
Abstract
Unsymmetric disulfides are prevalent in natural products and are essential in medicinal chemistry and materials science, but their robust synthesis poses significant challenges. In this paper, we report an expeditous transition-metal-free methodology for synthesizing unsymmetric disulfides through the addition of perthiyl radicals to alkenes. This study marks the use of generating perthiyl radicals by reacting SO2 with unactivated alkyl (pseudo)halides (Cl/Br/I/OTs). Various primary, secondary and tertiary alkyl (pseudo)halides substituted with different functional groups successfully function as suitable reactants. The formation of perthiyl radicals and their involvement in the reaction process are verified through mechanistic studies and DFT calculations. Overall, this method leverages readily available alkyl electrophiles and alkenes alongside SO2 in a single reaction setup to efficiently form both carbon-sulfur and sulfur-sulfur bonds simultaneously.
Similar content being viewed by others
Introduction
Disulfides are widely utilized across various disciplines, including biology1, food chemistry2, and pharmaceutical sciences3. Of particular note is the pharmaceutical industry’s effective employment of S-S bond linkers in formulating a range of therapeutics aimed at treating diabetes, alcoholism, and cancer, through their incorporation into antibody, small molecules and peptides (Fig. 1A)4,5,6,7,8.

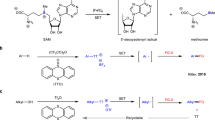
A Representative biorelevant disulfide compounds. B Strategies for unsymmetric disulfides construction and perthiyl radical generation. C This work: one-pot unsymmetrical disulfides synthesis via perthiyl radicals addition.
Traditional methods to prepare disulfides primarily rely on the disulfide exchange or the couplings of two sulfur-containing compounds. Contemporary strategies for disulfide synthesis often employ disulfurating reagent (RSS−LG) to forge C-SS bonds, with persulfide cations (RSS+) or persulfide anions (RSS−) serving as crucial intermediates9,10,11,12,13,14,15,16,17,18,19,20,21 (Fig. 1B). However, there have been no reports on a method for directly synthesizing unsymmetrical disulfides using perthiyl radicals (RSS•)22,23,24,25. To date, several methods have been explored for generating perthiyl radicals (Fig. 1B), and one of the earliest methods involved the thermal decomposition of tetrasulfides26,27,28,29. However, the reverse reaction, i.e., the dimerization of RSS•, occurs with minimal or no activation energy, resulting in low overall conversion30. An alternative approach involves carbon radical substitution on tetrasulfides31,32. Additionally, the hydrogen-atom-transfer (HAT) process emerges as a powerful and effective strategy for radical production, as the relatively weak RSS–H bond dissociation enthalpy (~70 kcal/mol) enables the oxidation of hydropersulfides to RSS• by mild oxidants33,34,35. Another potent and efficient strategy is photochemistry, which activates S-Cl bonds in RSSCl to form perthiyl radicals26. Unfortunately, these methods for generating perthiyl radicals often require stringent experimental conditions and can only be applied to the pre-synthesized polysulfides. Furthermore, the preparation of polysulfides is often complex and time-consuming. Hence, the development of a versatile and convenient approach for generating perthiyl radicals in a single reaction remains a significant priority. This will not only greatly simplify the process of preparing unsymmetric disulfides but also improve synthesis efficiency.
On the other hand, sulfur dioxide (SO2) is widely used in the pharmaceutical and organic chemical synthesis sectors36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66. In recent years, our research group has focused on the versatile chemical properties of SO2 and developed several synthetic methods in which are carbon radicals capture SO2 molecule to form sulfonyl radicals. Building on our previous work involving sulfonyl radicals, the objective of this study is to develop a general approach using SO2 as the sulfur source to obtain perthiyl radicals for the synthesis of unsymmetric disulfides from easily accessible starting materials in one pot. To achieve this desired reaction, our hypothesis is that a sulfonyl radical can be reduced to a sulfur radical, which can then capture the second SO2 molecule and undergo subsequent reduction to form the target perthiyl radical (Fig. 1B). Implementing of this protocol presents several challenges: (1) A suitable reducing agent which can effectively reduce sulfonyl radicals to sulfur radicals is in need. (2) The sulfur radicals have to capture SO2 specifically. (3) Perthiyl radicals are thermodynamically stable and readily form dimers, making the search for a suitable perthiyl radical receptor an intriguing task. (4) The question arises as to whether the perthiyl radicals would continue to capture one more SO2 molecule. (5) Sulfur radical addition products and perthiyl radical homocoupling products may be produced instead of the desired disulfides.
In this study, perthyl radicals are generated from easily accessible alkyl halides and SO2, enabling the one-step synthesis of unsymmetric disulfides. This process entails two instances of C–S bond formation and one instance of S–S bond formation (Fig. 1C).
Results
Reaction optimization
The disulfide reaction was initially investigated by studying the reaction between the model reactants 1-bromocyclopentane 1, ethyl acrylate 2 and ex-situ generated SO2 gas (from SOgen)36 (Table 1). The optimized conditions were: PhSiH3 (3.2 equiv) as reductant, HCOOLi (3.0 equiv) as the base, a 1:2.5:2.5 ratio of 1-bromocyclopentane to ethyl acrylate to SO2, 2.0 mL mixture of NMP-DMSO (1:1) as solvent and 40 °C as the reaction temperature. Under these conditions, product 3 was obtained in 72% isolated yield (entry 1, Table 1). The reaction did not occur in the absence of PhSiH3 or HCOOLi (entries 2–3). Substituting PhSiH3 with Et3SiH as the reductant resulted in a substantial reduction in the product yield (entry 4), while the use of other reductant sources such as PMHS and NaBH4 directly suppressed the reaction (entries 5–6). It was observed that the choice of base had a significant impact on the reaction. Replacing HCOOLi with K2CO3, DABCO, and HCOOK led to a dramatic decrease in yields (entries 7–9). In addition, two alternative SO2 surrogates and S8 were explored. Using S8 instead of SO2 resulted in product yields of 49% (entry 10). The use of DABSO resulted in a significantly reduced yield (entry 11), while the utilization of another inorganic SO2 surrogate, namely K2S2O5 (entry 12), completely hindered the conversion. Furthermore, the choice of solvent also influenced the reaction outcome. The use of DMSO or NMP resulted in product yields of only 60% and 43%, respectively (entries 13–14). Instead, a mixture of 1:1 DMSO and NMP was found to be the optimal solvent for the reaction.
Substrate scopes
With the optimal reaction conditions identified, we proceeded to explore the generality of the reaction. First, the scope with respect to the alkyl bromides was investigated (Fig. 2). We observed that both linear and α-branched primary alkyl bromides could be successfully converted into the corresponding disulfides (4-8). The reaction exhibited a high chemoselectivity for activating alkyl bromides over alkyl fluorides, as evidenced by the formation of product 9. In addition, the alkyl bromides bearing remote functional groups, such as benzyl ether and trimethylsilane, were found to be compatible with the reaction conditions (10,11). Moreover, alkyl bromides bearing aromatic rings, such as non-substituted and substituted phenyl and naphthyl rings, proved to be suitable substrates in the transformation (12-16). Notably, we found that heterocycle substituents, such as N-protected pyrrolidines, N-protected piperazine, thiophene, were well-tolerated on the alkyl bromides (17-19). Additionally, substrates containing unsaturated propargyl and allyl derivatives underwent the conversion smoothly, providing the desired products (20, 21). Next, secondary alkyl bromides were tested in the reaction. Apart from the model product 3, some secondary alkyl bromides with various carbon chain length could lead to the desired products (22-25). Interestingly, various secondary benzyl bromides were also suitable substrates, successfully providing products 26-28. In addition, two examples of α-carbonyl alkyl bromides went through the transformation and gave the target disulfides (29, 30). To our delight, tertiary alkyl bromides with large steric hindrance were also proved amenable for the reaction (31-33), indicating that this strategy had potential for the construction of hindered unsymmetrical disulfides.

Reaction conditions: alkyl bromides (0.2 mmol, 1.0 equiv), α, β-unsaturated esters (0.5 mmol, 2.5 equiv), SO2 (0.5 mmol, 2.5 equiv), PhSiH3 (0.64 mmol, 3.2 equiv), HCOOLi (0.6 mmol, 3.0 equiv), DMSO (1.0 mL) and NMP (1.0 mL), at 40 °C for 9 h under argon atmosphere. Isolated yields were reported. std. cond. = standard conditions.
Continuously, a series of alkyl iodides were employed in the reaction in place of the model reactant alkyl bromides (Fig. 3). Pleasingly, disulfide products 4, 7, 11, 12, 23, and 27 which could be prepared from primary alkyl bromides were also successfully prepared from alkyl iodides. Primary alkyl iodide with longer carbon chain was tolerated in the reaction, providing the target product 34. Two secondary alkyl iodides also went the reaction smoothly and produced the desired products 23 and 27. In the same way, alkyl chlorides were used in place of bromide analogs (Fig. 3). Alkyl chlorides containing terminal alkene, terminal alkyne or allyl group could afford the corresponding products 21, 35 and 36 respectively. Some secondary alkyl chlorides were also suitable substrates for this protocol (23, 26, 27). It was gratifying that pseudohalides were compatible with the reaction conditions. Various alkyl tosylates could be converted into the corresponding disulfides in 45%-69% yields (Fig. 3). For example, products 4, 12 were obtained again by using primary alkyl tosylates instead of the primary alkyl bromides. This protocol also had good tolerance for alkyl tosylates containing aromatic rings decorated with various substituents (37-41). Tosylate substrates containing heterocycles and terminal alkene were also compatible with this transformation (42-44). Both linear (45, 25, 22) and cyclic (3, 46, 47) secondary alkyl tosylates successfully underwent the desired transformation.

Reaction conditions: alkyl electrophile (0.2 mmol, 1.0 equiv), α, β-unsaturated esters (0.5 mmol, 2.5 equiv), SO2 (0.5 mmol, 2.5 equiv), PhSiH3 (0.64 mmol, 3.2 equiv), HCOOLi (0.6 mmol, 3.0 equiv), DMSO (1.0 mL) and NMP (1.0 mL), at 40 °C for 9 h under argon atmosphere. Isolated yields were reported. std. cond. = standard conditions.
We next began to investigate the scope of alkene derivatives for the reaction (Fig. 4). α, β-unsaturated esters (48-56) were all tolerated under the standard experiment conditions. Interestingly, excellent regioselectivity of the reaction was demonstrated by using vinyl acrylate as a reactant (55). To our delight, vinyl pyridine could be used as an effective perthiyl radical acceptor (57). α-aryl substituted α, β-unsaturated ester was also proved to be a suitable substrate, providing product 58 in moderate yield. Additionally, α, β-unsaturated amide could readily participate in the reaction (59). Furthermore, we aimed to showcase the practical applications of this strategy by utilizing pharmaceuticals and natural products as reaction substrates for modification purposes. As a results, disulfides derived from Diacetonefructose (60), Estrone (61), Paroxetine (62), L-Menthol (63) and DL-α-Tocopherol (64) were successfully prepared respectively.

Reaction conditions: alkyl bromides (0.2 mmol, 1.0 equiv), α, β-unsaturated esters (0.5 mmol, 2.5 equiv), SO2 (0.5 mmol, 2.5 equiv), PhSiH3 (0.64 mmol, 3.2 equiv), HCOOLi (0.6 mmol, 3 equiv), DMSO (1.0 mL) and NMP (1.0 mL), at 40 °C for 9 h under argon atmosphere. Isolated yields were reported. std. cond. = standard conditions.
Mechanistic investigations
To shed light on the nature of this reaction system, radical trapping experiments were carried out (Fig. 5A). When the reaction was conducted without ethyl acrylate, the TEMPO-carbon radical adduct compound 65 was detected. When TEMPO was added to the standard reaction in which SO2 was added, the target disulfide 3 was not observed and the S-centered radical was efficiently trapped by TEMPO (66). Moreover, when TEMPO was added after 20 min of the reaction, perthiyl radicals and sulfur radicals were generated and trapped, proved by the detection of 66 and 67. Next, the parallel kinetic isotope effect (KIE) experiments (from initial rate) showed that the kH/kD value was 4.43, and the competitive KIE was determined to be kH/kD = 2.57, indicating that the protonation step was involved in the turnover-limiting step (Fig. 5B). In the template reaction, we have identified two side products: disulfide compound 68 with a yield of 9% and trisulfide compound 69 with a yield of 5%. Subsequently, we performed a control experiment under standard reaction conditions, excluding the addition of ethyl acrylate. Dimers of disulfides 68, trisulfides 69, and tetrasulfides 70 were identified, with yields of 32%, 25%, and 18%, respectively. To gain more insight into the reaction mechanism, symmetrical disulfide 68, trisulfide 69, or tetrasulfide 70 was utilized in place of reactant 1 in reactions with reactant 2, all eventually generating product 3 in yields of 50%, 53%, and 45%, respectively. Additionally, trace amount of thioether 3” was detected in the reaction between compound 68 and ethyl acrylate. In the template reaction, 1.0 equivalent of SO2 was used instead of 2.5 equivalents, and as a result, trace amount of thioether product 3” was detected by GCMS. These results indicate the generation of sulfur radicals and demonstrate that there is an interconversion between the sulfur radicals and compound 68 throughout the reaction process (Fig. 5C).

A Radical-trapping experiments with TEMPO; B KIE experiments; C Intermediate experiments; D Proposed mechanism. std. cond. = standard conditions.
Proposed mechanism
On the basis of the initial mechanistic studies, a plausible reaction mechanism is proposed herein (Fig. 5D). Initially, the key species I may be generated from the interaction between alkyl bromide and a radical initiator which generated from the silane. Secondly, the alkyl radical I is trapped by SO2 to form the alkylsulfonyl radical species II, which would undergo fast deoxygenation, giving thiyl radical intermediate III, which has an equilibrium with compound 68. Intermediate III could capture an additional molecule of SO2, and then undergoes the same deoxygenation process to yield perthiyl radical IV. IV may either couple with III or undergo homocoupling to form compounds 69 and 70, respectively. This process is reversible. Then, perthiyl radical adds to 2 and generates V which then undergoes HAT with PhSiH3 to produce the disulfuration product 3.
DFT studies
To gain more in-depth insights into the reaction mechanism, density functional theory (DFT) calculations were conducted (Fig. 6). Silicon radical formation through PhSiH3 is the initiation step67,68,69. First of all, we found that the base additive (HCOO−) enables a much more facile reaction pathway. Its binding with PhSiH2 radical is exergonic by -2.8 kcal/mol, resulting in a new radical intermediate PhSiH2-HCOO•−, which is a quite strong reductant (Eox = −2.51 V versus NHE). In addition, the silane-formate complex, PhSiH3-HCOO−, exhibits a significantly elongated Si–H bond (1.57 angstrom, versus 1.48 angstrom in PhSiH3), indicating that it may act as a superstrong hydrogen donor. Then, the halogen-atom transfer (XAT) process between 1 and PhSiH2-HCOO•− resulting in the formation of alkyl radical (INT1) is determined to have a free energy barrier of 14.5 kcal/mol (TS1). Following that, SO2 is rapidly trapped by INT1 without encountering any energy barrier (Supplementary Fig. 8). We have calculated the barrier for the addition of INT2 to alkene, which is 16.6 kcal/mol. As the reduction of INT2 is barrierless, the addition will not form a competitive side product pathway. The hydrogen transfer from PhSiH3-HCOO− to INT2 is barrierless, resulting in the formation of INT3 and PhSiH2-HCOO•−. Then they immediately undergo a single electron transfer event, and the so-generated radical anion INT4 and PhSiH2OOCH rapidly re-associate to afford INT4’. Then, the INT4’ undergoes deoxygenation through the cleavage of the S–OH bond, hydrogen abstraction, and sequential radical elimination to produce sulfur radical (INT6) and poly(phenylsiloxanes), which may exist in linear or cyclic (INT5, detected by HRMS) forms. Its free energy profile has been explored in Supplementary Fig. 9. Next, we have calculated the barrier for the addition of INT6 to alkene, which is 13.8 kcal/mol, as well as determined that the homocoupling of INT6 occurs barrierlessly. However, as shown in the energy profile, once INT6 is transformed into INT7, it is barrierlessly reduced, furthermore, the energy barrier required for the irreversible transformation from INT6 to INT7 is 11.2 kcal/mol. As a result, the thioether product will not form. Considering that compound 68 may represent an important resting state during the reaction, we further investigated the relevant mechanisms of the transformation of compound 68, as shown in Supplementary Fig. 10. Subsequently, INT7 is reduced barrierlessly by PhSiH3-HCOO−, after which INT8 undergoes a single-electron transfer with a free energy barrier of 7.9 kcal/mol. And then, INT9 and PhSiH2OOCH rapidly re-associate to afford INT9’. Next, INT9’ undergoing deoxygenation is similar to that of transforming INT4’ into INT6, resulting in the formation of perthiyl radical (INT10), as shown in Supplementary Fig. 9. We have found that the association of INT10 with the third equivalent of SO2 unable to give any minimum on the potential energy surface, and thus further combination with SO2 is unrealistic (Supplementary Fig. 11). The perthiyl radical has the ability to undergo addition to ethyl acrylate (2), resulting in the formation of a carbon-centered radical (INT11) with a free energy barrier of 20.0 kcal/mol (TS2). Subsequently, this newly formed radical abstracts a hydrogen atom from PhSiH3-HCOO− through transition state TS3 with an activation free energy of 26.1 kcal/mol to yield the desired product 3 and PhSiH2-HCOO•− which participates in the propagating chain process. The overall barrier is 26.1 kcal/mol, as determined by the hydrogen abstraction of the radical INT11 via the transition state TS3. Overall, the pathway calculated by DFT is consistent with the assumption described in Fig. 5D.

All molecular geometries were optimized at the M06-2X/6-31 + G(d) level of theory using SMD model to account for solvation energies in DMSO.
In summary, we have developed a unified protocol for hydro-disulfuration of alkenes with alkyl electrophiles under mild conditions. This reaction uses readily abundant alkyl electrophiles and alkenes as starting materials, providing straightforward access to unsymmetrical disulfides with excellent functional group tolerance. It is demonstrated as an effective example of the synthesis of unsymmetrical disulfides by using SO2 as a divalent sulfur source. Phenylsilane serves both as the radical initiator and reductant. This research provides a convenient method for synthesis of unsymmetrical disulfides which are challenging to access. Further investigations will focus on expanding the perthiyl radical acceptor scope and finding widespread application for this disulfuration strategy.
Methods
General procedure for the synthesis of compounds 3-64: In an argon fulfilled glovebox, perbromothiophene 1,1-dioxide (219.8 mg, 0.51 mmol, 2.5 equiv), tetradecane (1.0 mL), were added into chamber B with a magnetic stirring bar, followed by addition of 1-methyl-4-vinylbenzene (59.3 mg, 0.5 mmol, 2.5 equiv). HCOOLi (0.6 mmol, 31.2 mg), alkyl electrophiles (-Cl/-Br/-I/-OTs) (0.2 mmol) and alkenes (0.5 mmol) were added into chamber A, then anhydrous NMP (1.0 mL) and DMSO (1.0 mL) was added into chamber A. Then, PhSiH3 (0.64 mmol, 79 μL) was added into chamber A by microsyringe. The two-chamber was sealed and removed out of the glovebox. The chamber B was allowed to stir at 100 °C using heating mantle with 600 rpm stirring speed for 10 min (chamber A was not allowed to stir). After then, the chamber A was allowed to stir at 40 °C using heating mantle with 300 rpm stirring speed for 9 h. Upon completion, the reaction was quenched with water (30 mL) and extracted with EA (30 mL). The extract was washed by brine (15 mL) and dried over anhydrous Na2SO4. After filtering, the filtrate was concentrated, and the residue was purified by flash silica gel column chromatography using petroleum ether/ethyl acetate as eluent to afford pure products 3-64.
Data availability
The authors declare that all data generated or analyzed during this study are included in this article and Supplementary Information. The energetics and Cartesian coordinates for all species are presented in the Supplementary Data 1. Data are also available from the corresponding author upon request.
References
Lee, M. H. et al. Disulfide-cleavage-triggered chemosensors and their biological applications. Chem. Rev. 113, 5071–5109 (2013).
Silva, F. et al. Short total synthesis of ajoene. Angew. Chem. Int. Ed. 57, 12290–12293 (2018).
Góngora-Benítez, M., Tulla-Puche, J. & Albericio, F. Multifaceted roles of disulfide bonds. peptides as therapeutics. Chem. Rev. 114, 901–926 (2014).
Beck, A., Goetsch, L., Dumontet, C. & Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 16, 315–337 (2017).
Zhao, B. & Burgess, K. TrkC-targeted kinase inhibitors and PROTACs. Mol. Pharm. 16, 4313–4318 (2019).
Naderi, N., Darmishonnejad, Z., Tavalaee, M. & Nasr-Esfahani, M. H. The effect of alpha-lipoic acid on sperm functions in rodent models for male infertility: a systematic review. Life Sci. 323, 121383 (2023).
Yang, L. P. H. Romidepsin. Drugs 71, 1469–1480 (2011).
Salvia, L. A. et al. Targeting neuroendocrine tumors with octreotide and lanreotide: Key points for clinical practice from NET specialists. Cancer Treat. Rev. 117, 102560 (2023).
Xiao, X., Feng, M. & Jiang, X. New design of a disulfurating reagent: facile and straightforward pathway to unsymmetrical disulfanes by copper-catalyzed oxidative cross-coupling. Angew. Chem. Int. Ed. 55, 14121–14125 (2016).
Xiao, X., Xue, J. & Jiang, X. Polysulfurating reagent design for unsymmetrical polysulfide construction. Nat. Commun. 9, 2191 (2018).
Xue, J. & Jiang, X. Unsymmetrical polysulfidation via designed bilateral disulfurating reagents. Nat. Commun. 11, 4170 (2020).
Qi, J., Wei, F., Huang, S., Tung, C.-H. & Xu, Z. Copper(I)-catalyzed asymmetric interrupted Kinugasa reaction: synthesis of α-thiofunctional chiral β-lactams. Angew. Chem. Int. Ed. 60, 4561–4565 (2021).
Zhang, Q., Li, Y., Zhang, L. & Luo, S. Catalytic asymmetric disulfuration by a chiral bulky three-component Lewis acid-base. Angew. Chem. Int. Ed. 60, 10971–10976 (2021).
Chen, X., Shao, W. & Deng, G.-J. Nickel-catalyzed deoxygenative disulfuration of alcohols to access unsymmetrical disulfides. ACS Catal. 14, 6451–6461 (2024).
Chen, S. et al. Sandmeyer-type reductive disulfuration of anilines. Org. Lett. 23, 7428–7433 (2021).
Bai, Z. et al. Synthesis of N-acyl sulfenamides via copper catalysis and their use as S-sulfenylating reagents of thiols. Nat. Commun. 13, 6445 (2022).
Zou, J. et al. Phthalimide-carried disulfur transfer to synthesize unsymmetrical disulfanes via copper catalysis. ACS Catal. 9, 11426–11430 (2019).
Ren, X. et al. Access to polysulfides through photocatalyzed dithiosulfonylation. Angew. Chem. Int. Ed. 62, e202302199 (2023).
Tian, Q. & Li, Y. Design of a bilateral disulfurating reagent for unsymmetrical polysulfidation. Angew. Chem. Int. Ed. 62, e202302861 (2023).
Asanuma, H., Kanemoto, K., Watanabe, T. & Fukuzawa, S.-I. N-(Morpholine-4-dithio)phthalimide: a shelf-stable, bilateral platform molecule enabling access to diverse unsymmetrical disulfides. Angew. Chem. Int. Ed. 62, e202219156 (2023).
Yu, Q., Zhang, X. & Jiang, X. Bilateral unsymmetrical disulfurating reagent design for polysulfide construction. Angew. Chem. Int. Ed. 63, e202408158 (2024).
Zhang, J. & Studer, A. Decatungstate-catalyzed radical disulfuration through direct C-H functionalization for the preparation of unsymmetrical disulfides. Nat. Commun. 13, 3886 (2022).
Wu, Z. & Pratt, D. A. Radical substitution provides a unique route to disulfides. J. Am. Chem. Soc. 142, 10284–10290 (2020).
Huang, H., Wu, Z. & Pratt, D. A. Photocatalytic C–S bond formation using N-thiophthalimide and N-perthiophthalimide derivatives. ACS Catal. 13, 13912–13919 (2023).
Wu, Z. & Pratt, D. A. A divergent strategy for site-selective radical disulfuration of carboxylic acids with trisulfide-1,1-dioxides. Angew. Chem. Int. Ed. 60, 15598–15605 (2021).
Burkey, T. J. et al. The tert-butylperthiyl radical. J. Org. Chem. 50, 4966–4967 (1985).
Kende, I., Pickering, T. L. & Tobolsky, A. V. The dissociation energy of the tetrasulfide linkage. J. Am. Chem. Soc. 87, 5582–5586 (1965).
Benson, S. W. Thermochemistry and kinetics of sulfur-containing molecules and radicals. Chem. Rev. 78, 23–35 (1978).
Hawari, J. A., Griller, D. & Lossing, F. P. Thermochemistry of perthiyl radicals. J. Am. Chem. Soc. 108, 3273–3275 (1986).
Chauvin, J.-P. R., Haidasz, E. A., Griesser, M. & Pratt, D. A. Polysulfide-1-oxides react with peroxyl radicals as quickly as hindered phenolic antioxidants and do so by a surprising concerted homolytic substitution. Chem. Sci. 7, 6347–6356 (2016).
Everett, S. A., Schoeneich, C., Stewart, J. H. & Asmus, K. D. Perthiyl radicals, trisulfide radical ions, and sulfate formation: a combined photolysis and radiolysis study on redox processes with organic di- and trisulfides. J. Phys. Chem. A 96, 306–314 (1992).
Wu, Z., Back, T. G., Ahmad, R., Yamdagni, R. & Armstrong, D. A. Mechanism of reduction of bis(2-hydroxyethyl) trisulfide by eaq- and.bul.CO-2. Spectrum and scavenging of RSS.bul. radicals. J. Phys. Chem. C 86, 4417–4422 (1982).
Lindahl, S. & Xian, M. Recent development of polysulfides: chemistry and biological applications. Curr. Opin. Chem. Biol. 75, 102325–102331 (2023).
Everett, S. A., Folkes, L. K., Wardman, P. & Asmus, K. D. Free-radical repair by a novel perthiol: reversible hydrogen transfer and perthiyl radical formation. Free Radic. Res. 20, 387–400 (1994).
Terryn, H., Tilquin, B. & Houée-Levin, C. Formation of perthiyl free radicals in irradiated glutathione. Res. Chem. Intermed. 31, 727–736 (2005).
Jia, X., Kramer, S., Skrydstrup, T. & Lian, Z. Design and applications of a SO2 surrogate in palladium-catalyzed direct aminosulfonylation between aryl iodides and amines. Angew. Chem. Int. Ed. 60, 7353–7359 (2021).
Chen, L. et al. Enantioselective four-component arylsulfonylcyanation of vinylarenes via the insertion of SO2 enabled by SOgen as SO2 surrogate. ACS Catal. 12, 10764–10770 (2022).
Qiu, G., Lai, L., Cheng, J. & Wu, J. Recent advances in the sulfonylation of alkenes with the insertion of sulfur dioxide via radical reactions. Chem. Commun. 54, 10405–10414 (2018).
Ye, S., Qiu, G. & Wu, J. Inorganic sulfites as the sulfur dioxide surrogates in sulfonylation reactions. Chem. Commun. 55, 1013–1019 (2019).
Zhang, J., Wang, P., Li, Y. & Wu, J. Asymmetric sulfonylation with sulfur dioxide surrogates: a new access to enantiomerically enriched sulfones. Chem. Commun. 59, 3821–3826 (2023).
Zeng, D., Wang, M., Deng, W. & Jiang, X. The same oxygenation-state introduction of hypervalent sulfur under transition-metal-free conditions. Org. Chem. Front. 7, 3956–3966 (2020).
Wang, M. & Jiang, X. The Same oxidation-state introduction of hypervalent sulfur via transition-metal catalysis. Chem. Rec. 21, 3338–3355 (2021).
Emmett, E. J. & Willis, M. C. The development and application of sulfur dioxide surrogates in synthetic organic chemistry. Asian J. Org. Chem. 4, 602–611 (2015).
Blum, S. P., Hofman, K., Manolikakes, G. & Waldvogel, S. R. Advances in photochemical and electrochemical incorporation of sulfur dioxide for the synthesis of value-added compounds. Chem. Commun. 57, 8236–8249 (2021).
Chen, G. & Lian, Z. Multicomponent reactions based on SO2 surrogates: recent advances. Eur. J. Org. Chem. 26, e202300217 (2023).
Chen, Y., Murray, P. R. D., Davies, A. T. & Willis, M. C. Direct copper-catalyzed three-component synthesis of sulfonamides. J. Am. Chem. Soc. 140, 8781–8787 (2018).
Huang, J. et al. Accessing chiral sulfones bearing quaternary carbon stereocenters via photoinduced radical sulfur dioxide insertion and Truce–Smiles rearrangement. Nat. Commun. 13, 7081 (2022).
Meng, Y., Wang, M. & Jiang, X. Multicomponent reductive cross-coupling of an inorganic sulfur dioxide surrogate: straightforward construction of diversely functionalized sulfones. Angew. Chem. Int. Ed. 59, 1346–1353 (2020).
Li, H. et al. Synthesis of multifluoromethylated γ-sultines by a photoinduced radical addition–polar cyclization. Angew. Chem. Int. Ed. 62, e202300159 (2023).
Lou, T. S.-B. et al. Chemoselective nickel electrocatalytic sulfinylation of aryl halides with SO2. Angew. Chem. Int. Ed. 61, e202208080 (2022).
Pedersen, P. S., Blakemore, D. C., Chinigo, G. M., Knauber, T. & MacMillan, D. W. C. One-pot synthesis of sulfonamides from unactivated acids and amines via aromatic decarboxylative halosulfonylation. J. Am. Chem. Soc. 145, 21189–21196 (2023).
Zhao, F. & Wu, X. Sulfonylation of bismuth reagents with sulfinates or SO2 through BiIII/BiV intermediates. Organometallics 40, 2400–2404 (2021).
Hethcox, J. C. et al. Nickel-catalyzed sulfonylation of aryl bromides enabled by potassium metabisulfite as a uniquely effective SO2 surrogate. Angew. Chem. Int. Ed. 62, e202217623 (2023).
Hou, X., Liu, H. & Huang, H. Iron-catalyzed fluoroalkylative alkylsulfonylation of alkenes via radical-anion relay. Nat. Commun. 15, 1480 (2024).
Qu, S., Li, X., Li, X. & Wang, L. Copper-catalyzed chemoselective (amino)fluorosulfonylation of hydrocarbons via intramolecular fluorine-atom transfer. ACS Catal. 14, 4318–4328 (2024).
Chen, Z. et al. Iron-mediated cyanoalkylsulfonylation/arylation of active alkenes with cycloketone oxime derivatives via sulfur dioxide insertion. Adv. Synth. Catal. 362, 3004–3010 (2020).
Zhou, N. et al. Copper-catalyzed multicomponent oxysulfonylation of alkenes with cyclobutanone oxime esters and hydroxamic acids via the insertion of sulfur dioxide. Adv. Synth. Catal. 364, 4020–4025 (2022).
Yang, Y., Xiao, F., Lin, J. & Xiao, J. The shuttle of sulfur dioxide: iridium/copper-cocatalyzed trifluoromethylfluorosulfonylation of alkenes. Adv. Synth. Catal. 365, 301–306 (2023).
Zhang, M. et al. Direct synthesis of sulfonamides via synergetic photoredox and copper catalysis. ACS Catal. 13, 11580–11588 (2023).
Zhang, H. et al. Photocatalytic fluorosulfonylation of aliphatic carboxylic acid NHPI esters. Org. Chem. Front. 9, 4854–4860 (2022).
Sun, Y., Li, C., Xi, J., Wei, Z. & Liao, W. Electrochemical tandem cyclization to access sulfonylated fused sultams via SO2 insertion with sodium metabisulfite. Org. Chem. Front. 10, 705–711 (2023).
Liang, X. et al. Visible-light-driven electron donor–acceptor complex induced sulfonylation of diazonium salts with sulfinates. Green. Chem. 23, 8865–8870 (2021).
Hofman, K., Liu, N. & Manolikakes, G. Radicals and sulfur dioxide: a versatile combination for the construction of sulfonyl-containing molecules. Chem. Eur. J. 24, 11852–11863 (2018).
Chen, Z., Liu, N., Bolte, M., Ren, H. & Manolikakes, G. Visible-light mediated 3-component synthesis of sulfonylated coumarins from sulfur dioxide. Green. Chem. 20, 3059–3070 (2018).
Liu, Y. et al. Visible-light promoted one-pot synthesis of sulfonated spiro [4,5] trienones from propiolamides, anilines and sulfur dioxide under transition metal-free conditions. Chem. Commun. 55, 12212–12215 (2019).
Zhu, H., Shen, Y., Wen, D., Le, Z. & Tu, T. Selective synthesis of ortho-substituted diarylsulfones by using NHC-Au catalysts under mild conditions. Org. Lett. 21, 974–979 (2019).
Toutov, A. A. et al. Silylation of C–H bonds in aromatic heterocycles by an earth-abundant metal catalyst. Nature 518, 80–84 (2015).
Liu, W. et al. Potassium tert-butoxide-catalyzed dehydrogenative C–H silylation of heteroaromatics: a combined experimental and computational mechanistic study. J. Am. Chem. Soc. 139, 6867–6879 (2017).
Zhao, S. & Mankad, N. P. Cu‐catalyzed hydroxymethylation of unactivated alkyl iodides with CO to provide one-carbon-extended alcohols. Angew. Chem. Int. Ed. 57, 5867–5870 (2018).
Acknowledgements
This work is supported by National Natural Science Foundation of China (22301192), Sichuan Science and Technology Program (2024NSFSC1124), West China Hospital 135 project (ZYYC23017) and Postdoctor Research Fund of West China Hospital, Sichuan University (2024HXBH027). The authors would like to thank Shiyanjia Lab (www.shiyanjia.com) for the support of DFT calculation.
Author information
Authors and Affiliations
Contributions
Z.L. and X.Z. conceived the idea and led the project. F.Z., X.H., and M.Z. designed, conducted, and analyzed the experiments. X.H., F.Z., and X.Z. wrote the paper and prepared the Supplementary Information. N.L. interpreted the DFT data. Q.W. assisted in collecting new compounds.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhiming Li, Zhen Wang, and Ke Yang for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, F., He, X., Zhou, M. et al. Generation of perthiyl radicals for the synthesis of unsymmetric disulfides. Nat Commun 16, 23 (2025). https://doi.org/10.1038/s41467-024-55310-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55310-x
