
Abstract
CD73, an ectoenzyme responsible for adenosine production, is often elevated in immuno-suppressive tumor environments. Inhibition of CD73 activity holds great promise as a therapeutic strategy for CD73-expressing cancers. In this study, we have developed a therapeutic anti-human CD73 antibody cocktail, HB0045. HB0045 is a 1:1 mixture of two humanized monoclonal IgG1 antibodies (mAbs), HB0038 and HB0039. The cocktail not only harnesses the advantages of its parental mAbs in enzyme inhibition but also shows a significantly greater capability of promoting T cell proliferation in vitro. Structural analyses show that HB0045 effectively locks the CD73 dimer in a “partially open” non-active conformation through a double lock mechanism. In various animal models of syngeneic and xenograft tumors, HB0045 inhibits tumor growth more potently than the single mAbs. Collectively, our findings provide functional and structural insights into the mechanism of a CD73-targeting antibody cocktail.
Similar content being viewed by others

Introduction
Immunotherapy targeting the PD-1/PD-L1 and CTLA-4 pathways of immune suppression has greatly advanced cancer therapy and yet benefits only a small fraction of patients1. To enhance and expand the scope of cancer immunotherapy, research has recently moved towards the identification of additional immunosuppressive pathways. The purinergic signaling pathway is one of such pathways being actively explored2. The purinergic signaling pathway involves the participation of extracellular ATP, ADP, and adenosine as the main signaling molecules for evolutionary conserved cell communication3. Two ectonucleotidases, NTPDase1/CD39, and ecto-5’-nucleotidase/CD73, play central roles in this pathway. Briefly, CD39 enzymatically converts extracellular ATP and ADP into AMP in a stepwise manner, while CD73 catalyzes the hydrolysis of AMP to adenosine4. The generation and accumulation of adenosine are believed to exert an immunosuppressive effect by impairing the cytotoxic antitumor immune response and counteracting the efficacy of immune checkpoint inhibitor therapies5,6. In addition to this, the CD73-adenosine axis has also been implicated in chemoresistance, tumor growth, metastasis, and tumor angiogenesis7,8,9. Furthermore, elevated expression of CD73 has been correlated with unfavorable clinicopathologic characteristics and shorter patient survival time10. Consistent with these findings, genetic and protein expression analyses have revealed up-regulation of CD73 in various human carcinomas, including breast, colon, pancreas, and lung11. Given its pivotal role in cancer development, the purinergic signaling pathway has emerged as a promising target for cancer immunotherapy12,13. Of note, in addition to high expression in tumor microenvironment, the Human Protein Atlas database showed that CD73 is also widely expressed in many normal tissues and cells such as lung, stomach, gallbladder, lymphatic endothelial cells, glandular epithelial cells, naive CD8 T cell, naïve B cells. The first-in-human study of the anti-CD73 monoclonal antibody (mAb) oleclumab, administered alone or in combination with the anti-PD-L1 mAb durvalumab in patients with advanced solid tumors, indicated that oleclumab ± durvalumab had a manageable safety profile, with no dose-limiting toxicities occurring during escalation. However, treatment-related adverse events were observed, the most common being fatigue (15%), diarrhea (9%), and rash (7%). Increased levels of AST, ALT, and blood bilirubin were also observed in some cases14.
Although antagonism to many key components of the purinergic signaling pathway including CD39, CD73, and downstream adenosine receptors have been explored, it is antagonism of CD73 through small-molecular inhibitors and blocking antibodies that have received the most attentions12. CD73 exists in the form of a homodimer and consists of a glycosylated N-terminal domain and a glycosylated C-terminal domain, connected by a highly flexible α-helical linker. The active catalytic pocket of CD73 is formed by N-terminal zinc ion binding sites and C-terminal AMP binding sites in a “closed conformation”15. Importantly, CD73 undergoes an extensive conformational switch between an open and a closed state. This conformational change is contingent upon the presence or absence of substrates at the active sites, indicating a dynamic nature of CD73’s enzymatic activity3. Upon binding to AMP via the C-terminal domain, CD73 undergoes a conformational change, adopting a closed active state where the N-terminal domain and zinc cofactors align with the AMP to initiate the catalytic process, ultimately leading to the production of adenosine. Subsequently, the N-terminal domain undergoes a significant lateral rotation, opening the substrate-binding site to release the product upon completion of the catalytic reaction3. Small-molecular inhibitors typically target the active catalytic pocket of CD73. On the other hand, blocking antibodies tend to bind to the intra-molecular or inter-molecular N-terminal chains of the CD73 homodimer, preventing it from adopting a “closed” conformation. The latter strategy is utilized by several therapeutic antibodies, including Oleclumab (MEDI9447), which is currently in clinical Phase 3 development by AstraZeneca16.
Recent studies have highlighted that anti-CD73 biparatopic antibodies exhibit enhanced enzyme inhibition and improved tumor control17,18. However, the molecular basis underlying the superiority of this approach over conventional mAbs remains unclear. In this work, we employ an antibody cocktail strategy, which has proven effective against SARS-CoV-2 infections19. Accordingly, we select two humanized anti-CD73 mAbs targeting distinct, non-overlapping epitopes to create a specific cocktail. Our findings suggest that, when compared to individual mAbs, the cocktail exhibits superior enzyme inhibition in vitro and more effective tumor control in vivo, regardless of the adaptive immune system presence. Furthermore, by combining cryo-electron microscopy (EM), hydrogen-deuterium exchange mass spectrometry (HDX-MS), mutagenesis techniques, and molecular simulations, we elucidate the structure of a ternary complex wherein a CD73 dimer is bound by the Fabs of HB0038 and HB0039, offering molecular insights into the enhanced efficacy of this cocktail.
Results
Identification and humanization of mAbs targeting CD73
By utilizing the hybridoma technique, we generated a substantial repertoire of ~3000 mAbs targeting CD73. These antibodies underwent thorough sequence liability identification, CD73-based binding assays, and enzymatic activity assays. Antibody sequences have been selected based on the fulfillment of the following criteria: (1) Absence of potential degradation sequence motifs, such as NG or DG, which are prone to deamidation of asparagine and isomerization of aspartic acid, respectively; (2) Capability to bind to both human and monkey CD73 antigens with comparable affinity, facilitating subsequent toxicological studies in monkeys; (3) The binding kinetics parameters satisfy specific criteria, with an equilibrium dissociation constant (‘binding constant’, KD) lower than 5 × 10−9 and a dissociation rate (‘off rate’, kd) lower than 5 × 10−3, as determined by surface plasmon resonance (SPR) assays; (4) Both the half-maximal effective concentration (EC50) for binding and the half-maximal inhibitory concentration (IC50) for enzymatic inhibition is below 100 nM; (5) The melting temperature exceeds 65 °C, and the temperature of aggregation is above 68°C by analyzing protein stability. The sequences that met these conditions were applied to epitope binning analysis for classification. From these extensive screening analyses, we successfully identified two highly promising therapeutic candidate mAbs, HB0038 and HB0039, alternatively termed 38 and 39, which specifically target the CD73 antigen derived from hybridoma cells (Supplementary Fig. 1A).
Given that CD73 antigen is expressed on various normal cell tissues as well as immune cells, it may cause antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity activities. Therefore, two mutations (Leu234A and Leu235A) were introduced into the heavy chain Fc regions of both HB0038 and HB003920. CD73 can also undergo cleavage, resulting in the release of a soluble form that circulates in the bloodstream. Recent studies have shown that certain CD73-specific antibodies can selectively inhibit the enzymatic activity of membrane-bound CD73 on cancer cells and their derived extracellular vesicles, without affecting the enzymatic activity of soluble CD7321,22. Of note, Oleclumab has been shown to inhibit both forms of CD73’s enzymatic activity16.
The binding and functional characterization of HB0038 and HB0039 targeting CD73
To evaluate the binding activity towards membrane-bound CD73, we initially generated CHO-K1 cells with high expression of human CD73. Subsequently, flow cytometry was utilized to assess their binding to CHO-K1-hCD73 cells. The EC50 values derived from the dose-response curves were 11.75 nM for HB0038 and 0.24 nM for HB0039 (Fig. 1A). Thus, the EC50 values of HB0038 and HB0039 differed by approximately 50-fold in a cell membrane-bound CD73 binding assay.

A Flow cytometry analysis of HB0038 and HB0039 binding to CHO-S cells stably overexpressing human CD73 protein. For clarity, “38” refers to HB0038 and “39” refers to HB0039 in subsequent references. Results represent two technical replicates from one of three independent experiments; data are presented as mean values ± SD. B Soluble CD73 activity inhibition by HB0038, HB0039, HB0045, Oleclumab, Mupadolimab, and AK119. Inhibition efficiency was calculated using Eq. 1 as described in the Methods section. Results represent three technical replicates from one of three independent experiments; data are presented as mean values ± SD. C Membrane-bound CD73 activity inhibition by HB0038, HB0039, HB0045, Oleclumab, and lgG control. Inhibition efficiency was calculated using Eq. 2 as described in the Methods section. mCD73: membrane-bound CD73. Results represent two technical replicates from one of three independent experiments; data are presented as mean values ± SD. D CD73 activity inhibition by HB0038, HB0039, and HB0045, CD73MIX indicates a mixture of soluble and membrane-bound CD73. Inhibition efficiency was calculated using Eq. 3 as described in the Methods section. Results represent two technical replicates from one of three independent experiments; data are presented as mean values ± SD. E, F Antibody-mediated proliferation in CD4+ and CD8+ T cells with dose-dependent HB0038, HB0039, or HB0045. Stim: anti-CD3 antibody + anti-CD28 antibody + IL-7. Workflow refers to Supplementary Fig. 2A. Results represent two technical replicates from one of three independent experiments; data are presented as mean values ± SD.
Potent inhibition of CD73 enzymatic activity is reflected by a combination of high maximum enzymatic inhibition at saturating antibody concentrations and low IC50 values. Our screening process revealed that HB0038 and HB0039 likely target distinct epitopes on CD73, suggesting a potential synergistic or additive effect through their combined usage. Thus, we decided to explore the potential differences between HB0038, HB0039, and their combined forms in terms of inhibiting both the soluble and membrane-bound CD73 enzymatic activity. HB0038 demonstrated a higher maximum enzymatic inhibition at saturating antibody concentrations and a lower IC50 value for soluble CD73 compared to HB0039 (Fig. 1B and Supplementary Fig. 1E). Conversely, HB0039 displayed a higher maximum enzymatic inhibition at saturating antibody concentrations and a lower IC50 value for membrane-bound CD73 when compared to HB0038 (Fig. 1C and Supplementary Fig. 1F). Although HB0038 and HB0039 exhibited different levels of enzymatic inhibition towards soluble and membrane-bound CD73, they each inhibited the growth of MC38 colorectal tumors—genetically engineered to overexpress human CD73–to a similar extent in mice with humanized CD73 and CD39 expression (Supplementary Fig. 1B–D). Upon investigating various combinations of HB0038 and HB0039, we found that all tested combinations exerted a similar effect (Supplementary Fig. 1E, F). However, considering the pragmatic considerations for industrial production, we chose to proceed with the 1:1 ratio, designated as HB0045, or alternatively represented as 45.
HB0045 combines parental antibodies HB0038 and HB0039 for superior enzymatic inhibition
In further detail, regarding the enzymatic inhibition of soluble CD73, HB0039, Oleclumab (generated in-house), Mupadolimab (anti-CD73 antibody developed by Corvus, generated in-house), and AK119 (anti-CD73 antibody developed by Akeso, generated in-house), exhibited a “hook effect” when present in molar excess relative to the CD73 homodimer, despite HB0039 showed obvious “hook effect” only at higher concentration (Fig. 1B and Supplementary Fig. 1E). This phenomenon has previously been reported with Oleclumab16, suggesting that a bivalent binding model is necessary to inhibit the function of soluble CD73 for these antibodies. Gratifyingly, both HB0038 and HB0045 did not exhibit the hook effect and demonstrated superior maximal enzymatic inhibition at higher concentrations, indicating that these two antibodies employed a different binding and regulatory mechanism towards soluble CD73 (Fig. 1B and Supplementary Fig. 1E). In terms of the enzymatic inhibition of membrane-bound CD73, HB0039, HB0045, and the benchmarked antibody Oleclumab displayed comparable IC50 values for inhibiting the enzymatic activity of membrane-bound CD73, which were 0.53 nM, 1.04 nM, and 0.41 nM, respectively. However, HB0038 exhibited a significantly higher IC50 value of 65.27 nM, indicating substantially poorer inhibitory efficacy compared to HB0039 and HB0045 (Fig. 1C).
Next, we investigated enzyme inhibition in the presence of both soluble and membrane-bound CD73, a condition that closely mimics physiological conditions23. In such a setting, we found that the maximum rates of enzymatic inhibition achieved by HB0038, HB0039, and HB0045 against mixed CD73 were ~88%, 56%, and 100%, respectively. The IC50 values for HB0038 and HB0045 inhibiting the enzymatic activity of the CD73 were 43 nM and 1.3 nM, respectively (Fig. 1D). Taken together, HB0045 harnessed the advantages of both its parental antibodies and achieved a significantly better inhibition effect in vitro.
HB0045 mitigates the inhibitory effects of the adenosine pathway on immune cell proliferation in vitro
The accumulation of adenosine in the tumor microenvironment is known to have immunosuppressive effects by interacting with adenosine A2A receptors on immune cells. Adenosine can be generated from ATP or AMP through the CD39 and CD73 axes. Blocking CD73 is believed to prevent adenosine accumulation and alleviate the inhibitory effects of adenosine on T-cell proliferation and differentiation24. To assess the impact of HB0045, a CD4+ and CD8+ T cell proliferation assay was conducted in the presence of ATP (Supplementary Fig. 2A). CD8+ and CD4+ T cells exhibited around 5 divisions when stimulated by anti-CD3 and anti-CD28 antibodies. However, the presence of ATP significantly inhibited T-cell proliferation due to the conversion of ATP to adenosine by CD39 and CD73-expressing cells in PBMCs. Strikingly, HB0045 demonstrated the ability to alleviate the ATP-mediated inhibition of CD8+ and CD4+ T cell proliferation. This was evidenced by an increased number and ratio of proliferating CD8+ and CD4+ T cells (Supplementary Fig. 2B). Intriguingly, proliferation of CD8+ and CD4+ T cells in response to AMP was observed exclusively in the HB0045-stimulated group but not in the groups treated with HB0038, HB0039, or Oleclumab, even at concentrations up to 1000 nM (Fig. 1E–F and supplementary Fig. 2C, D).
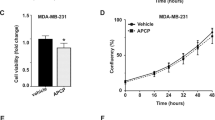
Independent of antibody-mediated enzymatic inhibition, antibody-mediated CD73 internalization has been reported to downregulate its levels on the cell surface, thereby inhibiting tumor growth11,25,26. To test whether HB0045 could mediate CD73 internalization, we used MDA-MB-231 cells, which have high endogenous CD73 expression, to assess CD73 levels. Interestingly, HB0045 exhibited significantly better efficiency than HB0038 and HB0039 alone (Supplementary Fig. 2E). However, it was difficult to evaluate internalization efficiency on CD4+ cells due to the low expression level of CD73 (Supplementary Fig. 2F, left panel). HB0045 and HB0039 demonstrated similar antibody internalization, while HB0038 showed very low antibody internalization on CD8+ cells (Supplementary Fig. 2F, right panel). These data suggest that HB0045-mediated internalization could efficiently decrease CD73 protein levels on the cell surface, particularly where CD73 is highly expressed, compared to its parental antibodies. However, this might not play a direct role in promoting T-cell proliferation. This mechanism may be even more relevant in the tumor microenvironment, where tumor cells with high CD73 expression are present.
Overall, these results provide compelling evidence that HB0045 can mitigate the inhibitory effects of the adenosine pathway on immune cells and stimulate immune cell proliferation.
HB0038 and HB0039 recognize different epitopes of CD73 in a non-competitive manner
To gain a comprehensive understanding of the mechanism of action employed by HB0038, HB0039, and HB0045, we utilized various complementary approaches to investigate their interactions with the CD73 homodimer. Initially, we performed HDX-MS to map the epitopes on the CD73 antigen that bind to HB0038 and HB0039. By comparing the exchange kinetics between free CD73 and CD73 complexed with HB0038, we identified two regions within the N-terminal domain of soluble CD73 (amino acids 143NIKAKGPLASQISGL157 and 178SKETPFLSNPGTNL191) and one region within the C-terminal domain of soluble CD73 (amino acids 381WNHVSM386). These regions displayed reduced deuterium uptake upon binding to HB0038. In contrast, only one region within the N-terminal domain of soluble CD73 (amino acids 159LPYKVLPVGDEVVG172) exhibited decreased deuterium uptake upon binding to HB0039 Fab (Supplementary Fig. 4). By mapping these epitope regions onto the crystal structure of CD73 (PDB code: 6TVG), we discovered that the binding region of HB0038 was in close proximity to the catalytic center of CD73, whereas the binding region of HB0039 was confined to the N-terminal region, distant from the catalytic center (Fig. 2A). This distinct binding pattern suggests that HB0038 and HB0039 may employ different mechanisms to inhibit the enzyme activity of CD73.

A Hydrogen-deuterium exchange (HDX) identified the epitopes of HB0038 and HB0039 on CD73. These were then mapped onto the CD73 structure (PDB code: 4H1Y) via PyMOL. B–K Time-resolved SPR analysis detailing affinity between HB0038 and CD73 wide type (WT) and its mutants. All CD73 residues identified by HDX analysis potentially involved in HB0038 binding were mutated to Ala individually and were assessed for their contribution to HB0038 binding. The SPR profile of HB0038 against specific CD73 WT and CD73 mutations in the HDX epitope study. L–Q As in (B–K), but between HB0039 and CD73.
To validate and refine the interface identified by HDX, we conducted alanine scanning mutagenesis of the residues within the epitopes of CD73 by SPR analysis. Both HB0038 and HB0039 antibodies had high binding affinities towards wide-type soluble CD73, with the equilibrium dissociation constant (KD) values of 3.59 × 10−10 and 5.71 × 10−10, respectively (Fig. 2B, L, and Supplementary Table 1). The introduction of mutations such as N143A, K145A, K147A, W381A, and N382A on CD73 significantly abolished the interaction between HB0038 and CD73 (Fig. 2C–K, and Supplementary Table 1). Similarly, mutations including L159A, K162A, and D168A on CD73 strongly attenuated the interaction between HB0039 and CD73. Furthermore, the V170A and K162A&V170A mutations almost completely abolished the interaction between HB0039 and CD73 (Fig. 2M–Q, and Supplementary Table 1). Taken together, the experimental results from the alanine scanning mutagenesis further confirmed the epitopes identified by HDX-MS and defined the main surfaces involved in HB0038 and HB0039 binding to CD73.
Negative staining EM analysis of the HB0038-HB0039-CD73 complex
To fully characterize the binding sites of HB0038 and HB0039 on CD73 and gain further structural insights into the mechanism by which these antibodies inhibit CD73 enzyme activity, we initially conducted negative staining EM analysis of the HB0038 (IgG)–HB0039 (IgG)–CD73 complex. We initially encountered several challenges as the samples precipitated during negative staining preparation and did not yield well-defined particles. We hypothesized that this phenomenon could be attributed to crosslinking between different particles via the (Fab)2 arms of either HB0038 or HB0039, as suggested by previous studies27.
To investigate this possibility, we then examined the inhibitory function of the isolated Fab from HB0038 or HB0039. Intriguingly, we discovered that the Fab of HB0038 could still partially inhibit CD73 activity. In contrast, the Fab of HB0039 completely lost its inhibitory activity (Fig. 3B), suggesting that HB0039 might exert its inhibitory function through an inter-dimer bridge. Given that the HB0038 Fab alone can retain its inhibitory function, we utilized the HB0038 (Fab)–HB0039 (IgG)–CD73 complex as samples for subsequent negative staining EM analysis.

A Visualization of the CD73 antigen (6TVG), Fab (1M71), and complete IgG (1IGY) docked within the electron density map derived from negative staining of the CD73-HB0039 IgG-HB0038 Fab complex. B Enzymatic assay results display the concentration-dependent inhibitory effects on soluble CD73 enzymatic activity by HB0038 IgG, HB0039 IgG, HB0038 Fab, and HB0039 Fab. Results represent two technical replicates from one of three independent experiments; data are presented as mean values ± SD.
In line with our hypotheses, the negative staining now showed that HB0038 (Fab)—HB0039 (IgG)-CD73 complex formed relative homogeneous particles. Interestingly, we observed that the HB0038 (Fab)—HB0039 (IgG)—CD73 complex could form a loop structure (Supplementary Fig. 5A). After multiple rounds of classification, we carefully selected relatively intact and well-defined particles for initial model building and subsequent refinement. Among the generated classes, Class 1 represented a classical “Y” shaped model of a full-length HB0039 IgG antibody. Notably, there were variations in the loop structure observed in different classes, including Class 2 and Class 3, with Class 2 being the predominant conformation (Supplementary Fig. 5B).
We successfully docked the structures of the CD73 dimer (PDB ID: 6TVG), the Fab structure representing the HB0038 Fab (PDB ID:1M71), and the full-length IgG structure representing the HB0039 IgG (PDB ID: 1IGY) into the EM density map of Class 2, resulting in the generation of a pseudo-atomic model for the HB0038 (Fab)–HB0039 (IgG)–CD73 complex (Fig. 3A). The docked structure revealed that two HB0039 IgG antibodies bound to two CD73 dimers, forming a loop structure. Simultaneously, the two HB0038 Fabs bound to the corresponding binding sites on the CD73 dimers. Notably, the loop structure observed in the HB0038 (Fab)—HB0039 (IgG)—CD73 complex was likely induced by the bivalent binding of the full-length HB0039 (IgG) to two different CD73 dimers simultaneously. Through its two Fab arms, HB0039 formed inter-dimer bridges between the two CD73 dimers, resulting in the formation of these oligomer particles. Of note, the flexibility of Fc made the HB0039 IgG’s segment indiscernible in the negative staining EM map. Drawing from this EM map of the HB0038 (Fab)–HB0039 (IgG)–CD73 (Fig. 3A), we provided a visual representation of the intact HB0038 (IgG)—HB0039 (IgG)—CD73 complex (Supplementary Fig. 5C), elucidating its propensity to precipitate.
Cryo-EM structural analysis combined with dynamic simulations unveils the cooperative mechanism of the HB0045 antibody cocktail
To gain further insights into the molecular mechanism underlying the inhibitory effect of HB0045 on CD73, we employed cryo-EM to solve the ternary structure of the CD73 extracellular domain complexed with HB0038 Fab and HB0039 Fab. The cryo-EM density map was refined to a resolution of 3.06 Å for model building and refinement (Supplementary Fig. 6 and Supplementary Table 2). In this structure, CD73 was observed as a dimer, with each monomer simultaneously interacting with one HB0038 Fab and one HB0039 Fab at distinct sites (Fig. 4A, B). The electron densities of CD73 and the fragment variable (Fv) domains of HB0038 and HB0039 were highly resolved (Supplementary Fig. 7A). However, the constant domain CH1 of the Fab was challenging to model accurately due to their inherent flexibility. In line with our HDX-MS findings, HB0038 exhibited simultaneous binding to the N-terminal and C-terminal regions surrounding the catalytic sites of CD73, while HB0039 was specifically bound to the N-terminal domain of CD73 alone (Fig. 4B).

A Cryo-EM visualization of the CD73-HB0038 Fab-HB0039-Fab complex. Within the CD73 dimer, one monomer is depicted in magenta while its counterpart is in purple. The fragment variable (Fv) domains of HB0039 are illustrated in varying shades of green, with one HB0038 Fv domain in blue (with the second in cyan). The CH1 domains for both HB0038 and HB0039 Fabs are represented in gray. B Cartoon representation of the CD73-HB0038 Fab-HB0039-Fab complex, colored as in (A). C Close-up views of the interaction interfaces: Interface I between HB0039 (in green) and CD73 (in magenta) and Interface II between HB0038 (in blue) and CD73. Interacting residues are depicted as sticks. D Comparative alignment of different CD73 conformations when bound to various antibodies (referenced antibodies omitted for clarity). CD73 conformations bound to TB38 and TB19 are shown in shades of cyan, while its conformation with HB0045 is represented in magenta. The fully open and fully closed conformation of CD73 were shown in gray and blue, respectively.
HB0039 interacts extensively with the N-terminal domain of CD73, spanning from R109 to N211, covering a significant surface area of ~1772 Å2. Notably, specific residues in HB0039, such as R30, S55, S57, K65, and Y104, engage in robust hydrogen bonding interactions with L132, G156, D168, E169, and Y110 on CD73 (Fig. 4C). HB0038, on the other hand, recognizes the regions that connect the N-terminal and C-terminal domains of CD73. One of the complementarity-determining region (CDR) loops of HB0038 inserts into a pocket between these two domains (Fig. 4C and Supplementary Fig. 7A). Of note, residues W50, Y103, and Y109 of HB0038 engage in hydrophobic interactions with L191, V192, F183, and P182 of CD73, respectively. Additionally, S52, S92, Y103, S106, and N108 of HB0038 form hydrogen bonds with E378, G148, E180, K147, and T189 of CD73, respectively (Fig. 4C). Through these intricate interactions, HB0045 locks CD73 in a distinct conformation referred to as a “partially open” state, which differs from the conformations bound by other antibodies or inhibitors (Fig. 4D and Supplementary Fig. 7C).
James E. et al. described a highly potent biparatopic CD73 antibody, with its two Fab arms originating from the parent antibodies TB19 and TB38, which have non-overlapping epitopes18 (Supplementary Fig. 7B). While they successfully obtained separate crystal structures of human CD73 in complex with both TB19 and TB38 Fabs, they could not elucidate how the combined action of TB19 and TB38 Fabs resulted in the enhanced potency of the biparatopic antibody18. To decipher the effect of HB0045 in comparison to HB0038 or HB0039, we employed molecular dynamic simulations. This approach allowed us to observe structural evolutions and specific shifts over time by measuring the root-mean-square deviation (RMSD) and determining the movement extent of particular residues through the root-mean-square fluctuation (RMSF). Armed with the ternary structure of the CD73-HB0038 (Fab)-HB0039 (Fab) complex, we conducted a molecular dynamics simulation study based on this structure. Our simulation experiments revealed that the CD73 backbone, when bound to the HB0045, exhibited the lowest RMSD compared to its apo form or when bound with either HB0038 or HB0039 (Supplementary Fig. 7D). Furthermore, the RMSF of the hinge region and the C-terminal domain of CD73, both crucial for catalytic activity, showed a significant reduction when bound with the HB0045 compared to other states (Supplementary Fig. 7E). Such findings suggest that the CD73-HB0045 complex adopts a conformation that is markedly more stable than the CD73-HB0038 or CD73-HB0039 complex.
These findings provide compelling structural evidence that the combination of HB0038 and HB0039 antibodies can effectively stabilize the CD73 dimer in a distinct and stable “partially open” conformation. Importantly, this conformation renders CD73 inactive for catalytic activity, leading to a potent inhibition of CD73 enzyme activity. By targeting different epitopes and working in concert, HB0038 and HB0039 antibodies achieve a remarkable inhibitory effect on CD73, highlighting the potential of this antibody combination as a therapeutic strategy.
The HB0045 antibody cocktail exhibits potent antitumor effects in various tumor models by regulating immunosuppression
Here we showed that the HB0045 antibody cocktail inhibits CD73 activity and also promotes T-cell proliferation in vitro. Additionally, through negative staining and Cryo-EM techniques, we determined that HB0045 exerts its inhibitory activity by locking CD73 into a “partially open” state. However, the extent to which HB0045 impacts tumor growth inhibition in vivo remains to be validated.
Triple-negative breast cancer (TNBC) is a subset of breast cancer characterized by the absence of hormone receptors and HER2 expression. TNBC is known for its aggressive nature, poor clinical outcomes, and limited treatment options28. TNBC’s response to treatment was investigated in a subcutaneous MDA-MB-231 xenograft model employing immunodeficient NPG mice, which are conducive to human PBMC cell transplantation (Fig. 5A). We evaluated tumor growth inhibition by comparing the efficacy of the HB0045 antibody cocktail to that of HB0038, HB0039, and Oleclumab. As expected, all anti-CD73 antibodies resulted in significantly smaller tumor volumes compared to the control IgG-treated group (Fig. 5B). Remarkably, HB0045 exhibited a significantly enhanced inhibition of tumor growth compared to its parental mAbs, HB0038 and HB0039. This result highlights the enhanced antitumor effects of combining HB0038 and HB0039 in the HB0045 antibody cocktail. Crucially, all three treatment groups (HB0045, HB0038, and HB0039) outperformed the Oleclumab group in terms of antitumor efficacy. This inhibitory effect on tumor weight was consistent with the observations from tumor volume measurements, while the overall weight of these mice did not change significantly (Fig. 5B and Supplementary Fig. 8F). As strong tumor inhibition was achieved at a dose of 3 mg/kg of HB0045, we performed an additional experiment with increased doses of Abs (5 mg/kg for HB0045, HB0038, HB0039, and Oleclumab). In this setting, HB0045 (5 mg/kg, equivalent to 2.5 mg/kg each of HB0038 and HB0039) displayed significant inhibition of tumor growth, whereas HB0038 or HB0039 at a dose of 5 mg/kg only achieved moderate inhibition of tumor growth (Supplementary Fig. 8E). The results reinforced our view that the HB0045 antibody cocktail has better tumor inhibition than either of two mAbs alone.

A Workflow illustrating CD73 antibody treatment across different tumors in PBMC-humanized NPG-immunodeficient mice. “I.P.” denotes intraperitoneal injections; “BIW” indicates administration twice weekly. It was created in BioRender. Zhifeng, Y. (2024) https://BioRender.com/r91t302. B Impact of varying anti-CD73 antibody doses on MDA-MB-231 tumor growth and weight in PBMC-humanized NPG mice. Once tumors reached an average size of 50 ~ 100 mm³, mice bearing tumors were grouped randomly into six segments (n = 8) and administered i.v with 1 × 107 PBMCs. Treatments were inclusive of the IgG negative control (3 mg/kg), in-house Oleclumab (1 mg/kg), HB0038 (1 mg/kg), HB0039 (1 mg/kg), and HB0045 at two dosages (1 mg/kg and 3 mg/kg). Treatment commenced on the same day with bi-weekly intraperitoneal antibody dosing for 4 weeks. P_volume (38 1 mg/kg vs 45 1 mg/kg) = 0.0087; P_volume (Oleclumab 1 mg/kg vs 45 1 mg/kg) <0.0001; P_volume (Oleclumab 1 mg/kg vs 45 3 mg/kg) <0.0001; P_weight (Oleclumab 1 mg/kg vs control 3 mg/kg) <0.0001; P_weight (Oleclumab 1 mg/kg vs 45 1 mg/kg) = 0.0141; P_weight (45 1 mg/kg vs 45 3 mg/kg) = 0.004. C NPG mice received a subcutaneous inoculation of BxPC-3 (5 × 106) in Matrigel on their right flank. Upon tumors achieving an average size of 68 mm³, mice with tumors were grouped randomly into four segments (n = 6) and administered i.v with 1 × 107 PBMCs. Treatments began on day one, with bi-weekly intraperitoneal antibody dosing spanning four weeks. P < 0.0001. D The HB0045 antibody cocktail (anti-CD73), coupled with the HB0025 antibody (anti-PD-L1/VEGF bispecific antibody) and HB0027 (anti-4-1BB antibody), demonstrated potent antitumor capabilities in the A375 melanoma models using PBMC-humanized NPG mice (n = 6). Unless otherwise specified, the concentration of antibody used in samples was 1 mg/kg. P_volume (Vehicle vs 45) < 0.0001; P_volume (45 vs 25 + 45) = 0.0013; P_volume (25 + 45 vs 25 + 45 + 27) = 0.008; P_weight (Vehicle vs 45) < 0.0001; P_weight (27 vs 25 + 45 + 27) = 0.0054. Prism was employed to compute P values for tumor volumes using two-way ANOVA coupled with Tukey’s multiple-comparison test. For tumor weights, P values were determined using a one-way ANOVA. *P < 0.05, **P < 0.01, ****P < 0.0001. Data are presented as mean values ± SEM.
Despite notable advancements in diagnosis and treatment methods, pancreatic cancer remains a highly aggressive gastrointestinal malignancy associated with a very poor prognosis29. Importantly, metabolic enzyme screenings have revealed that overexpression of CD73 in pancreatic ductal adenocarcinoma is associated with an immunosuppressive tumor microenvironment. Furthermore, pancreatic ductal adenocarcinoma with poor survival exhibits elevated expression of CD73 and increased interstitial adenosine levels, particularly in highly hypoxic microenvironments30. To investigate the potential anti-pancreatic cancer effect of the HB0045 antibody cocktail, we conducted an evaluation of the antitumor effects of HB0038, HB0039, and HB0045, using a subcutaneous BxPC-3 xenograft model in PBMC-humanized NPG mice. The results revealed that administration of HB0038, HB0039, and HB0045 at a dose of 5 mg/kg significantly reduced tumor volume compared to the negative control group at the experimental endpoint (Fig. 5C). The tumor growth inhibition (TGI) rates for tumor volume were 57.83%, 59.60%, and 69.77%, respectively, as corroborated by the tumor images. Of note, the inhibitory effect on tumor weight in the HB0038, HB0039, and HB0045 groups was different from that of the negative control group, without a significant change in the mice’s weight between groups (Fig. 5C and Supplementary Fig. 8G).
The combination of anti-PD-L1 and anti-CD73 antibodies has shown significant inhibition of EGFR-mutated non-small cell lung cancer tumor growth, accompanied by an increase in tumor-infiltrating CD8 + T cells and enhanced production of IFN-γ and tumor necrosis factor (TNF) by CD8 + T cells31. In contrast, neither anti-PD-L1 nor anti-CD73 antibody alone exhibited tumor growth inhibition in a xenograft mouse model31. Additionally, it has been reported that CD73 plays an inhibitory role in agonistic anti-4-1BB/CD137 Ab therapy, a member of the TNF receptor family. In agreement with this, the concurrent administration of anti-CD73 and anti-4-1BB antibodies synergistically increased the number of effector CD8 + T cells and promoted tumor regression32. Given that we have developed bispecific antibody, HB0025, targeting PD-L1/VEGFR33, and an anti-4-1BB-agonistic antibody, HB0027, we sought to investigate the antitumor effects of HB0045 in combination with these two antibodies in preclinical-animal models. In our study, administration of HB0025, HB0027, and HB0045 led to a significant reduction in tumor volume compared to the vehicle group at the experimental endpoint, with TGI rates of 57.48%, 51.08%, and 54.45%, respectively (Fig. 5D). Notably, the combination of HB0025 and HB0045, as well as the combination of HB0027 and HB0045, exhibited significantly enhanced inhibition of tumor expansion compared to HB0045 alone, with TGI values of 66.47% and 68.23%, respectively. Furthermore, the combination of HB0025, HB0027, and HB0045 further increased the TGI to 78.30%, and the difference was statistically significant compared to the two-drug combination groups (Fig. 5D). The weight of these mice did not change significantly (Supplementary Fig. 8H).
We also evaluated the pharmacokinetics properties of HB0038, HB0039, and HB0045 antibody cocktail. As shown in Supplementary Fig. 8I–K and Supplementary Table 3, at doses of 5 mg/kg, the half-lives (t1/2) of HB0038, HB0039, and HB0045 in immunodeficient NPG mice34, which lack T cells, B cells, and NK cells, were about 446 hours, 352 hours, and 434 hours, respectively. The Cmax of HB0038, HB0039, and HB0045 were about 46601, 45115, and 48896 ng/ml, respectively. These data indicated that the half-lives and maximum serum antibody concentrations were comparable between the antibody cocktail and its parental antibodies.
HB0045 directly inhibits tumor growth in immunodeficient mice models devoid of T cells, B cells, and NK cells
CD73 is widely recognized for its role as a key extrinsic immunomodulator, producing adenosine that exerts an immunosuppressive effect within the tumor microenvironment. Recent studies, however, have also highlighted its intrinsic properties within tumor cells, which are proving to be as significant as its extrinsic roles35.
An earlier study demonstrated that CD73 antibody therapy moderately inhibited tumor growth in the E0771 breast model, but not in the 4T1.1 model, when tested in severe-combined immunodeficient mice8. Further research involving genetic depletion of CD73 in various human cancer cell lines grown in immunodeficient mice has shown reductions in both tumor growth and metastatic potential36,37. Given these findings, we hypothesized that the effectiveness of CD73 inhibition may vary based on antibody efficacy and tumor model specifics. To this end, we decided to test our HB0045 antibody in a HepG2 liver cancer model using immunodeficient mice.
Our data indicate that HB0045 promotes the proliferation of T cells (Fig. 1E, F, Supplementary Fig. 2A–D). To investigate HB0045’s effect on tumor growth independent of these cells, we utilized an immunodeficient NCG mouse model, which also lacks T, B, and NK cells. We then established HepG2 human liver cancer cell-derived xenografts, within this model, to evaluate the anticancer activity of HB0045 (Fig. 6A). Treatment with HB0045 significantly alleviated tumor-associated weight loss (Fig. 6B), suppressed tumor growth (Fig. 6C), and extended overall mice survival rates (Fig. 6D). These results underscore HB0045’s potential to inhibit tumor progression even without T, B and NK cells, thereby emphasizing the critical role of CD73’s intrinsic properties in tumor cells. Of note, although NCG mice lack mature T, B, and NK cells, they retain myeloid cells. Therefore, we cannot exclude the possibility that CD73 may influence myeloid cells in the regulation of tumor growth.

A Treatment schematic depicting HB0045’s action against HepG2 subcutaneous tumor growth in immunodeficient NCG mice (n = 6). “I.P.” denotes intraperitoneal injections; “BIW” indicates administration twice weekly. It was created in BioRender. Zhifeng, Y. (2024) https://BioRender.com/r91t302. B Corresponding to (A), changes in body weight throughout the duration of HB0045 treatment. C Corresponding to (A), Trajectory of tumor volume during HB0045 treatment. D Corresponding to (A), Survival outcomes for mice following HB0045 administration, survival analysis was calculated using the Kaplan–Meier method and log-rank test. P = 0.0009. E Strategy for employing HB0045 to counteract pancreatic cancer in a PDX model (n = 6). It was created in BioRender. Zhifeng, Y. (2024) https://BioRender.com/r91t302. F In alignment with (E), monitoring of tumor volume, complemented by representative tumor images, throughout HB0045 treatment in the pancreatic cancer PDX model. P_volume (Vehicle vs 45 20 mg/kg)=0.0378; P_volume (Vehicle vs. 45 40 mg/kg) <0.0001. Prism was employed to compute P values for tumor volumes using two-way ANOVA coupled with Tukey’s multiple-comparison test. *P < 0.05, ***<0.001, ****P < 0.0001. Data are presented as mean values ± SEM.
Crucially, to assess the efficacy of HB0045 in a clinically relevant setting, we finally utilized patient-derived xenograft (PDX) model of pancreatic cancer in immunodeficient C-NKG mice, which are similar to NPG or NCG mice lacking mature T, B, and NK cells (Fig. 6E). Remarkably, HB0045 treatment resulted in dose-dependent tumor regression (Fig. 6F). Overall, the results from these experiments underscore the dual role of CD73 in both immunosuppression and tumor cell-intrinsic processes, and support the therapeutic potential of HB0045 in targeting CD73 for cancer treatment.
Discussion
In this study, we have developed an antagonistic CD73 antibody cocktail, HB0045, composed of HB0038 and HB0039. Our findings indicate that HB0038 binds selectively when the N-terminus and C-terminus of CD73 form a specific conformation. In contrast, HB0039 can be associated with CD73 irrespective of its conformation. We also found that HB0038 exhibited a superior inhibitory effect on soluble CD73 enzyme activity without the hook effect, while HB0039 showed a greater inhibitory effect on membrane-bound CD73 enzyme activity. Encouragingly, HB0045 retained the beneficial properties of both parental antibodies.
A number of studies have demonstrated that CD73 antibodies can alleviate ATP/AMP-mediated inhibition of T cell proliferation17,38,39,40. To evaluate whether HB0045 exhibits superior efficacy in promoting T cell proliferation compared to its parental antibodies, we used PBMCs, which comprise 45-70% T cells41, for testing. Our results indicated that HB0045 significantly enhanced T cell proliferation (Fig. 1E and F, and Supplementary Fig. 2C and D). Interestingly, Oleclumab, HB0038, and HB0039 alone showed minimal activity in this system. This could be attributed to the system’s threshold for restoring CD73-imposed suppression by anti-CD73 antibodies. HB0045 demonstrated superior immune restoration, likely due to enhanced enzyme inhibition and CD73 down-regulation through antibody-mediated internalization. Additionally, our T cell proliferation assays were conducted using human PBMCs containing myeloid cells. Myeloid cells are known to contribute to CD73-imposed immune suppression42. It is of interest to investigate whether HB0045 affects these immune cells to restore adenosine-mediated immune suppression in the future.
It is well established that the N-terminus and C-terminus of CD73 undergo dynamic rotations between open and closed conformations. From our assessments, we discerned that HB0045 enforces a “double lock” on CD73. Specifically, HB0039 stabilizes CD73 via inter-dimer bridges, crafting a loop structure (Fig. 3A). Concurrently, HB0038 can engage with both the N-terminus and C-terminus of CD73, adding a secondary locking mechanism that inhibits its mobility (Fig. 4). We postulate that this dual-locking strategy is pivotal for the higher efficacy of HB0045, when compared to HB0038 or HB0039 alone. Molecular simulations based on the cryo-EM structure revealed that HB0045 potently inhibits CD73 enzyme activity by stabilizing a unique “partially open” conformation of the CD73 dimer (Supplementary Fig. 7D, E).
Importantly, HB0045 exhibited enhanced enzymatic inhibition potency towards both soluble and membrane-bound CD73 compared to its individual parental antibodies, particularly when both soluble and membrane-bound CD73 was present (Fig. 1D). These improved in vitro activities translated into favorable in vivo anticancer effects in multiple animal models. Specifically, we observed an enhanced antitumor effect of HB0045 in both TNBC and pancreatic cancer models (Fig. 5B, C). Furthermore, we demonstrated that HB0045 exhibited tumor growth suppression in immune-deficient mouse models, which lack T cells, B cells, and NK cells. These findings highlight the potential significance of the tumor cell-intrinsic properties of CD73 in addition to its role in immune suppression. Of note, these immune-deficient mice still retain myeloid cells. Therefore, we cannot exclude the possibility that CD73 may influence myeloid cells in the regulation of tumor growth. In conclusion, we believe that HB0045 can inhibit tumor growth through mechanisms that are dependent or independent of T cells, B cells, and NK cells, as further evidenced by additional mouse data (Supplementary Fig. 8A–D).
In summary, our investigations collectively demonstrate that HB0045-an antibody cocktail targeting different CD73 epitopes and exhibiting distinct mechanisms of action- offers a promising approach for developing improved therapies for diseases associated with abnormal CD73 overexpression, such as various malignant tumors.
Methods
Ethics and permission
This study complies with all relevant ethical regulations. All in vivo studies were performed according to the Regulations for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees of Biocytogen Pharmaceutical (Beijing) Co., Pharmalegacy Co., and Nanjing Medical University, respectively. Figures 5A, 6A, E are created in BioRender. Zhifeng, Y. (2024) https://BioRender.com/r91t302. Supplementary Figs. 1A, B, and 2A created in BioRender. Zhifeng, Y. (2024); https://BioRender.com/g11k473.
Use of cell lines, mice, and human PBMC cells
The TNBC cell line MDA-MB-231 and human melanoma cell line A375 were obtained from the Chinese Academy of Sciences. The human pancreatic cancer cell line BxPC-3 was purchased from Cobioer Co., Ltd. The human hepatocellular carcinoma cell line HepG2 was purchased from ATCC. The mouse MC38 colorectal cancer cell line overexpressing human CD73 (B-hCD73 MC38) was purchased from Biocytogen Co., Ltd. The CHO-K1 cell line overexpressing human CD73 (CHO-K1-huCD73-3F4) was prepared in-house. No commonly misidentified cell lines were used in the study, and they were properly authenticated by commercial companies with STR profiling or Karyotyping assay. The immunodeficient Prkdcscid Il2rgnull (NPG) mice were purchased from Beijing Vitalstar Biotechnology Co., Ltd. The immunodeficient NOD-Prkdcem26Cd52Il2rgem26Cd22/NjuCrl (NCG) mice were purchased from GemPharmatech Co., Ltd. The immunodeficient NOD-Prkdcscid Il2rgem1/Cyagen (C-NKG) mice were purchased from Cyagen Biosciences Inc. The CD39/CD73 humanized (B-hCD73/hCD39) C57BL/6 mice were purchased from Biocytogen Co., Ltd. Mouse-related experiments were carried out with the approvals of Institutional Animal Care and Use Committee of Biocytogen Pharmaceutical (Beijing) Co. (IACUC Number: BAP-BJ-PS-01-2110147 for B-hCD73 MC38 tumor model in Fig. S1B), Pharmalegacy Co. (IACUC number: PL20210105-1 for MDA-MB-231, BxPC-3 and HepG2 tumor models in Figs. 5B, C and 6B respectively; PL220119-3 for A375 tumor model in Fig. 5D; PL230131-2 for MDA-MB-231 in Fig. S8A), and Nanjing Medical University (IACUC number: IACUC-1706003 for pancreatic PDX tumor model in Fig. 6F). These institutes had been fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). The tumor cell lines and mouse strains used in this study were listed in supplemental materials. All the operations and techniques used on these laboratory animals conformed with the rules of laboratory animal welfare and ethics and the “3 R principles”. All the laboratory animals received humanitarian care throughout the whole research process. For human PBMCs, ethics was approved by the Ethics Committee of Taicang First People’s Hospital (Approval Scheme No: 2022-SR-006).
Use of antibodies and proteins
The sequences of the anti-CD73 antibodies oleclumab, mupadolimab, and AK119 were obtained from patents US20180194858A1, US20190077873A1, and CN113527489A, respectively. Heavy and light chain genes were cloned into the expression vector derived from pcDNA3.1, expressed using the CHO-S or CHO-K1 system, and the proteins were purified by protein A chromatography (Cytiva). The sequences of anti-CD73 antibodies HB0038 and HB0039, anti-PD-L1/VEGFR bispecific antibody, anti-4-1BB antibody HB0027 were prepared in-house. IgG1 isotype control with L234A and L235A mutation at Fc domain was used. The wild-type CD73 antigen was purchased from ACRO Biosystems. The CD73 mutant antigens were prepared by Novoprotein Scientific Inc.
Generation, expression, and purification of HB0038, HB0039 and HB0045
BALB/c mice were immunized with recombinant human CD73 protein (Sino Biological, HPLC-10904-H08H), and spleen cells were isolated and fused with SP2/0 myeloma cells for generation of hybridomas according to standard methods43. Soluble and membrane-bound CD73-based binding assay and enzymatic activity inhibition functional assay, epitope binning assay were used to screen the hybridomas and rank hits order. Potential variable gene sequences were recovered from hybridomas using SMART 5’RACE kits (634859, Clontech Laboratories) according to manufacture methods, and chimeric antibodies with human IgG1 Fc-silent (L234A&L235A double mutation) were generated and characterized. The 3A9 and 124A9 clones with different binding epitopes were selected for humanization. Sequences of the heavy chain and light chain for humanization were designed independently, wherein the mouse amino acid sequences in the framework regions were replaced with the human germline template with the highest homology, and back mutations were made to maintain its binding potency equivalent to the corresponding chimera. Finally, the humanized anti-CD73 antibody for the 3A9 clone was named HB0038, and the humanized anti-CD73 antibody for the 124A9 clone was named HB0039.
HB0038, HB0039 were overexpressed in CHO-S or CHO-K1 cells, and the harvested cell culture fluid containing antibodies was purified by protein A column (Mab, SelectSuRe, Cytiva), then the purified antibodies were subjected to anion exchange chromatography (Poros XQ resin, Thermo Fisher) for further purification. The HB0045 antibody cocktail was prepared by mixing HB0038 and HB0039 in a ratio of 1:1.
Binding kinetics and affinity via SPR binding assay
The SPR study was performed using the Biacore 8 K™ system (Cytiva, Sweden). The sample compartment and flow cell temperature of the Biacore 8 K system was set at 25 °C and the data acquisition rate was 10 Hz. HB0038, HB0039 antibodies were captured on the Series S Sensor Chip Protein A (29127556, Cytiva) as ligands at about 60 response units (RU), and the wide-type (CD3-H52H7, ACRO Biosystems) or mutant CD73 antigens with a C-terminal 6XHis tag at an initial concentration of 100 nM were serially 2-fold diluted with the HBS-EP running buffer (BR100669, Cytiva) as analytes. The analytes were injected for 120 seconds (s) and dissociated for 600 s at a rate of 50 μL/min. After each cycle, the sensor surface was regenerated with 0.1 M glycine (BR100354, Cytiva), pH 1.5. Multi-cycle kinetics were applied to the assay. Binding kinetics were calculated using Biacore Insight Evaluation Software (Version 3.0.12.15655). The original data were evaluated in a 1:1 binding model with a fitted local kinetics model.
Cell membrane-bound CD73 binding assay
CHO-K1 cell line stably expressing full-length CD73 was constructed (CHO-K1-CD73-3F4) and used in FACS analyses for testing anti-CD73 Abs binding. For CD73 binding assay, CHO-K1-CD73-3F4 cells were seeded at 2 × 104 cells per well in a 96-well U plate. The plate was centrifuged at 300 × g for 3 min and discarded the supernatant. Cells were then suspended in serially diluted antibodies and incubated at room temperature for 30 min. The antibody was diluted serially in a three-fold ratio with an initial concentration of 200 nM. After that, cells were washed with 1% BSA (A600332-010, Sangon) in PBS (B320KJ, BasalMedia) twice, followed by staining with PE-conjugated antibody Goat Anti-Human Fcγ (109-115-098, Jackson) and incubated at room temperature for 15 min. The cells were then washed with 1% BSA in PBS again and analyzed by Flow cytometry. Four-parameter fitting was performed with the MFI vs antibodies working concentration using GraphPad Prism software, and the dose-effect relationship and EC50 were determined.
HDX detected by MS
5 mM purified antigen, antibody or complex of antigen and antibody (molar ration 1:1) were incubated in buffer (50 mM HEPES, pH 7.5, 50 mM NaCl, 5% glycerol, 4 mM MgCl2, 2 mM DTT) for 1 hour before the HDX reactions at 4°C. 4 μL samples were then diluted into 16 μL D2O on exchange buffer (50 mM HEPES, pH 7.5, 50 mM NaCl, 2 mM DTT) and incubated for various HDX time points (e.g., 0, 10, 60, 300, 900 s) at 4°C, then quenched by mixing with 20 μL of ice-cold 3 M guanidine HCl and 1% trifluoroacetic acid. Each quenched sample was immediately injected into the LEAP Pal 3.0 HDX platform. Upon injection, samples were passed through an immobilized pepsin column (2 mm × 2 cm) at 120 μL/min, and the digested peptides were captured on a C18 PepMap300 trap column (Thermo Fisher Scientific) and desalted. Peptides were separated across a 2.1 mm × 5 cm C18 separating column (1.9 μm Hypersil Gold, Thermo Fisher Scientific) with a linear gradient of 4%—40% CH3CN and 0.3% formic acid over 6 min. Sample handling, protein digestion, and peptide separation were conducted at 4°C. MS data were acquired using a Fusion Orbitrap mass spectrometer (Thermo Fisher Scientific) with a measured resolving power of 65,000 at m/z 400. HDX analyses were performed in triplicate, with single preparations of each complex of antigen and antibody. The intensity-weighted mean m/z centroid value of each peptide envelope was calculated and subsequently converted into a percentage of deuterium incorporation. Statistical significance for the differential HDX data is determined by an unpaired t-test for each time point, a procedure that is integrated into the HDX Workbench software44. Corrections for back-exchange were made based on an estimated 70% deuterium recovery, and accounting for the known 80% deuterium content of the deuterium exchange buffer.
The HDX data from all overlapping peptides were consolidated to individual amino acid values using a residue averaging approach. Briefly, for each residue, the deuterium incorporation values and peptide lengths from all overlapping peptides were assembled. A weighting function was applied in which shorter peptides were weighted more heavily and longer peptides were weighted less. Each of the weighted deuterium incorporation values was then averaged to produce a single value for each amino acid. The initial two residues of each peptide, as well as prolines, were omitted from the calculations45.
Blockade of recombinant soluble CD73 enzymatic activity
Anti-CD73 antibodies were diluted serially with 1× TM buffer (MB-059, Rockland) by a dilution factor of 1.75 with different starting concentration (Fig. 1B: 102 nM; Supplementary Fig. 1E: 133 nM), and the recombinant CD73 (CD3-H52H7, ACRO Biosystems) was also dissolved and diluted to 1 μg/ml with TM buffer, then the 40 ul diluted antibodies and 40 μl antigen were mixed, and added to a 96-well plate (136101, Thermo Fisher). The plate was incubated at 20°C for 30 min. The CD73 enzyme substrate was prepared with 200 μM ATP and 600 μM AMP in a 1:1 ratio by volume. 80 μL substrate solution was added to a 96-well plate and reacted for 10 minutes at 20 °C. Finally, 90 μL of CellTiter-Glo® solution (G7573, Promega) was added to the 96-well plates, and residual ATP was quantified. Luminescence was quantified on a micro-plate reader (Spectra Max i3, MD). Residual ATP versus Ab concentration was plotted on graphs using GraphPad Prism software. Antibodies IC50 were calculated via a four-parameter curve of enzyme activity inhibition. The percentage of soluble CD73 enzymatic activity inhibition was calculated as described below:
Blockade of membrane-bound CD73 enzymatic activity
Anti-CD73 antibodies were diluted serially by a dilution factor of five with different starting working concentrations (Fig. 1C and Supplementary Fig. 1F: 200 nM). CHO-K1 cells overexpressing transfected human CD73 were digested and resuspended, and the density was adjusted to 4.0 ~ 5.0 × 105 cells/mL with a complete medium. The cells were seeded to a 96-well plate at a volume of 100 μL per well, then centrifuged the plate under 500 × g for 5 min. The supernatants were discarded, and the remaining cells were suspended with the prepared dose range of anti-CD73 Abs at 100 μL/well for 20 mins at 37 °C. Later, the plate was centrifuged under 500 × g for 5 min, washed with 1× TM buffer (MB-059, Rockland) twice, and resuspended with 180 μM AMP (1930, Sigma) at 100 μL/well. After incubation for 1 hour the plate was centrifuged again under 500 × g for 5 min, 50 μL of supernatants were taken and transferred to a black 96-well plate. Then added 50 μL of 60 μM ATP solution (A6419, Sigma) and reacted for 15 min at 37 °C, residual ATP was quantified using the Cell Titer-GloLuminescent Cell Viability Assay (G7572, Promega). Emitted light was measured on an Enspire plate reader. Residual ATP versus Abs concentration was plotted on graphs, and EC50 (s) were calculated using GraphPad Prism software. The percentage of membrane-bound CD73 enzymatic activity inhibition was calculated as described below:
Simultaneous blockade of membrane-bound and soluble CD73 enzymatic activity
HB0045 was first diluted threefold for four concentrations, then 1.5-fold for eight concentrations, and finally threefold for two concentrations, with a starting working concentration of 648 nM. HB0038 and HB0039 were diluted serially by a dilution factor of three with a starting concentration of 600 nM. CHO-K1-huCD73-3F4 cells expressing human CD73 were digested and resuspended, and the density was adjusted to 1.2 × 106 cells/mL with a complete medium.
The cells were seeded to a 96-well plate at a volume of 100 μL/well, then centrifuged the plate under 500 × g for 5 min. The supernatants were discarded, and the remaining cells were suspended with the prepared anti-CD73 Abs at 25 μL/well. Later, added 25 μL of 0.075 μg/mL CD73 solution (CD3-H52H7, ACRO) to the plate and reacted for 20 min at 37 °C. Then added 25 μL of 240 μM AMP solution (1930, Sigma) and reacted for 60 min at 37 °C. Finally added 25 μL of 80 μM ATP solution (A6419, Sigma) and reacted for 15 min at 37 °C, residual ATP was quantified using the Cell Titer-Glo®Luminescent Cell Viability Assay (G7572, Promega). Emitted light was measured on an Enspire plate reader. Residual ATP versus Abs concentration was plotted on graphs and IC50 (s) were calculated using GraphPad Prism software. The enzymatic inhibition percentage of simultaneous blockade of membrane-bound and soluble CD73 (CD73mix) was calculated as described below:
HB0045 alleviated ATP-mediated inhibition of T-cell proliferation
Frozen human PBMCs (Milestone Biotechnologies) were thawed in a 37 °C water bath and washed once with PBS. PBMCs were then incubated with 5 μM Cell Trace Violet (C34557, Thermo Fisher Scientific) in PBS for 20 min at RT keeping away from light. During the incubation, cells were mixed by turning the tube upside down every 2–3 minutes. After incubation, PBMCs were collected by centrifugation and washed once with RPMI 1640 medium containing 10% FBS (10099141 C, GIBCO) and 1% Penicillin/Streptomycin (GIBCO). Then cells were counted and adjusted to 1 × 105 cells/well in the presence of 1 μg/ml anti-CD3 (317326, Biolegend), 1 μg/ml anti-CD28 (14-0289-82, Thermo Fisher Scientific) antibodies, and 10 ng/ml IL-7 (207-IL-010, R&D) in a 96-well plate (NUNC). 30 μg/ml HB0045 was incubated with PBMCs for 30 minutes before 200 μM ATP (A6419, Sigma) was added. After 4 days of incubation, the cells were collected by 500 × g centrifugation and incubated with 100 μg/ml APC-cy7 anti-Human CD4 (317418, Biolegend, 1:100) and 100 μg/ml BV510 anti-Human CD8 (344732, Biolegend, 1:100) antibodies on ice for 30 minutes and washed twice, then subject to FACS analysis (BD Canto II) of CD4+ or CD8+ T-cell proliferation after live/dead staining with Propidium iodide (P4170, Sigma).
Anti-CD73 antibodies alleviated AMP-mediated inhibition of T-cell proliferation
Frozen human PBMCs (Milestone Biotechnologies) were thawed in a 37 °C water bath and washed once with PBS. PBMCs were then incubated with 5 μM Cell Trace Violet (C34557, Thermo Fisher Scientific) in PBS for 20 min at RT keeping away from light. During the incubation, cells were mixed by turning the tube upside down every 2–3 minutes. After incubation, PBMCs were collected by centrifugation and washed once with RPMI 1640 medium containing 10% FBS (10099141 C, GIBCO) and 1%Penicillin/Streptomycin (GIBCO). Then cells were counted and adjusted to 1 × 105 cells/well in the presence of 1 μg/ml anti-CD3 (317326, Biolegend), 1 μg/ml anti-CD28 (14-0289-82, Thermo Fisher Scientific) antibodies, and 10 ng/ml IL-7 (207-IL-010, R&D) in 96-well plate (NUNC). HB0045, HB0038, HB0039, and Oleclumab antibodies were diluted serially by a dilution factor of 3 with starting concentration of 675 nM, then they were incubated with PBMC for 30 min, respectively. 800 μM AMP (HY-A0181A, MCE) was added later. After 4 days of incubation, the cells were collected by 500 × g centrifugation and incubated with 200 μg/ml FITC anti-human CD4 (317408, BioLegend, 1:100) and 25 μg/ml APC anti-human CD8 (344722, BioLegend, 1:200) antibodies on ice for 30 min (Fig. 1E, F), or 50 μg/ml APC anti-human CD4 (317416, BioLegend, 1:200) and 100 μg/ml BV510 anti-Human CD8 (344732, Biolegend, 1:100) antibodies on ice for 30 min (Supplemental Fig. 2C, D). Washed twice, then subject to FACS analysis (BD Canto II) of CD4+ or CD8+ T-cell proliferation after live/dead staining with Propidium iodide (P4170, Sigma).
Antibody-mediated internalization assay
The internalization of antibodies was detected with pH-sensitive fluorescent dyes displying “turn-on” fluorescent at acidic pH. Briefly, HB0038, HB0039, HB0045, and negative control antibodies were conjugated with pHAb dye according to manufacturer’s protocol (G9845, Promega). The human PBMCs were seeded to a 24-well plate with 2.0 × 105 cells/well, then incubated with 5 μg/mL pHAb labeled antibodies for 0, 2, 4, 6, 12, 24, and 48 hours at 37 °C. After PBMCs were harvested, 200 μg/ml FITC anti-hCD4 antibody (11-0049-42, Thermo Fisher Scientific, 5 μl/well) and 50 μg/ml eFluor anti-hCD8 antibody (48-0088-42, Thermo Fisher Scientific, 5 μl/well) were used for isolating CD4+ T cells and CD8+ T cells, respectively. Likewise, MDA-MB-231 cells were also treated in the same way. The mean fluorescence intensities (MFI) of antibody internalization in CD4+ T cells, CD8+ T cells and MDA-MB-231 cells were measured using flow cytometry (Thermo, Attune NxT). And four-parameter fitting was performed to calculate the internalization kinetics with the MFI vs time using GraphPad Prism software.
Single particle negative staining of CD73-HB0038 Fab-HB0039 IgG complex
40 nM protein sample was applied onto a glow-discharged carbon-coated copper grid. Grids were put into the TF20 EM (FEI Company, Eindhoven, The Netherlands) operated at 200 kV. Images were collected at a nominal magnification of ×62,000 (pixel size 1.8 Å) using a Gatan CCD camera. Processing was performed using cryoSPARC v3.3.1. Micrographs were performed whole micrograph CTF-collection using CTFfind4. Particles were picked using blob pick, extracted and aligned using 2D classification. Particles were further classified using Ab-initial reconstruction and homogeneous classes were further refined.
To build the model of CD73-HB0038 Fab -HB0039 IgG complex, the structure of CD73 complex (from PDB 6TVG), a fab structure (from PDB 1M71), and an IgG structure (from PDB 1IGY) was placed and rigid-body fitted into the negative staining reconstruction map using UCSF Chimera46.
Single particle cryogenic EM of CD73-HB0038 Fab-HB0039 Fab complex
A 3 µL protein sample (0.6 μM) was applied onto a glow-discharged holey carbon grid (Quantifoil R 1.2/1.3 300 M Au). The grid was blotted to a filter paper for 3 s with a blot force of 0 at 4°C and 100% humidity and plunge-frozen in liquid ethane using a Vitrobot MarkIV (Thermo Fisher Scientific, USA). Cryo-EM movies were collected with a 300 kV Titan Krios electron microscope (Thermo Fisher Scientific, USA) equipped with a K3 direct electron detector (Gatan, USA) in counting mode. All movies were automatically recorded using EPU at a magnification of 105 K, with a physical pixel size of 0.846 Å. Each 3.0 s movie was dose-fractionated into 50 frames and contained a total dose of 55 electrons per Å2. For the sample, a total of 5,933 images were collected with a defocus range from −1.0 µm to −2.4 μm, and the slit width of Gatan Quantum GIF energy filter (Gatan, USA) was set to be 20 eV.
All image processing was performed using cryoSPARC v3.3.1 unless indicated otherwise. All dose-fractioned movies were performed patch motion with cryoSPARC’s implementation. Patch CTF estimation, 2D classification, heterogeneous refinement, and homogeneous refinements were all performed in cryoSPARC. A total of 2,621,675 particles were auto-picked using a template picker and extracted with a binning factor of 2 (200-pixel box size, 1.692 Å per pixel). 802,460 particles were selected after 2D classification based on the complex integrity. This particle setting was used for Ab-Initio reconstruction with three classes, which were then used as 3D volume templates for heterogeneous refinement. A subset of 154,776 particles from the class showing clear structural features were used for re-centering and re-extraction of unbinned particles (0.846 Å per pixel, 400-pixel box size), then subjected to 3D classification, and 62,605 particles were selected. These particles were subjected to homogeneous refinements, non-uniform refinement, and local refinement giving rise to a 3.06 Å map of four fabs connecting CD73.
To build the model of the CD73-HB0038 Fab-HB0039 Fab complex, the structure of CD73 (from PDB 6TVG) and a fab structure (from PDB 1M71) was placed and rigid-body fitted into the cryo-EM map using UCSF Chimera. The model was manually built in Coot with the guidance of the cryo-EM map47 and in combination with real space refinement using CCPEM48.
Molecular simulations
The atomic structures of apo-CD73 and that bound with HB0038, HB0039, or HB0045 were obtained from the model of Cryo-EM structure. Each model contained a monomeric CD73 and was optimized by the addition of hydrogen bonds, capping of the free N and C terminal amino acids, and assignment of pKa value to the titratable side chains of amino acids at pH 7 + 0.5. The resulting complexes were solvated with the TIP3P water model in the orthorhombic box-shaped system with defined boundary conditions. For the solvent system, 0.15 M NaCl was added to the solution system which was also neutralized with Na+ or Cl-. The periodic boundary condition was used at 10 Å. The whole built system was further minimized for 100 ps, then the minimized system was conducted for 50 ns simulation at 310 K using the OPLS-2005 force field.
In vivo animal efficacy study
Drug efficacy assays on subcutaneous xenograft models in human PBMC-humanized Prkdcscid Il2rgnull (NPG) mice
For the evaluation of the drug efficacy on different tumor models in human PBMC-humanized Prkdcscid Il2rgnull (NPG) mice, human TNBC cells MDA-MB-231 were cultured in L-15 medium (11415-064, Gibco) supplemented with 10% fetal bovine serum (10270-106, Gibco) in a 37°C incubator without CO2. Human pancreatic cancer cells BXPC-3 were cultured in RPMI 1640 medium (10-040-CVR, Corning cellgro) supplemented with 10% fetal bovine serum (10270-106, Gibco) in a 37°C incubator containing 5% CO2. Human melanoma cells A375 were cultured in DMEM medium (10-013-CVR, GIBCO) supplemented with 10% fetal bovine serum (10270-106, Gibco) in a 37°C incubator containing 5% CO2. Mice were housed in the animal centre of Pharmalegacy Co. with specific pathogen-free (SPF) housing condition (light/dark cycle: 12 h/12 h; temperature: 20–26 °C; humidity: 40 ~ 70%). All animals were housed in the same room and randomly distributed into experimental and control groups (2–6 animals per cage). Before ten generations of continuous cell culture, MDA-MB-231 (5 × 106 cells/mouse), or BXPC-3 (5 × 106 cells/mouse), or A375 (3 × 106 cells/mouse) tumor cells in 100 μL of PBS solution were mixed with an equal volume of Matrigel, then inoculated subcutaneously into the right flank of the NPG mice. Every mouse in each group received 1 × 107 PBMC (100 μL) via the tail vein. When tumors grew to an average of 50–80 mm3, tumor-bearing mice were randomly divided into different groups according to tumor volume and body weight. Treatment started on the same day. Antibodies were dosed twice weekly for 4 weeks intraperitoneally. The longest axis (L) and the shortest axis (W) of the tumor were measured with callipers, and the tumor volume (TV) was measured using the formula: TV (mm3) = 0.5 × L × W2. The tumor volume was then used to calculate the tumor growth inhibition rate of TV (TGItv%) using the formula: TGItv% = 1–(T/C) × %, where T is the average relative tumor volume (RTV) of each treated group, and C is the average RTV of the vehicle control group. RTV is the ratio of tumor volume after administration to pre-dose. Animals should be euthanized by cervical dislocation after anesthesia if the diameter of the tumor reached 20 mm in either dimension or the tumor volume reached 3000 mm3.
Drug efficacy assays on a PDX pancreatic cancer model in immunodeficient NOD-Prkdcscid Il2rgem1/Cyagen (C-NKG) mice
The pancreatic cancer patient-derived (PDX) tumor sample (Sample number: 050125) was obtained from Personaloncology Co. The immunodeficient NOD-Prkdcscid Il2rgem1/Cyagen (C-NKG) mice (female, 5–8 weeks old, purchased from Cyagen Biosciences Inc.) were housed in the animal center of Nanjing Medical University with SPF housing condition (light/dark cycle: 12 h/12 h; temperature: 20–26 °C; humidity: 40~70%). The tumor tissue was pruned to the size of 2 × 2 × 2 mm3, and then inoculated subcutaneously into the right flank of the immunodeficient C-NKG mice. When the mean tumor volume reached about 100–120 mm3, a random stratified grouping method was used to start group administration according to tumor volume and animal weight. The mice were randomly divided into 4 groups (6 mice per group). The groups were administered with vehicle or doses of HB0045 (10, 20, 40 mg/kg) on the same day twice weekly for 4 weeks intraperitoneally. The tumor volume (TV) and tumor growth inhibition of TV (TGItv%) were calculated as described in the section above. Animals should be euthanized by cervical dislocation after anesthesia if the diameter of the tumor reached 20 mm in either dimension or the tumor volume reached 3000 mm3.
Drug efficacy assays on a subcutaneous HepG2 xenograft model in immunodeficient NOD-Prkdcem26Cd52Il2rgem26Cd22/NjuCrl (NCG) mice
Human hepatoma cells HepG2 used in this experiment were cultured in DMEM medium with 10% FBS in a 37 °C incubator containing 5% CO2. Before ten generations of continuous cell culture, 100 µL PBS containing about 5 × 106 HepG2 cells and equal volume Matrigel were mixed, then inoculated subcutaneously into the right flank of the NCG mice. The mice were anesthetized with 3–4% isoflurane before inoculation. When the tumor grew to an average of about 98.00 mm3, 12 mice were randomly divided into 2 groups (6 mice per group) based on tumor volume and body weight. The groups were administered with vehicle or HB0045 (3 mg/kg) on the same day. Animals should be euthanized if any one or more of the following conditions occurred during the experiment: (1) The diameter of the tumor reached 20 mm in either dimension, or the tumor volume reached 3000 mm3; (2) The weight loss of the mice reached 20% of its body weight at the start of treatment; (3) The tumor developed ulceration, necrosis or infection; (4) Animals showed abnormal movement or paralysis. When euthanizing mice, death was defined, and survival curves were plotted.
Drug efficacy assays on a subcutaneous human CD73 overexpressed MC38 (huCD73-MC38) xenograft model in CD39/CD73 humanized (B-hCD73/hCD39) C57BL/6 mice
The mouse MC38 colorectal cancer cells overexpressing human CD73 (B-hCD73 MC38) were cultured in DMEM complete growth medium (10-013-CVR, GIBCO) supplemented with 10% fetal bovine serum (FSP500, ExCell), 1% L-Glutamine, 1% NEAA, 1% Sodium pyruvate and 1% HEPES in a 37°C incubator containing 5% CO2. Mice were housed in the animal centre of Biocytogen Co., Ltd. with SPF housing conditions (light/dark cycle: 12 h/12 h; temperature: 20–26 °C; humidity: 40~70%). All animals were housed in the same room and randomly distributed into experimental and control groups (2–6 animals per cage). 100 µL PBS containing about 5 × 105 human CD73 overexpressed MC38 (huCD73-MC38) colorectal cancer cells were then inoculated subcutaneously into the right flank of the CD39/CD73 humanized (B-hCD73/hCD39) C57BL/6 mice. Treatment started when tumors grew to an average of 100–150 mm3. Antibodies of HB0038 antibody (10 mg/kg), and HB0039 antibody (10 mg/kg) were dosed twice weekly for 3 weeks intraperitoneally. The tumor volume (TV) and tumor growth inhibition of TV (TGItv%) were calculated as described in the section above. Animals should be euthanized by cervical dislocation after anesthesia if the diameter of the tumor reached 20 mm in either dimension or the tumor volume reached 3000 mm3.
Drug efficacy assays on subcutaneous MDA-MB-231 xenograft models in immunodeficient Prkdcscid Il2rgnull (NPG) mice with or without human PBMC
For the comparison of the drug efficacy on MDA-MB-231 tumor models in immunodeficient Prkdcscid Il2rgnull (NPG) mice with or without human PBMC, MDA-MB-231 (5 × 106 cells/mouse) tumor cells in 100 μL of PBS solution were mixed with an equal volume of Matrigel, then inoculated subcutaneously into the right flank of the NPG mice. Two groups of mice (N = 8) received 1 × 107 human PBMC (100 μL) via the tail vein on the day of inoculation, while the other two groups (N = 6) did not. When tumors grew to an average of 50–80 mm3, tumor-bearing mice were dosed with PBS or HB0045 (3 mg/kg) twice weekly for 4 weeks intraperitoneally. The tumor volume (TV) and tumor growth inhibition of TV (TGItv%) were calculated as described in the section above. Animals should be euthanized by cervical dislocation after anesthesia if the diameter of the tumor reaches 20 mm in either dimension or the tumor volume reaches 3000 mm3.
Pharmacokinetics studies in the immunodeficient Prkdcscid Il2rgnull (NPG) mice
These studies were performed in compliance with the IACUC at Pharmalegacy Co. (IACUC number: PL240229-1). A total of 18 immunodeficient Prkdcscid Il2rgnull (NPG) male mice were assigned to 6 groups (3/group) and received single intraperitoneal (IP) injection of HB0038 (1 mg/kg, 5 mg/kg), HB0039 (1 mg/kg, 5 mg/kg), and HB0045 (1 mg/kg, 5 mg/kg), respectively. Blood samples were collected before dosing and at 1, 8, 24 (Day 1), 72 (Day 3), 168 (Day 7), 240 (Day 10), and 336 (Day 14) hours post-dose for detection of serum concentrations of antibodies.
Validated ELISA assays were developed for the detection of antibodies in serum samples. Firstly, diluted AffiniPure Goat Anti-Human IgG, Fcγ Fragment Specific (109-005-170, Jackson) to 0.5 µg/mL with 1xPBS (A610100-0001, BBI) and added 100 µL per well into a 96-well plate. Incubated overnight at 4 °C. After washing, blocking buffer containing 3% BSA (abs9157, Absin) was added to each well, followed by adding serum samples, standard samples, and QC (quality control) samples, and incubating at room temperature for reaction. Then HRP-conjugated antibody Goat Anti-Human Fcγ (109-035-098, Jackson) and TMB substrate (PR1200, Solarbio) were added sequentially for signaling detection, which was acquired by reading absorbance at 450 nm. Response versus concentration for the calibration curve was fitted by a four-parameter logistic model.
Statistical analyses and graphics
The results of tumor volumes and tumor weights are presented as mean ± standard error of the mean, and the in vitro binding and functional results are presented as mean ± standard deviation (SD). p < 0.05 was considered to indicate statistical significance (*p < 0.05, **p < 0.01, ***p < 0.001 and **** p < 0.0001 using the PRISM software). The significant differences in the tumor volumes between multiple groups were analyzed by two-way ANOVA Tukey’s multiple-comparisons test, while the tumor weights between multiple groups were analyzed by one-way ANOVA. The significant differences in the tumor volumes and tumor weights between two specific groups in supplemental Fig. 8A, B were analyzed by two-sided t test. The significant differences in anti-CD73 antibody-mediated proliferation in CD4+ and CD8 + T cells between two specific groups were also analyzed by a two-sided t test. Survival analysis was calculated using the log-rank test by the Kaplan–Meier method.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Cryo-EM maps of the CD73-HB0038 Fab-HB0039 Fab data have been deposited in the EM data bank under accession code EMD-60281. Model of this complex was deposited in the Protein Data Bank under accession code 8ZNZ. HDX-MS data for CD73-HB0038 and CD73-HB0039 have been deposited in the PRIDE database with identifier PXD057867 and PXD057868, respectively. All other data are available in the article and its Supplementary files or from the corresponding author upon request. Source data are provided with this paper.
References
Kraehenbuehl, L., Weng, C. H., Eghbali, S., Wolchok, J. D. & Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 19, 37–50 (2022).
Cekic, C. & Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 16, 177–192 (2016).
Knapp, K. et al. Crystal structure of the human ecto-5’-nucleotidase (CD73): insights into the regulation of purinergic signaling. Structure 20, 2161–2173 (2012).
Moesta, A. K., Li, X. Y. & Smyth, M. J. Targeting CD39 in cancer. Nat. Rev. Immunol. 20, 739–755 (2020).
Vijayan, D., Young, A., Teng, M. W. L. & Smyth, M. J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 17, 765 (2017).
Young, A., Mittal, D., Stagg, J. & Smyth, M. J. Targeting cancer-derived adenosine: new therapeutic approaches. Cancer Discov. 4, 879–888 (2014).
Yan, A. et al. CD73 promotes glioblastoma pathogenesis and enhances its chemoresistance via A(2B) adenosine receptor signaling. J. Neurosci. 39, 4387–4402 (2019).
Stagg, J. et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl Acad. Sci. USA 107, 1547–1552 (2010).
Allard, B. et al. Anti-CD73 therapy impairs tumor angiogenesis. Int. J. Cancer 134, 1466–1473 (2014).
Morello, S. et al. Soluble CD73 as biomarker in patients with metastatic melanoma patients treated with nivolumab. J. Transl. Med. 15, 244 (2017).
Jin, R. et al. Dual mechanisms of novel CD73-targeted antibody and antibody-drug conjugate in inhibiting lung tumor growth and promoting antitumor immune-effector function. Mol. Cancer Ther. 19, 2340–2352 (2020).
Xia, C., Yin, S., To, K. K. W. & Fu, L. CD39/CD73/A2AR pathway and cancer immunotherapy. Mol. Cancer 22, 44 (2023).
Antonioli, L. et al. Switching off CD73: a way to boost the activity of conventional and targeted antineoplastic therapies. Drug Discov. Today 22, 1686–1696 (2017).
Bendell, J. et al. First-in-human study of oleclumab, a potent, selective anti-CD73 monoclonal antibody, alone or in combination with durvalumab in patients with advanced solid tumors. Cancer Immunol. Immunother. 72, 2443–2458 (2023).
Antonioli, L., Pacher, P., Vizi, E. S. & Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 19, 355–367 (2013).
Geoghegan, J. C. et al. Inhibition of CD73 AMP hydrolysis by a therapeutic antibody with a dual, non-competitive mechanism of action. MAbs 8, 454–467 (2016).
Gammelgaard, O. L. et al. Targeting two distinct epitopes on human CD73 with a bispecific antibody improves anticancer activity. J. Immunother. Cancer 10, e004554 (2022).
Stefano, J. E. et al. A highly potent CD73 biparatopic antibody blocks organization of the enzyme active site through dual mechanisms. J. Biol. Chem. 295, 18379–18389 (2020).
Weinreich, D. M. et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with Covid-19. N. Engl. J. Med. 384, 238–251 (2021).
Wang, X., Mathieu, M. & Brezski, R. J. IgG Fc engineering to modulate antibody effector functions. Protein Cell 9, 63–73 (2018).
Kellner, M. et al. A novel anti-CD73 antibody that selectively inhibits membrane CD73 shows antitumor activity and induces tumor immune escape. Biomedicines 10, 825 (2022).
Miller, R. A. et al. Anti-CD73 antibody activates human B cells, enhances humoral responses and induces redistribution of B cells in patients with cancer. J. Immunother. Cancer 10, e005802 (2022).
Turiello, R., Pinto, A. & Morello, S. CD73: a promising biomarker in cancer patients. Front. Pharm. 11, 609931 (2020).
Schneider, E. et al. CD73-mediated adenosine production by CD8 T cell-derived extracellular vesicles constitutes an intrinsic mechanism of immune suppression. Nat. Commun. 12, 5911 (2021).
Terp, M. G. et al. Anti-human CD73 monoclonal antibody inhibits metastasis formation in human breast cancer by inducing clustering and internalization of CD73 expressed on the surface of cancer cells. J. Immunol. 191, 4165–4173 (2013).
Hay, C. M. et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 5, e1208875 (2016).
Weissenberger, G., Henderikx, R. J. M. & Peters, P. J. Understanding the invisible hands of sample preparation for cryo-EM. Nat. Methods 18, 463–471 (2021).
Bianchini, G., Balko, J. M., Mayer, I. A., Sanders, M. E. & Gianni, L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 13, 674–690 (2016).
Mizrahi, J. D., Surana, R., Valle, J. W. & Shroff, R. T. Pancreatic cancer. Lancet 395, 2008–2020 (2020).
Zhao, J. et al. Overexpression of CD73 in pancreatic ductal adenocarcinoma is associated with immunosuppressive tumor microenvironment and poor survival. Pancreatology 21, 942–949 (2021).
Tu, E. et al. Anti-PD-L1 and anti-CD73 combination therapy promotes T cell response to EGFR-mutated NSCLC. JCI Insight 7, e142843 (2022).
Chen, S. et al. CD73 expression on effector T cells sustained by TGF-beta facilitates tumor resistance to anti-4-1BB/CD137 therapy. Nat. Commun. 10, 150 (2019).
Cui, X. et al. A novel bispecific antibody targeting PD-L1 and VEGF with combined anti-tumor activities. Front. Immunol. 12, 778978 (2021).
Zhou, Q., Facciponte, J., Jin, M., Shen, Q. & Lin, Q. Humanized NOD-SCID IL2rg-/- mice as a preclinical model for cancer research and its potential use for individualized cancer therapies. Cancer Lett. 344, 13–19 (2014).
Kowash, R. R. & Akbay, E. A. Tumor intrinsic and extrinsic functions of CD73 and the adenosine pathway in lung cancer. Front. Immunol. 14, 1130358 (2023).
Lupia, M. et al. CD73 regulates stemness and epithelial-mesenchymal transition in ovarian cancer-initiating cells. Stem Cell Rep. 10, 1412–1425 (2018).
Petruk, N. et al. CD73 facilitates EMT progression and promotes lung metastases in triple-negative breast cancer. Sci. Rep. 11, 6035 (2021).
Perrot, I. et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. 27, 2411–2425 e2419 (2019).
Zhou, Y. et al. Pharmacology, pharmacokinetics, and toxicity characterization of a novel anti-CD73 therapeutic antibody IBI325 for cancer immunotherapy. Int. J. Biol. Macromol. 229, 158–167 (2023).
Wurm, M. et al. A novel antagonistic CD73 antibody for inhibition of the immunosuppressive adenosine pathway. Mol. Cancer Ther. 20, 2250–2261 (2021).
Kleiveland, C. R. In: The impact of food bioactives on health: in vitro and ex vivo models (eds. K. Verhoeckx et al.) 161–167 (2015).
Li, J. et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 6, e1320011 (2017).
Zhang, C. Hybridoma technology for the generation of monoclonal antibodies. Methods Mol. Biol. 901, 117–135 (2012).
Pascal, B. D. et al. HDX workbench: software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom. 23, 1512–1521 (2012).
Song, B. et al. Ordered assembly of the cytosolic RNA-sensing MDA5-MAVS signaling complex via binding to unanchored K63-linked poly-ubiquitin chains. Immunity 54, 2218–2230 e2215 (2021).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D. Biol. Crystallogr. 60, 2126–2132 (2004).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D. Biol. Crystallogr. 67, 355–367 (2011).
Acknowledgements
This study is funded by Huaota Biopharmaceutical Co. Ltd. We gratefully acknowledge the support provided by the Chinese National Natural Science Foundation (Grant no. 32470827 and 32241021), and Guangzhou Science and Technology Project (Grant no. 2024A04J6351), and Guangdong Talent Project (Grant no. 2023ZT10Y154). We also appreciate the startup fund from the Guangzhou Institutes of Biomedicine and Health directed to L.W., as well as the Shanghai Rising-Star Program (Grant no. 23QB1400400) granted to J.G.X. We extend our thanks to Dr Xiaoli Xiong (GIBH) and the Wan lab for their insightful comments and proofreading of the manuscript.
Author information
Authors and Affiliations
Contributions
L.W., Y.Z., Xiangyang Z., and J.G.X. conceived the study. J.G.X., S.C., Y.H., and Xi Z. performed most experiments, except for those for the negative staining, cryo-EM sample preparation and data collection work, which was assisted by Z.C., M.W., X.Y., and H.Shi from Biortus Biosciences Co. Ltd. Y.W., L.Z., X.R., H.J, H.Y., X.Wei, Y.F, X.Chen, X.Cui, X.P., S.W, S.X., H.Shang, Y.P., W.X., H.L. Q.C., Z.Y., Y.X. and M.L. assisted J.G.X. with protein purification, writing–review and editing. J.C.Z., R.B., S.D., and J.He. analyzed part of cryo-EM data. Z.Y. and X.Wu drew most of the schematic diagrams. L.W., J.G.X., Y.Z., J. Huang, and C.L.C. wrote the manuscript, with assistance from Y.H. and Xiangyang Z. J.G.X. managed the project.
Corresponding authors
Ethics declarations
Competing interests
All authors except L.W., Y.H., Z.Y., J.C.Z., X.Wu, H.L., Z.C., M.W., X.Y., H. Shi, Y.X., R.B., S.D., J.He., J.Huang., and C.L.C. are employed by Huaota Biopharma. The author L.W., Y.H., Z.Y., J.C.Z., X.Wu, H.L., Z.C., M.W., X.Y., H. Shi, Y.X., R.B., S.D., J. He, J. Huang, and C.L.C. declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, JG., Chen, S., He, Y. et al. An antibody cocktail targeting two different CD73 epitopes enhances enzyme inhibition and tumor control. Nat Commun 15, 10872 (2024). https://doi.org/10.1038/s41467-024-55207-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55207-9
