
Abstract
Advances in improving the operational lifetime of highly efficient organic photovoltaic (OPV) and understanding photo-degradation mechanisms in molecular level are currently limited, especially on the promising inverted OPV, posing critical challenges to commercialization. Here, we demonstrate a radical scavenger (3-(3,5-Di-tert-butyl-4-hydroxyphenyl)propionic acid) capped ZnO (BHT@ZnO) nanoparticles as the electron transport layer providing effective surface oxygen vacancy passivation and reactive radical capture capability. Encouragingly, this BHT@ZnO-based empowered device achieves a record inverted OPV efficiency of 19.47% (Certificated efficiency: 18.97%). The devices demonstrate light soaking-free behavior, long-term stability under ISOS-D-1 (94.2% PCE retention after 8904 h in ambient) and ISOS-L-1 testing protocol (81.5% PCE retention after 7724 h in MPP). More importantly, we elucidate detailed degradation mechanism in OPV involving selectively catalytic degradation of donor and acceptor by superoxide and hydroxyl radicals, respectively, as well as the degradation pathway of polymer donor upon radiation exposure. Performance enhancement and mechanism comprehension provide strong support for the development of OPV technology.
Similar content being viewed by others
Introduction
Organic photovoltaic (OPV) has shown great potential for energy conversion in specific applications, such as transparent and wearable devices, due to properties like low-cost, lightweight, non-toxic, transparency, and flexibility1,2,3,4,5,6,7,8,9. Benefitting from the delicate optimization of polymer donor (e.g., PM6) and small molecular acceptor (e.g., Y6, BTP-eC9) system, the certificated power conversion efficiency (PCE) of state-of-the-art OPV has surpassed 19%10,11,12,13,14,15,16,17,18. Present champion PCE OPVs are all regular (p-i-n) structures based on PEDOT:PSS hole transport layer, which poses huge challenges to ambient stability due to deliquescence19. For the successful commercialization of OPV, addressing the lifetime-related issues in highly efficient OPV from both material- and device-level are crucial20,21,22,23,24,25,26,27.
Interlayer plays a vital role in both efficiency and stability of OPV. Inverted (n-i-p) OPVs have long been shown to be more promising in terms of device stability28, especially benchmark zinc oxide (ZnO) as bottom electron transport layers (ETL). However, for the state-of-the-art OPV with non-fullerene acceptors, the inverted devices have not realized the high expectation - they tend to exhibit relatively lower PCE (reported 18.75%29 and certified 16.76%30) compared to regular devices, and the stability is also unsatisfied31,32. This performance discrepancy is connected to the abundance of surface oxygen vacancies (VO+) on ZnO, leading to trap-assisted charge carrier recombination. In addition, the adsorption of hydroxyl ions, O2 and H2O molecules on VO+ results in unfavorable light-soaking problem33.
Currently, most research attributes the degradation of photovoltaic devices during operation to morphological instabilities in the active layer. However, it is important to note that this morphological evolution is likely to be rooted in the degradation of materials at the molecular level. Previous research has provided some evidence of the light-induced instability of highly performing non-fullerene acceptors (NFAs), which are catalyzed by reactive oxygen species (ROS), generated by ZnO34. Nonetheless, systematic understanding regarding the degradation mechanism of photoactive materials is seriously lacking. For instance, key questions including the priority of degradation behavior in high-performing donor-acceptor blending systems, the roles of O2, H2O, and interlayers in the photoactive layer decomposition, and the degradation pathway of photoactive materials remain poorly comprehended.
To tackle these issues, in this work, we designed radical scavenger 3-(3,5-Di-tert-butyl-4-hydroxyphenyl)propionic acid (BHT-acid) capped ZnO nanoparticles (BHT@ZnO NPs) to provide a multifunctional solution. The propionic acid groups in BHT-acid passivated the surface VO+ and removed the dangling hydroxyl ion in ZnO NPs, while the BHT moiety reduced the surface tension and captured the reactive radicals. Consequently, the inverted OPV based on BHT@ZnO demonstrated record efficiencies of 19.47% using the PM6:BTP-eC9:o-BTP-eC9 system. Equal excitingly, it exhibited long ISOS-D-1 and ISOS-L-1 lifetime with 94.2% PCE retention (with light-soaking-free behavior) after 8904 h of ambient storage without encapsulation and 81.5% PCE retention after 7724 h of maximum power point (MPP) tracking in real-world measurement.
More importantly, a deep comprehension of the active layer degradation mechanism under light radiation is achieved. Initially, we discovered an interesting phenomenon that under light illumination in N2 or air, the degradation trends of donor (PM6) and acceptor (BTP-eC9) are opposite. Specifically, BTP-eC9 shows a higher degradation rate in N2, while PM6 exhibits a higher degradation rate in the air on ZnO. Subsequently, we established the selectively catalytic degradation pathways of PM6 and BTP-eC9 by superoxide radicals (·O2−) and hydroxyl radicals (·OH), respectively. We then identified that the ·O2−-induced benzo[1,2-b:4,5-b’]dithiophene (BDT) oxidation is the main factor in PM6 degradation failure. Consequently, we confirmed that BHT@ZnO effectively suppressed the ROS, including ·O2− and ·OH, by passivating the surface defects (reducing ROS generation) and scavenging the generated ROS. This provided excellent protection for the active layer materials. Hence, this work not only achieved high efficiency and stable OPVs, expanded understanding of the active layer material degradation mechanism, but also provided guidance for future OPV materials and device design towards commercially viable OPV technology.
Result and discussion
Synthesis and characterization of the BHT@ZnO NPs
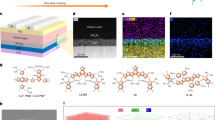
The synthesis of zinc oxide nanoparticles was carried out according to a known method in the literature as a control for comparison, while for the BHT@ZnO NPs, the surface modification of ZnO involved binding it with BHT-acid (Fig. 1a)35,36. The carboxylate group enables BHT to anchor on the surface of ZnO37,38. The BHT-liganded ZnO nanoparticles demonstrated high uniformity and an average size of approximately 8 nm based on transmission electron microscopy (TEM) images (Fig. 1b). Additionally, the lattice fringes of the (101) and (002) crystal plane were observed at space distances of approximately 0.246 and 0.261 nm. The formation of BHT@ZnO NPs was verified by X-ray Diffraction (XRD) as evidenced by the characteristic peaks that were assigned to ZnO with a wurtzite structure (Supplementary Fig. 1). The BHT@ZnO NPs exhibit superior dispersion in methanol compared to ZnO NPs. After being stored at low temperatures (4–8 °C) for over one month, the BHT@ZnO NPs remain clear and free of turbidity (Supplementary Fig. 2a). Additionally, the zeta potential of BHT@ZnO in methanol remains stable throughout the storage period (Supplementary Fig. 2b–d). This enhanced colloidal stability can be attributed to a reduction in OH groups on the surface of ZnO, as well as the steric hindrance provided by the tert-butyl groups in BHT, which effectively prevents the aggregation of the nanoparticles37.

a Synthesis route of ZnO and BHT@ZnO NPs; b TEM images of BHT@ZnO NPs; c FT-IR spectra of BHT-acid, ZnO NP and BHT@ZnO NPs; d XPS O 1 s signals of ZnO and BHT@ZnO NPs; e XPS Zn 2p signals of ZnO and BHT@ZnO NPs; f PL spectra of ZnO and BHT@ZnO NPs excited at 350 nm; g EPR of ZnO and BHT@ZnO NPs with or without light illumination of 5 min; h UPS spectra of ZnO and BHT@ZnO films deposited on ITO glass. Source data are provided as a Source Data file.
Fourier transform infrared spectroscopy (FT-IR) was used to determine the surface modification of BHT@ZnO (see Fig. 1c). Both BHT-acid and BHT@ZnO NPs exhibit a peak at 881 cm−1, indicating the presence of benzene ring. Peaks in the range of 2800–3000 cm−1 and 1100–1250 cm−1 (green area) correspond to alkyl groups (tert-butyl and methylene) and phenolic hydroxyl in both BHT-acid and BHT@ZnO, suggesting the presence of BHT moiety in BHT@ZnO NPs. The peaks observed at 3632 and 1708 cm−1 (star marked) indicate free hydroxyl O–H and C=O bond stretching vibration of the carboxylate group in BHT-acid, disappear in BHT@ZnO, indicating the formation of the carboxylate salt (COOZn). Additionally, a high-wavelength-number shift from 428 cm−1 to 454 cm−1 is observed after BHT capping, indicating the formation of a new Zn–O bond and the reduction of surface VO+.
To further identify the surface properties of BHT@ZnO, X-ray photoelectron spectroscopy (XPS) was employed39. The O 1s peak of ZnO NPs was assigned to different binding energies, including 530.8 eV for lattice oxygen (Zn–O), 531.51 eV for VO+, and 532.69 eV for surface-adsorbed oxygen (Fig. 1d)40,41,42. The stoichiometric ratio of all these peaks is summarized in Supplementary Table 1. The ratio of VO+ to lattice oxygen ratio of BHT@ZnO is 0.96, lower than that of ZnO (1.22), which indicates that BHT surface ligand modification passivates the surface defect of ZnO NPs. The higher amount of adsorbed oxygen on the surface of BHT@ZnO is attributed to the presence of oxygen in BHT moiety. Additionally, the Zn 2p peaks of BHT@ZnO NPs showed a lower-binding-energy shift (0.28 eV) than ZnO NPs, which is attributed to the reduced VO+ on BHT@ZnO NPs by intensive coordination between COO− and Zn2+ (Fig. 1e).
Under 350 nm excitation, ZnO NPs demonstrate strong photoluminescence (PL) emission at 564 nm, which arises from the defect-induced emission related to oxygen vacancies43. However, the photoluminescence emission from BHT@ZnO NPs shows a blue-shift at 551 nm and lower intensity, indicating the passivation of surface oxygen-vacancy-related defects by the carboxylic group in BHT moiety (Fig. 1f). Electron paramagnetic resonance (EPR) spectrum was used to further confirm the nature of defects in the nanoparticles (Fig. 1g). The spectrum revealed a signal at a g value of 1.958 in ZnO NPs, and the intensity is enhanced after light illumination, which is likely attributed to the presence of surface VO+ defects and generated ROS29,44,45,46. In contrast, the EPR signal in BHT@ZnO NPs significantly decreased with and without light, indicating the effective passivation by BHT moiety. And a combined multiple peaks were observed in the range of g = 2.00, which could be attributed to the BHT-radicals as the mediated production of the ROS captured by BHT radical scavenger47.
Compared to ZnO, BHT@ZnO thin film displays an approximately 5-nm red-shift in absorbance cutoff according to Supplementary Fig. 3. The derived Tauc plot indicates that the bandgap of BHT@ZnO has a minor decrease of 3.40 eV in contrast to 3.47 eV of ZnO. Furthermore, we executed ultraviolet photoelectron spectroscopy (UPS) to determine the work function (Fig. 1h). The work function of ZnO was reduced from −4.30 eV to −4.01 eV with the incorporation of the BHT moiety. As indicated by the density functional theory (DFT), this work function variation could be attributed to the dipole of the surface ligands. Supplementary Fig. 4a shows that the HOMO is predominantly situated in the electron-rich polyphenol portion, while the LUMO is distributed throughout the molecule. As a result, the electron-state density distribution induces a dipole moment of 2.38 Debye, which up-shifts the fermi-level of nanoparticles (Supplementary Fig. 4b)48. This work function variation in BHT@ZnO is expected to offer better energy alignment and reduce the Schottky barrier in the ETL/acceptor interface, which is advantageous for charge transport39.
The surface properties of the nanoparticles are also studied. Atomic force microscopy (AFM) revealed that both ZnO and BHT@ZnO thin films had smooth and uniform surfaces, with root-mean-square (RMS) roughness values of 2.82 and 3.43 nm, respectively (Supplementary Fig. 5). Contact angle measurements were taken to calculate the surface tension. Both water and ethylene glycol exhibited higher contact angles on BHT@ZnO film than on ZnO film (Supplementary Fig. 6). According to Supplementary Table 2, the calculated surface tension of BHT@ZnO film was found to be lower (35.00 mN·N−1) than ZnO films (50.96 mN·N−1). This observation can be attributed to the presence of tert-butyl groups in BHT moiety that prevent water molecule adsorption by BHT@ZnO.
Photovoltaic performance
The inverted OPV devices were fabricated with the configuration of ITO/ZnO (BHT@ZnO)/active layer/MoO3/Ag (Fig. 2a and Supplementary Fig. 7)49. The chemical structures of materials in this work are presented in Supplementary Fig. 8. The BHT-acid is not an ideal ETL for OPV without ZnO (Supplementary Fig. 9). Figure 2b shows the current density versus voltage (J–V) characteristics of OPV under simulated AM 1.5G solar illumination with different ETL. Further details on the performance of the devices are outlined in Table 1. The PCE of the device based on ZnO ETL was found to be 17.43% with an open-circuit voltage (VOC) of 0.840 V, a short-circuit current density (JSC) of 27.13 mA·cm−2, and a fill factor (FF) of 76.49%. When BHT@ZnO was used instead of ZnO as ETL, the champion PCE was enhanced to 18.48% with VOC of 0.848 V, JSC of 27.46 mA·cm−2, and FF of 79.36% after optimization (as shown in Supplementary Table 3). External quantum efficiency (EQE) characterization was used to prove the photon response of the OPV (as shown in Fig. 2c). The calculated integral current densities were found to be 26.6 and 26.3 mA·cm−2 for BHT@ZnO-based and ZnO-based OPV, respectively, which is consistent with the measured JSC. The BHT@ZnO-based device exhibited improved EQE within the range of 500–900 nm, suggesting better charge transport and collection abilities, even under the same optical transmission of ETL. Regarding the higher VOC value in the BHT@ZnO-based device, we conducted a VOC loss analysis. As shown in Supplementary Fig. 10 and Supplementary Table 4, the lower VOC in the control ZnO-based device stems from the more serious non-radiative recombination loss, which should be related to the intrinsic surface defects of ZnO without BHJ treatment (will be discussed later).

a Device structure of inverted OPV; b J–V curves of inverted OPVs based on ZnO and BHT@ZnO ETL, Inset: the histogram of devices based on ZnO and BHT@ZnO ETL; c EQE spectra of ZnO and BHT@ZnO ETL; d Ambient storage stability and light-soaking effect after storage; e J–V curves variation during light soaking; f Light-dependent VOC variation of fresh and aged devices; MPP tracking of g unencapsulated device and h encapsulated device in ambient condition based on ZnO and BHT@ZnO ETL. Source data are provided as a Source Data file.
Under dark conditions, we investigated the J-V characteristics of devices based on different electron transport layers (ETLs) and found that BHT@ZnO-based device exhibited reduced reversed saturated (leakage) current under reversed bias in comparison to ZnO-based device (Supplementary Fig. 11a). To further understand the charge transport and collection ability, the relationship between photocurrent density ( Jph) and effective voltage (Veff) was analyzed50. As seen in Supplementary Fig. 11b, charge dissociation/transport probability (P) increased from 95.70% to 98.36% when modified with BHT moiety, indicating enhanced charge transport of BHT@ZnO.
The ideality factor (n) fitting from light-dependent VOC was 1.16 and 1.04 for ZnO and BHT@ZnO, respectively (Fig. 2f). The decreased n value represented suppressed trap-assisted recombination, which resulted from the reduced surface defects in ETL. To further verify, transient photocurrent (TPC) and transient photovoltage (TPV) techniques were measured to evaluate the recombination (Supplementary Fig. 12). Compared to the ZnO-based device, the BHT@ZnO-based device showed accelerated charge extraction (0.42 μs versus 0.54 μs) and longer carrier lifetime (0.80 μs versus 0.43 μs), consistent with the reduced trap-assist recombination. Furthermore, electrochemical impedance spectroscopy (EIS) was conducted with a bias equal to VOC to investigate the interfacial resistance51. From the fitted equivalent-circuit model, the ZnO-based device had series resistance (Rs) and recombination resistance (Rrec) of 62.57 Ω and 1762 Ω, respectively, while the BHT@ZnO-based device had Rs and Rrec of 68.26 Ω and 2605 Ω, respectively (Supplementary Fig. 13). This reflects the reduced trap-assisted recombination in the BHT@ZnO devices, which is attributed to the surface passivation of ZnO nanoparticles by BHT.
Device stability
The stability of OPV devices based on ZnO and BHT@ZnO NPs under various aging conditions was assessed. Initially, the photovoltaic performance of unencapsulated devices was tested to determine the decay rate when aged in ambient conditions (room temperature, 20-30% relative humidity). We observed an interesting yet abnormal phenomenon of the light-soaking effect on OPV J-V testing. The PCE variations are provided in Fig. 2d, while detailed parameters are summarized in Supplementary Fig. 14. The freshly prepared devices using both ETLs exhibit no light-soaking effect, but they manifest a growing light-soaking effect after exposure to ambient conditions. And longer aging time exacerbates the light-soaking effect. In ZnO-based devices, as the ambient condition aging time increases, not only the first scan tested PCE drops dramatically, but the continued testing time needed for reaching stable state also increases significantly. Specifically, after approximately 96 h of storage at ambient conditions, the ZnO-based device exhibits a 61.5% retention rate of its initial PCE at its first J–V scan (without soaking). Thereafter, the device’s PCE gradually improves to 90.1% of the initial PCE after soaking under AM 1.5G (100 mW·cm−2) for about 292 s. Then, after 6840 h storing, the ZnO-based device shows a PCE lower than 10% of its initial value in the first J–V scan. The PCE is improved to 70.8% after soaking for 1380 s. The J–V curves variation of the ZnO-based device during the soaking time is provided in Fig. 2e. The device presents an initial S-shape during the initial few J-V scans. Three parameters including VOC, JSC, and FF are significantly increased by illumination. In contrast, the devices based on BHT@ZnO show barely light-soaking effects when even exposed to ambient conditions for the long term (Supplementary Fig. 15). After 7176 h storing, the device based on BHT@ZnO still shows 88.3% of initial PCE in its first J–V scan, then slightly increases to 92.8% after soaking for 34 s.
Metal oxide-based inverted OPV devices without encapsulation have been observed to exhibit a light-soaking effect that should be connected to the adsorption of O2 and H2O, leading to electron extraction from the conducting band of the metal oxide and the formation of a depletion region and upward band bending. This upward band bending impedes electron extraction at the ZnO/bulk heterojunction (BHJ) or ZnO/ITO interfaces52. During the light-soaking process, the photogenerated hole in ZnO recombines with oxygen adsorption, desorbing them and facilitating improved performance. It also suggests that the significantly suppressed light-soaking effect in BHT@ZnO-based devices can be attributed to the surface passivation of VO+, which blocks the adsorption of O2 or H2O and prevents the formation of a depletion region and upward band bending53.
Although the PCE would be partially recovered after light soaking, the devices based on ZnO still show serious degradation after ambient aging. The detailed parameters of the PCE degradation after ambient storage and light soaking recovered are provided in Supplementary Fig. 16. Specifically, a significant burn-in loss is observed which shows a PCE decay of 21% in the first 600 h. After storing at ambient dark conditions for over 8784 h, only around 72.9% of the initial PCE was retained in the devices based on ZnO NPs. In contrast, the devices based on BHT@ZnO retain about 92.8% of the initial PCE after over 8904 h. The rapid PCE decay is mainly associated with VOC and FF. To figure out the charge recombination behavior of the fresh and aged devices, the VOC variation versus light intensity was measured after light soaking (Fig. 2f). The aged device based on ZnO shows a higher ideality factor n of 1.42 than the fresh device (1.16) which suggest severer trap-assisted recombination occurs after aging at ambient condition. It indicates that the oxygen adsorption/desorption on the ZnO surface is not fully reversible. The oxygen adsorption would not fully desorb from the VO+ after long-term ambient aging, even under long time illumination. In contrast, the n value of the BHT@ZnO-based fresh device exhibits a smaller n of 1.04 and slightly increases to 1.10 after aging which indicates reduced trap-assist recombination.
To prove the oxygen adsorption behavior, both the aged ZnO and the aged BHT@ZnO in ambient conditions for 7 days were performed with XPS measurement (Supplementary Fig. 17 and Supplementary Table 1). Compared to the fresh ZnO film, the aged ZnO showed a higher content of the surface-adsorbed oxygen from ambient conditions. While the surface-adsorbed oxygen content of the fresh and aged BHT@ZnO are very similar, which demonstrates the modification of the BHT moiety can shield the VO+, blocking the oxygen adsorption.
The devices were also aging under light exposure (100 mW·cm−2 white LED) with/without encapsulation at open-circuit conditions. After aging at ambient conditions for about 78 h, the ZnO-based device without encapsulation retains only 35% PCE of the pristine value, while the counterpart of the BHT@ZnO-based device is over 70% (Supplementary Fig. 18). The light-soaking behavior was also observed in unencapsulated ZnO-based devices after aging under illumination in ambient (Supplementary Fig. 19). In addition, the encapsulated devices based on BHT@ZnO also show a significant improvement in the PCE retention increase to 85% after 1440 h 1-sun aging, vs. 52% in ZnO case (Supplementary Fig. 20). Besides, the device lifetime was also tested at UV illumination. Under the UV exposure in N2, the ZnO-based devices showed rapid degradation with only 56% PCE retention after 45 h, which is much lower than the counterpart of BHT@ZnO-based devices (83%) (Supplementary Fig. 21).
The operational stability of the devices was further tracked under MPP with continuously simulated 1-Sun illumination using LED arrays in ambient conditions (Supplementary Fig. 22). As shown in Fig. 2g and Supplementary Fig. 23, the device based on ZnO ETL without encapsulation exhibited rapid PCE degradation, decreasing to about 60% of the initial value within 60 min. When ETL is BHT@ZnO, the OPV device showed stable PCE output, retaining over 75% of the initial value after 240 min of MMP aging without encapsulation. Meanwhile, encapsulated devices were tested in MPP conditions (Fig. 2h and Supplementary Fig. 24). The device based on ZnO NPs had serious PCE decay, with the PCE decreasing to lower than 80% of the initial PCE after 580 h. By contrast, the BTH@ZnO-based device retained 85.04% of the initial PCE after aging for 2207 h. This demonstrates that the BTH capping could effectively protect the ZnO from ambient pressures and enhance the operational stability of the OPV devices.
Morphology variation
Grazing-incidence wide-angle x-ray scattering (GIWAXS) was used to detect the evolution of the BHJ layers based on ZnO and BHT@ZnO before and after aging under light illumination in ambient conditions. The images showed that both the ZnO/BHJ and BHT@ZnO/BHJ exhibited a face-on orientation with a π-π stacking (010) peak with a Q vector of 1.785 Å−1 along the profile in the out-of-plane (OOP) direction and a lamellar stacking (100) peak with a Q vector of 0.295 Å−1 along the profile in the in-plane (IP) direction (Fig. 3a). The fresh BHJ based on ZnO and BHT@ZnO ETL exhibited similar crystalline coherence length (CCL) of 18.60 Å and 18.18 Å, respectively (Supplementary Table 5). After aging under UV illumination, obvious decay for both the π-π stacking peak and lamellar stacking peak was observed in the ZnO-based BHJ film (Fig. 3b, c). The CCL of the BHJ decreased significantly to 15.93 Å, indicating serious morphology variation/crystallinity degradation of BHJ after aging, which may originate from photoactive material decomposition. In contrast, BHT@ZnO-based BHJ exhibited mitigated decay in both the π-π stacking peak and lamellar stacking peak, with the CCL of 17.67 Å after aging, a slight drop from 18.18 Å in fresh (Fig. 3d). Furthermore, The AFM analysis revealed the morphological changes in the BHJ after exposure to UV. Surface morphology was examined at different locations during various aging stages to systematically analyze the evolution of the active layer. UV-vis absorption measurements of the BHJ were conducted simultaneously to assist analysis (Supplementary Fig. 25). Initially, distinct nanofiber structures and smooth surfaces with RMS roughness values of 2.00 nm and 1.76 nm were observed in the BHJ coated on ZnO and BHT@ZnO, respectively (Fig. 3e, f). As the aging process progressed, the absorption intensity of the BHJ gradually decreased, which will be discussed in detail in the subsequent section. Concurrently, the fibrous morphology of the BHJ began to fade, eventually transitioning into a spherical agglomeration on ZnO after 240 h of UV exposure (Supplementary Fig. 26). In contrast, the BHJ exhibited a relatively moderate decay in surface morphology on BHT@ZnO ETL, maintaining the nanofiber structure at the same aging stage (Supplementary Fig. 27). The GIWAXS pattern and AFM image variation confirmed that BHT modification effectively suppresses the degradation of the active layer under light illumination.

a 2D GIWAXS patterns; 1D GIWAXS profiles in b OOP direction and c IP direction; d Calculated CCL values of active layers based on fresh and aged ZnO and BHT@ZnO, respectively; e Height images measured from AFM and f phase images measured from AFM. Source data are provided as a Source Data file.
Radical-induced selective catalytic degradation of polymer/NFA
The trace amount of impurity in the active layer can cause defects to form, significantly affecting the performance of the photovoltaic system. Therefore, it is crucial to comprehend the molecular-level degradation of the active layer54. UV-vis absorption spectra of the active layer after aging were measured. According to Fig. 4a, the PM6:BTP-eC9 active layer coated on neat glass exhibited remarkable chemical stability, with unchanged absorption for both the donor (PM6) and acceptor (BTP-eC9) under continuous UV radiation in N2 (O2 < 10 ppm, H2O < 10 ppm). When coated on ZnO, the absorption of PM6 remained almost unchanged, while the absorption of BTP-eC9 was noticeably weakened, remained ca. 86% of initial intensity after UV radiation in N2 as displayed in Fig. 4b. It can be inferred that when coated on ZnO ETL, the decay of BTP-eC9 is due to oxidation by hydroxyl radicals (HO·) generated from the adsorbed hydroxyl ion (OH−) on the ZnO surface34. The aging profile of the blending film suggests that the polymer donor PM6 is not oxidized by HO·, implying that HO· selectively oxidizes BTP-eC9.

The active layer coated on glass, ZnO and BHT@ZnO under UV illumination (365 nm, 6 W) in a–c N2 (O2 content <10 ppm, H2O content <10 ppm); d–f Dry air (O2 content ≈ 19%, relative humidity ≈ 1%); g–i Ambient (O2 content ≈ 19%, relative humidity ≈ 60–70%); j–l Summary of the absorption peak intensity for PM6 and BTP-eC9. Source data are provided as a Source Data file.
When aging on glass at dry air (O2 content ≈ 19%, relative humidity ≈ 1%), the absorption decay of the active layer was severer than under UV/N2 conditions. However, opposite to UV/N2, the degradation of PM6 was stronger than BTP-eC9 under UV/dry air. According to Fig. 4d, PM6 suffered from an obvious absorption decrease (80% of initial intensity after 240 h), even if without any ETL. We speculated the strong degradation of PM6 under UV/ambient was due to being oxidized by superoxide anion radical (·O2−), which could be generated from O2/UV condition55,56. Moreover, moderate decay of BTP-eC9 was observed (95% of initial intensity after 240 h). The same samples were also aged in humid air (relative humidity ≈ 60–70%) and showed similar absorption decay (75% and 94% retention for PM6 and BTP-eC9 after 240 h aging) (Fig. 4g). It indicated that H2O in air may not participate in active layer decay process under UV without ETL. So H2O-participated transformation from ·O2− to HO· would not occur in this stage55. In addition, the pure BTP-eC9 showed higher UV resistance to pure PM6 (Supplementary Fig. 28). In this case, we concluded that the oxidation of ·O2− to PM6 is much higher than BTP-eC9. In other words, the ·O2− exhibited selective oxidization to PM6 rather than BTP-eC9.
When aging on ZnO in air, both the PM6 and BTP-eC9 suffered severer absorption decay than their counterparts without ETL (Fig. 4e, h). As we mentioned ·O2− and HO· were the culprits for PM6 and BTP-eC9 degradation, respectively, severer absorption decay means more ·O2− and HO· generated due to the presence of ZnO/UV radiation. Specifically, the electron in the valence band of ZnO was excited to the conducting band. The O2 and H2O adsorbed on the surface VO+ of ZnO could extract the electron in the conducting band and hole in the valence band, then generate extra ·O2− and HO·, respectively. Furthermore, the BTP-eC9 experienced stronger absorption decay in humid air compared to dry air, suggesting that H2O-participated HO· generation could occur with the existence of ZnO ETL (Fig. 4h).
When replacing ZnO with BHT@ZnO, both the PM6 and BTP-eC9 exhibited higher absorption retentions at the same aging states (Fig. 4j–l), which indicates the ·O2− and HO· were suppressed with the existence of BHT@ZnO. A similar phenomenon was observed in active layers on ZnO and BHT@ZnO samples when illuminated with white LED light (Supplementary Fig. 29). To exclude the thermal stress during the photobleaching process under UV illumination, the devices underwent aging at 80 °C in dark conditions and under both nitrogen and ambient atmospheres (Supplementary Fig. 30). The absorption characteristics of both PM6 and BTP-eC9 remained consistent before and after continuous heating, suggesting that the influence of thermal stress on molecular stability could be effectively eliminated.
Based on this, EPR was employed to detect the generation of HO· and ·O2− under UV/N2 and UV/ambient, respectively, to prove our speculation. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) was used as the radical trap. As observed in Supplementary Fig. 31, ZnO did not generate any radicals under dark conditions in both N2 and ambient. However, ZnO showed notable HO· signals under UV/N2, and ·O2− signals under UV/ambient (Fig. 5a, b). In BHT@ZnO samples, both the HO· and ·O2− signals were suppressed. This could be attributed to two reasons as follows. First, the passivated VO+ on the BHT@ZnO surface decreased the adsorption of residual hydroxyl ion, O2 and H2O (in ambient), thus decreasing the source of HO· and ·O2−. Secondly, the radical scavenging ability of BHT could effectively capture the radicals, cutting off the degradation process of photoactive materials of OPV. Besides, to further demonstrate the radical scavenging capability of the BHT, BHT-acid was added into the Fenton’s reagent with a concentration of 0.5 mg/ml. In Fenton’s reagent, HO· was generated in H2O solution, while ·O2− was generated in methanol solution57. The details of Fenton’s reagent can be found in SI. According to Supplementary Fig. 32, the intensities of EPR signals for both HO· and ·O2− based on samples with BHT are obviously lower than the sample without BHT during the whole reaction. Apparently, the reduction of the ROS is scavenged by the BHT.

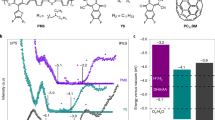
EPR spectra of the DMPO extracted from ZnO and BHT@ZnO films in a UV/N2 and b UV/air; MALDI-TOF-TOF-MS spectra of c 3BDDBDT and d 3BDTBDD after aging at UV/air for 168 h; FT-IR spectra of fresh and aged e 3BDTBDD and f 3BDDBDT samples; g Possible degraded product of BDT-BDD copolymers catalyzed by ·O2−; h Fitted peaks of PM6 absorption spectra aging under UV/air with different time. Dash lines were obtained from the experiment, and colored areas were fitted peaks: pink area indicated a strong aggregated (ordered packing) phase, blue area indicated an amorphous phase; i Dihedral angle between BDT (BDT1O, DBT2O) and BDD unit extracted from DFE simulations. Source data are provided as a Source Data file.
Degraded products of polymer/NFA active layer system
The degradation products of the NFA were first investigated by matrix-assisted laser desorption/ionization time-of-flight/time-of-flight mass (MALDI-TOF/TOF-MS) spectroscopy and provided in Supplementary Fig. 33. The degradation of non-fullerene acceptors primarily occurs at the linkage between the fused-ring core and the terminal isocyanate (IC) groups, specifically through vinyl group oxidation, which leads to bond cleavage. The oxidation of the vinyl group is mainly catalyzed by ·OH radical. Additionally, degradation also affects the side alkyl chains58. However, the other key component - polymer donor is yet to be determined. To determine the degradation path of the polymer donor PM6 in the presence of ·O2−, it is necessary to address the challenge of characterizing the molecular variation of the polymer itself, which is hampered by its undefined structure, including molecular weight and number of repeating units. To overcome this limitation, we employed two oligomers, namely 3BDDBDT and 3BDTBDD, both of which have well-defined structures, to simulate the degradation process of PM6 (or PBDB-T)59,60. These oligomers serve as simplified versions of PM6 and are found to exhibit poor photostability (Supplementary Fig. 34). To analyze the oxidation products of the two oligomers after aging under UV/air, we employed MALDI-TOF/TOF-MS spectroscopy (Fig. 5c, d). In the spectra, we observed distinct peaks corresponding to the initial molecular weights (m/z) of 1791.4 for 3BDDBDT and 1760.5 for 3BDTBDD (blue area). Furthermore, we observed a series of peaks with m/z values of 1807.4 (1791.4 + 16), 1823.4 (1791.4 + 32), and 1855.4 (1791.4 + 64) in the 3BDDBDT film. Similarly, m/z values of 1776.5 (1760.5 + 16), 1792.5 (1760.5 + 32), and 1824.5 (1760.5 + 64) were observed in the 3BDTBDD film. These peaks indicate that the aged products of both oligomers exhibited an increased molecular weight that corresponded to multiples of 16. To further confirm the oxidation position, the degradation process of both monomers (BDT and BDD) oligomers (3BDDBDT and 3BDTBDD) has been detected by FT-IR. First, both monomers are easily oxidized, in which the new signals in aged monomers belong to the S=O bond (ca. 1050 and 1200 cm−1), C=O (ca. 1710 cm−1) bond, and OH group (ca. 3500 cm−1) (Supplementary Fig. 35)61. In the case of oligomers, similar FT-IR signals in these ranges have also been observed. However, the oxidation of 3BDTBDD is much faster than 3BDDBDT (Fig. 5e, f and Supplementary Fig. 36). 3BDTBDD with two BDT units on both sides shows a higher oxidation rate, indicating the BDT unit (including the fused ring and side chains) is more prone to oxidation due to the higher electron density when consisting of the electron push–pull alternative structures. Previous reports also proved that the sulfur in the large conjugated fused rings (e.g., benzothiophene) is easier to oxidize than thiophene62,63. Based on these findings, we provide the possible oxidized products of the BDD-BDT copolymers (PBDB-T and PM6) in Fig. 5g. It should be noted that while the side groups in PM6 may undergo oxidation, the photobleaching in PM6 is primarily attributed to the oxidation of the main conjugated backbone chain (i.e., BDT unit). These compounds are the products of thiophene oxidized by ·O2− or ·O2H64,65.
Although the BDT (n)oxide (BDTnO) (n = 1–4) contained matter may not be the final decomposed product, it still plays a decisive role in material properties. On the one hand, the oxidation process from thiophene in BDT to the sulfoxide or sulfone would completely convert the electron-donating property to an electron-accepting property. This would lead to the destruction of electron donor/acceptor alternative units in the PM6 backbone and the loss of the intramolecular charge transfer process65,66,67. On the other hand, the sp2 hybridization in thiophene changes to sp3 hybridization in oxidized thiophene, weakening the π-π stacking of donor materials by increased intermolecular steric hindrance. The reduced π-π stacking was already observed in the GIWAXS measurement. Consequently, these preliminarily aged BDTO-contained products significantly impact the properties of initial donor materials, which greatly deteriorates the photovoltaic performance.
For further correlating the molecular packing variation by PM6 oxidation, the absorption spectra of pure PM6 thin films with different aging states were investigated (Fig. 5h). The peak of 1.97 eV indicated the order packing phase while 2.11 eV indicated the amorphous phase68,69. The order phase exhibited an area ratio of 13.2%, 5.16%, and 2.95% for fresh, 23 h, and 67 h aged PM6, respectively (Supplementary Table 6). The decreased order phase and increased amorphous phase of PM6 should be attributed to the deteriorated conjugated plane in the polymer main chain.
Notably, recent research revealed that the stronger backbone twist occurred in bleached PM6 film after UV/ambient conditions24, but the origin of this backbone twist is still unknown. Here we calculated the dihedral angle between BDT and BDD in BDT-BDD, BDT1O-BDD, and BDT2O-BDD molecules, respectively, by DFT simulations to study the change of geometry structures in the PM6 backbone (Supplementary Fig. 37). The dihedral angle between BDT/BDD moieties is 5.01°, while the oxide products, BDT1O-BDD and BDT2O-BDD exhibited larger dihedral angle of 19.05° and 13.79° (Fig. 5i). It indicated the enlarged dihedral angle inside the backbone of PM6 was due to the generate of BDTnO (n = 1–4). This enlarged dihedral angle (twisted backbone) also deteriorates the conjugated plane and converts the order phase in PM6 to an amorphous phase. Our work is consistent with previous simulated results and provided more and deeper evidence for explaining this UV/ambient induced backbone twisting of PM624. For future donor materials designing, it is necessary to explore other electron donor monomers for replacing the BDT unit aiming to improve the stability at the molecule level.
Figure 6 presents a summarized schematic diagram illustrating the radical generation and scavenging processes involved in the stability issue of the active layer under light illumination and the function of BHT@ZnO. First, in the absence of ZnO, UV illumination of O2 leads to the generation of ·O2− (Process 1). Second, when ZnO is excited by UV, O2 adsorbed in VO+ extracts the electrons in the conducting band and generates additional ·O2− (Process 2). PM6, the donor material, is easily oxidized by ·O2−. Additionally, UV excitation of ZnO allows H2O adsorbed in VO+ to extract the hole in the valence band and generate HO· (Process 3). Lastly, the adsorbed hydroxyl ion in VO+ extracts the hole in the valence band of ZnO and generates extra HO· (Process 4). The strongly oxidizing HO· selectively decomposes non-fullerene acceptors. The adsorption of O2 and H2O from the external environment by VO+ contributes to the light-soaking effect and aggravates the material's deterioration. For the BHT@ZnO ETL, the carboxyl groups effectively passivate VO+, and two hydrophobic tert-butyls in BHT prevent the adsorption of O2 and H2O, thereby blocking Processes 2 and 3. The residual hydroxyl ions are also neutralized by BHT-acid, blocking Process 4. Moreover, BHT efficiently scavenges the inevitably generated HO· and ·O2−, thus preventing the decomposition of both donors and acceptors.

Left (red area): Four potential mechanisms of ROS generation in ZnO under light illumination, along with the degradation pathways of both the BDT-based donor and NFA small molecule acceptor catalyzed by ·O2− and ·OH, respectively. Right (blue area): Surface passivation of ZnO by BHT and its effects on reducing O2/H2O adsorption and scavenging ROS.
Finally, to prove the versatility of BHT@ZnO, three systems including PM6:Y6, PM6:L8-BO, and PM6:BTP-eC9:o-BTP-eC9 have been employed as active layers in inverted OPV70. As seen in Fig. 7a–c and Table 1, the devices based on BHT@ZnO performed better PCE of 17.52% and 18.45% of landmark systems PM6:Y6 and PM6:L8-BO, respectively, which were obviously higher than the ZnO-based devices (16.49% and 17.38%). Moreover, we applied a ternary system PM6:BTP-eC9:o-BTP-eC9 which is reported by our group with a high efficiency approaching 20% (regular structure) into inverted structure70. The corresponding inverted BHT@ZnO-based device exhibited a record-high efficiency of 19.47% with a VOC of 0.859 V, JSC of 28.39 mA·cm−2, and FF of 79.87%. The device was certificated by Enli Tech Optoelectronic Calibration Lab (ISO/IEC 17025:2017 accredited Calibration Lab) with an efficiency of 18.97% (Supplementary Fig. 38). Noted that the efficiency gap between inverted and regular devices has been decreased to ~0.4% based on BHT@ZnO ETL. More excitingly, the device based on BHT@ZnO exhibited excellent stability under MPP tracking (Fig. 7d and Supplementary Fig. 39). After continuously aging at MPP under light exposure for over 7724 h, the BHT@ZnO-based inverted OPV demonstrating an excellent lifetime with 81.5% PCE retention. To the best of our knowledge, it is the first to achieve such a long measured (rather than extrapolated) lifetime of OPV with a high certified PCE of 19%, including regular and inverted OPV devices (Fig. 7e and Supplementary Table 6).

J–V curves of OPV devices based on a PM6:Y6, b PM6:L8-BO, and c PM6:BTP-eC9:o-BTP-eC9 systems; d Lifetime of inverted OPV based on PM6:BTP-eC9:o-BTP-eC9 active layer and BHT@ZnO ETL under MPP tracking; e Statistical figure of PCE vs. T80 of OPV. Source data are provided as a Source Data file.
Discussion
In summary, a multifunctional BHT@ZnO nanoparticle was designed and synthesized as an ETL for inverted OPV to simultaneously enhance efficiency and stability. BHT@ZnO exhibited decreased surface energy and reduced surface defects compared to unmodified ZnO. This modification resulted in a remarkable PCE of 19.47% in inverted OPVs. Notably, the inverted OPVs showed light-soaking-free behavior and excellent stability under ISOS-D-1 (94.2% PCE retention after 8904 h ambient storage without encapsulation) and ISOS-L-1 testing protocol (81.5% PCE retention after 7724 h MPP tracking). Additionally, we discovered and elucidated the detailed degradation mechanism of the active layer systems, which involved the selective catalysis of PM6 and BTP-eC9 by superoxide and hydroxyl radicals, as well as the oxidation products of BDT-based donor materials upon radiation exposure. Furthermore, we demonstrated the ability of BHT@ZnO as an ETL to suppress the superoxide and hydroxyl radicals, thereby preventing the degradation of the active layer molecules. This study provides a deeper understanding of the degradation mechanism of active layer materials, offers a potential solution for ETL improvement, and has significant implications for the future design of active layer materials.
Methods
Materials
All materials are purchased from commercial suppliers: PM6, BTP-eC9, L8-BO, Y6 (Solarmer Energy Inc.), TCB (Tokyo Chemical Industry Co., Ltd.), Zn(OAc)2·2H2O, KOH, BHT-acid (Sigma Aldrich). Other materials were purchased from Sigma Aldrich. All reagents and solvents are used directly without further purification.
BHT@ZnO nanoparticles synthesis
First, Zn(OAc)2·2H2O (2.963 g) was dissolved in methanol (125 ml) at 65 °C for 10 min. After fully dissolved, KOH solution (1.335 g in 65 ml methanol) was added dropwise for 15 min under vigorous stirring. Then the reaction was kept at 65 °C for 4 h and then stood still for 3 h to fully precipitate. The obtained ZnO nanoparticles were washed twice with methanol. After that, the obtained ZnO nanoparticles reacted with BHT-acid (0.05 g) at room temperature for 6 h to finish the reaction. Finally, the obtained BHT@ZnO was washed twice with methanol dispersed in methanol to form the ink.
Device fabrication and characterization
First, the patterned ITO-coated glass was cleaned by detergent, de-ionized water, acetone, and isopropyl alcohol (IPA) for 40 min under ultrasonication sequentially. The substrate then was dried in N2 flow and treated with UV ozone for 20 min. The prepared ZnO and BHT@ZnO NPs dispersion were coated on ITO and thermal annealed on a hot plate at 130 °C for 20 min. Then TCB (10 mg/ml) in the PM6:BTP-eC9, PM6:Y6, and PM6:L8-BO (D:A = 1:1.2) with the total concentration of 17 mg/ml dissolved in chloroform were coated on the samples, followed by thermal annealing on a hot plate at 100 °C for 10 min. For the ternary system, the ratio of PM6:BTP-eC9:o-BTP-eC9 is 1:1.05:0.15 with a total concentration of 16 mg/ml in chloroform with DIB additive (12.5 mg/ml). All the precursor solution would be heated to 60 °C for an hour to fully dissolve the solutes then cooled to room temperature before use. After coating, the active layer experienced a process of thermal annealing at 100 °C for 5 min. 5-nm MoO3 and 100-nm Ag were then vacuum deposited as the electrode. All devices were tested with a metal mask whose area is ~6.1 mm2. The J–V curves of the OPV were tested by Keithley 2400 source meter and solar simulator (SS-F7-3A, Enli Tech. Co., Ltd., Taiwan) along with AM 1.5 G spectra whose intensity was corrected by a standard silicon solar cell at 100 mW/cm2. The EQE was measured by a certified incident photon to electron conversion (IPCE) equipment (QE-R) from Enli Technology Co., Ltd. The lifetime of the device was tested by MPP tracking equipment (Enli Technology Co., Ltd.).
EPR measurement
EPR measurements were carried out in ambient conditions on an ADANI SpinscanX spectrometer, operating at 100 kHz field modulation using the 150 ut modulation amplitude. For the trapping of OH radical, the ZnO was immersed in DMPO anhydrous toluene solution (50 mM) in N2 under dark or illuminated by AM 1.5 G (100 mW·cm−2) for 20 min. The sample was drawn by capillary which was furtherly sealed by tweezers after heating by an alcohol lamp. The capillary was placed into the quartz tube and then placed into the EPR cavity. For the trapping of O2 radical, the prepared ETL were immersed in DMPO methanol solution (50 mM) in ambient conditions under dark or illuminated by AM 1.5 G (100 mW·cm−2). The next steps were similar to the trapping of OH radicals.
Other characterizations
UV-vis absorption spectra and transmittance spectra were carried out by Varian Cary® 300 UV-Vis spectrophotometers (Agilent Technologies). MALDI-TOF/TOF-MS was carried out by the Bruker UltrafleXtreme. Atomic force microscopic (AFM) images were acquired using a Bruker Dimension Icon in tapping mode. GIWAXS measurements were carried out with a Xeuss 2.0 SAXS/WAXS laboratory beamline using a Cu X-ray source (8.05 keV, 1.54 Å) and Pilatus3R 300K detector. The incidence angle is 0.2°. TPV was conducted under 1 sun condition by illuminating the device with a white light-emitting diode and set at the open-circuit condition. TPC was set to the short-circuit condition in the dark. The output signal was collected by a Keysight oscilloscope. The contact angles were tested by a DSA-100 (KRUSS Germany) contact angle meter.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding author on request. The main data supporting the findings of this study are available within the published article and its Supplementary Information and source data files. Additional data are available from the corresponding author on request. Source data are provided with this paper.
References
Cheng, P., Li, G., Zhan, X. & Yang, Y. Next-generation organic photovoltaics based on non-fullerene acceptors. Nat. Photonics 12, 131–142 (2018).
Yan, C. et al. Non-fullerene acceptors for organic solar cells. Nat. Rev. Mater. 3, 18003 (2018).
Zhao, Y. et al. Achieving sustainability of greenhouses by integrating stable semi-transparent organic photovoltaics. Nat. Sustain. 6, 539–548 (2023).
Cui, Y. et al. Single-junction organic photovoltaic cells with approaching 18% efficiency. Adv. Mater. 32, 1908205 (2020).
Jiang, Y. et al. An alcohol-dispersed conducting polymer complex for fully printable organic solar cells with improved stability. Nat. Energy 7, 352–359 (2022).
Qin, F. et al. Robust metal ion-chelated polymer interfacial layer for ultraflexible non-fullerene organic solar cells. Nat. Commun. 11, 4508 (2020).
Liu, X., Zhong, Z., Zhu, R., Yu, J. & Li, G. Aperiodic band-pass electrode enables record-performance transparent organic photovoltaics. Joule 6, 1918–1930 (2022).
Sun, Y. et al. Flexible organic photovoltaics based on water-processed silver nanowire electrodes. Nat. Electron. 2, 513–520 (2019).
Huang, J. et al. Intrinsically stretchable, semi-transparent organic photovoltaics with high efficiency and mechanical robustness via a full-solution process. Energy Environ. Sci. 16, 1251–1263 (2023).
Lin, Y. et al. An electron acceptor challenging fullerenes for efficient polymer solar cells. Adv. Mater. 27, 1170–1174 (2015).
Xu, Y. et al. A new conjugated polymer that enables the integration of photovoltaic and light-emitting functions in one device. Adv. Mater. 33, 2101090 (2021).
Wang, J., Xue, P., Jiang, Y., Huo, Y. & Zhan, X. The principles, design and applications of fused-ring electron acceptors. Nat. Rev. Chem. 6, 614–634 (2022).
Li, C. et al. Non-fullerene acceptors with branched side chains and improved molecular packing to exceed 18% efficiency in organic solar cells. Nat. Energy 6, 605–613 (2021).
He, Z. et al. Single-junction polymer solar cells with high efficiency and photovoltage. Nat. Photonics 9, 174–179 (2015).
Wang, J. et al. Physical insights into non-fullerene organic photovoltaics. Nat. Rev. Phys. 6, 365–381 (2024).
Yuan, J. et al. Single-junction organic solar cell with over 15% efficiency using fused-ring acceptor with electron-deficient core. Joule 3, 1140–1151 (2019).
Fu, J. et al. 19.31% binary organic solar cell and low non-radiative recombination enabled by non-monotonic intermediate state transition. Nat. Commun. 14, 1760 (2023).
Fu, J. et al. Eutectic phase behavior induced by a simple additive contributes to efficient organic solar cells. Nano Energy 84, 105862 (2021).
Kawano, K. et al. Degradation of organic solar cells due to air exposure. Sol. Energy Mater. Sol. Cells 90, 3520–3530 (2006).
Du, X. et al. Efficient polymer solar cells based on non-fullerene acceptors with potential device lifetime approaching 10 years. Joule 3, 215–226 (2019).
Liu, Z.-X. et al. Molecular insights of exceptionally photostable electron acceptors for organic photovoltaics. Nat. Commun. 12, 3049 (2021).
Park, S. et al. Progress in materials, solution processes, and long-term stability for large-area organic photovoltaics. Adv. Mater. 32, 2002217 (2020).
Li, Y. et al. Lifetime over 10000 hours for organic solar cells with Ir/IrO(x) electron-transporting layer. Nat. Commun. 14, 1241 (2023).
Wang, Y. et al. The critical role of the donor polymer in the stability of high-performance non-fullerene acceptor organic solar cells. Joule 7, 810–829 (2023).
Cheng, P. & Zhan, X. Stability of organic solar cells: challenges and strategies. Chem. Soc. Rev. 45, 2544–2582 (2016).
Xu, X. et al. Interface-enhanced organic solar cells with extrapolated T(80) lifetimes of over 2 years. Sci. Bull. 65, 208–216 (2020).
Hong, L. et al. Simultaneous improvement of efficiency and stability of organic photovoltaic cells by using a cross-linkable fullerene derivative. Small 17, 2101133 (2021).
Shrotriya, V., Li, G., Yao, Y., Chu, C.-W. & Yang, Y. Transition metal oxides as the buffer layer for polymer photovoltaic cells. Appl. Phys. Lett. 88, 073508 (2006).
Xin, Y. et al. Multiarmed aromatic ammonium salts boost the efficiency and stability of inverted organic solar cells. J. Am. Chem. Soc. 146, 3363–3372 (2024).
Zhu, C. et al. Tuning the electron-deficient core of a non-fullerene acceptor to achieve over 17% efficiency in a single-junction organic solar cell. Energy Environ. Sci. 13, 2459–2466 (2020).
Pan, W. et al. An efficiency of 14.29% and 13.08% for 1 cm2 and 4 cm2 flexible organic solar cells enabled by sol–gel ZnO and ZnO nanoparticle bilayer electron transporting layers. J. Mater. Chem. A 9, 16889–16897 (2021).
Wang, Y. et al. New method for preparing ZnO layer for efficient and stable organic solar cells. Adv. Mater. 35, 2208305 (2023).
Qiu, Y., Peng, R., Shi, J., Chen, Z. & Ge, Z. Revealing and eliminating the light-soaking issue in metal oxide-based inverted organic solar cells. Adv. Funct. Mater. 33, 2300831 (2023).
Liu, B. et al. Visible light–induced degradation of inverted polymer:nonfullerene acceptor solar cells: initiated by the light absorption of ZnO layer. Sol. RRL 5, 2000638 (2020).
Beek, W. J., Wienk, M. M., Kemerink, M., Yang, X. & Janssen, R. A. Hybrid zinc oxide conjugated polymer bulk heterojunction solar cells. J. Phys. Chem. B 109, 9505–9516 (2005).
Marczak, R. et al. Communication via electron and energy transfer between zinc oxide nanoparticles and organic adsorbates. J. Phys. Chem. C. 113, 4669–4678 (2009).
Chen, X. et al. Functional ligand-decorated ZnO nanoparticles as cathode interlayers for efficient organic solar cells. ACS Appl. Energy Mater. 5, 1291–1297 (2021).
Yao, K., Chen, L., Chen, Y., Li, F. & Wang, P. Interfacial nanostructuring of ZnO nanoparticles by fullerene surface functionalization for “annealing-free” hybrid bulk heterojunction solar cells. J. Phys. Chem. C. 116, 3486–3491 (2012).
Li, S. et al. Achieving over 18 % efficiency organic solar cell enabled by a ZnO-based hybrid electron transport layer with an operational lifetime up to 5 years. Angew. Chem. Int. Ed. Engl. 61, e202207397 (2022).
Liu, X. et al. Fluidic manipulating of printable zinc oxide for flexible organic solar cells. Adv. Mater. 34, 2106453 (2022).
Yao, K. et al. Fullerene-anchored core-shell ZnO nanoparticles for efficient and stable dual-sensitized perovskite solar cells. Joule 3, 417–431 (2019).
Liu, B.-W. et al. ZnO surface passivation with glucose enables simultaneously improving efficiency and stability of inverted polymer: non-fullerene solar cells. Chin. J. Polym. Sci. 40, 1594–1603 (2022).
Wei, J. et al. Silane-capped ZnO nanoparticles for use as the electron transport layer in inverted organic solar cells. ACS Nano 12, 5518–5529 (2018).
Chen, D. et al. Influence of defects on the photocatalytic activity of ZnO. J. Phys. Chem. C. 118, 15300–15307 (2014).
Hu, L. et al. Significant enhancement of illumination stability of nonfullerene organic solar cells via an aqueous polyethylenimine modification. J. Phys. Chem. Lett. 12, 2607–2614 (2021).
Yan, L. et al. Simultaneous UV and air stability improvement of organic solar cells enabled by carbon-coated zinc oxide as cathode buffer layer. Cell Rep. Phys. Sci. 4, 101654 (2023).
Alkhorayef, M. et al. Evaluation of dose uncertainty in radiation processing using EPR spectroscopy and butylated hydroxytoluene rods as dosimetry system. Radiat. Phys. Chem. 141, 50–56 (2017).
Kroupa, D. M. et al. Tuning colloidal quantum dot band edge positions through solution-phase surface chemistry modification. Nat. Commun. 8, 15257 (2017).
Li, G., Chu, C.-W., Shrotriya, V., Huang, J. & Yang, Y. Efficient inverted polymer solar cells. Appl. Phys. Lett. 88, 253503 (2006).
Huang, J. et al. Significant influence of halogenation on the energy levels and molecular configurations of polymers in DTBDT-based polymer solar cells. Mater. Chem. Front. 3, 1244–1252 (2019).
Romero, B. et al. Characterization of Organic and Perovskite Solar Cells by Impedance Spectroscopy Vol. 11095 OPP (SPIE, 2019).
Wilken, S., Parisi, J. & Borchert, H. Role of oxygen adsorption in nanocrystalline ZnO interfacial layers for polymer–fullerene bulk heterojunction solar cells. J. Phys. Chem. C. 118, 19672–19682 (2014).
Kumar, R., Al-Dossary, O., Kumar, G. & Umar, A. Zinc oxide nanostructures for NO(2) gas-sensor applications: a review. Nanomicro Lett. 7, 97–120 (2015).
Che, Y. et al. Design of furan-based acceptors for organic photovoltaics. Angew. Chem. 62, 202309003 (2023).
D’Orazio, J., Jarrett, S., Amaro-Ortiz, A. & Scott, T. UV radiation and the skin. Int J. Mol. Sci. 14, 12222–12248 (2013).
van der Pol, T. P. A. et al. Origin, nature, and location of defects in PM6:Y6 organic solar cells. Adv. Energy Mater. 13, 2300003 (2023).
Pignatello, J. J., Oliveros, E. & MacKay, A. Advanced oxidation processes for organic contaminant destruction based on the fenton reaction and related chemistry. Crit. Rev. Environ. Sci. Technol. 36, 1–84 (2006).
Jiang, P. et al. On the interface reactions and stability of nonfullerene organic solar cells. Chem. Sci. 13, 4714–4739 (2022).
Xia, H. et al. Structure evolution from D-A-D type small molecule toward D-A-D-A-D type oligomer for high-efficiency photovoltaic donor materials. Dyes Pigm. 186, 108950 (2021).
Xia, H. et al. A–D–A–D–A-type oligomer versus A–D–A-type small molecule: synthesis and advanced effect of the D–A repeat unit on morphology and photovoltaic properties. ACS Appl. Energy Mater. 5, 3146–3155 (2022).
Gola, A., Bernardi, A., Pasut, G. & Musial, W. The influence of initiator concentration on selected properties of thermosensitive poly(acrylamide-co-2-acrylamido-2-methyl−1-propanesulfonic acid) microparticles. Polymers 13, 996 (2021).
Otsuki, S. et al. Oxidative desulfurization of light gas oil and vacuum gas oil by oxidation and solvent extraction. Energy Fuels 14, 1232–1239 (2000).
Zhang, B. et al. Catalytic oxidation of thiophene and its derivatives via dual activation for ultra-deep desulfurization of fuels. J. Catal. 287, 5–12 (2012).
Dell, E. J. & Campos, L. M. The preparation of thiophene-S,S-dioxides and their role in organic electronics. J. Mater. Chem. 22, 12945–12952 (2012).
Lei, T. et al. Highly efficient non-fullerene organic solar cells using 4,8-Bis((2-ethylhexyl)oxy)benzo[1,2-b:4,5-b′]dithiophene-based polymers as additives. Macromolecules 51, 4032–4039 (2018).
Pappenfus, T. M. et al. Synthesis and electronic properties of oxidized benzo[1,2-b:4,5-b’]dithiophenes. J. Org. Chem. 79, 9408–9412 (2014).
Zhen, S. et al. Efficient red/near-infrared fluorophores based on benzo[1,2-b :4,5-b′]dithiophene 1,1,5,5-tetraoxide for targeted photodynamic therapy and in vivo two-photon fluorescence bioimaging. Adv. Funct. Mater. 28, 1706945–1706960 (2018).
An, K. et al. Mastering morphology of non-fullerene acceptors towards long-term stable organic solar cells. Nat. Commun. 14, 2688 (2023).
Clark, J., Silva, C., Friend, R. H. & Spano, F. C. Role of intermolecular coupling in the photophysics of disordered organic semiconductors: aggregate emission in regioregular polythiophene. Phys. Rev. Lett. 98, 206406 (2007).
Fu, J. et al. Rational molecular and device design enables organic solar cells approaching 20% efficiency. Nat. Commun. 15, 1830 (2024).
Acknowledgements
Prof. Li acknowledges the support by the Research Grants Council of Hong Kong (Project Nos GRF 15307922, 15221320, C7018−20G, C4005−22Y), RGC Senior Research Fellowship Scheme (SRFS2223-5S01), Hong Kong Innovation and Technology Commission (ITC-MHKJFS MHP/020/23), the Hong Kong Polytechnic University (Sir Sze-yuen Chung Endowed Professorship Fund (8-8480), RISE (Q-CDBK), PRI (QCD7X), G-SAC5). Prof. Lee thanks the support of the Research Grants Council of Hong Kong, General Research Fund (Project No. C1009−17E).
Author information
Authors and Affiliations
Contributions
J.H., J.F., and B.Y. contributed equally to this work. G.L. and J.H. conceived the idea. J.H. and G.L. designed the experiments. J.H. and B.Y. synthesized the ZnO and BHT@ZnO. J.H. and J.F. carried out the device fabrication, lifetime measurement, and the majority of the characterization. H.X. provided the 3BDDBDT and 3BDTBDD. T.C. and T.W.B.L. carried out the MALDI-MS measurement. Y.L. carried out the DFT simulation. Z.R. carried out the PL measurement, and Q.L. carried out the FT-IR measurement. K.L. carried out the AFM measurement. G.Z. and H.Y. carried out the EQE-EL measurement. H.L. and X.L. carried out the GIWAXs measurement. Z.G. and C-S.L. carried out the UPS and XPS measurements. B.Y. and Y.P. carried out the EPR measurement. H.T.C. analyzed the data. J.H. wrote the manuscript. G.L. revised the manuscript and supervised the project. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Weiwei Li who co-reviewed with Chengyi Xiao, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, J., Fu, J., Yuan, B. et al. 19.5% Inverted organic photovoltaic with record long-lifetime via multifunctional interface engineering featuring radical scavenger. Nat Commun 15, 10565 (2024). https://doi.org/10.1038/s41467-024-54923-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54923-6
This article is cited by
-
Multifunctional interface engineering enables efficient and stable inverted organic photovoltaics
Nature Communications (2025)
-
Improved damp heat and thermal cycling stability of organic solar cells
Nature Energy (2025)
-
Stretchable Polymer Solar Cells Beyond 15% Efficiency: Strategies, Achievements, and Outlook
Chemical Research in Chinese Universities (2025)
