
Abstract
Nipah virus (NiV) is a non-segmented, negative-strand (NNS) RNA virus, belonging to Paramyxoviridae. The RNA polymerase complex, composed of large (L) protein and tetrameric phosphoprotein (P), is responsible for genome transcription and replication by catalyzing NTP polymerization, mRNA capping and cap methylation. Here, we determine the cryo-electron microscopy (cryo-EM) structure of fully bioactive NiV L-P polymerase complex at a resolution of 3.19 Å. The L-P complex displays a conserved architecture like other NNS RNA virus polymerases and L interacts with the oligomerization domain and the extreme C-terminus region of P tetramer. Moreover, we elucidate that NiV is naturally resistant to the allosteric L-targeting inhibitor GHP-88309 due to the conformational change in the drug binding site. We also find that the non-nucleotide drug suramin can inhibit the NiV L-P polymerase activity at both the enzymatic and cellular levels. Our findings have greatly enhanced the molecular understanding of NiV genome replication and transcription and provided the rationale for broad-spectrum polymerase-targeted drug design.
Similar content being viewed by others

Introduction
NNS RNA virus, also called Mononegavirales, consists of 11 virus families according to the 2019 taxonomy, with a single negative-sense RNA genome1. Paramyxoviridae is a member of the order Mononegavirales, including many deadly human pathogens such as measles virus (MeV), mumps virus (MuV), human parainfluenza virus 1–5 (HPIV1-5), and NiV2. NiV, a highly pathogenic zoonotic agent, causes acute febrile encephalitis and severe respiratory syndrome in humans3,4,5. NiV outbreaks have been reported in Malaysia, Singapore, Bangladesh, Philippines and India, among which the most recent outbreak occurred in the southern state Kerala of India in September 20236. The person-to-person transmission contributes to 40–75% fatality rate and even up to 92% during the outbreak in the Tangail district of Bangladesh7,8,9.
NiV genome contains six transcriptional units encoding nucleoprotein (N), phosphoprotein (P), matrix protein (M), fusion protein (F), glycoprotein (G), and large protein (L). These genes are transcribed in a sequentially attenuated order with the consensus gene-start (GS) and gene-end (GE) sequences10,11,12. L protein is a multifunctional enzyme carrying out all activities necessary for genome transcription and replication, and the essential cofactor P tethers L to nucleocapsid template, an antisense viral RNA (vRNA) encapsidated by N1. The correlation between L-P interaction and RNA synthesis activity has been verified for other NNS RNA viruses13,14,15.
Like other NNS RNA virus polymerases, NiV L includes three enzymatic domains: RNA dependent RNA polymerase (RdRp) domain, capping domain that catalyze mRNA capping by polyribonucleotidyltransferase (PRNTase) activity, dual-specificity methyltransferase domain (MT), and two structural domains: connector domain (CD), C-terminal domain (CTD). NiV P contains N-terminal domain (NTD), oligomerization domain (OD) and C-terminal domain (CTD) which are connected through long linkers. The RdRp encounters vRNA 3’ end by recognizing the bipartite promoters and catalyzes NTP polymerization within the active center16. The PRNTase domain possesses a HR motif which is conserved in most of the NNS RNA viruses and is predicted to have the PRNTase activity rather than the conventional guanyl transferase (GTase) activity17,18,19,20,21. In other NNS RNA viruses, the MT catalyzes the methylation of nascent mRNA firstly at the 2’-O, followed by the N7 position, an opposite order to common eukaryotic methylation reaction22,23,24,25, but how NiV L methylates the cap structure of mRNA needs to be verified.
Despite the persistent attempts, there are not many vaccines and drugs approved for clinical use in human against Nipah virus infection except for a neutralizing antibody (m102.4)26. Several henipavirus G-based vaccines, and the nucleoside analog drugs remdesivir and favipiravir have been shown efficient in animal model27. Recently, an allosteric inhibitor targeting the RNA polymerase, GHP-88309, was described as a promising broad-spectrum drug candidate against most paramyxovirus, but the replicon data shows NiV is resistant to this compound28. Therefore, the structural information of NiV RNA polymerase is in urgent need for the development of efficient antiviral drugs.
Here, we focus on the NiV replication machine and obtain the high-resolution cryo-EM structure of L-P polymerase complex. The structure illustrates the conserved RdRp and PRNTase domains of L, coupled with a compact POD tetramer and one PXD in L-bound state. For the first time, the interaction sites between NiV L and P are elucidated including numerous salt-bridges and hydrogen-bonds, which could be employed as a potential drug target to hinder virus propagation. We have showed the structural basis of the resistance of NiV L to the allosteric inhibitor GHP88309 and demonstrate that the non-nucleotide drug suramin can well inhibit NiV polymerase activity at both the enzymatic and cellular levels. Collectively, these findings will enhance the molecular understanding of NiV genome transcription and replication and promote the development of anti-NiV therapeutics.
Results
Recombinant NiV L-P complex protein is biologically active
We co-expressed the NiV L-P protein in High Five insect cells with pFastbacDual expression vector. The purified protein sample was confirmed as a complex of full-length L (250 kDa) and P (80 kDa) by size-exclusion-chromatography (SEC) and SDS-PAGE analysis (Fig. 1a). To verify the biological function, we measured the RNA polymerizing and capping activity using 96-nt 3’ vRNA promoter and 57-nt gene start (GS) sequence of N gene, respectively (Fig. 1b, c). The result demonstrates that NiV L-P polymerase complex can transcribe the 96-nt RNA in a de novo initiation manner and produce two kinds of bands, a ~12-nt abortive product and a longer product that is nearly 96-nt full-length. (Fig. 1b). The generation of the abortive product might be due to the use of a naked RNA template, instead of NP-coated RNA, as it has been demonstrated in vesicular stomatitis virus (VSV, another NNS RNA virus) that full processivity of the polymerase requires the template-associated N protein13. Furthermore, the L-P complex protein can generate an obvious GpppRNA product using the 57-nt GS RNA with 5’-triphophophate group and α32P-GTP as the substrates, indicating that PRNTase domain in NiV L-P protein is structurally correct and functional (Fig. 1c). We also showed that NiV L-P complex protein can transfer the methyl group from 3H-labeled S-adenosyl methionine (SAM) to Gppp-RNA with 4-fold activity higher than the ppp-RNA (Fig. 1d). The weaker ability of ppp-RNA methylation is consistent with the rule of 2’-O methylation preceding N7 methylation which has been testified in HMPV and VSV23,25. Taken together, these results indicated that the recombinant NiV L-P polymerase complex is catalytically active, and is suitable for further structural analysis.

a Size-exclusion chromatography and SDS-PAGE profiles of NiV L-P complex. Molecular weights (in kilodaltons, kDa) of protein maker are shown on the left, and the L and P bands are indicated on the right. At least three independent experiments were performed with similar results. b De novo transcription activity of NiV L-P complex. The 3’-end 96 nt sequence of NiV genome was chemically synthesized and used as the template. The micrograph was representative of three independent experiments using different protein preparations, all of which showed similar results. c Capping activity of NiV L-P complex. The 5’-triphosphorylated mRNA sequence of 5’UTR of N gene was generated by in vitro transcription using T7 RNA polymerase and used as the capping substrate. Data are representative of two independent experiments using different protein preparations, and both experiments showed similar results. d Methyltransferase activity of NiV L-P complex. The cap structure at the 5’-end of Gppp-RNA was formed by the Vaccinia virus capping enzyme, and Gppp-RNA was a better substrate for methyltransferase than ppp-RNA. The data represented mean values (histograms) ±s.d (error bars) of three independent experiments (n = 3). Source data are provided as a Source Data file. NiV Nipah virus.
Overall structure of NiV L-P polymerase complex
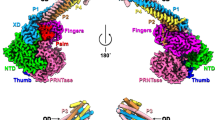
By cryo-EM method, the structure of L-P polymerase complex was determined to a resolution of 3.19 Å (Supplementary Fig. 1, Supplementary Table 1). The visible structural regions include the RdRp (residues 1–969) and PRNTase (residues 970-1452) domains of L protein, the oligomerization domain (POD) (residues 510–580) and the extreme C-terminal domain (PXD) (residues 652-709) of P protein (Fig. 2a). The RdRp-PRNTase core structure displays a conventional ring-like architecture like other NNS RNA virus polymerases29,30,31,32,33,34,35,36,37. The CD-MT-CTD region is not observed in our structure, probably due to its high flexibility, which was similar to the respiratory syncytial virus (RSV), human metapneumovirus (HMPV) and Ebola virus (EBOV) L polymerase structure29,32,33,34,36. POD is modeled as a four-helix bundle with each monomer comprising a long parallel helix, while the density of N-terminal caps is not well enough to build the model (Fig. 2b, c). This structure is in accordance with previously reported structure of tetramerization domain of NiV P (PDB: 4N5B)38. PXD is assembled by three helices with the same length to form a globular fold, and only one copy of PXD can be visualized owing to its interaction with L. The linker regions and PNTD are not structurally characterized because these regions are intrinsically disordered.

a Schematic diagram of the domain architecture of NiV L protein and phosphoprotein. The cryo-EM density map (b) and atomic model (c) of the NiV L-P complex. The L protein could be divided into five regions, including RdRp, PRNTase, CD, MTase and CTD. We could only observe the densities of RdRp (NTD, orange; fingers, blue; palm, red; thumb, green) and PRNTase (cyan) and build the model. The density of N-terminal cap of P protein was weak (gray) and could not be used to build an atomic model. The majority regions of the phosphoproteins were missing in our density map, only OD and one XD could be observed. The unmodelled structures were shown in white with a dotted outline. RdRp RNA-dependent RNA polymerase, NTD N-terminal domain, CD connector domain, MTase methyltransferase, CTD C-terminal domain, OD oligomerization domain, XD extreme C-terminal domain.
Among NNS RNA viruses, the P protein shares the lowest sequence similarity and NiV possesses the longest P protein39. We compared the NiV L-P complex structure with the closely related HPIV5 L-P complex structure and distantly related EBOV L-VP35 complex structure. We found that the orientation of POD tetramer binding to the L is variable and NiV L-P complex possesses the most tilted binding orientation (Supplementary Fig. 2a). We speculated that this should be caused by the difference in interaction details between the OD domain and L protein, and the different tilted binding orientation might be related to the movement of L-P polymerase along the viral RNP40. The POD tetramer structure is highly similar among these three viruses (Supplementary Fig. 2b). Of note, POD tetramer of NiV L-P complex adopts a relatively loose organization compared to the unbound POD tetramer (Supplementary Fig. 3). This rationalizes its versatile function as an adaptor during genome replication and transcription.
The RdRp-PRNTase core structure of L protein
The NiV L RdRp-PRNTase core structure most resembles the core structures of Newcastle disease virus (NDV) and HPIV5 L polymerase with root-mean-square deviation (RMSD) values of 1.43 and 1.64 Å, respectively (Supplementary Fig. 4). The RdRp domain adopts a standard right-hand configuration with the palm, fingers, thumb subdomains, appended by a substantial N-terminal subdomain (Fig. 3a). This configuration is highly conserved among almost all the RNA and DNA polymerases41. The catalytic center of RdRp can be further assigned into six conserved sequence motifs (A-F). Motifs A-E are located in the palm subdomain, while motif F, also called “fingers loop”, was found in the fingers subdomain (Fig. 3b).

a The NiV RdRp contains a classical right-handed structure including fingers (blue), palm (red) and thumb (green) subdomains. b The catalytic motifs of RdRp including motifs A-F. c Structural comparison between NiV and NDV PRNTase domain. Both structures could be overlaid well, giving an RMSD value of 1.29 Å, the conserved motifs A’-E’ were labeled with different colors in the structure of NDV PRNTase. d The structure of NiV PRNTase domain, the motif B’ and motif D’ were not observed. e Conformation changes of the priming loop and supporting helix during the polymerase switching from initiation to elongation state. RMSD root-mean-square deviation, VSV Vesicular stomatitis virus, NDV Newcastle disease virus, EBOV Ebola virus.
According to the sequence conservation of NNS RNA viruses, the PRNTase domain contains five key motifs (motifs A’-E’)42 (Fig. 3c). The motif B’ (GXXT) and motif D’ (HR) are responsible for GDP binding and covalent pRNA attachment, respectively42,43,44,45. In the NiV L-P structure, they are not observed due to the flexible conformation (Fig. 3d). Likewise, the omitted priming loop (1275-1289) is also due to its flexibility, suggesting a non-initiation state (Fig. 3e). We further compared the active site of NiV L RdRp domain with those from other NNS RNA viruses including VSV, NDV and EBOV, and found that NiV L-P polymerase structure solved here may represent an elongation state (Fig. 3e).
Using the actively transcribing influenza polymerase structure (PDB: 6QCX) to model an elongating NiV polymerase complex could help to identify the main channels of L protein, including template entry/exit channels, product exit channel and NTP substrate entry channel (Supplementary Fig. 5)46. The interface between RdRp and PRNTase domain is surrounded by basic residues and forms a positively charged groove, from which the RNA template passes through the fingers loop and arrives at the active site. A negatively charged channel within the PRNTase domain aside in the opposite of interface regulates the template exit. The NTP substrate entry channel is located in the RdRp domain near the binding site of PXD.
Structural basis of NiV L resistance to the allosteric GHP-88309 inhibitor
Previous study has indicated that NiV is resistant to the allosteric GHP-88309 inhibitor due to a histidine at position 1165 of L protein which is homologous to the LY1106H resistance substitution in MeV and SeV, and introduction of LH1165Y substitution into NiV L can confer susceptibility to GHP-8830928. To elucidate the resistance mechanism, we prepared the NiV L-P mutant protein with H1165Y substitution and solved the cryo-EM structure of NiV LH1165Y-P mutant polymerase complex to a resolution of 3.01 Å (Supplementary Fig. 6). We carefully examined the interaction network around H1165 and found that H1165 could form a hydrogen bond with the negatively charged residue E922 in the thumb subdomain of L protein. Although the density of the side chain of E922 was weak in the wildtype NiV L-P structure, we could ascertain the conformation at a contour level of 0.4 using a sharpened map with a B-factor value of −100. (Fig. 4a, b and Supplementary Fig. 7). By contrast, in the NiV LH1165Y-P mutant structure, the residue Y1165 will not form such interaction, and the density of the side chain of residue E922 becomes clear and rotates towards the GHP-88309 binding pocket (Fig. 4b and Supplementary Fig. 7). The residue E922 is highly conserved in different paramyxoviruses (Fig. 4c) and was shown to be critical for the binding of GHP-88309 inhibitor28. We superimposed the HPIV3 L-P structure with NiV L-P structure and found that the side chain of residue E863 of HPIV3 L adopts a similar orientation to that of the residue E922 of NiV LH1165Y mutant (Fig. 4d). Therefore, we deduced that the formation of hydrogen bond interaction between H1165 and E922 will destroy the binding of residue E922 to the GHP-88309 inhibitor, and then result in the natural resistance of NiV to the inhibitor. Further sequence alignment showed that Hendra virus (HeV) L also possesses a histidine (H) at the same position, whereas the henipaviruses Langya virus (LayV) and Mojiang virus (MojV) L polymerases have a tyrosine (Y) at that position that is similar to MeV and HPIV3 (Fig. 4c). It indicates that the HeV will have natural resistance to the GHP-88309 inhibitor, while the LayV and MojV will not have such resistance.

a Superimposition of structures of NiV wildtype and LH1165Y mutant polymerase. b Close-up of the mutant site. In the WT polymerase, the H1165 could form a hydrogen bond with the side chain of E922 and make the side chain flip to get away from the interface of RdRp and Capping domains which will lose the interaction with the GHP-88309. While, for the LH1165Y mutant polymerase, the side chain of Y1165 could not form a hydrogen bond with the E922 remaining toward the binding sites of GHP-88309. The structure of wildtype NiV L protein was colored by domains as Fig. 2. The structure of the H1165Y mutant was shown in white. c Sequence alignment of the L protein of paramyxovirus. The sequence alignment was performed using T-coffee73 (https://tcoffee.crg.eu/) and the figure was prepared by ESPript 3.074 (https://espript.ibcp.fr). d Structural comparison of different polymerases in the mutant site. In the structure of HPIV3, the conformation of E863 is like that of NiV LH1165Y mutant polymerase. HeV Hendra virus, LayV Langya virus, MojV Mojiang virus, MeV measles virus, HPIV3 Human parainfluenza virus 3.
Inhibition of NiV L polymerase activity by suramin drug
Suramin has been reported to target the viral polymerase of EBOV, norovirus and SARS-CoV-229,47,48. Here, we show that suramin is also a potent inhibitor of the NiV L-P polymerase complex. Addition of 100 μM suramin can almost completely abolish the de novo activity of the NiV L-P complex in an in vitro enzymatic assay (Fig. 5a). We further demonstrated that suramin could inhibit NiV RNP activity in the cell replicon system, with a half maximal effective concentration (EC50) of about 16.78 μM (Fig. 5b and Supplementary Fig. 8). These results suggested that suramin could target the NiV replication machinery in the cellular environment. Then, we performed the molecular docking analysis to investigate the inhibition mechanism. The results showed that suramin could be docked well in the NTP entry channel with a docking score of -10.039 (Fig. 5c). The interactions involved intensive hydrogen bonds, π-π and π-cation stacking interactions provided by polar amino acids (Fig. 5d). At the binding interface, residues E291, K542, R551, K724, N833, and K893 are conserved in all the NNS RNA viruses, suggesting the broad-spectrum inhibition of suramin against different NNS RNA viruses, by occluding substrates from entering the active site29.

a Inhibition activity of suramin against NiV L-P complex at the enzymatic level. Data are representative of two independent experiments using different protein preparations, and both experiments showed similar results. b Inhibition activity of suramin against NiV L-P complex in replicon cell. The suramin displayed obvious inhibitory effects on NiV polymerase in vitro and in vivo. Each point represents mean values ± s.d. (error bar) of three independent experiments using different cell preparations. c Molecular docking of suramin in NiV L protein. The structure was shown as surface and colored by domains. Suramin could bind at the NTP entry channel and prevent the substrates from entering the active site to inhibit the polymerase activity. d The interactions between NiV L protein and suramin in the docking structures. Several polar residues could form hydrogen bonds, π-π, and π-cation interactions with the suramin. The hydrogen bonds were indicated by dashed yellow lines and the π-π and π-cation interactions were indicated by dashed black lines. Source data are provided as a Source Data file.
Interaction between L and P
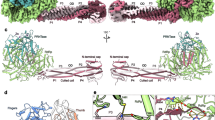
NiV P interacts with L predominantly through C-terminus of POD (562-573) and N-terminus of PXD (660-669), which involves a number of Van deer Waals forces (VDW) and electrostatic interactions (Supplementary Table 2). The L-P complex structure illustrates that the POD and PXD associate with the fingers, palm subdomains and NTD of L (Fig. 6a and Supplementary Table 2).

a Overall structure of NiV L-P complex. The L protein and phosphoprotein were colored by domains and protomers as in Fig. 2. The primary interaction interfaces of NiV L and P were distributed at three sites and defined as I, II, and III, representing three distinct regions of the P protein including the long helix, the end of POD, and PXD. Key atomic interactions between L protein and phosphoprotein at sites I (b), II (c), and III (d). The key interaction residues are shown as sticks and colored by elements. Hydrogen bonds are represented by yellow dashed lines.
Although the oligomerization domains of four P protomers adopt almost the same conformation, they exhibit various interactions with L. P1 makes extensive contacts with L, including four hydrogen bonds between H570, S565 in P1 and Y389, H423 in L (Fig. 6b and Supplementary Table 2). P2 binds to L via 64 atom to atom contacts in total and five hydrogen bonds are formed by I576, I578 and G580 in P2 and L387, K385 and D384 in L (Fig. 6c and Supplementary Table 2). P3 and P4 locate away from L without any interaction between them, and we speculate that these two protomers play a role in stabilizing the P tetramer or coupling with host factors. One PXD protomer directly contacts with L near the NTP entry channel mostly relying on the helix α1, whereas the helix α3 makes few contacts and the helix α2 is solvent exposed. Residues D657, S660, D662, R669, T670 and H671 in helix α1 are involved in the hydrogen bond and salt bridge interactions with H313, H320, D339, N346 and L300 in L, respectively (Fig. 6d and Supplementary Table 2). Previous study has revealed that Hendra virus (HeV) NTAIL binds PXD in the hydrophobic groove composed of helix α2 and helix α349, and NiV likely follow the same pattern in view of high sequence identity (L, 87%; P, 66%). This observation implicates that PXD bridges L to nucleocapsid through two non-overlapping regions.
Discussion
The transcription and replication of genome RNA depends on a viral polymerase for currently known RNA viruses, which puts the polymerase in a critical position for anti-virus drug targets. Within NNS RNA viruses, several polymerase structures have been solved, including VSV, RSV, HMPV, HPIV3, HPIV5, NDV, RABV and EBOV29,30,31,32,33,34,35,36,45. Here, for the first time, we determined the high-resolution cryo-EM structure of NiV L-P complex. Superimposing NiV L-P structure to those from other NNS RNA viruses reveals a relatively conserved architecture of L protein but various conformations of P protein in different NNS RNA virus families, resulting in the distinct interaction between L and P.
As an indispensable functional entirety, there have been many explorations about the interaction network between L and P. Previous mutational analysis work has indicated two distinct trends: for paramyxoviruses and pneumoviruses, such as NDV, HPIV3, HPIV5, MeV, SeV and RSV, the contact site in L-P complex was mapped to LNTD with PXD50,51,52,53,54,55, and a bipartite interaction of LNTD with POD and PXD has been reported in MeV56; on the other hand, for rhabdoviruses such as VSV and RABV, the LCTD and PNTD are essential for L-P interaction57,58,59. The resolved cryo-EM L-P polymerase complex structures have certified these biochemical characterizations. In VSV and RABV L-P complex structures, the PNTD associates with LCTD through three separate segments, which results in fixing the relative positions of the RdRp-PRNTase core and CD-MT-CTD modules30,45. The L-P complex structures of RSV, EBOV and HMPV demonstrated that each P promoter has distinct conformation and contacts unique locations of L allowing POD wraps around RdRp domain of L in an asymmetric tentacular arrangement, and in addition to this contact site, there is an extensive interface of PCTD and LNTD32,33,34. NDV L-P polymerase structure has the most similarity to NiV L-P structure, in which POD serves as a tetrameric helical bundle that directly interacts with L-RdRp and a single PXD localizes near the NTP substrate entry channel of L34. Sequence analysis within Henipavirus genus reveals the residues involved in L-P interaction are highly conserved (Supplementary Fig. 9). Therefore, based on this structural information of NiV L-P interaction, we can design broad-spectrum antiviral drugs against diverse henipaviruses by blocking the L-P interaction. For example, with the help of artificial intelligence method, we can design a small peptide drug that mimics the three-helices bundle PXD binding to L but with a much higher affinity to compete with the PXD binding60,61. Although the determined structure of NiV L protein showed high similarities with other NNS RNA virus polymerases, we found that NiV L protein contained a very long specific insert sequence in the palm domain (Supplementary Fig. 10a). However, we could not observe the density of this insert in our determined structure probably due to its flexibility. Then, we predicted the full-length NiV L structure using Alphafold 262, which showed that this insert element was located between the supporting helix and motif A (Supplementary Fig. 10). The supporting helix would insert to the polymerase active site and provide a buttress for the initiation of RNA synthesis, and then it would get away from the active site during elongation. Thus, we speculated that the specific insert of the NiV L protein would play a critical role in recognizing RNA promoter elements to regulate the initiation and elongation processes, which need to be verified in future studies.
Previous study has showed that a non-nucleotide inhibitor GHP-88309 had been proven to possess an unusual broad-spectrum antiviral activity against HPIV3, SeV, and MeV, probably by impairing the de novo polymerase initiation at the promoter28. It was shown to bind the intersection between the thumb subdomain of RdRp and PRNTase as an allosteric inhibitor, and various paramyxoviruses except NiV were predicted to be susceptible28. Interestingly, NiV shows natural resistance to GHP-88309 because of a histidine at L1165 position homologous to the Y1106H resistance mutation found in MeV and SeV. This can be explained by the wildtype and H1165Y-mutant-containing NiV L-P complex structures solved here. The residue H1165 can form a hydrogen bond with E922 and drag the sidechain away from the GHP-88309 binding site. As a result, residue E922 would lose the ability to interact with GHP-88309, corresponding to E863 in MeV28, therefore resulting in drug resistance. In addition to the GHP-88309 inhibitor, we also examined the inhibition activity of another non-nucleotide drug suramin, and the results showed that suramin can well inhibit the polymerizing activity of NiV L-P polymerase complex, probably through a similar mechanism of inhibiting NTP substrate entry, that was previously observed for EBOV L-VP35 polymerase29.
In summary, we determine the structure of NiV L protein in complex with tetrameric P protein and capture an elongation state with the supporting helix and priming loop unordered. Of note, we elucidate the structural basis of NiV resistance to the allosteric GHP-88309 inhibitor and provide a rationale for the design of GHP-88309-like allosteric against the highly pathogenic NiV. We also demonstrate that suramin could inhibit NiV L-P activity, suggesting the feasibility of developing broad-spectrum non-nucleoside antiviral drugs by targeting the NTP substrate entry channel of L protein, to treat the infection of NNS RNA viruses including NiV and EBOV.
Methods
Gene cloning
NiV-L (protein_id=NP_112028.1) and NiV-P (protein_id=NP_112022.1) genes were codon-optimized for expression in High Five cells (High5) and were cloned into pFastBacTMDual vector. The L gene with N-terminal Twin Strep tag was inserted downstream of the polyhedrin promoter, while the P gene with C-terminal 8× His tag was inserted downstream of the p10 promoter.
Protein expression and purification
The High5 cells were collected by centrifugation (830 g, 10 min) at 48 h (h) post-infection and were sonicated in lysis buffer (50 mM Tris-HCl, 300 mM NaCl, 10% glycerol, 1 mM Tris (2-carboxyethyl) phosphine (TCEP), pH 8.5). The supernatant was filtered with 0.22 μm cut-off filter following centrifugation (17,300 × g, 1 h), and then was loaded onto Strep Trap column (GE Healthcare, 5 mL). The target proteins were eluted with lysis buffer supplemented with 2.5 mM desthiobiotin. Finally, the proteins were further purified using size-exclusion chromatography (GE Healthcare, SuperoseTM 6 10/300) equilibrated with lysis buffer. The targeted proteins were concentrated and stored at −80 °C until use. For the LH1165Y-P mutant, the expression and purification processes were the same as above.
In vitro transcription
The 5’-triphosphorylated mRNA sequence of N gene 5’UTR (57 nt, 5’-pppAGGAACCAAGACAAACACUUUUGGUCUUGGUAUUGGAUCCUCAAGAAAUAUAUCAUC-3’) was synthesized by T7 RNA polymerase in vitro. The template for T7 RNA polymerase transcription is two completely complementary oligo-DNAs, which were heated at 72 °C for 10 min followed by slowly cooling to room temperature. The T7 promoter sequence (−17 to −1) was added to the 5’ terminus of 5’UTR sequence: 5’- TAATACGACTCACTATAAGGAACCAAGACAAACACTTTTGGTCTTGGTATTGGATCCTCAAGAAATATATCATC-3’, and the other complementary chain is 5’-GATGATATATTTCTTGAGGATCCAATACCAAGACCAAAAGTGTTTGTCTTGGTTCCTTATAGTGAGTCGTATTA-3’. The product was purified through DNase digestion, isopropanol precipitation, and ethanol precipitation, and validated by electrophoresis using 20% polyacrylamide gel containing 7 M urea (urea-PAGE) stained with Gel-Red.
RdRp activity assay and inhibition activity by suramin
To verify whether the purified NiV L-P complex harbors the polymerization activity, 1 μM L-P complex, 1 mM NTP, 1 μCi [α-32P]-GTP and 0.5 μM promoter (96 nt, 5’-AGGAUCCAAUACCAAGACCAAAAGUGUUUGUCUUGGUUCCUAAGUUUUAAAAAUACGUUAUAUAUUUUAACGUAUCCAUAUUCUCCCUUGUUUGGU-3’) were incubated in reaction buffer containing 20 mM Tris-HCl, pH 7.5, 10 mM KCl, 6 mM MgCl2, 2 mM DTT, 0.5% (v/v) Triton X-100, 10% (v/v) DMSO at 30 °C for 3 h. For the inhibition activity by suramin assay, we added a series of dilutions of suramin into the reaction system similar to the above. The reaction was quenched with the addition of formamide (Coolaber) and boiled at 95 °C for 5 min. The products were separated by 20% polyacrylamide gels containing 7 M urea in 0.5 X TBE buffer. Images were taken by exposing the gels on a storage phosphor screen and read with a Typhoon scanner (GE Healthcare).
Capping activity assay
For the capping activity assay, 0.3 μM NiV L-P complex was mixed with 5 μM 5’-triphosphorylated oligo-RNA (5’-pppAGGAACCAAGACAAACACUUUUGGUCUUGGUAUUGGAUCCUCAAGAAAUAUAUCAUC-3’), 2 μCi [α-32P]-GTP in capping reaction buffer containing 50 mM Tris-HCl, pH 8.0, 5 mM KCl, 1 mM MgCl2, 1 mM DTT at 30 °C for 3 h. The reaction was stopped by adding formamide and heating at 95 °C for 5 min. The capped products were separated using 15% gels containing 9 M urea in 0.5 X TBE buffer. The gels were exposed on the storage phosphor screen and recorded with a Typhoon scanner (GE Healthcare).
MTase activity assay
For the methyltransferase assay of NiV L-P complex, 15 μM RNA, 2 μM SAM, 0.9 μM protein, and 0.5 μCi 3H-SAM were incubated in MTase activity buffer (50 mM Tris-HCl, pH 8.5, 1 mM DTT) at 30 °C for 7 h. The assay includes two kinds of RNA substrates for comparison: ppp-RNA described above and its capped product, Gppp-RNA, generated by the Vaccinia virus capping enzyme (ThermoFisher). The reaction was stopped by adding 10 μL isopropanol for precipitation overnight. The next day, the supernatant was removed through centrifugation (13,700 × g, 15 min), and the RNA pellet was dissolved with 40 μL NaCl (300 mM) before adding 1 mL ULTIMA GOLD (Perkin Elmer) scintillation fluid. Finally, the enzyme activity was calculated by detecting the 3H-methyl signal transferred to RNA products using MicroBeta LumiJET (Perkin Elmer). All the MTase assay experiments were performed in triplicate.
Inhibition activity of suramin in NiV cell replicon system
To assess the inhibitory effect of suramin against NiV RNP activity in the cell, we incubated HEK293T cells with a serial dilution of suramin at 37 °C and 5% CO2 for 1 h. Subsequently, the cells were co-transfected with pCAGGS-L, pCAGGS-NP, pCAGGS-P, pCAGGS-T7 polymerase and a reporter plasmid encoding GFP flanked by T7 promoter, 5’- and 3’-terminal UTR sequences using Lipofectamine 3000 (Invitrogen). The NiV RNP activity was evaluated by the expression level of GFP, which was calculated by CellVoyager CQ1 (Yokogawa) after 48 h.
Cryo-EM sample preparation and data collection
To prepare the cryo-EM sample of wildtype NiV L-P complex, the purified proteins were diluted to ~0.8 mg/ml using glycerol-free buffer (50 mM Tris, 300 mM NaCl, pH8.5) to reduce the concentration of glycerol. A liquid of 3 μL protein solution was applied into glow-discharged Quantifol (R 1.2/1.3) grid with 100 % humidity at 4 °C. The grid was blotted for 3 s and plunged into the liquid ethane using FEI Vitribot Mark IV. The well-prepared cryogenic specimens were loaded into a 300 kV Titan Krios (Thermo Fisher Scientific) transmission electron microscope equipped with a post-column energy filter (Gantan) with a silt width of 20 eV. The images were recorded with K2 direct detector camera using supper-resolution counting mode with a calibrated pixel size of 1.36 Å. Each exposure was performed with a dose rate of 10 e−1pixel−1s−1 and an accumulative dose of 60 e−1 Å2 which fractionated into a movie stack of 30 frames. The defocus range of this dataset was −1.0 to −2.2 μm.
For NiV L H1165Y-P complex, the protein was diluted to ~0.5 mg/mL and loaded onto the glow-discharged Nitai grid (R1.2/1.3) following the same protocol for the wildtype complex. Movies were collected with a K3-subunit detector using the super-resolution counting mode with a pixel size of 0.82 Å. The exposure was performed with a dose rate of 20 e−1pixel−1s−1 and an accumulative dose of 50 e−1 Å2 for each micrograph, which fractionated into 30 frames. The defocus range of this dataset was roughly −1.5 to −2.0 μm.
Image processing
The stacked frames were aligned using MotionCor263 and the initial contrast transfer function (CTF) values were estimated by CTFFIND4.164. Particles were auto-picked using Laplacian-of-Gaussian bolb detection in Relion365 and then imported into the cryoSPARC66 for classification and reconstruction. For the wildtype NiV L-P complex, a total of 8,775,174 particles were selected and extracted from 5503 micrographs with a small box size (160 pix ×160 pix) and subjected to several rounds of 2D classification which resulted in a clean dataset of 2,344,070 particles. These particles were used for three rounds of heterogeneous refinement and the best particles with clear structure features were selected and re-extracted with a larger box size (300 pix ×300 pix) to perform non-uniform refinement which yielded a final density map of 3.19 Å resolution (Supplementary Fig. 1).
For NiV L H1165Y-P complex, a total of 1,191,521 particles were selected and extracted from 1050 micrographs with a box size of 256 pix × 256 pix and subjected to 2D classification. After several rounds of 2D classification, 320,318 particles were selected for two rounds of heterogeneous refinement. The best particles were selected and re-extracted with a large box size (420 pix × 420 pix) for non-uniform refinement which yielded a density map 3.15 Å resolution. To polish the resolution, dose weighted micrographs were generated using MotionCor2 with first two and last thirteen frames discarded. A final round of local refinement was performed, which generated a density map of 3.01 Å resolution (Supplementary Fig. 6). The local resolution distribution was estimated using ResMap67. The EM density maps were sharpened using DeepEMhancer68.
Model building
The initial models of NiV L and P protein were predicted using AlphaFold262 and rigidly docked into the density map of NiV L-P complex. The model was manually adjusted for local fit in COOT69 and refined against the density map in real space using PHENIX70 with the secondary structural restraints and Ramachandran restraints were applied. The model quality was assessed by MolProbity71. The structural figures were prepared using ChimeraX72 or Pymol (https://pymol.org/).
Molecular docking
The structure of NiV L protein was generated by deleting P protein in wildtype NiV L-P complex and used as the template structure for molecular docking. The sdf file of suramin was downloaded from the Protein data bank (PDB). The residues around the NTP entry channel were selected to generate the grid box. The protein and ligands were prepared and processed with standard routines. The molecular docking analysis was performed using Schrodinger suites-Maestro 11.5.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM density maps and atomic coordinates have been deposited to the Electron Microscopy Data Bank (EMDB) and the PDB, respectively. The accession numbers are listed as follows: NiV L-P complex (9IR3, EMD-60799), NiV LH1165Y-P complex (9IR4, EMD-60800). Source data are provided with this paper.
References
Liang, B. Structures of the Mononegavirales Polymerases. J. Virol. 94, e00175–00120 (2020).
Cox, R. & Plemper, R. K. The paramyxovirus polymerase complex as a target for next-generation anti-paramyxovirus therapeutics. Front. Microbiol. 6, 459 (2015).
Hossain, M. J. et al. Clinical presentation of nipah virus infection in Bangladesh. Clin. Infect. Dis. 46, 977–984 (2008).
Goh, K. J. et al. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N. Engl. J. Med. 342, 1229–1235 (2000).
Aditi & Shariff, M. Nipah virus infection: A review. Epidemiol. Infect. 147, e95 (2019).
Srivastava, S. et al. Recent Nipah virus outbreak in India: lessons and imperatives. Ther. Adv. Infect. Dis. 10, 20499361231208535 (2023).
Aljofan, M. Hendra and Nipah infection: emerging paramyxoviruses. Virus Res. 177, 119–126 (2013).
Lo, M. K. & Rota, P. A. The emergence of Nipah virus, a highly pathogenic paramyxovirus. J. Clin. Virol. 43, 396–400 (2008).
Arankalle, V. A. et al. Genomic characterization of Nipah virus, West Bengal, India. Emerg. Infect. Dis. 17, 907–909 (2011).
Fearns, R. & Plemper, R. K. Polymerases of paramyxoviruses and pneumoviruses. Virus Res. 234, 87–102 (2017).
Sugai, A., Sato, H., Yoneda, M. & Kai, C. Gene end-like sequences within the 3’ non-coding region of the Nipah virus genome attenuate viral gene transcription. Virology 508, 36–44 (2017).
Kuo, L., Fearns, R. & Collins, P. L. Analysis of the gene start and gene end signals of human respiratory syncytial virus: quasi-templated initiation at position 1 of the encoded mRNA. J. Virol. 71, 4944–4953 (1997).
Morin, B., Rahmeh, A. A. & Whelan, S. P. J. Mechanism of RNA synthesis initiation by the vesicular stomatitis virus polymerase. EMBO J. 31, 1320–1329 (2012).
Noton, S. L., Deflubé, L. R., Tremaglio, C. Z. & Fearns, R. The Respiratory Syncytial Virus Polymerase Has Multiple RNA Synthesis Activities at the Promoter. Plos Pathog. 8, e1002980 (2012).
Chattopadhyay, S. & Banerjee, A. K. Phosphoprotein, P of human parainfluenza virus type 3 prevents self-association of RNA-dependent RNA polymerase, L. Virology 383, 226–236 (2009).
Magoffin, D. E., Halpin, K., Rota, P. A. & Wang, L. F. Effects of single amino acid substitutions at the E residue in the conserved GDNE motif of the Nipah virus polymerase (L) protein. Arch. Virol. 152, 827–832 (2007).
Ogino, T., Yadav, S. P. & Banerjee, A. K. Histidine-mediated RNA transfer to GDP for unique mRNA capping by vesicular stomatitis virus RNA polymerase. Proc. Natl Acad. Sci. 107, 3463–3468 (2010).
Nishio, M. et al. Human parainfluenza virus type 2 L protein regions required for interaction with other viral proteins and mRNA capping. J. Virol. 85, 725–732 (2011).
Cortese, C. K., Feller, J. A. & Moyer, S. A. Mutations in domain V of the Sendai virus L polymerase protein uncouple transcription and replication and differentially affect replication in vitro and in vivo. Virology 277, 387–396 (2000).
Ogino, M., Ito, N., Sugiyama, M. & Ogino, T. The Rabies Virus L Protein Catalyzes mRNA Capping with GDP Polyribonucleotidyltransferase Activity. Viruses 8, 144 (2016).
Barik, S. The structure of the 5’ terminal cap of the respiratory syncytial virus mRNA. J. Gen. Virol. 74, 485–490 (1993).
Ogino, T., Kobayashi, M., Iwama, M. & Mizumoto, K. Sendai virus RNA-dependent RNA polymerase L protein catalyzes cap methylation of virus-specific mRNA. J. Biol. Chem. 280, 4429–4435 (2005).
Rahmeh, A. A., Li, J., Kranzusch, P. J. & Whelan, S. P. Ribose 2’-O methylation of the vesicular stomatitis virus mRNA cap precedes and facilitates subsequent guanine-N-7 methylation by the large polymerase protein. J. Virol. 83, 11043–11050 (2009).
Testa, D. & Banerjee, A. K. Two methyltransferase activities in the purified virions of vesicular stomatitis virus. J. Virol. 24, 786–793 (1977).
Paesen, G. C. et al. X-ray structure and activities of an essential Mononegavirales L-protein domain. Nat. Commun. 6, 8749 (2015).
Playford, E. G. et al. Safety, tolerability, pharmacokinetics, and immunogenicity of a human monoclonal antibody targeting the G glycoprotein of henipaviruses in healthy adults: a first-in-human, randomised, controlled, phase 1 study. Lancet Infect. Dis. 20, 445–454 (2020).
Lo, M. K. et al. Potent in vitro activity of beta-D-4’-chloromethyl-2’-deoxy-2’-fluorocytidine against Nipah virus. Antivir. Res. 175, 104712 (2020).
Cox, R. M. et al. Orally efficacious broad-spectrum allosteric inhibitor of paramyxovirus polymerase. Nat. Microbiol. 5, 1232–1246 (2020).
Yuan, B. et al. Structure of the Ebola virus polymerase complex. Nature 610, 394–401 (2022).
Horwitz, J. A., Jenni, S., Harrison, S. C. & Whelan, S. P. J. Structure of a rabies virus polymerase complex from electron cryo-microscopy. Proc. Natl Acad. Sci. USA 117, 2099–2107 (2020).
Abdella, R., Aggarwal, M., Okura, T., Lamb, R. A. & He, Y. Structure of a paramyxovirus polymerase complex reveals a unique methyltransferase-CTD conformation. Proc. Natl Acad. Sci. USA 117, 4931–4941 (2020).
Gilman, M. S. A. et al. Structure of the Respiratory Syncytial Virus Polymerase Complex. Cell 179, 193–204 e114 (2019).
Cao, D. et al. Cryo-EM structure of the respiratory syncytial virus RNA polymerase. Nat. Commun. 11, 368 (2020).
Pan, J. et al. Structure of the human metapneumovirus polymerase phosphoprotein complex. Nature 577, 275–279 (2020).
Xie, J. et al. Structural basis for dimerization of a paramyxovirus polymerase complex. Nat. Commun. 15, 3163 (2024).
Peng, Q. et al. Molecular mechanism of de novo replication by the Ebola virus polymerase. Nature 622, 603–610 (2023).
Cong, J. et al. Structure of the Newcastle Disease Virus L protein in complex with tetrameric phosphoprotein. Nat. Commun. 14, 1324 (2023).
Bruhn, J. F. et al. Crystal structure of the nipah virus phosphoprotein tetramerization domain. J. Virol. 88, 758–762 (2014).
Rockx, B., Winegar, R. & Freiberg, A. N. Recent progress in henipavirus research: molecular biology, genetic diversity, animal models. Antivir. Res. 95, 135–149 (2012).
Whitehead, J. D. et al. Structure of the N-RNA/P interface indicates mode of L/P recruitment to the nucleocapsid of human metapneumovirus. Nat. Commun. 14, 7627 (2023).
Peersen, O. B. A Comprehensive Superposition of Viral Polymerase Structures. Viruses 11, https://doi.org/10.3390/v11080745 (2019).
Neubauer, J., Ogino, M., Green, T. J. & Ogino, T. Signature motifs of GDP polyribonucleotidyltransferase, a non-segmented negative strand RNA viral mRNA capping enzyme, domain in the L protein are required for covalent enzyme-pRNA intermediate formation. Nucleic Acids Res. 44, 330–341 (2016).
Lij, J., Rahmeh, A., Morelli, M. & Whelan, S. P. J. A conserved motif in region V of the large polymerase proteins of nonsegmented negative-sense RNA viruses that is essential for mRNA capping. J. Virol. 82, 775–784 (2008).
Ogino, T. & Banerjee, A. K. The HR motif in the RNA-dependent RNA polymerase L protein of Chandipura virus is required for unconventional mRNA-capping activity. J. Gen. Virol. 91, 1311–1314 (2010).
Liang, B. et al. Structure of the L Protein of Vesicular Stomatitis Virus from Electron Cryomicroscopy. Cell 162, 314–327 (2015).
Kouba, T., Drncova, P. & Cusack, S. Structural snapshots of actively transcribing influenza polymerase. Nat. Struct. Mol. Biol. 26, 460–470 (2019).
Yin, W. C. et al. Structural basis for inhibition of the SARS-CoV-2 RNA polymerase by suramin. Nat. Structural Mol. Biol. 28. https://doi.org/10.1038/s41594-021-00570-0 (2021).
Mastrangelo, E. et al. Structure-Based Inhibition of Norovirus RNA-Dependent RNA Polymerases. J. Mol. Biol. 419, 198–210 (2012).
Communie, G. et al. Atomic resolution description of the interaction between the nucleoprotein and phosphoprotein of Hendra virus. PLoS Pathog. 9, e1003631 (2013).
Cevik, B. et al. The phosphoprotein (P) and L binding sites reside in the N-terminus of the L subunit of the measles virus RNA polymerase. Virology 327, 297–306 (2004).
Parks, G. D. Mapping of a region of the paramyxovirus L protein required for the formation of a stable complex with the viral phosphoprotein P. J. Virol. 68, 4862–4872 (1994).
Holmes, D. E. & Moyer, S. A. The phosphoprotein (P) binding site resides in the N terminus of the L polymerase subunit of sendai virus. J. Virol. 76, 3078–3083 (2002).
Malur, A. G., Choudhary, S. K., De, B. P. & Banerjee, A. K. Role of a highly conserved NH(2)-terminal domain of the human parainfluenza virus type 3 RNA polymerase. J. Virol. 76, 8101–8109 (2002).
Sourimant, J. et al. Fine mapping and characterization of the L-polymerase-binding domain of the respiratory syncytial virus phosphoprotein. J. Virol. 89, 4421–4433 (2015).
Smallwood, S., Ryan, K. W. & Moyer, S. A. Deletion analysis defines a carboxyl-proximal region of Sendai virus P protein that binds to the polymerase L protein. Virology 202, 154–163 (1994).
Du Pont, V., Jiang, Y. & Plemper, R. K. Bipartite interface of the measles virus phosphoprotein X domain with the large polymerase protein regulates viral polymerase dynamics. PLoS Pathog. 15, e1007995 (2019).
Morin, B., Liang, B., Gardner, E., Ross, R. A. & Whelan, S. P. J. An In Vitro RNA Synthesis Assay for Rabies Virus Defines Ribonucleoprotein Interactions Critical for Polymerase Activity. J. Virol. 91, https://doi.org/10.1128/JVI.01508-16 (2017).
Rahmeh, A. A. et al. Critical phosphoprotein elements that regulate polymerase architecture and function in vesicular stomatitis virus. Proc. Natl Acad. Sci. USA 109, 14628–14633 (2012).
Chenik, M., Schnell, M., Conzelmann, K. K. & Blondel, D. Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J. Virol. 72, 1925–1930 (1998).
Shi, Y. Drug development in the AI era: AlphaFold 3 is coming! The Innovation 5, https://doi.org/10.1016/j.xinn.2024.100685 (2024).
Huang, T. & Li, Y. Current progress, challenges, and future perspectives of language models for protein representation and protein design. Innovation 4, 100446 (2023).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, https://doi.org/10.7554/eLife.42166 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014).
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 66, 12–21 (2010).
Goddard, T. D. et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Notredame, C., Higgins, D. G. & Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 (2000).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Acknowledgements
We thank all the staff members at the center for Biological Imaging (CBI), Institute of Biophysics (IBP), Chinese Academy of Science (CAS) for assistance with data collection. This study was supported by the Major Project of Guangzhou National Laboratory (GZNL2024A01010 to Y.S.), National Key Research and Development Program of China (2021YFC2300700 to Y.S. and 2023YFC230690 to Q.P.), National Natural Science Foundation of China (NSFC) (32192452 to Y.S. and 82472250 to Q.P.) and Beijing Life Science Academy (BLSA: 2023000CA0030 to Y.S.).
Author information
Authors and Affiliations
Contributions
Y.S. conceived the study. Y.D., M.J. and Q.L. purified the protein samples and conducted biochemical studies. Q.P. prepared the cryo-EM specimens and collected data. Q.P. conducted the image processing and reconstruction. J.Q. and Q.P. built the atomic models. Y.S., Q.P., Y.D., M.J. and Q.L. analyzed the structure. Q.P., M.J., Y.B. and Y.S. wrote the manuscript. All authors participated the discussion and manuscript editing. Y.S. supervised all of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Peng, Q., Dong, Y., Jia, M. et al. Cryo-EM structure of Nipah virus L-P polymerase complex. Nat Commun 15, 10524 (2024). https://doi.org/10.1038/s41467-024-54994-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54994-5
This article is cited by
-
A deep learning and molecular modeling approach to repurposing Cangrelor as a potential inhibitor of Nipah virus
Scientific Reports (2025)
-
Cryo-EM structures of Nipah virus polymerases and high-throughput RdRp assay development enable anti-NiV drug discovery
Nature Communications (2025)
-
Structure of the measles virus ternary polymerase complex
Nature Communications (2025)
-
Structural basis of Nipah virus RNA synthesis
Nature Communications (2025)
-
In Silico Discovery of Tanshinone I, a Diterpenoid from Salvia miltiorrhiza (Danshen), as a Dual Antiviral Natural Inhibitor Targeting Conserved Allosteric Sites of HMPV and HRSV RNA-Dependent RNA Polymerases
Journal of Pharmaceutical Innovation (2025)
