of the PPXD domain of SecA during ATP hydrolysis and their important role in substrate translocation. While the present study did not observe the direct relationship between substrate translocation and conformational changes, it sets the stage for future work to visualize these dynamics, leading to a more nuanced understanding of the molecular dynamics involved in protein translocation, an ambitious and crucial goal for the field.
Methods
Protein preparation and protein translocation assay
The SecYAEG complex is composed of T. thermophilus SecY(L2V, R252G)-(GGSG)4-SecA1–939(C648S)-His10, SecE, and SecG; and the SecYA(ΔPPXD)EG complex is composed of SecY(L2V, R252G)-(GGSG)4-SecA1–230-GGSG-SecA363–939(C648S)-His10, SecE, and SecG. These complexes were expressed in E. coli BL21(DE3) harboring pAK22, which encodes SecG, and either pTT610 or a pTT610-modified plasmid, which encodes SecY-SecA, SecE, and SecG. After solubilization from the membrane fraction using n-Dodecyl-β-maltoside (GLYCON, #D97002), the complexes were purified through successive chromatography steps: Ni-NTA agarose (QIAGEN, #30250), Hi-Trap SP HP (Cytiva, # 17115101), and Superdex 200 (Cytiva, #28990944)4,59.
The preprotein E. coli proOmpA(L59)-sfGFP and proOmpA(L59-3Q) that were purified in a former study59 were used. preMglB (Q163C, K195C)-sfGFP, and prePhoA-sfGFP were purified by the same method as proOmpA(L59)-sfGFP. To prepare preproteins E. coli preMglB(Q163C, K195C)- and prePhoA-EFIEGR-H6-IDEEQKLISEEDLLRKR, the encoding DNA sequences were inserted into pBluescript SK(-), and E. coli TYE055 cells66 were transformed with the resulting plasmids. The transformants were cultivated at 30 °C to an A600 of approximately 1.0 in LB medium supplemented with 50 μg/mL ampicillin and 20 μg/mL chloramphenicol. Preprotein expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside for 2 h. The cells were sedimented by centrifugation, resuspended with buffer A (50 mM Tris-HCl pH 8.0, 1 M NaCl, 8 M Urea, 10 mM β-mercaptoethanol), and disrupted by sonication with a Q500 Sonicator (QSONICA). After successive centrifugations (6500 × g for 20 min and 100,000 × g for 30 min), the supernatant was passed through a HisTrap HP column (Cytiva, # 17524701) equilibrated with buffer A containing 20 mM imidazole-HCl pH 8.0. The column was washed with the same buffer, and the bound proteins were eluted with buffer A containing 200 mM imidazole-HCl pH 8.0 and then precipitated with 7.5% (final concentration) trichloroacetic acid. After centrifugation (18,000 × g for 4 min), the precipitates were washed with acetone and collected by centrifugation, dried, resuspended with buffer C (50 mM Tris-HCl pH 8.0, 8 M Urea, 10 mM β-mercaptoethanol), and placed in a Hi-Trap Q HP column (Cytiva, # 17115301) equilibrated with buffer C. The bound preproteins were washed with buffer C and eluted with buffer C containing 0–250 mM NaCl. Protein translocation was performed by mixing SecYEG- and SecYAEG-reconstituted liposomes with the purified preproteins. Proteins translocated into the liposomes were detected using protease resistance as an indicator59.
HS-AFM observation
HS-AFM observations were performed using a laboratory-developed instrument67. For the AFM cantilever, we employed the Olympus BL-AC10DS-A2, a cantilever characterized by a spring constant of approximately 0.1 N/m, resonating frequencies ranging from 400 to 600 kHz, and a quality factor (Q-value) of about 2 in aqueous environments. The cantilever’s native apex lacks the requisite sharpness for high-resolution AFM imaging; hence, amorphous carbon was deposited to construct a columnar protrusion on the apex via focused electron beam exposure in a scanning electron microscope. The apex’s sharpness was subsequently refined by plasma etching in argon gas, thus ensuring the acuity necessary for high-resolution imaging.
SecYAEG and SecYA(ΔPPXD)EG were reconstituted into nanodiscs composed of E. coli lipid (Avanti) and membrane scaffold protein (MSP1D1): The mixture of ~10 mg/ml SecYAEG complex, ~8 mg/ml MSP1D1, and ~8 mg/ml E. coli phospholipids (Avanti) was gently mixed at 4 °C for 1 h. Then, DDM was removed by Bio-Beads SM2 (Bio-Rad). The resulting nanodiscs were further purified using Superdex 20060. For HS-AFM imaging, molecules reconstituted in nanodiscs were diluted in buffer O (20 mM Tris-HCl pH 7.0, 2 mM MgCl2) to approximately 5 nM and a 2 µl aliquot of the sample was then dispensed onto a freshly cleaved mica substrate. After incubation at room temperature for 5 min, the mica surface was rinsed thoroughly with buffer O to remove any residual unbound molecules.
In the HS-AFM observations of the complex of SecYAEG-ND and substrate proteins, such as proOmpA-sfGFP, substrate proteins were initially diluted in a buffer solution (20 mM Tris-HCl pH 7.0, 8 M Urea, 50 mM NaCl, 12% glycerol, 2 mM MgCl₂). The high urea concentration denatured the folded proOmpA to the polypeptide chain, while the robust folding of sfGFP allowed it to maintain a globular structure. Subsequently, SecYAEG-ND and proOmpA-sfGFP were pre-mixed at a 1:1 molar ratio in a solution containing 5 mM DTT, 2 mM MgCl2, 12% glycerol, and 20 mM Tris-HCl pH 7.0. This mixture was incubated on ice and then loaded onto a freshly cleaved mica surface. Following a 5-minute incubation on the mica surface, the surface was rinsed with buffer O. For real-time observation of protein translocation by SecYAEG-ND, ATP was added to buffer O to a final concentration of 5 mM before imaging.
The time dependency of substrate elongation uses the following equation: Substrate translocation rate = {(Length of the substrate extending from the SecYEG-ND side) + (Total distance of SecYAEG structure: 10 nm)}/(Time from ATP addition to membrane translocation confirmation). To ensure uniform distribution of ATP throughout the 70 μl observation volume, the observation buffer was immediately mixed by pipetting after ATP addition. This forced mixing distributes ATP within a few seconds, which is negligible compared to the minute-scale time of the substrate translocation. This estimation of the translocation rate was based on measurements from five particles. In experiments observing the conformational changes of SecA shown in Fig. 3a, the SecYAEG-ND/proOmpA-sfGFP complex was incubated on mica for 30 min after the addition of ATP to 5 mM. For observations of the conformational changes under various nucleotide states shown in Fig. 4a, a mixture of SecYAEG-ND and proOmpA-sfGFP was prepared in a solution containing 5 mM ATP, 5 mM DTT, 2 mM MgCl2, 20 mM Tris-HCl pH7.0, and 12% glycerol, and then incubated at 37 °C for 60 min. The mixture was then applied to a mica substrate and incubated further for 5 min, after which the sample was rinsed with buffer O containing various nucleotides or nucleotide analogs, as well as AlCl3, NaF, or NaH₂PO₄. This was followed by HS-AFM imaging within buffer O.
MD simulation of the SecYAEG-nanodisc complex
The structures of fused proteins SecY-A and SecY-A(ΔPPXD), which are the same constructions used for HS-AFM observation, were predicted using AlphaFold2 2.1.068. The obtained Rank 0 and 4 models for SecY-A were corresponding to SecA-open and SecA-closed forms, respectively. The SecYAEG and SecYA(ΔPPXD)EG structures were modeled by additionally docking SecE and SecG of a SecYEG model (PDB ID 5AWW) after structural alignment with SecY coordinates of the Rank 0 model for SecY-A and SecY-A(ΔPPXD), respectively, followed by the energy minimization using Phenix69 to remove atomic clashes. The MSP1D1 nanodisc was modeled using the CHARMM-GUI Nanodisc Builder70, where the lipids are composed of POPE and POPG at the ratio of 3:1. The SecYAEG and SecYA(ΔPPXD)EG models were embedded near the center of the MSP1D1 nanodisc, where the overlapping lipids were removed to maintain the lipid ratio and experimental area per lipid of each lipid in the nanodisc. The orientations of SecYAEG and SecYA(ΔPPXD)EG models with respect to the nanodisc were determined based on the OPM database (PDB ID 5AWW [https://opm.phar.umich.edu/proteins/2952]71. The MD simulation system was solvated with a 150 mM NaCl solution. All acidic amino acids in the system were deprotonated. The system size is 161 × 161 × 161 Å3, and the total number of atoms is ~423,000. The CHARMM C36m force field parameters were used for proteins and membranes72. We performed three MD simulations with different initial velocities or initial configurations for each model (Supplementary Fig. 2a). Each system was gradually equilibrated using 10,000-step energy minimization, followed by equilibration in the NVT and NPT ensembles for 25 ns using the positional restraints on the protein and membranes. The production runs were carried out with a time step of 3.5 fs73 in the NPT ensemble at T = 303.15 K and P = 1 atm, where the Bussi thermostat and barostat were employed74. A cut-off distance of 12.0 Å was used for the non-bonded interaction calculations, and the particle-mesh Ewald method was employed for the long-range electrostatic interaction calculations. All MD simulations were performed using GENESIS ver. 2.075,76. The RMSD changes of the protein with respect to the initial structure showed similar trends in the three independent simulations for each model, indicating that the complex structure does not significantly depend on the initial velocities or configuration (Supplementary Figs. 2b and c). In the SecYA(ΔPPXD)EG and SecYAEG systems, the RMSD fluctuated around 10 Å and 6 Å, respectively, after 150 ns, indicating that both systems reached equilibrium. After the equilibration (before 150 ns) and production runs (after 150 ns), we selected the final snapshots from the longest production runs as the simulation models for the AFM analysis. The structure of SecYA(ΔPPXD)EG at 504 ns was used for Fig. 1. The SecYAEG model at 756 ns, with the SecY-A region replaced by the AF2 Rank 4 model, was the closed model in Fig. 3 and Supplementary Fig. 5. The SecYAEG model at 756 ns, with the SecA region replaced by the crystal structure of SecA (PDB ID 2IPC), was the wide-open model in Figs. 1 and 3, and Supplementary Fig. 5.
Construction of simulated AFM images
Based on collision simulations between the atomic coordinates of protein structures and model AFM probes, we generated simulated AFM images. The coordinates of SecYAEG-ND or SecYA(ΔPPXD)EG, optimized by AlphaFold and MD simulation as described above, were positioned to be probed by AFM from the circumferential orientation of the nanodisc. The AFM probe was modeled in a conical shape with a 10-degree cone angle and a tip radius set to 1.0 nm. To approximate the spatial resolution of actual HS-AFM images, a low-pass filter with a cutoff spatial frequency of 0.5 nm−1 was applied to the generated simulated AFM images. These simulated AFM images were produced with custom HS-AFM data analysis software, developed using Igor Pro-9 (WaveMetrics).
Comparison between actual HS-AFM and simulated AFM images
As a first step, the coordinate as an initial position was set to a side-on orientation in which the nanodisc is perpendicular to the X-Y plane. The simulated AFM images were created by rotation through 5 degrees with respect to the major axis of the nanodisc. Prior to image correlation for the observed image, the Sec molecule image was cropped as a region of interest. To set the same resolution with simulated images, image interpolation was applied for the observed image. In taking image correlation, the region of interest having the same size as the observed image was applied to the simulated image, and this region was shifted by 1 pixel each step along the X-Y direction of the simulated image. The image correlation score was taken for each shifted region in one image and the highest correlation score was adopted as the score of the simulated image. By iterating this step from 0 to 360° in increments of 5 degrees, we obtained image correlation scores for 72 simulated images for one HS-AFM image.
This calculation step was applied for three HS-AFM images of SecYA(ΔPPXD)EG-ND and the image correlation values were averaged for each angle to obtain the angle where all three correlation values were maximized. The angle with the highest value was adopted as the observed angle during imaging.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data needed to evaluate the conclusions in the paper are provided in the main text, including the main Figures, and the Supplementary Information. The generated protein structures in PDB format and Source data for the graphs are available in the Source data file. Further analytical and interpretive support are available from the corresponding author upon request. Source data are provided with this paper.
Code availability
Analysis code for image correlation between HS-AFM data and simulated data is available on Zenodo (https://doi.org/10.5281/zenodo.14064983).
References
Rapoport, T. A., Li, L. & Park, E. Structural and mechanistic insights into protein translocation. Annu. Rev. Cell Dev. Biol. 33, 369–390 (2017).
Tsirigotaki, A., De Geyter, J., Šoštaric, N., Economou, A. & Karamanou, S. Protein export through the bacterial Sec pathway. Nat. Rev. Microbiol. 15, 21–36 (2017).
Crane, J. M. & Randall, L. L. The Sec System: protein export in Escherichia coli. EcoSal Plus 7, ESP-0002-2017 (2017).
Tanaka, Y. et al. Crystal structures of SecYEG in lipidic cubic phase elucidate a precise resting and a peptide-bound state. Cell Rep. 13, 1561–1568 (2015).
Economou, A. & Wickner, W. SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell 78, 835–843 (1994).
Tsukazaki, T. Structure-based working model of SecDF, a proton-driven bacterial protein translocation factor. FEMS Microbiol. Lett. 365, fny112 (2018).
Tsukazaki, T. et al. Structure and function of a membrane component SecDF that enhances protein export. Nature 474, 235–238 (2011).
Kumazaki, K. et al. Structural basis of Sec-independent membrane protein insertion by YidC. Nature 509, 516–520 (2014).
Samuelson, J. C. et al. YidC mediates membrane protein insertion in bacteria. Nature 406, 637–641 (2000).
Paetzel, M. Structure and mechanism of Escherichia coli type I signal peptidase. Biochim. Biophys. Acta 1843, 1497–1508 (2014).
Troman, L. & Collinson, I. Pushing the envelope: the mysterious journey through the bacterial secretory machinery, and beyond. Front. Microbiol. 12, 782900 (2021).
Plummer, A. M. & Fleming, K. G. From chaperones to the membrane with a BAM! trends. Biochem. Sci. 41, 872–882 (2016).
Miyazaki, R. et al. Inner membrane YfgM-PpiD heterodimer acts as a functional unit that associates with the SecY/E/G translocon and promotes protein translocation. J. Biol. Chem. 298, 102572 (2022).
Tomasek, D. et al. Structure of a nascent membrane protein as it folds on the BAM complex. Nature 583, 473–478 (2020).
Wu, R. et al. Plasticity within the barrel domain of BamA mediates a hybrid-barrel mechanism by BAM. Nat. Commun. 12, 7131 (2021).
Doyle, M. T. et al. Cryo-EM structures reveal multiple stages of bacterial outer membrane protein folding. Cell 185, 1143–1156.e13 (2022).
Miyazaki, R., Watanabe, T., Yoshitani, K. & Akiyama, Y. Edge-strand of BepA interacts with immature LptD on the β-barrel assembly machine to direct it to on- and off-pathways. Elife 10, e70541 (2021).
Daimon, Y. et al. Reversible autoinhibitory regulation of Escherichia coli metallopeptidase BepA for selective β-barrel protein degradation. Proc. Natl Acad. Sci. USA 117, 27989–27996 (2020).
Shahrizal, M. et al. Structural basis for the function of the β-barrel assembly-enhancing protease BepA. J. Mol. Biol. 431, 625–635 (2019).
Bryant, J. A. et al. Structure-function characterization of the conserved regulatory mechanism of the Escherichia coli M48 metalloprotease BepA. J. Bacteriol. 203, e00434–20 (2020).
Narita, S.-I., Masui, C., Suzuki, T., Dohmae, N. & Akiyama, Y. Protease homolog BepA (YfgC) promotes assembly and degradation of β-barrel membrane proteins in Escherichia coli. Proc. Natl Acad. Sci. Usa. 110, E3612–E3621 (2013).
Hanada, M., Nishiyama, K. I., Mizushima, S. & Tokuda, H. Reconstitution of an efficient protein translocation machinery comprising SecA and the three membrane proteins, SecY, SecE, and SecG (p12). J. Biol. Chem. 269, 23625–23631 (1994).
Zimmer, J., Nam, Y. & Rapoport, T. A. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature 455, 936–943 (2008).
Ma, C. et al. Structure of the substrate-engaged SecA-SecY protein translocation machine. Nat. Commun. 10, 2872 (2019).
Li, L. et al. Crystal structure of a substrate-engaged SecY protein-translocation channel. Nature 531, 395–399 (2016).
Erlandson, K. J. et al. A role for the two-helix finger of the SecA ATPase in protein translocation. Nature 455, 984–987 (2008).
Bauer, B. W., Shemesh, T., Chen, Y. & Rapoport, T. A. A ‘push and slide’ mechanism allows sequence-insensitive translocation of secretory proteins by the SecA ATPase. Cell 157, 1416–1429 (2014).
Erlandson, K. J., Or, E., Osborne, A. R. & Rapoport, T. A. Analysis of polypeptide movement in the SecY channel during SecA-mediated protein translocation. J. Biol. Chem. 283, 15709–15715 (2008).
Bauer, B. W. & Rapoport, T. A. Mapping polypeptide interactions of the SecA ATPase during translocation. Proc. Natl Acad. Sci. USA 106, 20800–20805 (2009).
Gupta, R., Toptygin, D. & Kaiser, C. M. The SecA motor generates mechanical force during protein translocation. Nat. Commun. 11, 3802 (2020).
Dong, L. et al. Structural basis of SecA-mediated protein translocation. Proc. Natl Acad. Sci. USA 120, e2208070120 (2023).
Banerjee, T., Zheng, Z., Abolafia, J., Harper, S. & Oliver, D. The SecA protein deeply penetrates into the SecYEG channel during insertion, contacting most channel transmembrane helices and periplasmic regions. J. Biol. Chem. 292, 19693–19707 (2017).
Osborne, A. R., Clemons, W. M. Jr & Rapoport, T. A. A large conformational change of the translocation ATPase SecA. Proc. Natl Acad. Sci. USA 101, 10937–10942 (2004).
Hunt, J. F. et al. Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science 297, 2018–2026 (2002).
Gelis, I. et al. Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell 131, 756–769 (2007).
Vandenberk, N. et al. The preprotein binding domain of SecA displays intrinsic rotational dynamics. Structure 27, 90–101.e6 (2019).
Ernst, I. et al. Large conformational changes of a highly dynamic pre-protein binding domain in SecA. Commun. Biol. 1, 130 (2018).
Catipovic, M. A., Bauer, B. W., Loparo, J. J. & Rapoport, T. A. Protein translocation by the SecA ATPase occurs by a power-stroke mechanism. EMBO J. 38, e101140 (2019).
Allen, W. J. et al. Two-way communication between SecY and SecA suggests a Brownian ratchet mechanism for protein translocation. Elife 5, e15598 (2016).
Corey, R. A. et al. ATP-induced asymmetric pre-protein folding as a driver of protein translocation through the Sec machinery. Elife 8, e41803 (2019).
Ahdash, Z. et al. HDX-MS reveals nucleotide-dependent, anti-correlated opening and closure of SecA and SecY channels of the bacterial translocon. Elife 8, e47402 (2019).
Allen, W. J., Watkins, D. W., Dillingham, M. S. & Collinson, I. Refined measurement of SecA-driven protein secretion reveals that translocation is indirectly coupled to ATP turnover. Proc. Natl Acad. Sci. USA 117, 31808–31816 (2020).
Allen, W. J. et al. Rate-limiting transport of positively charged arginine residues through the Sec-machinery is integral to the mechanism of protein secretion. Elife 11, e77586 (2022).
Krishnamurthy, S. et al. Preproteins couple the intrinsic dynamics of SecA to its ATPase cycle to translocate via a catch and release mechanism. Cell Rep. 38, 110346 (2022).
Kusters, I. et al. Quaternary structure of SecA in solution and bound to SecYEG probed at the single molecule level. Structure 19, 430–439 (2011).
Chatzi, K. E., Sardis, M. F., Economou, A. & Karamanou, S. SecA-mediated targeting and translocation of secretory proteins. Biochim. Biophys. Acta 1843, 1466–1474 (2014).
Gouridis, G. et al. Quaternary dynamics of the SecA motor drive translocase catalysis. Mol. Cell 52, 655–666 (2013).
Or, E., Boyd, D., Gon, S., Beckwith, J. & Rapoport, T. The bacterial ATPase SecA functions as a monomer in protein translocation. J. Biol. Chem. 280, 9097–9105 (2005).
Seinen, A.-B., Spakman, D., van Oijen, A. M. & Driessen, A. J. M. Cellular dynamics of the SecA ATPase at the single molecule level. Sci. Rep. 11, 1433 (2021).
Chada, N. et al. Single-molecule observation of nucleotide induced conformational changes in basal SecA-ATP hydrolysis. Sci. Adv. 4, eaat8797 (2018).
Karner, A. et al. Tuning membrane protein mobility by confinement into nanodomains. Nat. Nanotechnol. 12, 260–266 (2017).
Sanganna Gari, R. R. et al. Direct visualization of the E. coli Sec translocase engaging precursor proteins in lipid bilayers. Sci. Adv. 5, eaav9404 (2019).
Frauenfeld, J. et al. Cryo-EM structure of the ribosome-SecYE complex in the membrane environment. Nat. Struct. Mol. Biol. 18, 614–621 (2011).
Dalal, K., Chan, C. S., Sligar, S. G. & Duong, F. Two copies of the SecY channel and acidic lipids are necessary to activate the SecA translocation ATPase. Proc. Natl Acad. Sci. USA 109, 4104–4109 (2012).
Alami, M., Dalal, K., Lelj-Garolla, B., Sligar, S. G. & Duong, F. Nanodiscs unravel the interaction between the SecYEG channel and its cytosolic partner SecA. EMBO J. 26, 1995–2004 (2007).
Koch, S. et al. Lipids activate SecA for high affinity binding to the SecYEG complex. J. Biol. Chem. 291, 22534–22543 (2016).
Taufik, I., Kedrov, A., Exterkate, M. & Driessen, A. J. M. Monitoring the activity of single translocons. J. Mol. Biol. 425, 4145–4153 (2013).
Tsukazaki, T. Structural basis of the Sec translocon and YidC revealed through X-ray crystallography. Protein J. 38, 249–261 (2019).
Sugano, Y., Furukawa, A., Nureki, O., Tanaka, Y. & Tsukazaki, T. SecY-SecA fusion protein retains the ability to mediate protein transport. PLoS One 12, e0183434 (2017).
Haruyama, T. et al. Single-unit imaging of membrane protein-embedded nanodiscs from two oriented sides by high-speed atomic force microscopy. Structure 27, 152–160.e3 (2019).
Müller, D. J., Janovjak, H., Lehto, T., Kuerschner, L. & Anderson, K. Observing structure, function and assembly of single proteins by AFM. Prog. Biophys. Mol. Biol. 79, 1–43 (2002).
Ando, T., Uchihashi, T. & Kodera, N. High-speed AFM and applications to biomolecular systems. Annu. Rev. Biophys. 42, 393–414 (2013).
Uchihashi, T., Iino, R., Ando, T. & Noji, H. High-speed atomic force microscopy reveals rotary catalysis of rotorless F₁-ATPase. Science 333, 755–758 (2011).
Fessl, T. et al. Dynamic action of the Sec machinery during initiation, protein translocation and termination. Elife 7, e35112 (2018).
Winkler, K. et al. Interaction of the motor protein SecA and the bacterial protein translocation channel SecYEG in the absence of ATP. Nanoscale Adv. 2, 3431–3443 (2020).
Yoshihisa, T. & Ito, K. Pro-OmpA derivatives with a His6 tag in their N-terminal ‘translocation initiation domains’ are arrested by Ni2+ at an early post-targeting stage of translocation. J. Biol. Chem. 271, 9429–9436 (1996).
Ando, T. et al. High-speed AFM and nano-visualization of biomolecular processes. Pflug. Arch. 456, 211–225 (2008).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D. Struct. Biol. 75, 861–877 (2019).
Qi, Y., Lee, J., Klauda, J. B. & Im, W. CHARMM-GUI nanodisc builder for modeling and simulation of various nanodisc systems. J. Comput. Chem. 40, 893–899 (2019).
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Jung, J. et al. Optimized hydrogen mass repartitioning scheme combined with accurate temperature/pressure evaluations for thermodynamic and kinetic properties of biological systems. J. Chem. Theory Comput. 17, 5312–5321 (2021).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Jung, J. et al. GENESIS: a hybrid-parallel and multi-scale molecular dynamics simulator with enhanced sampling algorithms for biomolecular and cellular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 5, 310–323 (2015).
Kobayashi, C. et al. GENESIS 1.1: A hybrid-parallel molecular dynamics simulator with enhanced sampling algorithms on multiple computational platforms. J. Comput. Chem. 38, 2193–2206 (2017).
Acknowledgements
We thank Kayo Abe for secretarial assistance, Kunihito Yoshikaie for technical support, and Yuji Sugita and Yoshiki Tanaka for useful suggestions. The MD simulations were carried out on Fugaku. This work was supported by JSPS/MEXT KAKENHI (Grant Nos. JP21H05157, JP24K03035 to T.M., Grant No. JP23K14146 to H.K., Grant Nos. JP22K15075, JP20K15733 to M.I., Grant Nos. JP22K15061, JP22H05567 to R.M., Grant Nos. JP21H000393, JP24K01309 to T.U., and Grant Nos. JP22H02567, JP22H02586, JP21H05155, JP21H05153, JP21K19226, JP21KK0125 to T.T.); PRESTO (JPMJPR20E1 to M.I.) and K Program (JPMJKP23H2 to T.T.) from the Japan Science and Technology Agency (JST); MEXT as “Program for Promoting Researches on the Supercomputer Fugaku” (Development and application of large-scale simulation-based inferences for biomolecules JPMXP1020230119); HPCI project (hp230209, hp240215, hp240277 to T.M.); MEXT Promotion of Development of a Joint Usage/ Research System Project: Coalition of Universities for Research Excellence Program (CURE) (Grant Number JPMXP1323015482); Natural Science Foundation of Shanghai (24ZR1403800 to M.I.); and private research foundations (The Chemo-Sero-Therapeutic Research Institute, Naito Foundation, Takeda Science Foundation, the Institute for Fermentation (Osaka), and Yamada Science Foundation) to T.T.
Author information
Authors and Affiliations
Contributions
T.U. and T.T. conceptualized the study. S.I., H.K., T.H., Y.S., R.M., M.I., and T.T. prepared the protein samples and analyzed their activity. Y.K., W.N., Y.N., T.H., and T.U. performed HS-AFM observations. T.M. performed MD simulations. Y.K., T.M., T.U., and T.T. analyzed the data, discussed its validity, and wrote the manuscript. T.M., T.U., and T.T. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Johannes Preiner, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kanaoka, Y., Mori, T., Nagaike, W. et al. AFM observation of protein translocation mediated by one unit of SecYEG-SecA complex. Nat Commun 16, 225 (2025). https://doi.org/10.1038/s41467-024-54875-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54875-x
