
Abstract
Reliable molecular property prediction is essential for various scientific endeavors and industrial applications, such as drug discovery. However, the data scarcity, combined with the highly non-linear causal relationships between physicochemical and biological properties and conventional molecular featurization schemes, complicates the development of robust molecular machine learning models. Self-supervised learning (SSL) has emerged as a popular solution, utilizing large-scale, unannotated molecular data to learn a foundational representation of chemical space that might be advantageous for downstream tasks. Yet, existing molecular SSL methods largely overlook chemical knowledge, including molecular structure similarity, scaffold composition, and the context-dependent aspects of molecular properties when operating over the chemical space. They also struggle to learn the subtle variations in structure-activity relationship. This paper introduces a multi-channel pre-training framework that learns robust and generalizable chemical knowledge. It leverages the structural hierarchy within the molecule, embeds them through distinct pre-training tasks across channels, and aggregates channel information in a task-specific manner during fine-tuning. Our approach demonstrates competitive performance across various molecular property benchmarks and offers strong advantages in particularly challenging yet ubiquitous scenarios like activity cliffs.
Similar content being viewed by others

Introduction
Empowered by the advancement of machine learning techniques, molecular machine learning has shown its great potential in computational chemistry and drug discovery1,2. The data-driven protocol allows the model to infer biochemical behaviors from simple representations like SMILES sequence3 and molecular graph, enabling fast identification of drug candidates via rapid screening of vast chemical spaces4, as well as prediction of binding affinity, toxicity, and other pharmacological properties5,6. These advancements significantly accelerate the drug discovery procedures, saving time and efforts from the traditional wet-lab experiments7,8. However, it is fundamentally challenging to learn an effective and robust molecular representation via machine learning, limited by the expensive gathering of precise biochemical labels and the complexity underlying the structure-property relationships (SPR). With data scarcity, models may become overly adapted to specific structural patterns within the training molecules, making it fail to generalize to the broader chemical space. In addition, the challenge of “activity cliffs”9 in drug discovery, where minor changes in molecular structure significantly alter the biological activity, further impose obstacles in developing accurate Quantitative SPR (QSPR) models6,10,11,12. Activity cliffs refer to the concept where structurally similar molecules may exhibit significantly different biological activities. Understanding activity cliffs is crucial for drug discovery, as it enables more efficient lead optimization and enhances predictive modeling with better identification and development of potential drug candidates13,14.
Inspired by the success of the pretrain-finetune workflow in computer vision15 and natural language processing16,17, various methods in molecule self-supervised learning (SSL)18,19,20,21,22,23,24,25 have emerged. In the self-supervised setting, machine learning models are pre-trained to learn generic molecular representations by optimizing the performance on pre-defined tasks on large-scale unannotated molecule data. These tasks are designed in a way such that solving them requires identification of important structural patterns and understanding of rudimentary chemical knowledge. Existing molecule SSL methods can be mainly classified into two categories: predictive and contrastive. Predictive learning18,19,20,26,27,28 aims to predict structural components given contexts at different levels, which mainly focuses on intra-data relationship. These methods often follow the conventional pipeline of reconstructing the molecular information from masked inputs. Contrastive learning20,22,23,29,30,31,32, initially proposed in computer vision33, aims to learn the inter-data relationship by pulling semantic-similar data samples closer and pushing semantic-dissimilar samples apart in the representation space. Note that this idea aligns well with common heuristics in chemistry, where structurally similar molecules are likely to exhibit similar physicochemical and biological properties. Most works attempt to generalize the same SSL methods across multiple domains (e.g., social networks and molecular graphs), whereas it is unsure whether the same learning schemes are compatible to all settings. Recently, several studies have pointed out that existing SSL methods may fail to learn effective molecular representation. RePRA34 was proposed to measure the representations’ potential in solving activity cliffs and scaffold hopping35. These are challenging yet ubiquitous tasks in drug discovery that requires the model to understand subtle chemical knowledge behind SPR. Experiments showed that most pre-trained representations perform worse than molecular fingerprints. The work in36 explored various molecular SSL methods and observed that some pre-training strategies can only bring marginal improvement, while some may induce negative transfer. Meanwhile, studies have also highlighted the incompetence of several pre-trained representations in predicting binding potency under activity cliffs6,12,37.
This work aims to enhance molecular representation learning that encodes robust and generalizable chemical knowledge. We start by identifying the two major drawbacks in existing methods: Firstly, in contrastive learning, the conventional formulations of the semantic-similar/dissimilar (i.e., positive/negative) samples are not well-tailored for molecular graphs. Most graph contrastive methods generate positive samples via graph perturbation, such as node/edge addition/deletion20,21,21,22,23. However, when applied on molecular graphs, chemical validity may be easily challenged. Molecules may also lose essential characteristics by perturbing important motifs (e.g., breaking an aromatic ring), shifting the “semantics” distant away. The negative samples (i.e., different molecules) are often treated equally, which essentially neglects the molecule structural relationship and the presence of specific molecular components; Second, almost all existing works attempt to learn a context-independent molecular representation space, aiming to generalize to various applications. However, this contradicts the fact that molecular properties are often context-dependent, from both the physical (e.g., surrounding environments) and biological (e.g., interaction with proteins) perspectives. In other words, it remains uncertain whether the same SSL tasks could align well with diverse downstream tasks of distinct properties in fine-tuning, thereby leading to the learning gap.
To approach the aforementioned challenges, we introduce a prompt-guided multi-channel learning framework for molecular representation learning. Each of the k channels, guided by a specific prompt token, is responsible for learning one dedicated SSL task. Essentially, the pre-trained model is able to learn k distinct representation spaces. During fine-tuning, a prompt selection module aggregates k representations into a composite representation and uses it for the downstream molecular property predictions. This involves determining which information channel is most relevant to the current application, thereby making the representation context-dependent. We later show how this composite formulation is more resilient to label overfitting and manifests better robustness. In addition, we design the pre-train tasks to form an interpolation from a global view to a local view of the molecular structures. Besides leveraging the global molecule contrastive learning and the local context prediction19, we introduce the task of scaffold contrastive distancing, highlighting the fundamental role of scaffolds in affecting molecular characteristics and behaviors. Since scaffolds are often treated as starting points for new compound design, scaffold distancing aims to map molecules with similar scaffolds (generated via scaffold-invariant perturbations) closer in the representation space. Additionally, it pushes molecules with different scaffolds apart, where the distance margin is computed adaptively based on structure composition difference. Note that scaffold distancing tackles the partial but core view of the molecules. The overall framework is pre-trained using ZINC1538, and evaluated on 7 molecular property prediction tasks in MoleculeNet5 and 30 binding potency prediction tasks in MoleculeACE6. Learning to leverage information from different channels for different applications, our method surpasses various representation learning baselines in both benchmarks. More importantly, our method is shown to handle the challenge of activity cliffs more effectively, whereas competing approaches are more susceptible to the negative transfer, leading to a substantial performance decline. This suggests that these methods may rely more on surface-level patterns even after pre-training or are more susceptible to knowledge forgetting during fine-tuning, causing them to struggle with challenging problems that require a nuanced understanding of chemical knowledge. On the contrary, our learned representation demonstrates enhanced ability in preserving pre-trained knowledge during fine-tuning, offering improved transferability and robustness compared to other baselines. Case study shows that our method has the potential to identify crucial patterns that contribute to activity cliffs, even when relying solely on topological information.
Results
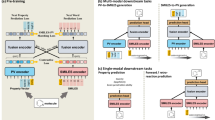
The proposed framework (Fig. 1) comprises three major components that differ from the conventional pretrain-finetune paradigm on molecules: (1) The prompt-guided multi-channel learning, (2) contrastive learning with adaptive margin, and (3) scaffold-invariant molecule perturbation. It demonstrates effectiveness on both the molecular property prediction5 and binding potency prediction6 benchmarks, offering enhanced robustness and interpretability.

a The prompt guided pretrain-finetune framework. For each downstream task, the model is optimized additionally on the prompt weight selection, locating the best pre-trained channel compatible with the current application. b Molecule contrastive learning (MCD), where the positive samples \({G}_{i}^{{\prime} }\) from subgraph masking is contrasted against negative samples Gj by an adaptive margin. c Scaffold contrastive distancing (SCD), where the positive samples \({G}_{i}^{{\prime} }\) from scaffold-invariant perturbation is contrasted by against negative samples Gj by an adaptive margin. d Context prediction (CP) channel consists of masked subgraph prediction and motif prediction tasks. e Prompt-guided aggregation module, which conditionally aggregates atom representations into molecule representation by prompt token. It is realized via a multi-head attention with prompt embedding hp being the query.
Prompt-guided multi-channel learning
We introduce a prompt-guided multi-channel learning framework for molecular representation learning, as shown in Fig. 1a. Essentially, the molecular graphs will first go through a unified encoder module, and then diverge into k different channels, each of which is responsible for learning distinct SSL tasks. For each channel, a prompt token pi is utilized to distinguish levels of molecule representation. This is realized via a prompt-guided readout operation39, which aggregates atom representations conditionally into molecule representation given the prompt token (Fig. 1e). Our experiment involves three learning channels, which are molecule distancing, scaffold distancing, and context prediction. Each channel focuses on a unique aspect of the molecular structure, enabling molecular representation learning from a set of hierarchical viewpoints, from a global view (i.e., entire molecule), a partial view (i.e., core structure), and down to a local view (i.e., functional groups).
We now briefly introduce the three learning channels. (i) Molecule distancing (Fig. 1b) is achieved using a variant of triplet contrastive loss40. It considers triplet of molecule samples {anchor, positive, negative}, where negative (i.e., dissimilar) samples are pushed apart against the anchor and positive (i.e., similar) samples by a margin α. On top of this, we propose the adaptive margin α( ⋅ ), as detailed below. It introduces another level of distancing constraints based on the structural similarity of molecule composition. We follow the work of Molecular Contrastive Learning of Representations (MolCLR)21 and generate positive samples via molecule subgraph masking. (ii) Scaffold distancing (Fig. 1c) is a contrastive learning task that focuses on scaffold differences. Molecule scaffolds are viewed as the foundation for a range of biologically active molecules. They play a crucial role in drug discovery and medicinal chemistry by providing a starting point for compound designs with desired pharmacological properties. In other words, molecules with similar scaffolds are more likely to possess similar physical (e.g., solubility, lipophilicity) and biological (e.g., conformational property when interacting with a protein) characteristics, and thereby sharing similar semantics. Scaffold distancing contrasts the scaffold-invariant molecule perturbations, as detailed below, against molecules with different scaffolds using the adaptive margin loss. (iii) Context prediction (Fig. 1d) involves masked subgraph prediction and motif prediction, which are also adopted in GROVER19. For each molecular graph, a random subgraph (i.e., a center atom and its one-hop neighbors) is masked out, and the model aims to reconstruct the subgraph based on its surrounding structures. Motif prediction aims to predict the existence of functional groups within the molecule. This learning channel mainly focuses on the local view of the molecule by identifying the existence of substructure and functional groups, while molecule distancing and scaffold distancing focus more on the global view and partial view, respectively. Each channel has its own readout layer, termed prompt-guided node aggregation (Fig. 1e). Presumably, the distribution of atom importance should be different across channels (Supplementary Figs. 7–9 and Supplementary Note 6). The learned channel-wise representations exhibit a high correspondence to the associated structural features (Supplementary Fig. 1 and Supplementary Note 1) and support hierarchical iterative clustering of the chemical space (Supplementary Fig. 2 and Supplementary Note 2). To further improve the robustness of the learned knowledge under this framework, we incorporate two regularization tricks on the intra-channel node aggregation and the inter-channel alignment. For the molecule and scaffold channels, the aggregation attentions are more encouraged to span across all atoms and scaffold atoms, respectively. Meanwhile, a small set of supervised tasks is utilized to regularize the composite representation under three prompt weight presets, hence improving the channel alignment. A comprehensive ablation study is provided in Supplementary Fig. 10 and Supplementary Note 7.1.
In the fine-tuning stage, the model is initialized with the same molecule encoder and prompt-guided aggregation modules, along with the learned parameters from pre-training. The parameters in the aggregation modules are fixed during fine-tuning as we aim to use it as a pooling layer independent of the downstream applications. In addition, a prompt-tuning module τθ( ⋅ ) is introduced to determine which channel is most relevant to the current application. It essentially learns a task-specific prompt distribution. The prompt weights are utilized to linearly combine the channel-wise information into a composite molecule representation, which is then used for the task-specific prediction. We discover that this approach is more effective than simply concatenating the representations (Supplementary Fig. 11 and Supplementary Note 7.2). We initialize the prompt weights by choosing the candidate which leads to the smoothest quantitative structure-property landscape (i.e., lowest roughness index)41 of the composite representation. More details are provided in the Method section.
Contrastive learning with adaptive margin
We further introduce the adaptive margin loss, a variant of the triplet loss40, that supports the contrastive learning in the first two channels (molecule distancing and scaffold distancing). In the conventional triplet loss, the representation distance between the anchor Gi and negative (i.e., semantic-dissimilar) sample Gj needs to be at least by margin α larger than the distance between the anchor Gi and positive (i.e., semantic similar) sample \({G}_{i}^{{\prime} }\). Note that this margin remains the same for any triplet considered. However, when applied to molecule triplets, it neglects the known structural relationship between molecules (e.g., co-existence of functional groups). To learn a more fine-grained molecule representation space, we propose to adaptively compute the molecule triplet margin based on the Tanimoto similarity between molecule fingerprints. As shown in Fig. 1b and c, the adaptive margin αMCD(.) considers the molecule structural similarity between Gi and Gj, while αSCD(.) considers the scaffold structural similarity between s(Gi) and s(Gj). Another issue with the conventional triplet loss is that it imposes no constraint on the representation space beyond the margin. It means that the actual representation distances are not necessarily to be well correlated with the computed margin, even if the margin constraints are fully satisfied. This is further elaborated the example in Supplementary Fig. 3. Therefore, we include a secondary term into the adaptive margin loss by considering the structural relationship among the anchor and different negative samples. Detailed formulation of the adaptive margin loss is included in Method section. With careful consideration of existing structural similarity, the learned representation space would better capture molecule relationships in a fine-grained representation space. A performance drop is observed when the adaptive margin loss is replaced with the conventional margin loss (Supplementary Fig. 10 and Supplementary Note 7.1).
Scaffold-invariant molecule perturbation
To generate semantic-similar samples (i.e., positive) for scaffold contrastive distancing, we propose to perturb only the terminal side chains of the molecule. In other words, the molecule scaffold (i.e., core structure) is preserved. This is done by first identifying the side chains and then performing fragment replacement based on a candidate fragment pool. To avoid significant alterations in molecule characteristics, we restrict the amount of changes to be fewer than five atoms. For simplicity reason, we consider the Bemis-Murcko framework as the scaffold. Figure 1c shows a sample perturbation with scaffolds highlighted in blue. In this example, the benzene ring is the identified scaffold, and either the carboxylic ester group or the carbonyl group is perturbed by another functional groups. Note that such perturbation is not limited to atom-level or bond-level editing, but also motif-level.
Molecular property prediction
To demonstrate the effectiveness of our approach, we first evaluate it on seven challenging classification datasets from MoleculeNet5, which is a large-scale curated benchmark that covers multiple molecular property domains (e.g., physiology, biophysics). The scaffold splits scheme18 is applied. The performance is evaluated using the ROC-AUC value. Each experimental result is averaged over three different runs following the prior works19,21,25. To demonstrate the effectiveness of our framework across different model architectures, we pre-train both a graph neural network GIN42 and a graph transformer GPS43, termed OursGIN and OursGPS, respectively. We compare our methods with twelve competitive molecular representation learning baselines, which cover a wide variety of pre-train SSL techniques, model architectures, and input representations (i.e., sequence, graph, geometry). Specifically, we first consider four contrastive baselines, including GraphLoG22, D-SLA23, MolCLR21, and KANO24. These methods also share the commonality of using GNNs to encode molecular graphs. Besides, we include six predictive baselines, including Hu et al.18, GROVER19, MoLFormer28, KPGT44, GEM25, and Uni-Mol27. MoLFormer serves as a strong baseline for sequence-based models, while GEM and Uni-Mol are strong baselines for 3D geometry models. We further incorporate GraphMVP45 and ImageMol46 as two multi-task learning baselines whose pre-training tasks cover both contrastive and predictive learning. Last but not least, we train the GIN and GPS models from scratch. Table 1 shows the performance comparison results. Our methods improve over the no-pretrain setting (i.e., GIN and GPS) by 12.6% and 8% ROC-AUC in average, respectively. When compared to the other SSL methods, our approach reaches state-of-the-art performances on BBBP, Clintox, BACE and SIDER datasets, while remaining highly competitive in the rest of the tasks. Our overall ROC-AUC score is 0.8% higher than the second best method (Uni-Mol). Notably, Uni-Mol leverages 3D geometric information and is an order of magnitude larger (in term of model parameters) than both OursGIN and OursGPS.
Binding potency prediction
We consider MoleculeACE6 as the second evaluation benchmark. It consists of 30 datasets retrieved from ChEMBL47. Each dataset contains binding potency measures (e.g., Ki value) of molecules against a macromolecular target. These datasets mainly focus on the structure-property relationships (SPR), where the phenomenon of activity cliffs is amplified. Activity cliffs refer to the cases where small changes in molecular structure significantly alters its biological activity, and understanding them is crucial for optimizing lead compounds and designing new molecules with desired activities. The phenomenon is also counter-intuitive in machine learning, as the machine learning model tends to make similar predictions given similar inputs. We take R-squared as the evaluation metric since relative binding potency ranking is more important than absolute prediction errors (e.g., RMSE) in the real world.
Figure 2a shows the performance of thirteen methods on the MoleculeACE benchmark under stratified splits6 (Supplementary Table 2). The multi-layer perceptron (MLP) is trained using the ECFP4 fingerprint. We also compare the average performance with respect to the model sizes (i.e., number of parameters) in Fig. 2b, as indicated by its x-axis and the size of the dots, and types of input representations (blue = 1D sequence, red = 2D topological graph, and yellow = 3D geometry). In general, OursGPS ranks in the second place, but with a significantly smaller model size than KPGT. Besides, almost all methods fail to surpass MLP with fingerprints. MolCLR and GraphLoG also show negative transfer compared to the GIN model. It demonstrates the incompetence of existing methods in learning the nuances of chemical knowledge behind the structural-property relationship, regardless of the input representations, pre-training strategies, and the model sizes6,12. One of the reasons behind KPGT’s strong performance could be attributed to the fact that the molecule fingerprint and descriptors are heavily embedded within the model. In addition, KPGT is pre-trained using the ChEMBL database47, such that over 99% of the testing molecules in MoleculeACE are already exposed to the model during the pre-training stage. In contrast, our models only have seen less than 5% of the testing molecules during the pre-training stage. The plot also suggests that there are no clear advantages of molecular representation learning using either 1D sequence or 3D geometry, especially when comparing the performance with Uni-Mol and MoLFormer.

a The violin plot illustrates model performance across 30 binding potency prediction tasks in MoleculeACE6. Performance on each dataset is averaged across three independent runs using stratified splits. The asterisk (*) indicates that the model is trained from scratch (no pre-train). The ordering of the methods from left to right is sorted by the average performance. The width of each violin represents the density of data points, with wider sections indicating a higher concentration of values. The minima and maxima are displayed by the lower and upper ends of the violin. The middle solid white line represents the median value. b Average model performance across 30 binding potency prediction tasks with respect to model sizes (also indicated by the size of the dots). The dashed line follows the performance of the multi-layer perceptron (MLP), which serves as the baseline comparison between traditional fingerprints and deep molecular representations. c Relationship between model performance and the presence of activity cliffs in each dataset measured by the roughness of molecular property landscape. Performance on each dataset is averaged across three independent runs using random splits. The size of the datasets is indicated by the size of the dots. The slope of the fitted line of each method indicates the correlation. OursGPS is used in this analysis.
We further study the model performance in relation to the presence of activity cliffs in each dataset, which is measured by the roughness index (ROGI)41 with respect to the ECFP4 fingerprints and potency labels. A smaller ROGI value indicates a smoother structure-property landscape, hence less activity cliffs. Since ROGI is a quantitative SPR (QSPR) metric that analyzes the overall chemical space, it does not account for any distribution shift between chemical spaces. Hence, its correlation with model performance would be easily obscured by any distribution shift among training, validation, and testing sets (Supplementary Fig. 14). For this reason, all experiments in this work that involves QSPR analysis are performed using random splits, following the work in41. As shown in Fig. 2c and Supplementary Table 3, the model performance is negatively correlated with landscape roughness for all methods, which is consistent with the results in41. Compared to the MLP with fingerprint and KPGT, our method is shown to be more robust against tasks with rough structure-property landscapes. In addition, the plot also indicates that our method performs better under data scarcity, where the size of dots represents the size of datasets.
Representation robustness
To study the robustness of the learned molecular representation, we propose to probe the fine-tuning process and evaluate the shift in the representation space. Essentially, the shift captures how much pre-trained chemical knowledge is distorted during fine-tuning. We examine the representation space of both the training and validation molecule set at five training timestamps. To clarify, the term “learned representation” refers to the numerical embedding used for the final prediction layer. We choose CHEMBL237_Ki, one of the largest binding potency prediction dataset in MoleculeACE, for the downstream target. For fair comparison, this analysis is performed among OursGIN, GraphLoG, and MolCLR, as these methods only differ by the SSL strategies for pre-training, while using the same pre-train dataset, GNN architecture and hyperparameters. In Fig. 3, each column represents a training timestamp, and each row pair represents the visualization of representation space in training and validation sets. The coloring represents the normalized potency labels for illustrative purposes. We also report four additional metrics that capture the representation characteristics along the training process: 1. Roughness Index (i.e., ROGI)41 captures the landscape roughness of molecular property given a representation. 2. Rand Index48 measures the proximity between fingerprint (ECFP4) clustering and representation clustering, serving as a proxy for the amount of structural information encoded within the current representation space. A higher Rand Index indicates a greater amount of structural knowledge being captured by the representation. 3. Cliff-noncliff Distance Ratio indicates the generalizability of the representation towards the activity cliffs. In short, cliff matched molecule pairs (MMPs) (i.e., similar molecules with different labels) should be more distant away compared to the non-cliff MMPs (i.e., similar molecules with similar labels). It is calculated as the ratio of the average distance between cliff MMPs to that of non-cliff MMPs. We also show three molecules on the plot for illustrative purposes, with one cliff MMP (indicated by the red arrow) and one non-cliff MMP (indicated by the blue arrow). 4. At last, we report the validation R-squared as the performance measure. Here are the three main takeaways from Fig. 3:
-
Our composed representation yields the lowest ROGI value to start with. In other words, our pre-trained knowledge is more transferable to the target application. This is also shown by our rapid convergence rate in the validation set, reaching a validation R-squared of 0.676 at epoch 10 (Supplementary Fig. 5 and Supplementary Note 4).
-
Our representation preserves the chemical knowledge learned from pre-training better than others, leading to less overfitting and better robustness. Since representations are optimized towards the property labels, the Rand index drops continuously for all methods. It means that the encoded information gradually shifts from being structure-oriented to label-oriented. However, our method has the lowest drop in Rand index of 0.072, compared to the drop of 0.09 by GraphLoG and 0.181 by MolCLR. The visualization also shows that the representations of MolCLR begin to overfit to the labels starting from epoch 10, resulting in the loss of substantial structural relationships between molecules. This explains its low ROGI value and validation R-squared along the training process.
-
Our representation seems to exhibit a better understanding of the nuances of chemical knowledge in activity cliffs. Our average cliff-noncliff distance ratio in the validation set is always above one and larger than that of GraphLoG and MolCLR. As illustrated by the triplet samples, the red arrow is longer than the blue arrow across fine-tuning epochs, while the closeness of cliff and non-cliff pairs in space (i.e., structurally similar) is maintained. For MolCLR, even though the red arrow can be much longer than the blue one, the molecules are distant away. It means that MolCLR fails to capture the structural similarity aspect of the activity cliffs. A detailed examination of the distance histogram is visualized in Supplementary Fig. 13.

The dynamics of molecule representation space of three methods at five finetune timestamps on dataset CHEMBL237_Ki (n = 2602). For each row pair, a 2D view of the representation space of both the training (top) and validation set (bottom) are visualized. The color map represents the normalized binding potency of each data point from lowest to highest within the dataset. Rand index and roughness index (ROGI) are reported for training, while R-squared value and cliff-noncliff distance ratio are reported for validation. The gray circles correspond to the clustering assignment using ECFP4 fingerprint. The red arrow indicates the distance of a cliff molecule pair, while the blue arrow indicates that of a non-cliff molecule pair.
To further understand why our composed representation is more resilient to the representation shift and can better preserve pre-trained knowledge, we present more analysis on three diverse datasets, along with the individual channel-wise representation behavior during fine-tuning. We choose CHEMBL237_Ki and CHEMBL262_Ki as the representative regression-based datasets of different scales, and BBBP as the typical classification-based dataset. The Rand index of representation clustering difference between the initial and the current timestamp is computed as a proxy (slightly different than before) for the representation shift. A smaller Rand index indicates a larger shift. As illustrated in Fig. 4, channels with the highest prompt weights often exhibit the largest shift, and vice versa. This is reasonable because these channels contribute more to the optimization against the labels. Conversely, even though low-weighted channels contribute less to the optimization, they are more likely to preserve the pre-trained knowledge. As a result, the composed representation derived from channel aggregation exhibits a certain level of resilience to representation shifts, making it potentially more robust than other methods. The probing analysis under the few-shot learning settings also reveals similar patterns (Supplementary Fig. 6 and Supplementary Note 5).

Detailed analysis of the representation shift during fine-tuning across three datasets. The dashed lines (top row) correspond to the representation shift (approximated by the Rand index) for each individual channel, while the solid lines represent the overall method. The bottom row displays the optimized prompt weights distributed over channels.
Activity cliffs analysis
We present a deeper analysis into the potential of our method in understanding the activity cliffs. To be more specific, we evaluate the relationship between the model explanations generated by the GNNExplainer49 and the predicted binding mode between the ligands and the protein pockets by AutoDock Vina50. Note that this analysis merely serves as a proof of concept, such that our representation has the potential of capturing influential and well-established factors in binding affinities. However, the fundamental limitation of utilizing topological information only is unavoidable, which we will discuss in Conclusion.
As shown in Fig. 5, visualized by PyMOL51, two series of compounds sharing the same scaffolds are potential inhibitors of glycogen synthase kinase-3 beta (GSK3β). The molecule activity cliff pairs, determined by the formulation in6, are compounds < a1, a2 > , < a1, a3 > , < b1, b2 > , and < b1, b3 > . The explanations of both our and MolCLR’s predictions are compared. In Fig. 5a, the potential intra-molecular halogen-bonding contact between the chlorine atom and hydrazone in compound a1, as indicated by the green dashed line, is disfavored for the inter-molecular hydrogen-bonding contact between the backbone carbonyl of the active site VAL-135 and hydrazone of the compound52. As shown by our model explanation, compared to the compound a2 and a3, the chlorine atom of compound a1, along with its associated benzene ring, contribute less to the overall binding affinity prediction. It aligns well with the predicted binding mode.

Evaluate the bioactivity binding mode and the atom importance captured by our model on a (a) 5-phenyl-4-phenyldiazenyl-1,2-dihydropyrazol-3-one series and a (b) N-phenyl-4-pyrazolo[1,5-b]pyridazin-3-ylpyrimidin-2-amine series. The binding modes of compound a1 and compound b1 in the active sites of GSK3β, with Protein Data Bank (PDB) ID: 3L1S, are predicted by AutoDock Vina50 and visualized by PyMOL51. The key hydrogen bonds between compounds and the active sites are highlighted by yellow dash lines, while the green dash line refers to the intra-molecular halogen bond. The color intensity on each atom indicates its respective contribution to the prediction of our method, computed from a GNNExplainer49.
The predicted binding mode in Fig. 5b shows that the orientation of the substituent (i.e., the alkoxy group in compound b1 at the 6 position of pyrazolo[1,5-b]pyridazine) will cause steric clash with PHE-67 in the G-rich loop of GSK3β, thereby leading to a loss of potency53,54. In contrast, the alkoxy groups at the 3’ and 5’ positions have no clear contact with the protein pocket. Remarkably, using only the topological information, our model can capture the importance of the influential alkoxy group at 6 position. In addition, the absence of the alkoxy group at 5’ position (from compound b2 to b3) does not affect the overall atoms’ contribution to the predicted potency. MolCLR performs equally good in terms of compound b2 and b3, but it fails to capture the influential alkoxy group in compound b1.
Discussion
In this work, we propose a multi-channel learning framework for molecular representation learning, which aims to encode and utilize robust chemical knowledge that is generalizable to diverse downstream applications. Each channel is dedicated to learning unique self-supervised tasks, focusing on distinct yet correlated global and local aspects of the molecule. Specifically, MCD captures the molecule’s global similarity, SCD highlights the fundamental role of the scaffold in affecting molecular characteristics, and CP targets the composition of functional groups. During fine-tuning, the model is able to identify which channel-wise representation is most relevant to the current application, thereby making the composite representation context-dependent.
One limitation of this framework is the need for a more effective prompt weight optimization mechanism. The initialization of prompt weights using roughness index can lead to sub-optimal performance. Since roughness index is a global QSPR metric that targets the overall chemical space, it does not account for any distribution shift between training and testing sets. This is the same for the other QSPR measures as well (e.g., SALI55, SARI56). As a result, the final representation performance may be less correlated with the initial roughness value under designated splits. This also explains the performance gap between Fig. 2a, b.
There are several interesting directions for future research. One promising direction is to incorporate different input representations into the framework. By merely leveraging topological molecular structure, the model is unable to differentiate molecular components with different conformations (e.g., functional groups’ orientation or atom’s chirality), which could significantly alter biochemical behaviors (see Supplementary Note 9 for further discussion). Besides, there exist other advanced data-driven techniques for studying the structural-activity relationship (SAR) that might be compatible with our framework. For example, Molecular Anatomy57 argues that the network clustering from scaffold fragmentation and abstraction allows high quality SAR analysis. Such investigations aim to transfer knowledge from cheminformatics to machine learning models, potentially improving both model interpretability and robustness. More importantly, while our method has immediate implications for drug discovery, its molecular representation robustness further shed lights on its promising potential in other sub-fields of chemistry, such as materials science and environmental chemistry.
Methods
Graph neural networks (GNNs)
A graph G = (V, E) is defined by a set of nodes V and edges E. In the molecular graph, each node denotes an atom, and the edge denotes the chemical bond. Let hv be the representation of node v, and hG be the representation of the graph G. Modern GNNs follow the message-passing framework, such that node representations are updated iteratively via neighborhood aggregation:
where N(v) is the neighborhood of node v, k denotes the layer index in a multi-layer GNN structure, and euv denotes the edge connecting two nodes u and v. The initialization of \({{{{\bf{h}}}}}_{v}^{0}\) comes from the predefined node features xv. The aggregate function integrates neighborhood information into the current node representation. The update function takes the updated node representation and the node representation from the previous k − 1 layer and performs operations like concatenation or summation. After the iterative updates, a permutation-invariant pooling operation is performed to get the representation for the entire graph G: \({{{{\bf{h}}}}}_{g}={{{\rm{READOUT}}}}({{{{\bf{h}}}}}_{v}^{k}| v\in V)\). There are various number of options for the readout operation, including simple operations of mean and max, and advanced differentiable approaches like DiffPool58 and GMT59.
Roughness index
To analyze the structure-property relationship (SPR) within a chemical space, various quantitative metrics are proposed (e.g., SALI55, SARI56, ROGI41). Even though the formulations are different, they all aim to capture the relationship between the representation difference and the property difference, which is also known as the molecular property landscapes. In this work, we mainly rely on ROGI value for both model training and experimental analysis. It is computed by first clustering the chemical space with different distance threshold t. For each cluster assignment, the weighted standard deviation σt is calculated over the property labels of each cluster prototype. As the distance threshold increases, σt will decrease monotonically from its initial value to zero (i.e., single cluster). If there are more similar molecules with similar labels, σt will decrease slowly, and vice versa. Eventually, roughness index is formulated as below:
The ROGI value has been shown to be strongly correlated with other metrics (e.g., SARI), as well as the representation performance on the given dataset41. One of the main advantages of using ROGI is that it is a generalized QSPR metric applicable to a wide range of molecular representations, including both fingerprints and neural representations, while other metrics like SARI are tailored only for molecular fingerprints. This allows us to compute landscape roughness on pre-trained molecular representations (Supplementary Fig. 4 and Supplementary Note 3) and perform deeper QSPR analysis.
Prompt-guided aggregation
Instead of using the same readout operations as the conventional GNNs, we adopt the prompt-guided aggregation, which is achieved using the multi-head attention. As shown in Fig. 1e, the embedding of the prompt token hp is treated as the attention query, while the representations hx of nodes/atoms x, are viewed as the keys and values. The resulted prompt-aware graph representation \({{{{\bf{h}}}}}_{g}^{p}={\sum }_{x}{\alpha }_{x}{{{{\bf{v}}}}}_{x}\), where the attention weight for each node x is computed as \({\alpha }_{x}=\,{{\mbox{softmax}}}\,({\{{{{\bf{q}}}}\cdot {{{{\bf{k}}}}}_{x}/\sqrt{{d}_{k}}\}}_{x})\), and q = Wqhp, kx = Wkhx, and vx = Wvhx. Here {Wq, Wk, Wv} represent parameters in the linear transformations, and \(\sqrt{{d}_{k}}\) is the scaling factor. Essentially, the prompt-aware \({{{{\bf{h}}}}}_{g}^{p}\) is aggregated from the weighted average of linear projection of hx.
Adaptive margin contrastive loss
Contrastive learning is a technique widely used in self-supervised learning, which aims to group semantic similar samples closer while pushing dissimilar samples distant apart in the latent representation space. In this work, we adopt the triplet loss40 to formulate the contrastive learning. It considers triplets of data samples: the anchor Gi, the positive (i.e., semantic-similar) sample \({G}_{i}^{{\prime} }\), and the negative (i.e., semantic-dissimilar) sample Gj. Its formulation is shown below.
where \({{{{\bf{h}}}}}_{{g}_{i}}\) denotes the latent representation for sample Gi, and \({{{{\bf{h}}}}}_{{g}_{i}}^{{\prime} }\) being the representation for its augmentation. The function d( ⋅ , ⋅ ) measures the L2 distance between two vectors. In general, this objective enforces the pair-wise distancing difference between \( < {G}_{i},{G}_{i}^{{\prime} } > \) and < Gi, Gj > to be at least α margin. However, as we discuss before, this formulation can only lead to a coarse-grained representation space, while neglecting the existing structural relationship among molecules (e.g., shared rings or functional groups). Also, this formulation does not pose any constraints on the representation space beyond the margin (Supplementary Fig. 3). Therefore, we propose to add an additional contrastive formulation with adaptive margin among negative samples and the anchor.
where α1( ⋅ ) and α2( ⋅ ) are the adaptive margin functions. Let \({{{{\bf{z}}}}}_{{g}_{i}}\) be the conventional structural features of the sample Gi. We use the ECFP4 fingerprint60, which hashes circular atom neighborhoods into fixed-length binary strings, to represent the structural features. In molecule contrastive distancing, the adaptive function \({\alpha }_{1}({G}_{i},{G}_{j})={\alpha }_{{{{\rm{offset}}}}}\times (1-\,{{\mbox{sim}}}\,({{{{\bf{z}}}}}_{{g}_{i}},{{{{\bf{z}}}}}_{{g}_{j}}))\), and \({\alpha }_{2}({G}_{i},{G}_{j},{G}_{k})={\alpha }_{{{{\rm{offset}}}}}\times (\,{{\mbox{sim}}}\,({{{{\bf{z}}}}}_{{g}_{i}},{{{{\bf{z}}}}}_{{g}_{j}})-\,{{\mbox{sim}}}\,({{{{\bf{z}}}}}_{{g}_{i}},{{{{\bf{z}}}}}_{{g}_{k}}))\), where sim( ⋅ , ⋅ ) denotes the Tanimoto similarity. The scaffold contrastive distancing has the same formulation, except that the molecule sample G is replaced by its scaffold s(G). Note that the formulation now considers quadruplet of data samples \( < {G}_{i},{G}_{i}^{{\prime} },{G}_{j(\ne i)},{G}_{k(\ne j\ne i)} > \). Even though the theoretical complexity is increased from O(N2K) to O(N3), we can perform fixed-size random sampling with respect to the computed similarity differences in α2( ⋅ ). Quadruplets are also dropped if the computed values are negative. Eventually, the optimization goal is to minimize the loss summation as \(\min {{{{\mathcal{L}}}}}_{adaptive}=\min {\sum }_{i,j,k}\, {\ell }_{i,j(\ne i),k(\ne j\ne i)}\).
Regularization
To further improve the robustness of the pre-trained model, we incorporate two regularization schemes for the intra-channel node aggregation and the inter-channel alignment. The former strategy aims to ensure that all atoms contribute to MCD, while only the scaffold atoms contribute to SCD. This is accomplished by a smooth L1 loss between the attention score and the atom importance matrix. We realize that without any regularization, the aggregation module may rely on specific structural patterns to perform the SSL tasks, causing the attention distribution to skew towards certain substructures. This is also known as the shortcut learning61 in deep learning models. As a result, it is possible to feed an incomplete view of the molecule to the property prediction layer (i.e., atoms that receive low attention across all channels) without the attention regularization. The latter regularization encourages better alignment of representation spaces. Since the three channels are learned separately via different tasks, the numeric values of representations at a given position may not be aligned. It means that the linear combination of representations before any fine-tuning may not be meaningful. To encourage the composite representation space to be better defined (e.g., subtracting \({{{{\bf{h}}}}}_{g}^{[MCD]}\) with \({{{{\bf{h}}}}}_{g}^{[SCD]}\) could represent the fragments beyond scaffold), we use a set of supervised tasks (e.g., predicting molecular weight and logP), along with the corresponding prompt weight presets, to regularize the channel alignment. These tasks mainly predict the molecule/scaffold descriptors using the composite representation. For example, the prompt weight preset for predicting the molecular weight would be [0.45, 0.1, 0.45], while the preset for predicting the weight of the scaffold would be [0.1, 0.45, 0.45]. We multiply all the regularization losses by a factor of 0.1 to prevent the regularization from dominating the learned representation.
Prompt-guided multi-channel learning
The overall multi-channel learning framework is inspired by the work in62. At the pre-train stage, the MCD and SCD channels perform the contrastive learning using the adaptive margin loss. Subgraph masking is used to generate positive samples for MCD. A subgraph is defined by a central atom along with its one-hop neighbors. The masking is performed at the attribute level, ensuring that the topological structure is retained. Meanwhile, the CP channel learns masked subgraph prediction as a multi-label classification task and motif prediction as a regression task: \({{{{\mathcal{L}}}}}^{{{{\rm{CP}}}}}={{{{\mathcal{L}}}}}_{{{{\rm{CE}}}}}+{{{{\mathcal{L}}}}}_{{{{\rm{SmoothL1}}}}}^{fg}\), where CE stands for cross-entropy loss, SmoothL1 for smooth L1 loss, and fg for normalized functional group descriptors. Overall, the framework is optimized using the three channel losses along with the regularization losses:
At the finetune stage, the pre-trained model parameter are used to initialize the model. Besides, we introduce an additional prompt selection module to combine representations from different channels into the task-specific (i.e., context-dependent) composite representation. Essentially, it learns the relevance between different pre-trained molecular knowledge and the downstream application, hence bridging the gap between pre-training and fine-tuning objectives. To incorporate task-specific SPR information into the model at an early stage of fine-tuning, we propose to initialize the prompt weights from computing the roughness index (ROGI)41. In short, the initial prompt weights should lead to a composite representation with the lowest ROGI value (i.e., smoothest quantitative structure-property landscape) with respect to the current application. A low ROGI value indicates smoother landscape, hence better modellability. We use a simple Bayesian optimization pipeline to find the best initialization with the lowest ROGI value on the training set. Essentially, the input parameters of the Bayesian optimization are k − 1 learnable scalers, where k is the number of channels. The black-box utility function first computes the distribution over k values by treating the input scalars as logits, and calculates the composite representation as well as its corresponding roughness index. We utilize the Quasi MC-based batch Expected Improvement as the acquisition function. After the initialization, the entire model except for the prompt-guided aggregation module is optimized towards the molecular property labels. The parameters in the aggregation modules are fixed during fine-tuning as we aim to use it as a pooling layer independent of the downstream applications. As shown in Supplementary Figs. 7–9, the node attention scores within the aggregation module reflect the expected atom importance. For example, the SCD’s aggregation attention captures mostly the scaffold atoms. We further discover that the learned prompt weights exhibit patterns that are well-aligned with the necessary chemical knowledge for solving the downstream tasks (Supplementary Fig. 12, Supplementary Table 1, and Supplementary Note 8).
Experimental setup
We pre-train our framework using the molecules from ZINC1538, which is an open-sourced database that contains 2 million unlabeled drug-like compounds. We use CReM63, an open-sourced molecule mutation framework, for the scaffold-invariant molecule perturbation. To be more specific, we first use RDKit64 to identify the Bemis-Murcko scaffold of molecules, as well as the connection sites (i.e., atom indices) between the scaffold and the side chains. The CReM algorithm then takes these indices and performs fragment replacement using an external fragment pool. Perturbation results are further filtered by chemical validity and the maximum number of changed atoms allowed. At last, we are able to successfully compute perturbations for 1,882,537 out of 2 million molecules. These molecules form our final pre-train database. In the contrastive-based pre-training, we consider five positive samples for each anchor molecule. In molecule distancing, we randomly apply subgraph masking five times on the original molecule. In scaffold distancing, we randomly sample five scaffold-invariant perturbations. In terms of the model architecture, we follow the same architecture setup as the graph neural network in18,21,22,23, which is a 5-layer Graph Isomorphism Network (GIN)42 with hidden dimension size equals to 300. In addition, we also pre-train a GPS Graph Transformer model43 with the same number of layer and hidden dimension size. We use the same molecule feature sets as works in19,24. During fine-tuning, we utilize the scaffold splits method18 for MoleculeNet5, with a train, validation, and test ratio of 8:1:1. On the other hand, we apply the stratified splits for MoleculeACE6, as proposed by itself, with a train, validation, and test ratio of 8:1:1.
Baselines
We consider twelve baseline pre-training methods in our experiments. 1. GraphLoG22 achieves a hierarchical prototypical embedding space with the conventional graph perturbation techniques; 2. D-SLA23 proposes the discrepancy learning to refine the embedding space with the conventional graph perturbation techniques; 3. MolCLR21 utilizes the NT-Xent33 contrastive loss with molecule perturbation techniques, including atom/bond editing and subgraph masking; 4. KANO24 introduces the knowledge graph prompting techniques to augment molecular graph, while the augmentations are also learned in the contrastive manner. 5. MoLFormer28 performs large scale of masked language modeling on SMILES sequence using a linear attention Transformer. 6. Hu et al.18 adopts masked attribute prediction and molecular subgraph prediction; 7. KPGT44 proposes the Line Graph Transformer on molecular graphs and performs knowledge prediction. It takes a masked molecular graph, molecule fingerprint, and molecule descriptors as input, and aims to reconstruct the molecule fingerprint and descriptors during pre-training. 8. GROVER19 proposes the GTransformer architecture and applies context prediction learning on molecular graphs; 9. GEM25 encodes the 3D geometric information of molecules and predicts the bond angle and atom distance. 10. Uni-Mol27 also incorporates 3D information of the molecule and pre-trains a SE(3) Transformer65 using 3D position recovery and masked atom prediction. We further include 11. GraphMVP45, which performs both contrastive and predictive learning between 2D toplogical and 3D geometric information of the molecule. 12. ImageMol46, which leverages visual information of molecule images for molecular property prediction. We also train the GIN42 and GPS43 models from scratch for comparison. We reproduce the performance of MolCLR, GROVER, GEM, KPGT, MoLFormer, and KANO on the MoleculeNet benchmark using their provided checkpoints and code repositories. Note that the reported results of GROVER, KPGT, MoLFormer and KANO in their original papers are evaluated under the balanced scaffold splits, which is different than the deterministic scaffold splits used in this work and the rest of the baseline methods. Both splits aim to hold out a set of molecules with scaffolds that are not seen during training. The deterministic scaffold splits prioritizes the satisfaction of the specified split ratio, while the balanced scaffold splits prioritizes the balance of scaffold frequency across the training, validation, and test sets. We use the deterministic scaffold splits in our experiments primarily because most of the baselines also employ it. All other aspects, including hyperparameter choices for both the models and the training setup, remain consistent with the default configurations of these baseline methods for replicating the results. For the MoleculeACE benchmark, we leverage their corresponding repositories, default configurations, and the provided model checkpoints of these baseline methods and fine-tune each task using either MoleculeACE’s stratified splits or random splits.
Representation space probing
We propose to analyze the dynamics of representation space during fine-tuning to evaluate the representation robustness and generalizability. To be more specific, we probe the mapping of the representation space at five different training timestamps (epoch 0, epoch 10, epoch 20, epoch 50, epoch 100). The 2D view is constructed via the T-SNE66 dimension reduction technique. Besides the visualization, we also report four additional metrics that capture the representation characteristics. We report the ROGI value and Rand index in the training set, plus the R-squared value and cliff-noncliff distance ratio in the validation set. The Rand index is calculated from two clustering assignments. The first clustering is formed using k-means clustering with the molecule ECFP4 fingerprint (radius=2, nBits=512). The second clustering is formed using k-means clustering with the representation at the current timestamp. We set the same number of clusters for these two clusterings. In terms of the cliff-noncliff distance ratio, we first identify the cliff and the noncliff molecule pairs using the same principle in MoleculeACE6. Then, we compute and average the representation distance of each pair. At last, the cliff-noncliff ratio is formed by normalizing their average distance.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All relevant data supporting the key findings of this study are publicly available. All datasets utilized in this paper can be found at https://github.com/yuewan2/MolMCLor the Zenodo repository at https://doi.org/10.5281/zenodo.1401043667. This includes the processed pre-train dataset of ZINC15, the MoleculeNet benchmark (originally from https://github.com/deepchem/deepchem) and the MoleculeACE benchmark (originally from https://github.com/molML/MoleculeACE). The reference protein structure for 3L1S used in this study is available in the Protein Data Bank under accession code 3L1S. Source data are provided with this paper.
Code availability
The source code of this work can be accessed via the GitHub repository at https://github.com/yuewan2/MolMCLor the Zenodo repository at https://doi.org/10.5281/zenodo.1401043667.
References
Goh, G. B., Hodas, N. O. & Vishnu, A. Deep learning for computational chemistry. J. Comput. Chem. 38, 1291–1307 (2017).
Ramsundar, B. et al. Deep Learning for the Life Sciences (O’Reilly Media, 2019).
Weininger, D. Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988).
Gentile, F. et al. Artificial intelligence–enabled virtual screening of ultra-large chemical libraries with deep docking. Nat. Protoc. 17, 672–697 (2022).
Wu, Z. et al. Moleculenet: a benchmark for molecular machine learning. Chem. Sci. 9, 513–530 (2018).
van Tilborg, D., Alenicheva, A. & Grisoni, F. Exposing the limitations of molecular machine learning with activity cliffs. J. Chem. Inf. Model. 62, 5938–5951 (2022).
Hay, M., Thomas, D. W., Craighead, J. L., Economides, C. & Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 32, 40–51 (2014).
Dowden, H. & Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 18, 495–496 (2019).
Pérez-Villanueva, J., Méndez-Lucio, O., Soria-Arteche, O. & Medina-Franco, J. L. Activity cliffs and activity cliff generators based on chemotype-related activity landscapes. Mol. Divers. 19, 1021–1035 (2015).
Stumpfe, D., Hu, Y., Dimova, D. & Bajorath, J. Recent progress in understanding activity cliffs and their utility in medicinal chemistry. J. Med. Chem. 57, 18–28 (2014).
Sheridan, R. P. et al. Experimental error, kurtosis, activity cliffs, and methodology: What limits the predictivity of quantitative Structure-Activity relationship models? J. Chem. Inf. Model. 60, 1969–1982 (2020).
Deng, J. et al. A systematic study of key elements underlying molecular property prediction. Nat. Commun. 14, 6395 (2023).
Stumpfe, D. & Bajorath, J. Exploring activity cliffs in medicinal chemistry. J. Med. Chem. 55, 2932–2942 (2012).
Dimova, D., Heikamp, K., Stumpfe, D. & Bajorath, J. Do medicinal chemists learn from activity cliffs? a systematic evaluation of cliff progression in evolving compound data sets. J. Med. Chem. 56, 3339–3345 (2013).
Deng, J. et al. Imagenet: A large-scale hierarchical image database. In Proc. CVPR 2009 (2009).
Devlin, J., Chang, M.-W., Lee, K., and Toutanova, K. BERT: Pre-training of deep bidirectional transformers for language understanding. In Proc. NAACL-HLT 2019, 4171–4186 (2019).
Radford, A. et al. Language models are unsupervised multitask learners. OpenAI blog 1, 9 (2019).
Hu, W. et al. Strategies for pre-training graph neural networks. In Proc. ICLR 2020 (2020).
Rong, Y. et al. Self-supervised graph transformer on large-scale molecular data. In Proc. NeurIPS 2020 (2020).
Xia, J. et al. Mole-BERT: Rethinking pre-training graph neural networks for molecules. In Proc. ICLR 2023 (2023).
Wang, Y., Wang, J., Cao, Z. & Barati Farimani, A. Molecular contrastive learning of representations via graph neural networks. Nat. Mach. Intell. 4, 279–287 (2022).
Xu, M., Wang, H., Ni, B., Guo, H. & Tang, J. Self-supervised graph-level representation learning with local and global structure. PMLR 139, 11548–11558 (2021).
Kim, D., Baek, J. & Hwang, S. J. Graph self-supervised learning with accurate discrepancy learning. In Proc. NeurIPS 2022 (2022).
Fang, Y. et al. Knowledge graph-enhanced molecular contrastive learning with functional prompt. Nat. Mach. Intell. 5, 542–553 (2023).
Fang, X. et al. Geometry-enhanced molecular representation learning for property prediction. Nat. Mach. Intell. 4, 127–134 (2022).
Kim, D. & Oh, A. How to find your friendly neighborhood: Graph attention design with self-supervision. In Proc. ICLR 2021 (2021).
Zhou, G. et al. Uni-mol: A universal 3d molecular representation learning framework. In Proc. ICLR 2023 (2023).
Ross, J. et al. Large-scale chemical language representations capture molecular structure and properties. Nat. Mach. Intell. 4, 1256–1264 (2022).
Zhu, Y. et al. Graph contrastive learning with adaptive augmentation. In Proc. WWW 2021, 2069-2080 (2021).
Veličković, P. et al. Deep graph infomax. In Proc. ICLR 2019 (2019).
Xu, D., Cheng, W., Luo, D., Chen, H. & Zhang, X. Infogcl: Information-aware graph contrastive learning. In Proc. NeurIPS 2021 (2021).
You, Y., Chen, T., Shen, Y. & Wang, Z. Graph contrastive learning automated. PMLR 139, 12121–12132 (2021).
Chen, T., Kornblith, S., Norouzi, M. & Hinton, G. A simple framework for contrastive learning of visual representations. In Proc. ICML 2020 (2020).
Zhang, Z., Bian, Y., Xie, A., Han, P. & Zhou, S. Can pretrained models really learn better molecular representations for ai-aided drug discovery? J. Chem. Inf. Model. 64, 2921–2930 (2024).
Böhm, H.-J., Flohr, A. & Stahl, M. Scaffold hopping. Drug Discov. Today Technol. 1, 217–224 (2004).
Sun, R., Dai, H. & Yu, A. W. Does gnn pretraining help molecular representation? In Proc. NeurIPS 2022 (2022).
Wu, F. et al. Instructbio: A large-scale semi-supervised learning paradigm for biochemical problems. Preprint at http://arxiv.org/abs/2304.03906 (2023).
Sterling, T. & Irwin, J. J. Zinc 15 - ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015).
Liu, Z., Yu, X., Fang, Y. & Zhang, X. Graphprompt: Unifying pre-training and downstream tasks for graph neural networks. In Proc. WWW 2023 (2023).
Schroff, F., Kalenichenko, D. & Philbin, J. FaceNet: A unified embedding for face recognition and clustering. In Proc. CVPR 2015 (2015).
Aldeghi, M. et al. Roughness of Molecular Property Landscapes and Its Impact on Modellability. J. Chem. Inf. Model. 62, 4660–4671 (2022).
Xu, K., Hu, W., Leskovec, J. & Jegelka, S. How powerful are graph neural networks? In Proc. ICLR 2019 (2019).
Rampášek, L. et al. Recipe for a General, Powerful, Scalable Graph Transformer. In Proc. NeurIPS 2022 (2022).
Li, H. et al. A knowledge-guided pre-training framework for improving molecular representation learning. Nat. Commun. 14, 7568 (2023).
Liu, S. et al. Pre-training molecular graph representation with 3d geometry. In Proc. ICLR 2022 (2022).
Zeng, X. et al. Accurate prediction of molecular properties and drug targets using a self-supervised image representation learning framework. Nat. Mach. Intell. 4, 1004–1016 (2022).
Gaulton, A. et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–D1107 (2012).
Hubert, L. & Arabie, P. Comparing partitions. J. Classif. 2, 193–218 (1985).
Ying, Z., Bourgeois, D., You, J., Zitnik, M. & Leskovec, J. Gnnexplainer: Generating explanations for graph neural networks. In Proc. NeurIPS 2019 (2019).
Trott, O. & Olson, A. J. Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010).
DeLano, W. L. et al. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr 40, 82–92 (2002).
Arnost, M. et al. 3-aryl-4-(arylhydrazono)-1h-pyrazol-5-ones: Highly ligand efficient and potent inhibitors of gsk3β. Bioorg. medicinal Chem. Lett. 20, 1661–1664 (2010).
Tavares, F. X. et al. N-phenyl-4-pyrazolo [1, 5-b] pyridazin-3-ylpyrimidin-2-amines as potent and selective inhibitors of glycogen synthase kinase 3 with good cellular efficacy. J. Med. Chem. 47, 4716–4730 (2004).
Stevens, K. L. et al. Synthesis and evaluation of pyrazolo [1, 5-b] pyridazines as selective cyclin dependent kinase inhibitors. Bioorg. medicinal Chem. Lett. 18, 5758–5762 (2008).
Guha, R. & Van Drie, J. H. Structure–activity landscape index: identifying and quantifying activity cliffs. J. Chem. Inf. Model. 48, 646–658 (2008).
Peltason, L. & Bajorath, J. Sar index: Quantifying the nature of structure-activity relationships. J. Med. Chem. 50, 5571–5578 (2007).
Manelfi, C. et al. "Molecular Anatomy”: a new multi-dimensional hierarchical scaffold analysis tool. J. Cheminform. 13, 54 (2021).
Ying, Z. et al. Hierarchical graph representation learning with differentiable pooling. In Proc. NeurIPS 2018 (2018).
Baek, J., Kang, M. & Hwang, S. J. Accurate learning of graph representations with graph multiset pooling. In Proc. ICLR 2021 (2021).
Morgan, H. L. The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service. J. Chem. Documentation 5, 107–113 (1965).
Geirhos, R. et al. Shortcut learning in deep neural networks. Nat. Mach. Intell. 2, 665–673 (2020).
Wang, Z. et al. Multi-level protein structure pre-training via prompt learning. In Proc. ICLR 2023 (2023).
Polishchuk, P. CReM: chemically reasonable mutations framework for structure generation. J. Cheminform. 12, 28 (2020).
Landrum, G. et al. rdkit/rdkit: 2024_03_3 (q1 2024) release. https://doi.org/10.5281/zenodo.11396708 (2024).
Fuchs, F. B., Worrall, D. E., Fischer, V. & Welling, M. Se(3)-transformers: 3d roto-translation equivariant attention networks. In Proc. NeurIPS 2020 (2020).
van der Maaten, L. & Hinton, G. Visualizing data using t-sne. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Wan, Y., Wu, J., Hou, T., Hsieh, C.-Y. & Jia, X. Multi-channel learning for integrating structural hierarchies into context-dependent molecular representation. https://zenodo.org/record/14010436 (2024).
Acknowledgements
We have no specific acknowledgments to report for this work.
Author information
Authors and Affiliations
Contributions
Y.W. proposed and implemented the methodology, and conducted the computational experiments. J.W. carried out the chemical analysis of the model’s predictions. T.H., C.H., and X.J. provided guidance throughout the project. Y.W. and J.W. led the paper writing and all authors contributed.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Bruno Junior Neves, Zaiqing Nie, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wan, Y., Wu, J., Hou, T. et al. Multi-channel learning for integrating structural hierarchies into context-dependent molecular representation. Nat Commun 16, 413 (2025). https://doi.org/10.1038/s41467-024-55082-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55082-4
This article is cited by
-
AI-enabled scientific revolution in the age of generative AI: second NSF workshop report
npj Artificial Intelligence (2025)
