
Abstract
Hyperactivation of ribosome biogenesis (RiBi) drives cancer progression, yet the role of RiBi-associated proteins (RiBPs) in breast cancer (BC) is underexplored. In this study, we perform a comprehensive multi-omics analysis and reveal that assembly and maturation factors (AMFs), a subclass of RiBPs, are upregulated at both RNA and protein levels in BC, correlating with poor patient outcomes. In contrast, ribosomal proteins (RPs) do not show systematic upregulation across various cancers, including BC. We further demonstrate that the oncogenic activation of a top AMF candidate in BC, DCAF13, enhances Pol I transcription and promotes proliferation in BC cells both in vitro and in vivo. Mechanistically, DCAF13 promotes Pol I transcription activity by facilitating the K63-linked ubiquitination of RPA194. This process stimulates global protein synthesis and cell growth. Our findings uncover a modification of RPA194 that regulates Pol I activity; this modification is dysregulated in BC, contributing to cancer progression.
Similar content being viewed by others

Introduction
Hyperactivation of RiBi results in excessive ribosome production, which can lead to genome instability1 and impaired translational fidelity2, ultimately contributing to the development and progression of various cancers3,4. RiBi is a complex, multistep process involving approximately 200 proteins5. These RiBPs facilitate the folding, processing, and modification of rRNA precursors (pre-rRNA), culminating in the production of mature 18S, 28S, and 5.8S rRNAs. They also play a crucial role in assembling these mature rRNA into pre-40S and pre-60S subunits6 within the nucleolus. Following their formation, pre-40S and pre-60S subunits are exported to the cytoplasm for further maturation. In the cytoplasm, the 40S and 60S subunits are assembled into the mature 80S ribosome, which is capable of translating mRNAs. Despite the recognized role of RiBi dysregulation in cancer, the specific alterations and potential contributions of various RiBPs in BC remain largely unexplored.
Aberrant increases in the size and number of the nucleoli, a consequence of hyperactive RNA polymerase I (Pol I) transcription, were linked to cancer a century ago and continue to serve as markers of poor prognosis in various cancer types7. Several oncogenic signaling pathways, such as MYC8, RAS/MAPK9, and PI3K/AKT10, contribute to the activation of Pol I transcription in cancer cells. Given that Pol I transcription serves as a rate-limiting step of RiBi11, its hyperactivation leads to enhanced RiBi activity, resulting in an excessive production of ribosomes within cells. Inspired by the important role of Pol I in cancer development and progression, several small molecules have been identified for targeting Pol I. These molecules have shown effectiveness in eradicating cancer cells12, with some even progressing to clinical trials13. However, despite the important role of Pol I transcription in diverse physiological and pathological processes, the mechanisms regulating its activity remain largely elusive.
In this research, we systematically investigate the association between RiBPs and BC, a cancer with the highest global prevalence14. Our study unveils a pervasive upregulation of AMFs in BC, and their upregulation is significantly associated with poor prognosis among BC patients. Surprisingly, we find that BC does not require a systematic upregulation of RPs, despite the expected excess of ribosomes in BC. Furthermore, we demonstrate that a top BC-associated AMF candidate, DCAF13, plays a pivotal role in promoting the proliferation of BC cells. It accomplishes this by orchestrating both Pol I transcription and pre-rRNA processing. Specifically, DCAF13 mediates K63-linked ubiquitination of RPA194, the largest subunit of Pol I15, thereby stimulating transcription. Our results highlight the crucial role that dysregulated pre-rRNA processing pathway plays in both hyperactivated RiBi and malignant cellular growth.
Results
Multi-omics analysis of RiBPs uncovers differential roles of AMFs and RPs in BC
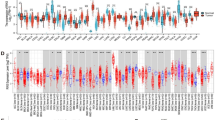
To investigate the contributions of dysregulated RiBi machinery in RiBi hyperactivation and BC, we compiled a comprehensive list of 300 RiBPs (Supplementary Data 1) involved in RiBi regulation from recent studies by Dörner et al. 16, Singh et al. 17 and the REACTOME database (v73)18. This list included 85 RPs and 215 AMFs (Supplementary Data 1, Supplementary Fig. 1a). Utilizing multi-omics data from TCGA19,20, we systematically investigate the dysregulation of these RiBPs across various cancer types. Consistent with the general hyperactivation of RiBi process across cancers21, we observed that 85.7% (1403/1638) of expression alterations in RiBPs were characterized by upregulation (Fig. 1a and Supplementary Fig. 1b–e). Intriguingly, while RiBi hyperactivation might suggest an expected stoichiometric upregulation of various RPs, our data revealed that at least one-third of the examined cancer types, including BC, did not exhibit a systematic upregulation of RPs (Fig. 1a, b). In contrast, AMFs were broadly upregulated across different cancer types (Fig. 1a, b). These observations indicate two distinct RiBP remodeling patterns across cancers, reflecting the differential regulation of RPs and AMFs by distinct signaling pathways within cells22. Furthermore, proteomics data in BC showed that the protein expression level of AMFs was systematically elevated, whereas most RPs remained largely unaltered (Fig. 1c, d).

a Patterns of RiBP dysregulation across 15 different cancer types. Variations in color intensity correspond to the log2-fold change (log2FC) in expression levels. The upper bar plot illustrates the frequency with which each RiBP is dysregulated across the cancer types examined. A mutation heatmap reveals the proportion of BC patients with RiBP mutations. b Bar graphs depicting the percentage of significantly up-regulated AMFs (blue) and RPs (red) across 15 cancer types. c Heatmap of RiBPs proteome profiles in BC patients (Tumor n = 133, Normal n = 18, from CPTAC project70). d Stacked bar chart showing the counts of up-regulated, unchanged and down-regulated RPs and AMFs as inferred from differential proteomic analysis. e The mutation frequency of core-AMFs (n = 94), known oncogenes (n = 206), tumor suppressor genes (TSGs, n = 220), and transcription factors (TFs, n = 1353) in BC patients. f–g Boxplots illustrating the log2FC in the mRNA expression level of core-AMFs across different tumor grades (Grade 1, n = 183; Grade 2, n = 632; Grade 3, n = 253) (f) and clinical subtypes (HR, n = 657; HER2, n = 153; TN, n = 170) (g) compared to normal in BC patients. h Kaplan3-Meier survival analysis of TCGA-BC dataset (high n = 546, low n = 540) based on predictive survival using 113 core-AMFs gene signatures as determined by gene expression levels. i Gene silencing effects of 113 core-AMFs using open-source shRNA data in 16 BC cell lines and 200 other cancer cell lines. Distribution of log2 ratios between final cell populations and initial DNA pool for core-AMFs, known oncogenes, known TSGs, and non-cancer genes. e–g, i, Box plots show the distribution of data across groups. The central line within the box represents the median value. The upper and lower edges of the box represent the 75th and 25th percentiles, respectively (Interquartile Range, IQR). The whiskers extend from the box to the maximum and minimum values within 1.5 times the interquartile range (IQR), with any data points beyond this range considered as outliers. Data shown as mean ± SEM. Statistical analysis between all groups was conducted by two-sided Wilcoxon rank-sum test (e–g, i) or two-sided Log-rank (Mantel-Cox) test (h).
Next, we focused our investigation on understanding the role of AMFs in BC, which has the highest number of dysregulated AMFs among cancers lacking systematic upregulation of RPs (Fig. 1b). AMFs that consistently upregulated in over half of the TCGA cancer types we investigated were categorized into a core group, termed ‘core-AMFs’, and 113 such core-AMFs were identified (Supplementary Data 1). Notably, core-AMFs showed a higher frequency of copy number variation (CNV) gains and fewer mutations than known tumor suppressor genes (TSGs) in BC (Fig. 1e, and Supplementary Fig. 1f). Additionally, they exhibited a comparable amplification and mutation rate to known oncogenes and TFs (Fig. 1e, Supplementary Fig. 1f). Elevated expression levels of core-AMFs also correlated significantly with higher tumor grade (Fig. 1f) and more aggressive BC subtypes (Fig. 1g). Importantly, elevated expression levels of core-AMFs, but not of RiBPs that remained unaltered in their expression in BC, correlated with poorer patient prognosis in BC (Fig. 1h, Supplementary Fig. 1h).
To investigate the impact of core-AMFs on cancer cell proliferation, we analyzed RNAi data from short hairpin RNA (shRNA) screens of 14,222 genes in 16 BC and 200 other cancer cell lines23. The core-AMFs exhibited significant inhibition of cell proliferation upon silencing, comparable to well-characterized oncogenes and higher than TSGs and non-cancer genes (Fig. 1i). These results suggest that core-AMFs have strong pro-growth functions in BC cells. To further validate the biological functions of AMFs in BC cell lines, we selected three core-AMFs: DCAF13 (DDB1 And CUL4 Associated Factor 13), DKC1 (Dyskerin Pseudouridine Synthase 1), and RPP40 (Ribonuclease P/MRP Subunit P40), based on their intersection across both the TCGA and Chia-Ho cohorts. These genes were not only significantly upregulated in BC but also ranked among the top 10 most survival-associated AMFs in both cohorts (Fig. 2a-c, Supplementary Fig. 1g and Supplementary Fig. 2a, b). Among them, DCAF13 is a known component of the Small Subunit (SSU) processome24 and shares orthology with Sof1, a yeast protein implicated in rRNA processing within yeast25; DKC1 is the pseudouridine synthase present in all H/ACA small nucleolar ribonucleoproteins (snoRNPs)26; RPP40 is a 40 kDa subunit of ribonuclease MRP, exhibiting multifunctional roles beyond its involvement in rRNA processing27. Using the surface sensing of translation (SUnSET) assay, a radioactivity-independent method for monitoring new protein synthesis28, we demonstrated that downregulating these core-AMFs resulted in reduced global protein synthesis rates in different BC cell lines (Fig. 2d and Supplementary Fig. 2c–e). Upon depletion of these core-AMFs (Supplementary Fig. 2f, g), we observed substantial reductions in cell proliferation (Fig. 2e and Supplementary Fig. 2h, i) and colony growth (Fig. 2f and Supplementary Fig. 2j) across different BC cells, signifying their roles in cancer promotion. Moreover, the knockdown of these three core-AMFs in normal cell line MCF10A (Supplementary Fig. 2k), which exhibits a lower expression level of these factors compared to cancer cell lines, resulted in a less pronounced growth inhibition effect than that observed in cancer cells (Fig. 2g). This result suggests that cancer cells are more dependent on AMFs than normal cells. Collectively, our data revealed the pivotal role of AMFs in driving cancer, possibly through the activation of RiBi.

a The top 10 BC prognosis-associated RiBPs from the TCGA-BC and Chia-Ho cohorts. The upper bar chart displaying the -log10 p-value for these RiBPs, derived from Kaplan-Meier overall survival analysis. The middle and lower heatmap showing the p-value and associated log2FC for these RiBPs, respectively. b Violin plots illuminating the differential mRNA expression levels of DCAF13, DKC1, and RPP40 between BC and normal tissue samples. c Kaplan-Meier overall survival curves elucidating the prognostic significance of DCAF13, DKC1, and RPP40 mRNA expression levels in BC patients. d A SUnSET assay executed on MDA-MB-231 cells following the knockdown of each of the three aforementioned AMFs. On the right is the corresponding statistical graph (n = 3 biological replicates). e, f CCK8 assay (e) and colony formation assay (n = 3 biological replicates) (f) administered to MDA-MB-231 cells after knockdown of DCAF13, DKC1, and RPP40, respectively. g Colony formation assay performed on MCF10A cells following the knockdown of DCAF13, DKC1, and RPP40, respectively. The corresponding statistical graph is shown on the right (n = 3 biological replicates). b Box plots show the distribution of data across groups. The central line within the box represents the median value. The upper and lower edges of the box represent the 75th and 25th percentiles, respectively (Interquartile Range, IQR). The whiskers extend from the box to the maximum and minimum values within 1.5 times the interquartile range (IQR), with any data points beyond this range considered as outliers. a–d, f, g Data shown as mean ± SEM. P-values of survival analysis were calculated using the two-sided Log-rank (Mantel-Cox) test, p-value of differential expression analysis were calculated using the Likelihood Ratio Test, and subsequently adjusted using the Benjamini-Hochberg (BH) method to obtain False Discovery Rate (FDR) value (a). Statistical significance was calculated by two-sided Wilcoxon rank-sum test (b), two-sided Log-rank (Mantel-Cox) test (c), two-tailed unpaired Student’s t-test (d, f, g). Source data are provided as a Source Data file.
DCAF13 is the most amplified RiBP with prognostic value for BC
Among the three core-AMFs subjected to functional analysis, DCAF13 emerges as the most survival-associated (Fig. 2a-c). Additionally, it stands out as the most frequently amplified RiBP in BC (Fig. 3a and Supplementary Fig. 3a; CNV gain observed in 17% of patients). Therefore, we selected DCAF13 for further functional analysis. DCAF13, primarily localized in the nucleolus, is pivotal in the processing and maturation of pre-rRNA29, playing a critical role in the assembly and maturation of ribosomes. Due to the extensive and important functions of DCAF13, we performed a thorough exploration to better understand its roles and underlying molecular mechanisms as a core-AMF. RNA immunoprecipitation (RIP) assay coupled with real-time quantitative reverse transcription PCR (qRT-PCR), and ChIRP30 assay demonstrated that DCAF13 binds to pre-rRNA in vivo (Fig. 3b–e), supporting that DCAF13 is indeed an AMF. Notably, there was a significant positive correlation between DCAF13 mRNA expression level and copy number variations (p-value = 6.89e-270, Fig. 3f), implying that the upregulation of DCAF13 in BC might, at least in part, be attributable to its amplification. Consistently, DCAF13 amplification was associated with unfavorable overall survival in BC patients (p-value = 0.003211, Fig. 3g), mirroring the upregulation of DCAF13 mRNA in BC (Fig. 2b, c). To investigate whether the elevated mRNA expression of DCAF13 results in the accumulation of DCAF13 proteins in BC, we performed an immunohistochemistry (IHC) assay on 140 matched BC samples. Our results revealed a significant increase in DCAF13 protein levels in BCs compared to adjacent normal breast tissues (p-value < 0.0001, Fig. 3h, i). Moreover, Kaplan-Meier analysis further confirmed a significant correlation between higher DCAF13 protein expression in BC and shorter overall survival of patients (p-value = 0.0443, Fig. 3j).

a Heatmap depicting the amplification frequency for three core-AMFs, CUL4-DDB1-DCAF13 complex genes, and other DCAF family members. b Western blot analysis of DCAF13 levels to validate efficiency of DCAF13 capture in RIP samples. c qRT-PCR assays confirming enrichment of U3 and pre-rRNA, but not ACTIN mRNA in DCAF13 RIP products (n = 3 biological replicates). d qRT-PCR assays in the upper panel demonstrating enrichment of U3 and pre-rRNA (n = 3 biological replicates). The lower panel displaying localization of ChIRP probes in on pre-rRNA. e ChIRP assays indicating successful capture of DCAF13 and DDX21 by both pre-rRNA and U3 probes, but not GAPDH. f Scatter plot illustrating correlation between DCAF13 mRNA expression level and CNV. X-axis denotes CNV segment value, y-axis denotes DCAF13 mRNA expression level, dot colors indicate CNV type. g Kaplan-Meier overall survival analysis for CNV in BC. h IHC staining of DCAF13 in tumor and normal tissues. i Paired box plot showing the H-score among paired normal and tumor patient tissues (n = 140 per group). j Kaplan–Meier overall survival analysis of IHC rating scores in BC. k-n Association between DCAF13 mRNA expression and clinicopathological factors: age (Age <= 40 years, n = 116; Age > 40 years, n = 1788) (k), tumor size (Size <10 cm, n = 37; Size >= 10 cm, n = 1847) (l), tumor grade (Grade 1, n = 165; Grade 2, n = 740; Grade 3, n = 927) (m), and molecular subtypes (HR, n = 1369; HER2, n = 236; TN, n = 299) (n). i, k–n Box plots show the distribution of data across groups. The central line within the box represents the median value. The upper and lower edges of the box represent the 75th and 25th percentiles, respectively (Interquartile Range, IQR). The whiskers extend from the box to the maximum and minimum values within 1.5 times the interquartile range (IQR), with any data points beyond this range considered as outliers. Scale bars,100μm (h). c, d, f, g, i–n Data shown as mean ± SEM. Statistical tests: two-tailed unpaired Student’s t-test (c, d, i), two-sided Pearson and Spearman correlation coefficient (f), two-sided Log-rank (Mantel-Cox) test (g, j), two-sided Wilcoxon rank-sum test (k–n). Source data are provided as a Source Data file.
Correlating DCAF13 mRNA expression with the clinicopathological status of patients with BCs, we observed a significant association between high DCAF13 mRNA expression and younger patient age (p-value = 0.00066, Fig. 3k), larger tumor size (p-value = 0.016, Fig. 3l), higher tumor grade (Fig. 3m), and more aggressive subtypes (Fig. 3n). Importantly, elevated DCAF13 expression served as an independent prognostic factor for BCs in a multivariate Cox regression analysis (p-value = 0.00021, Supplementary Fig. 3b). These results suggested that DCAF13 is associated with the development and progression of BC. Analysis of DCAF13 mRNA expression in both normal and cancer tissues from various cancer types within TCGA, including BC, revealed a consistent elevation of DCAF13 expression in cancerous tissues when compared to their normal counter (Supplementary Fig. 3c). This finding suggested a conserved cancer-promoting function of DCAF13 across various cancer types.
DCAF13 promotes RiBi by coordinating Pol I transcription and pre-rRNA processing
Next, we verified the DCAF13’s capacity to promote RiBi by overexpressing it in cancer cells. The SUnSET assay showed that ectopic expression of DCAF13 substantially increased the abundance of newly synthesized proteins in two BC cell lines (Fig. 4a), suggesting an increase in RiBi rates. To decipher how DCAF13 promotes RiBi, we performed a co-immunoprecipitation (co-IP) assay using an antibody targeting endogenous DCAF13. The association proteins were subjected to silver staining (Fig. 4b) and mass spectrometry (MS) (Fig. 4c). Notably, our complex enrichment analysis revealed that DCAF13-binding proteins showed a prominent enrichment in the SSU processome responsible for 18S rRNA processing24, indicating DCAF13’s essential role in this complex17 (Fig. 4c, Supplementary Data 3). We also found a significant enrichment of proteins linked to the Pol I complex in the co-IP study (Fig. 4c), suggesting that DCAF13 may coordinate both Pol I transcription and pre-rRNA processing. In light of the rate-limiting role of Pol transcription in the RiBi process31, our subsequent investigations centered on DCAF13’s regulatory role in Pol I transcription.

a Overexpression of DCAF13 promotes the synthesis rate of nascent proteins in MDA-MB-231 and MDA-MB-468 cells. b Silver staining assay identifying proteins interacting with DCAF13 following Immunoprecipitation with anti-DCAF13 antibody. c Complex enrichment analysis of DCAF13-interacting proteins, with log10 p-value calculated using Benjamini linear step-up correction method. d IF assays detecting the localization of DCAF13 and RPA194 in MDA-MB-231 cells treated with DMSO, ActD, or transfected with siRNA targeting to DCAF13. e IF assays of full-length DCAF13 or truncated DCAF13 using anti-HA antibody to visualize its localization. f 5-Ethynyl uridine (EU) assays of MDA-MB-231 cells transfected with either a scramble or siDCAF13. g, h qRT-PCR assay detecting the RNA levels of DCAF13 and nascent pre-rRNA after knocking down (siD13) (g) or overexpressing (oeD13) (h) DCAF13 in MDA-MB-231 cells (n = 3 biological replicates). i IF assays in MDA-MB-231 cells treated with DMSO, ActD, or transfected with siDCAF13, siDDB1, and siCUL4A, with simultaneously stained for UBF and NPM. j RNA-fluorescence in situ hybridization (RNA FISH) assay of MDA-MB-231 cells treated with DMSO or ActD using a Cy3-U3 DNA probe, co-staining with NPM and UBF antibodies, to label U3 and nucleolar markers concurrently. k-l Co-IP assay of MDA-MB-231 cells using either anti-IgG, anti-DCAF13 (k), or anti-RPA194 antibody (l), and analyzed by Western blotting with indicated antibodies. Scale bars, 5μm (d–f, i, j). g, h Data shown as mean ± SEM. Statistical significance was calculated by two-tailed unpaired Student’s t-test. Source data are provided as a Source Data file.
To confirm DCAF13’s influence on Pol I activity, we performed immunofluorescence (IF) assays, which demonstrated the colocalization of DCAF13 with RPA194 (Fig. 4d). The RNA-binding domain SOF1 deletion, but not the WD40 domain32, disrupted DCAF13’s nucleolar localization (Fig. 4e and Supplementary Fig 4a). We then used 5-ethynyl uridine (EU) labeling to examine new RNA synthesis33. DCAF13-depleted cells exhibited markedly reduced nucleoli-associated EU signals compared to controls (Fig. 4f). Consistently, qRT-PCR analysis revealed a decrease in pre-rRNA levels upon DCAF13 knockdown (Fig. 4g) and an increase following its overexpression (Fig. 4h). To further validate the regulation of DCAF13 on Pol I activity, we constructed an inducible degradation system by knocking in FKBP12F36V into the N-terminal of DCAF13 using CRISPR/Cas9 technology34 (Supplementary Fig 4b). This modification allows the rapid degradation of FKBP12F36V-3×FLAG-tagged DCAF13 within 30 min (Supplementary Fig 4c). As DCAF13 levels decreased, we observed a corresponding reduction in Pol I activity (Supplementary Fig 4d). Notably, DCAF13 depletion led to segregation of nucleolar components and the formation of ‘nucleolar caps’35, a hallmark of Pol I inhibition36, similar to Actinomycin D (ActD) treatment effects37 (Fig. 4i and Supplementary Fig 4e–g). Intriguingly, following ActD treatment, DCAF13 is displaced from the nucleolus, whereas its binding RNA U3 remains within the nucleolus (Fig. 4d, j). This suggests that the localization of DCAF13 in the nucleolus may be associated with pre-rRNA, rather than with U3. These results collectively demonstrate that DCAF13 boosts RiBi by enhancing Pol I transcription.
In terms of the mechanism underlying DCAF13’s regulation of Pol I transcription, we evaluated its interaction with Pol I complex subunits. Western blot analysis of anti-DCAF13 immunoprecipitants revealed the highest binding affinity between RPA194 and DCAF13 among Pol I subunits (Fig. 4k and Supplementary Fig. 4h). Additional co-IP and western blot analysis of anti-RPA194 immunoprecipitants confirmed this interaction (Fig. 4l). SiRNA-resistant SFB-RPA194 alone failed to restore normal protein synthesis and cellular proliferation when both DCAF13 and RPA194 were knocked down (Supplementary Fig. 4i, j), underlying the dependency of RPA194’s function on DCAF13. Collectively, these results suggest that DCAF13 may regulate Pol I transcription by interacting with RPA194.
DCAF13 and USP36 dynamically modulate K63-linked ubiquitination on RPA194, essential for Pol I activity
Given that DCAF13 can function within a CUL4-DDB1-DCAF13 E3 ligase complex38, we sought to investigate whether Pol I transcription is regulated by the E3 ligase activity of DCAF13. Strikingly, depleting either CUL4A or DDB1 phenocopies the effects observed with both DCAF13 depletion and ActD treatment, resulting in the formation of ‘nucleolar caps’ (Fig. 4i and Supplementary Fig. 4e–g). This led us to hypothesize that the CUL4A-DDB1-DCAF13 ubiquitin ligase complex could target RPA194 for ubiquitination. In line with this, depletion of endogenous DCAF13 resulted in a decrease in ubiquitin-conjugated RPA194 levels (Fig. 5a). Conversely, overexpression of DCAF13 increased these levels (Fig. 5b), providing evidence for DCAF13-dependent ubiquitination of RPA194 in vivo. DCAF13 includes WD40 and SOF1 domains. The SOF1 domain is associated with the nucleolar localization of DCAF13, while the WD40 domain is responsible for interacting with the DDB1 component of the Cullin4-RING E3 ligase (CRL4) complex39 and substrate recognition and binding40. Depleting either domain abrogates the ubiquitination activity of DCAF13 (Fig. 5c) and inhibit pre-rRNA synthesis (Supplementary Fig. 5a).

a, b Western blot analysis of the ubiquitination levels of RPA194 in MDA-MB-231 cells subjected to DCAF13 knockdown (a) or overexpression (b), followed by HA-Ub transfection for 48 hours. c Western blot analysis of the ubiquitination levels of RPA194 in HeLa cells co-transfected with full-length DCAF13 or domain-depleted DCAF13, along with HA-Ub. d Western blot analysis of the ubiquitination levels of RPA194 in MDA-MB-231 cells transfected with either scramble or siDCAF13 for 24 hours, followed by a subsequent 24-hour transfection with HA-ubiquitin (Lys48 or Lys63-specific). e Western blot analysis of K63-linked ubiquitination levels of RPA194 in HeLa cells subjected to serum starvation and subsequent serum activation. f Western blot analysis of ubiquitination levels of RPA194 in HeLa cells transfected with either scramble or siUSP36 for 24 hours, followed by an additional 24-hour transfection with HA-K48 or HA-K63. g Co-IP assays on HeLa cells transfected with pKO-SFB-USP36 to explore interactions between RPA194 and USP36. h IF assays detecting the localization of USP36 and RPA194 in HeLa cells treated with DMSO, ActD, or transfected with siUSP36. Scale bars, 5μm (h). Source data are provided as a Source Data file.
To identify the specific type of ubiquitin linkage that DCAF13 adds to RPA194, we employed ubiquitin mutants that exclusively permit K48 or K63 linkages by introducing lysine residues at only one of these positions. Cells expressing only K63 ubiquitin, where K63 was the sole available lysine residue, exhibited substantially higher levels of ubiquitin-conjugated RPA194 compared to cells expressing K48 ubiquitin (Fig. 5d). Importantly, DCAF13 knockdown dramatically diminished the levels of K63-linked, but not K48-linked, ubiquitin-conjugated RPA194 (Fig. 5d). We further examined whether K63-linked ubiquitination levels on RPA194 were correlated with Pol I activity, which is known to be modulated by serum starvation and refeeding41. Interestingly, serum-starved cells exhibited a noticeable decrease in the levels of K63-linked ubiquitination on RPA194, which was rapidly restored upon serum refeeding (Fig. 5e). Moreover, the level K63-linked ubiquitination in RPA194 also decreased after cells were subjected to ActD treatment (Fig. 5e). Collectively, our results strongly suggest that DCAF13 regulates Pol I activity by promoting K63-linked ubiquitination of RPA194.
Ubiquitin signaling is a dynamic process that finely tunes protein activity by the coordinated actions of E3 ligases and deubiquitinating enzymes (DUBs)42. To identify DUBs involved in regulating K63-linked ubiquitination of RPA194, we analyzed immunofluorescence data for 54 DUBs sourced from The Human Protein Atlas database, antibody company websites, and published works43,44,45,46,47,48,49 (Supplementary Fig. 5b). Of these, only USP36 and USP51 were specifically localized to the nucleolus (Supplementary Fig. 5b). Ectopic expression of USP36-GFP and USP51-GFP confirmed their nucleolar localization (Supplementary Fig. 5c). Notably, only USP36 overexpression can effectively remove K63-linked ubiquitination from RPA194, whereas USP51 cannot (Supplementary Fig. 5d). Prior studies in yeast have shown that the nucleolus-localized USP36 can remove K48-linked ubiquitination from RPA19450, thereby stabilizing it. In human cells, USP36 has been shown to remove K48-linked ubiquitination induced by BMH-21 from RPA19412. We confirmed the interaction between USP36 and RPA194 in human cells using reciprocal immunoprecipitation (Fig. 5g and Supplementary Fig. 5e, f). Remarkably, the knockdown of USP36 resulted in a notable elevation in the levels of K63-linked ubiquitin-conjugated RPA194, without affecting its K48-linked counterpart (Fig. 5f and Supplementary Fig. 6a, b), while overexpression of USP36 effectively removed K63-linked ubiquitination from RPA194 (Supplementary Fig. 6c, d). These results suggest USP36 may mainly be responsible for the regulation of K63-linked ubiquitination level on RPA194 when cell under normal cellular condition. Unexpectedly, USP36 depletion led to the formation of ‘nucleolar caps’ (Fig. 5h), highlighting the important role of proper deubiquitination of RPA194 in regulating of Pol I transcription.
DCAF13 mediates K63-linked ubiquitination at K1180 and K1184 sites on RPA194
To demonstrate that DCAF13 promotes Pol I transcription by enhancing K63-linked ubiquitination of RPA194, we sought to identify the ubiquitination sites on RPA194 mediated by DCAF13. First, we expressed RPA194 segment plasmids in Escherichia coli, isolated and purified the corresponding proteins, and conducted an in vitro GST pull-down assay to detect the binding region of RPA194 with DCAF13. We found that DCAF13 exhibits high affinity for the N-terminal fragment of RPA194, and also shows considerable affinity for its C-terminal fragment (Fig. 6a, Supplementary Fig. 7a), suggesting the ubiquitination sites may be located on these two sections of RPA194. Leveraging mass spectrometry data from Kim et al. 51, we identified seven potential ubiquitination sites within this C-terminal fragment and five in the N-terminal fragment (Supplementary Fig. 7a) for further verification. Intriguingly, mutations of all potential ubiquitination sites in the C-terminal, but not the N-terminal, of RPA194 led to the formation of ‘nucleolar caps’, indicating the RPA194 mutant has an impaired function in rDNA transcription (Fig. 6b)36. Mutation analysis showed mutation of K1180/1184, positioned closely on the surface of Pol I (Fig. 6d), lead to a significant reduction in K63-linked ubiquitination levels (Fig. 6c). Ectopic expression of DCAF13 significantly enhanced the K63-linked ubiquitination of WT RPA194, but not of the K1180/1184 R mutant (Fig. 6e). Moreover, in contrast to several other lysine sites located on the periphery of the pol I complex, such as K1101R and K1574R, only the K1180/1184 R mutation in RPA194 spontaneously induced the formation of ‘nucleolar caps’ (Supplementary Fig. 7b), which was consistent with the results obtained in EGFP-FBL Knock-in Hela cells (Fig. 6f and Supplementary Fig. 7c). These findings indicate that DCAF13 mediates the K63-linked ubiquitination at the K1180/1184 sites on RPA194.

a GST pull-down assay using purified GST, GST-RPA194-N, GST-RPA194-M, GST-RPA194-C and DCAF13 protein in MDA-MB-231 cell lysates, and employing anti-GST beads as the bait, demonstrated direct binding region. b After transfection with FN-RPA194-WT, FN-RPA194-Nmut and FN-RPA194-Cmut plasmid respectively, the Hela cells were directly subjected to IF experiments. The subcellular localization and morphology of the different forms of RPA194 were then observed under confocal microscopy. c After transfecting the RPA194 single point mutant plasmid and HA-K63 in Hela cells, the ubiquitination levels of different mutant form of RPA194 were analyzed by western blot. d The spatial structure of the Pol I complex mapped by ChimeraX, where K1101, K1180, K1184 and K1574 are located on the periphery of the complex. e Western blot analysis of the effect about over-expression of DCAF13 on K63-linked ubiquitination of RPA194 wild type and mutant. f IF assay of EGFP-FBL knock-in Hela cells after transfected with Ruby3-RPA194-WT or Ruby3-RPA194-K1180/1184 R. Scale bars, 5 μm (b, f). Source data are provided as a Source Data file.
DCAF13-mediated K63-linked ubiquitination of RPA194 promotes Pol I transcription
We next investigated how the K63-linked ubiquitination of RPA194 mediated by DCAF13 regulates Pol I activity. While K63-linked ubiquitination has been shown to induce targeted protein degradation52, our data revealed that neither the depletion nor ectopic expression of DCAF13 affected the protein stability of RPA194 (Fig. 7a and Supplementary Fig. 8a-c). Additionally, neither the knockdown nor the ectopic expression of USP36 influenced the protein expression of RPA194 (Supplementary Fig. 8d, e). Furthermore, the depletion of DCAF13 had no impact on the protein expression levels of various Pol I subunits or the interaction among them (Fig. 7b). These results suggest that K63-linked ubiquitination does not regulate the Pol I activity by modulating the protein stability of RPA194 or Pol I holoenzyme assembly.

Western blot analysis of RPA194 levels in MDA-MB-231 cells transfected with either scramble control or siDCAF13 and subsequently treated with 10 μg/mL cycloheximide (CHX) for specified durations. b Western blot analyses detecting POLR1B, POLR1C, POLR1D, and POLR1E levels in MDA-MB-231 cells transfected with either scramble control or siDCAF13, following immunoprecipitation with an anti-POLR1A antibody. c Chromatin immunoprecipitation sequencing (ChIP-Seq) assay identifying RPA194 enrichment on rDNA in MDA-MB-231 cells transfected with either scramble control or siDCAF13. Read normalization was accomplished using Counts Per Million (CPM) via deepTools; the structure of 18S, 5.8S, and 28S rRNA genes is depicted below the profile. d ChIP-qPCR analysis detecting the binding of TAF1C to rDNA in MDA-MB-231 cells transfected with scramble control or siDCAF13 (n = 3 biological replicates). e ChIP-qPCR analysis detecting the binding of Pol I to rDNA in Hela cells transfected with EV, FN-RPA194-WT or FN-RPA194-K1180/1184 R (n = 3 biological replicates). f Western blot analysis of the levels of K63-linked ubiquitination of RPA194 in HeLa cells transfected with either scramble control or siCK2. g qRT-PCR assays measuring RNA levels of CK2 and pre-rRNA in HeLa cells transfected with siCK2 (n = 3 biological replicates). h Western blot analysis of the levels of K63-linked ubiquitination of RPA194 in HeLa cells transfected with an oe-DCAF13 plasmid and/or siCK2. d, e, g Data shown as mean ± SEM. Statistical significance was calculated by two-tailed unpaired Student’s t-test. Source data are provided as a Source Data file.
Since the Pol I transcription is inhibited when DCAF13 is depleted, we investigated the rDNA occupancy of Pol I in MDA-MB-231 cells by using chromatin immunoprecipitation followed by high-throughput DNA sequencing (ChIP-Seq) upon DCAF13 knockdown. Our results showed that DCAF13 depletion significantly reduces the binding of Pol I to the non-promoter region of rDNA (Fig. 7c), while its occupancy at the promoter region remains unaffected. This lack of effect on Pol I recruitment to the promoter suggests that DCAF13 might not be involved in the regulation of pre-initiation complex (PIC) formation41. Consistently, ChIP-qPCR assays showed that DCAF13 knockdown did not affect the occupancy of TAF1C at the rDNA promoter region (Fig. 7d). Furthermore, the K1180/1184 R RPA194 mutant showed significantly reduced occupancy than WT RPA194 in non-promoter rDNA regions, but similar occupancy at the promoter (Fig. 7e). Collectively, these findings suggest that DCAF13-mediated K63-linked ubiquitination of RPA194 is required for the Pol I transcription, it does not influence the recruitment of Pol I to the promoter region.
Pol I transcription is regulated by casein kinase 2 (CK2)53. Notably, when CK2 was knocked down (Supplementary Fig. 8f) or inhibited with the CK2-specific inhibitor CX-494554, both the levels of K63-linked ubiquitination on RPA194 (Fig. 7f and Supplementary Fig. 8g) and pre-rRNA levels were significantly reduced (Fig. 7g and Supplementary Fig. 8h). Furthermore, the over-ubiquitination of RPA194 induced by ectopic expression of DCAF13 was completely abrogated when CK2 was either absent or its activity inhibited by CX-4945 (Fig. 7h and Supplementary Fig. 8i). These results suggest that CK2 plays a critical role in regulating DCAF13-mediated K63-linked ubiquitination of RPA194.
DCAF13 promotes BC tumor growth both in vitro and in vivo
Finally, we sought to validate the cancer-promoting function of DCAF13. In transient overexpression experiments involving two distinct BC cell lines (Supplementary Fig. 9a), we found that ectopic expression of DCAF13 significantly enhanced the proliferation of both MDA-MB-231 and MDA-MB-468 cells (Fig. 8a, b). Importantly, the knockdown of RPA194 could be rescued by reintroducing WT RPA194, but not by K1180/1184 R mutant of RPA194 (Fig. 8c), highlighting the important role of DCAF13-mediated K63-linked ubiquitination at K1180/1184 on RPA194 in driving the proliferation of BC cells. Flow cytometry analysis demonstrated that DCAF13-depleted cells exhibited elevated levels of the G2/M population, accompanied by a corresponding reduction in the G1 population in different BC cells (Fig. 8d and Supplementary Fig. 9b). In addition, DCAF13 knockdown induced massive apoptosis in two BC cell lines (Fig. 8e and Supplementary Fig. 9c). These observations firmly establish the role of DCAF13 in promoting the proliferation of BC cells by accelerating cancer cell cycle progression and suppressing apoptosis.

a, b CCK8 assay (a) and colony formation assay (b) conducted on MDA-MB-231 and MDA-MB-468 cells following the overexpression of DCAF13 (n = 3 biological replicates). c Colony formation assay of MDA-MB-231 cells transfected with either siNC or siRPA194 alone, and included cells transfected with the EV, FN-RPA194-WT, or FN-RPA194-K1180/1184 R plasmid respectively in combination with siRPA194 (n = 3 biological replicates). d, e Flow cytometry analyses assessing cell cycle alterations (d) and apoptotic events (e) in MDA-MB-231 and MDA-MB-468 cells after knocking down DCAF13 (n = 3 biological replicates). f Representative images depicting resected tumors harvested from BALB/c nude mice, which were inoculated with MDA-MB-231 cells stably expressing either the Empty Vector (control), WT DCAF13, shNC (control), or shDCAF13 plasmids. g, h Longitudinal measurements of tumor volumes collected at predetermined intervals (n = 8/group for EV/oe-DCAF13, n = 9/group for shNC/shDCAF13). i The weights of the excised tumors recorded at the designated end point of the study (n = 6/group for EV/oe-DCAF13, n = 7/group for shNC/shDCAF13). j Schematic model of this study drawn by eBioart of Guangzhou Guorenqidian Shengwukeji Co., Ltd. DCAF13 facilitates K63-linked ubiquitination of RPA194, which can be removed by USP36. The elevated expression of DCAF13 in BC leads to hyper-ubiquitination of RPA194 and boosts the transcription activity of Pol I, promoting BC progression through increasing protein synthesis rates. a–e, g–i, Data shown as mean ± SEM. Statistical significance was calculated by two-tailed unpaired Student’s t-test. Source data are provided as a Source Data file.
We further sought to investigate the impact of DCAF13 on tumor growth in vivo. To achieve this, we subcutaneously injected MDA-MB-231 cells with stable integration of Tet-inducible shRNA or ectopically expressed DCAF13 into nude mice. Notably, the induction of doxycycline (dox) led to the depletion of DCAF13, significantly inhibiting the proliferation of these cells in vivo (Fig. 8f, h, i). Conversely, the ectopic expression of DCAF13 strikingly enhanced the tumorigenic potential of these cells in vivo (Fig. 8f, g, i). Collectively, our results unequivocally demonstrate the ability of DCAF13 to promote breast tumor growth both in vitro and in vivo.
Discussion
Hyperactivation of RiBi is known to promote cancer, yet the specific alterations in RiBi machinery in cancer remain unclear. Our study uncovers a systematic remodeling of RiBPs across different cancer types and identifies a close association between the AMFs and BC. Significantly, we showed that the top candidate AMF, DCAF13, promotes the proliferation of BC cells by activating Pol I transcription. We demonstrated that DCAF13 facilitates Pol I transcription by introducing K63-linked ubiquitination to its largest subunit, RPA194 (Fig. 8j). Our study revealed a cancer-related modification on RPA194, which holds promise as a potential therapeutic target.
Although the RiBi hyperactivation in cancer suggests a need for increased RPs, we found that in over one-third of the cancers studied, most RPs weren’t significantly upregulated. This unexpected finding is reminiscent of prior findings indicating an abundant supply of various RPs readily available for ribosome assembly in normal cells, with RiBi rates dependent on the rate of rRNA synthesis55. Therefore, it is possible that in these cancer types with normal RP expression, the cells from which they originate may possess a sufficient quantity of RPs to generate an adequate number of ribosomes, thereby supporting cancer development and progression. Our findings suggest that in these cancers with normal RP expression, the vulnerability may lie in the rRNA generation system, making it a potential target for intervention.
The role of AMFs, especially core-AMFs, in cancer development and progression is not yet fully understood. Our research reveals that DCAF13, a core-AMF, promotes BC cell proliferation by activating Pol I transcription, a rate-limiting step in RiBi. The hyperactivation of RiBi has been linked to genome instability, proteotoxic stress, decreased translation fidelity, and alter mRNA translation patterns, all of which contribute to both the initiation and progression of various cancers21. Mendillo et al. showed that enhanced RiBi can induce the activation of heat-shock factor1 (HSF1)-dependent transcriptional program, which may drive cancer progression56. It appears that different AMFs influence cancer via unique mechanisms. For instance, overexpression of core-AMF FBL in BC cells triggers rRNA hyper-methylation, resulting in the generation of ‘onco-ribosomes’ characterized by impaired translational fidelity and increased internal ribosome entry site (IRES)-dependent translation initiation of key oncogenes57. In Hepatocellular carcinoma (HCC), another core-AMF, TRMT112, is upregulated, leading to elevated N6-methyladenosine (m6A) modifications on 18S rRNA. This modification causes the ribosomes to selectively translate mRNAs involved in fatty acid metabolism, ultimately contributing to the progression of HCC58. Our study and others suggest that AMF upregulation not only boosts ribosome production to meet the needs of rapidly proliferating malignant cells but also reconstructs the ribosomes into ‘onco-ribosomes’. These specialized ribosomes accelerate cancer cell evolution, enabling quick adaption to varying conditions2.
Although several AMFs are known to regulate Pol I activity59, the specific mechanisms remain elusive. In this study, we identified a core-AMF, DCAF13, plays a crucial role in promoting Pol I transcription by introducing K63-linked ubiquitination to RPA194, its largest subunit. Notably, when Pol I transcription is inhibited, K63-linked ubiquitination diminishes, causing the redistribution of Pol I complex to the ‘nucleolar caps’. This behavior markedly contrasts with that of Pol II, which, despite structural and functional similarities60, undergoes rapid K48-linked ubiquitination modification and subsequent degradation when its transcription stalls61. This divergence in behavior may be attributed to their unique cellular roles. Pol I is primarily responsible for transcribing a single gene, the rDNA gene, within the nucleolus, making it easier for cells to recycle Pol I upon restoring transcription. Pol II, on the other hand, faces more challenging conditions as it transcribes a large number of genes, each with different transcription control mechanisms, and its transcription occurs in dispersed locations throughout the nucleus. Consequently, when Pol II’s transcription activity is inhibited, degrading Pol II becomes a more efficient and safer way for cells to facilitate its reuse.
In this study, we demonstrate that DCAF13-mediated K63-linked ubiquitination at K1180 and K1184 on RPA194 is essential for Pol I transcription. The K1180/1184 R mutation in RPA194 leads to the formation of ‘nucleolar caps’, serving as a marker of rRNA transcriptional impairment. Additionally, K63-linked ubiquitination of RPA194 is observed, albeit to a lesser extent, in MCF10A cells. These findings suggest that ubiquitination at the K1180/1184 residues of RPA194 can occur constitutively in all cell types and is crucial for the proper functioning of Pol I transcription. Furthermore, we observe a corresponding increase in the K63-linked modification of RPA194 upon serum-induced activation of Pol I transcription. This discovery represents a significant advancement in the field of Pol I transcription, as it underscores the critical role of RPA194 ubiquitination in regulating transcriptional activity, providing new insights into the molecular mechanisms governing rRNA synthesis.
In summary, our study highlights the important role of AMFs in promoting cancer, potentially achieved through the RiBi activation. These insights suggest that AMFs could serve as promising targets for cancer therapy.
Methods
Study approval
This research complies with all relevant ethical regulations. The Immunohistochemical experiments using tissue samples from patients in this study were approved by the medical ethics committee of Sun Yat-Sen Memorial Hospital, and written informed consent to publish clinical information potentially identifying individuals was obtained from all patients. All animal experiments were conducted according to protocols approved by the Institutional Animal Care and Use Committee of Sun Yat-Sen University in agreement with the Declaration of Helsinki.
Plasmid construction
Full-length and truncated variants of DCAF13 were amplified via PCR using cDNA reverse-transcribed from RNA isolated from HeLa cells. These amplified products were subcloned into the pCDH-CMV expression vector. For RNA interference studies, oligonucleotides specifically targeting DCAF13 were engineered into the pLKO-Tet-On shRNA vector. A scrambled shRNA plasmid served as a negative control. Of the shRNAs evaluated, two sequences (5’-GGAAUCUAACUCAGCGGAATT-3’ and 5’-GCAGUGCUUGAUGUGGAUUTT-3’) exhibited optimal DCAF13 knockdown efficiency. To achieve the overexpression of full-length RPA194 and USP36, PCR-amplified fragments from HeLa cell-derived cDNA were first subcloned into the pDONR221 entry vector. These were subsequently transferred into a destination vector equipped with an SFB tag via Gateway Technology (Invitrogen, Camarillo, CA, USA). The pmRuby3-B23 and pmNeongreen-RPA194 expression vectors were kindly provided by Prof. Lingling Chen (Shanghai Institutes of Biological Sciences, Chinese Academy of Sciences). Additionally, HA-tagged ubiquitin constructs (HA-Ub, HA-Ubk48, and HA-Ubk63) were generously gifted by Prof. Ronggui Hu (Institute of Biochemistry and Cell Biology, SIBS, CAS). The expression plasmids FN-RPA194 and Ruby3-RPA194 were obtained by using ClonExpress Ultra One Step Cloning Kit (Vazyme) to modify the pmNeongreen-RPA194 plasmid. The corresponding site mutation plasmids were obtained via Hieff Mut™ Multi Site-Directed Mutagenesis Kit (Yeasen). All primers are synthesized in Guangzhou IGE Biotechnology Co., Ltd.
Cell culture, cell transfection and lentiviral Infection
Human cell lines MDA-MB-468, MDA-MB-231, MCF10A, HMLE, HeLa, and HEK293Twere all procured from ATCC. Among them, MDA-MB-468, MDA-MB-231, HeLa, and HEK293T cells cultured in DMEM (Vigonob) supplemented with 10% fetal bovine serum (Lonsera) at 37 °C with 5% CO2, while MCF10A and HMLE cells cultured in DMEM/F12 (GIBCO) which contains 5% Horse Serum (Invitrogen), 20 ng/mL EGF (SinoBiological), 0.5 mg/mL Hydrocortisone (MCE), 100 ng/mL Cholera Toxin (Sigma) and 10 µg/mL Insulin (Beyotime) at 37 °C with 5% CO2. Plasmid transfections were performed using Viafect (Promega). For siRNA transfections, Lipofectamine RNAiMAX reagent (Invitrogen) was used following its specified protocol. The relevant siRNA sequences are provided in Supplementary Data 5. To establish stable cells through lentiviral infection, the destination vectors were packaged into lentiviruses using the packaging plasmids pMD2.G (RRID: Addgene_12259) and psPAX2 (RRID: Addgene_12260) by transfecting them into 293 T cells. Subsequently, MDA-MB-231 cells were infected with the lentiviral supernatant, and selection was carried out using media containing puromycin (2 µg/mL).
Construction of the instant-inducible degradation system (dTAG)
The FKBP12F36V TAG was inserted into the target gene using the CRISPR-Cas9 system, followed by the addition of the TAG deactivator to rapidly degrade the target protein. Specifically, sgRNA with high N-terminal cleavage efficiency for the target gene was selected and incorporated into the pX330-U6-sgRNA-CBH-Cas9-P2A-EGFP vector. The dTAG donor vector was constructed by homologous recombination, selecting homologous sequences of approximately 20 bp from regions 1-5 bp upstream and downstream of the Cas9 protein cleavage site. The constructed Cas9 vector, dTAG donor vector, and paired PITCH sgRNA vector (which linearizes the donor vector to enhance homologous recombination repair efficiency) were transfected into cells in a 6 cm dish at ratios of 800 ng, 1200 ng, and 800 ng, respectively. After 48 hours, the cells were digested and plated into a 10 cm dish. Following a week of selection with puromycin-containing medium, the medium was replaced with standard culture medium. Once monoclonal colonies became visible, they were selected and verified by Western Blot. The target protein in the constructed cells was observed to be approximately 15 kDa larger than the wild type.
Construction of stable EGFP-FBL knock-in Hela cells
The EGFP tag was inserted into the target gene using the CRISPR-Cas9 system. SgRNA with high N-terminal cutting efficiency for the target gene was selected and constructed into the pX330A-1×2 Cas9 vector. Homologous sequences of approximately 20 bp were chosen from regions 1-5 bp upstream and downstream of the Cas9 protein cleavage site, and the EGFP donor vector was constructed using homologous recombination. The constructed Cas9 vector, Donor vector, and matching PITCH sgRNA vector were transfected into cells in a 6 cm dish at a ratio of 800 ng, 1200 ng, and 800 ng, respectively. After 48 hours, the cells were digested and plated into a 10 cm dish. Following a week of selection in puromycin-containing medium, the medium was replaced with standard culture medium. When monoclonal colonies became visible to the naked eye, they were selected for further verification by Western Blot and immunofluorescence (IF). The target protein in the constructed cells was observed to be 35 kDa larger than the wild type, and green fluorescence was detected under a fluorescence microscope.
RNA isolation and quantitative RT-PCR
Total RNA from each cultured cell line or cells treated differently was extracted using Trizol Reagent (Invitrogen) following the manufacturer’s protocol. For qRT-PCR, cDNA synthesis was performed using PrimeScript RT Enzyme Mix I (TAKARA) with RT Primer Mix and 5X PrimeScript Buffer 2 after gDNA Eraser treatment (TAKARA). QPCR was carried out using TB Green Premix Ex Taq II (TAKARA) and the CFX96TM real-time system (BIO-RAD). The relative expression of various gene sets was normalized to actin mRNA. The primer sequences for qRT-PCR used in this study are listed in Supplementary Data 6.
Western blot analysis
Immunoblots were conducted using whole-cell lysates prepared with RIPA buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% NP40, 0.5% sodium deoxycholate, 0.5 mM DTT) supplemented with protease and phosphatase inhibitors (APExBIO). The lysates were then boiled in 5x SDS loading buffer, resolved on an SDS-PAGE assay, and transferred to PVDF membranes (Millipore). Subsequently, the membranes were blocked with 5% milk in PBST and incubated with primary antibodies overnight at 4 °C. The specific antibodies used are listed in Supplementary Data 4. For detection, secondary antibodies conjugated to horseradish peroxidase, anti-rabbit IgG (Transgene, 1:10,000 dilution), or anti-mouse IgG (Transgene, 1:10,000 dilution) were employed. Protein band signals were visualized using the ECL Western Blotting Substrate (Vazyme).
Protein synthesis assay (SUnSET)
Proteins were labeled using a modified protocol based on the SUnSET method28. MDA-MB-231 and MDA-MB-468 cells were transfected with their corresponding plasmids or siRNAs and cultured for a period of 48 hours. Following this, the culture medium was replaced with complete medium supplemented with 10 µg/mL of puromycin, and cells were incubated at 37 °C for an additional 15 minutes to facilitate the end-labeling of newly synthesized proteins. Post-incubation, cells were lysed using RIPA buffer, and total protein extracts were subjected to Western blotting. The newly synthesized proteins were identified using an antibody against puromycin.
Immunohistochemistry
Tissue samples from patients listed in Supplementary Data 2 were fixed with formalin and paraffin-embedded, followed by antigen retrieval achieved by immersing slides in Sodium Citrate Antigen Retrieval Solution and heating in the microwave at high-grade until close to boiling, then reducing to mid-grade for 10 minutes. The sections were subsequently stained with anti-DCAF13 antibody (ab195121, Abcam, 1:200 dilution) overnight at 4 °C and labeled with an HRP-conjugated secondary antibody at room temperature for 1 hour. The reactions were visualized using diaminobenzidine (DAB) as the chromogenic substrate. Sections were examined under an Olympus IX71 microscope, and images were acquired using NIS-Elements F 4.60.00 software. Based on the combined score of DCAF13 intensity and the percentage of positive cells, cases were classified into two groups: DCAF13 low-expression (cases with a sum of DCAF13 intensity and % of positive cells scores ≤ 3) and DCAF13 high-expression (cases with a sum of DCAF13 intensity and % scores ≥ 4).
Cell viability assay (CCK8)
For cell viability assay, we determined with Cell Counting Kit 8 (CCK8, Fudebio). Under specified conditions, 1000 cells per well were inoculated into the 96-well plates and allowed to adhere overnight. CCK8 reagent (10% in corresponding medium) was added on the first day, the third day and the fifth day respectively, and after incubation for two hours, the absorbance of the cells was measured at 450 nm by Multifunctional Microplate Detector (TECAN), the data was analyzed to reflect the proliferative activity of the cells.
Clonogenic assay
For clonogenic assay, under the indicated conditions, 500 cells per well were seeded into 6-well plates and allowed to adhere overnight. The cells were cultured in medium containing 10% FBS for 7-14 days, and then fixed using 4% PFA for 15 minutes followed by 0.1% crystal violet staining for 15 minutes. The clones stained with crystal violet were washed to clean the residual floating color, dried, photographed with the Elispot (AID) and counted for analysis.
Apoptosis assay
Both cells and supernatant were harvested and washed once with PBS, Subsequently, samples were stained with Annexin V-FITC and propidium iodide (PI) for flow cytometric analysis using a CytoFLEX instrument (Beckman, RRID: SCR_019627).
Cell-cycle analysis
Following disassociation with trypsin, cell pellets were obtained and washed once with PBS. The cells were then fixed in 75% ice-cold ethanol overnight. After fixation, the cells were washed twice with PBS, treated with RNase A, and stained with propidium iodide (0.05 mg/mL) for 15 minutes. Subsequently, the PI-labeled samples were analyzed using a flow cytometer (CytoFLEX, Beckman, RRID: SCR_019627).
Xenograft tumor growth
All BALB/c nude mice (6-week-old, female) were sourced from the Laboratory Animal Center at Sun Yat-Sen University. All mice were housed in cages containing aspen chip bedding in rooms under standard conditions in accordance with the institutional guidelines of the Animal Care and Use Committee of Sun Yat-Sen University. For the subcutaneous xenograft model, MDA-MB-231 cells, either stably overexpressing or exhibiting knockdown of DCAF13, were subcutaneously implanted into these mice at a density of 1,000,000 cells per mouse. Upon attaining a tumor volume of 100 mm³ in the group with inducible shDCAF13, doxycycline was administered intraperitoneally (1 μg/mL in 100 μL) to induce sustained DCAF13 knockdown. Tumor dimensions were then recorded every 48 hours. A similar bi-daily measurement protocol was followed for the overexpression group after observable subcutaneous tumor formation. In alignment with ethical protocols, tumor growth was monitored for a period of 4-5 weeks, following which the tumors were excised for volumetric and mass measurements to facilitate subsequent analyses. A total of 40 mice were used, with 10 mice in each group.
Protein extraction and enzyme digestion
The MDA-MB-231 cells were divided into two groups (normal rabbit IgG for control and anti-DCAF13 antibody for the experimental group), with three replicates per group and two biological replicates, resulting in a total of 12 samples.
MDA-MB-231 cells were lysed using NETN buffer (20 mM Tris–HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, and 0.5% Nonidet P-40) supplemented with protease and phosphatase inhibitors (Bimake) for 30 minutes. Supernatants were then clarified at 21,000 × g to remove debris, followed by incubation with the antibody (Normal rabbit IgG or anti-DCAF13 antibody) for 12 hours at 4 °C. On the second day, Dynabeads™ Protein G (Invitrogen) were added and incubated at 4 °C for 4 hours. The beads were washed five times with NETN buffer, and beads-bound proteins were subsequently eluted. Half of the eluate was retained for western blotting and silver staining analysis, while the remaining portion was used for mass spectrometry sample preparation. As for pre-treatment of mass spectrometry samples, the beads-bound proteins were washed twice with 50 mM NH4CO3, and proteins were reduced with 5 mM dithiothreitol (Sigma) at 50 °C for 30 minutes. Proteins were alkylated with 10 mM iodoacetamide (MCE) at 25 °C for 45 minutes in the dark. Samples were digested with trypsin (Promega) at 1:50 enzyme-to-protein ratio. After 16–18 h of digestion, trypsin-digested samples were eluted with 0.1% TFA and vacuum-dried. After the peptide is redissolved, C18 Spin Columns (OASIS) were used to purify the peptide to be detected. The demineralized peptides were drained by vacuum centrifuge concentrator and used for subsequent mass spectrometry detection.
LC-MS/MS
Ten micrograms of peptide mixture were dissolved in 20 μL of 0.1% formic acid in water and monitored on a liquid chromatography-tandem mass spectrometry detection (LC-MS/MS) system consisting of nanoflow liquid chromatography column (Easy nLC 1200 system, Thermo Fisher) coupled to an Orbitrap Fusion mass spectrometer with a nanoelectrospray ion source. Label-free quantification was performed. Briefly, the peptide mixture was loaded onto a 150-mm column (75 μm × 150 mm, C18 Tip, 11.9 μm, New Objective) using buffer A (0.1% formic acid) and separated by buffer B (0.1% formic acid in 80% acetonitrile in water) at a flow rate of 300nL/min (0-3 min, 4–8% B; 3–45 min, 8–28% B; 45–53 min, 28–38% B; 53–55 min, 38–100% B; 55–60 min, 100% B). Following the 4D-Faims method, the mass spectrometer cycle was programmed to collect one full MS scan followed by 10 data-dependent MS/MS scans. The MS scans (300-2000 m/z range, 4e5 AGC, 50 ms maximum ion time) were collected at a resolution of 60,000 at m/z 200 in profile mode, and the standard resolution with Faims voltages (ON) of -45V and -60V was obtained. The MS/MS spectra (1.6 m/z isolation width with 0.5 m/z offset, 30% collision energy, auto AGC target, and 50 ms maximum ion time) were acquired at a resolution of 15,000 at m/z 200 in centroid mode. Dynamic exclusion was set to exclude previously sequenced precursor ions for 60 s within a 10-ppm window. Precursor ions with + 1 and + 7 or higher charge state were excluded from sequencing. Proteome Discoverer 2.4(PD) software with SEQUEST and Mascot search engine (version 2.3.01) was used for the quantification analysis of the original mass spectrometry data. The maximum number of missing cleavage sites was 3. The precursor mass tolerance was ± 10 ppm, and the fragment mass tolerance was ± 0.6 Da. The static modification was carbamidomethyl (carbamidomethyl/+ 57.021 Da), and the dynamic modifications were oxidation (oxidation/+ 15.995 Da), and acetylation (acetyl/+ 42.011 Da). The false discovery rates (FDRs) at the protein and peptide levels were both set to 0.05%. Missing values in individual experiments are denoted by ‘Not a Number (NaN)’. Log10 values were calculated for the intensities of each mean value, and log2 ratios of the quantified values to intensity were compared.
Complex enrichment analysis
Leveraging the results from database searching of using Proteome Discoverer 2.4 (PD) software, we first removed potential contaminants, such as keratins. Next, we applied a stringent filter to include only those results where the peptide spectrum match (PSM) count in the IP group was at least five times higher than in the input group. The filtered data were then subjected to complex enrichment analysis using the CPDB online tool. Additionally, we conducted separate analyses of the Pol I and SSU complexes. The results of the enrichment analysis were further processed by applying a -log10 transformation to the p-value, which were then organized and visualized in a bar chart to display the enrichment outcomes.
Co-immunoprecipitation
For co-immunoprecipitation, cells were lysed using NETN buffer. A tenth of the cell lysates was saved as the input sample, while the remaining lysates were divided into two parts for overnight incubation with indicated antibody. Following this, Dynabeads™ Protein G (Invitrogen) were added to each sample and incubated for 4 hours. After five washes with NETN buffer, the samples were boiled in protein loading buffer and subjected to Western blotting.
Denaturing immunoprecipitation
Cells were washed with ice-cold PBS and lysed with denatured lysis buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 40 mM DTT, and 5% glycerol) and boiled at 95 °C for 15 minutes. The lysates were then diluted 10-fold with NETN buffer and centrifuged at 15,000 × g for 10 minutes at 4 °C. Then the supernatants were incubated with the indicated antibody for 4 hours at 4 °C, and then with protein A/G magnetic beads (MCE) for 2 hours at 4 °C, followed by washing with NETN buffer five times and boiling in protein loading buffer. The bound proteins were separated by SDS-PAGE and subjected to Western blotting.
Silver staining
For silver staining analysis, equal protein aliquots from endogenous immunoprecipitants, isolated using either normal rabbit IgG or anti-DCAF13 antibody from MDA-MB-231 cells, were subjected to 10% SDS-PAGE. The gel was then stained utilizing the Rapid Silver Stain for Mass Spectrometry kit (Beyotime) in accordance with the manufacturer’s guidelines.
GST pull-down assay
After transformation, the prokaryotic expression plasmid was introduced into Escherichia coli and cultured overnight at 37 °C. On the following day, IPTG (0.2 mM) was added to induce protein expression at 37 °C for 4 hours. The cultures were then centrifuged at 3200 × g for 15 minutes at 4 °C. The supernatant was discarded, and 20 mL of NETN buffer was added to the bacterial pellet. The bacteria were lysed using sonication, and the lysate was centrifuged at 3200 × g to remove debris. The supernatant was incubated with GST agarose beads (Transgene) at 4 °C for 4 hours. The beads were washed thoroughly with NETN buffer, and 1/10th of the sample was taken as input. Concurrently, 1 mL of NETN lysate buffer was added to a 10 cm plate of adherent cells, which were incubated and lysed on ice for 30 minutes. The lysate was centrifuged, and the supernatant was collected and incubated with pre-treated GST agarose beads for an additional 4 hours. After washing the beads, 100 µL of NETN buffer was added to resuspend the beads, and protein loading was prepared along with the input sample. Finally, the samples were denatured at high temperature and analyzed by western blotting.
EU assay
MDA-MB-231 cells were pretreated with 100 μM 5-Ethynyl Uridine (5-EU, Click Chemistry Tools) for 2 hours to label nascent pre-rRNA. Subsequently, the cells were fixed with 4% paraformaldehyde at room temperature for 15 minutes and permeabilized with 0.5% Triton X-100 for 5 minutes. The cells were then incubated with AFDye 488 Azide (Click Chemistry Tools) in a reaction buffer (100 mM Tris, pH 8.5/1 mM CuSO4/50 μM fluorescent azide/100 mM ascorbic acid) at room temperature for 30 minutes. After incubation, the nuclei were counterstained with DAPI, and images reflecting the ability of biosynthesis of nascent pre-rRNA were acquired using a laser scanning confocal microscope (Zeiss LSM 800 with airyscan).
Immunofluorescence (IF)
Cells cultured on glass coverslips underwent fixation in 4% paraformaldehyde for 15 minutes and were then permeabilized using 0.5% Triton X-100 for an additional 5 minutes. Following a 30-minute block in 5% bovine serum albumin (BSA), the cells were exposed to the designated primary antibodies during an overnight incubation at 4 °C. The samples were then rinsed thrice in PBST and stained with the appropriate secondary antibodies at room temperature for 1 hour. Post-DAPI staining, cellular imaging was conducted using a Zeiss LSM 800 confocal microscope (RRID: SCR_015963).
ActD treatment
To induce nucleolar transcriptional suppression and facilitate the formation of a characteristic ‘nucleolar cap’, cells were treated with actinomycin D (ActD, Selleck) at a low concentration. Specifically, the original culture medium was replaced, and cells were incubated in medium supplemented with 50 ng/mL ActD for one hour. Subsequently, immunofluorescence was carried out using a relevant nucleolar marker to characterize and visualize the ‘nucleolar cap’.
RNA in situ hybridization
To detect U3, RNA FISH was conducted following a previously described method with an in vitro transcribed Cy3-labeled antisense probe. In brief, cells were fixed with 4% PFA for 15 minutes and permeabilized with 0.5% Triton X-100 for 5 minutes. Subsequently, denatured Cy3-labeled FISH probes were incubated with the cells in hybridization buffer (50% formamide in 2xSSC) at 37 °C overnight. The U3 probe sequence is provided in Supplementary Data 6. Following hybridization, the nuclei were counterstained with DAPI, and the U3 signal was visualized directly using a laser scanning confocal microscope (Zeiss LSM 800 with airyscan).
To visualize U3, NPM, and UBF1 simultaneously, cells were co-stained with primary antibodies against NPM and UBF1 after the incubation with RNA probe.
The subsequent steps were carried out as mentioned above for IF to visualize the colocalization of U3, NPM, and UBF1.
Serum starvation and activation
For serum starvation treatment, we utilized a low-glycemic medium (GIBCO) distinct from DMEM and cultured cells without serum for 24 hours and 48 hours respectively. To activate pre-rRNA transcription, serum activation was performed by adding normal high-glucose DMEM medium containing 20% serum after 48 hours of serum starvation, and the cells were further cultured for 24 hours.
RNA immunoprecipitation (RIP)
MDA-MB-231 cells were subjected to UV irradiation (254 nm, 2,000 kJ) in cold PBS. After cross-linking, the cells were lysed with RIPA buffer containing protease and RNasin inhibitors, and the mixture was incubated on ice for 30 minutes. The lysates were then sonicated using Bioruptor Plus (Diagenode). After sonication, the lysates were precleared with Dynabeads™ Protein G for 1 hour at 4 °C. Subsequently, the protein-RNA complexes were immunoprecipitated for 4 hours at 4 °C using anti-DCAF13 antibody, with normal rabbit IgG added as a control. The immunoprecipitated complexes were then incubated with Dynabeads™ Protein G overnight. Following this, the beads-bound RNA was isolated using phenol and chloroform, and qRT-PCR was performed as described above. The primer sequences used in this study are listed in Supplementary Data 6.
Chromatin isolation by RNA purification (ChIRP)
The ChIRP assay was conducted following the previously described method62 Cells were fixed with 3% formaldehyde (Sigma) to form the chromatin/protein complexes. Subsequently, the cells were lysed with lysis buffer (0.05 M Tris-HCl pH7.0, 0.1 mM EDTA, 1% SDS) containing protease and RNasin inhibitors and sheared using Bioruptor Plus. Biotin-labeled RNA probes, which were pre-synthesized, were added to the cell lysates to incubate and hybridize with them overnight at 37 °C. The probe sequences can be found in Supplementary Data 6. Next, equal volume of C-1 beads (Invitrogen) was added into the protein mixture and incubated at 37 °C for 30 min. The RNA and bound proteins were then extracted separately, with RNA being eluted and subjected to qRT-PCR analysis using the primers listed in Supplementary Data 6. The proteins were analyzed separately using Western blotting.
Chromatin immunoprecipitation followed by high-throughput DNA sequencing (ChIP-Seq)
Nuclei were isolated from fixed MDA-MB-231 cells using 1% formaldehyde (10 minutes, RT) and quenched with 0.125 M glycine for 5 minutes. After centrifugation, the cells were resuspended in lysis buffer and subjected to sonication with Bioruptor to yield 250-500 bp fragments. The samples were then incubated overnight with the respective antibodies, and the protein-DNA complexes were captured on protein A/G beads (Invitrogen). Subsequently, the immunoprecipitated samples were collected in elution buffer and subjected to reversal of the crosslink overnight at 65 °C. The protein was digested by incubating with protein K for 1 hour at 55 °C, and RNaseA was added and incubated at 37 °C for 30 minutes to remove the RNA. DNA was then purified for High-throughput sequencing.
Protein structure visualization and analysis
Protein structure visualization and analysis were conducted using UCSF ChimeraX (version 1.7.1). First, the PDB file for human RNA polymerase I (PDB ID: 8A43) was loaded. The specific lysine residues in the RPA194 protein structure (chain A) were selected and displayed in their current visual representation. The display style was adjusted as needed using the graphical interface, and the color was modified either through the ‘color’ command or the corresponding options in the interface. These display adjustments facilitated clear visualization of the spatial positioning of individual lysine residues within the protein complex, with K1101, K1180, K1184, and K1574 located on the periphery of the complex. This approach allowed for effective 3D visualization of both the protein structure and its lysine residues, aiding in subsequent analysis.
Human gene catalog
We downloaded a part list of 198 RiBP genes from Reactome database (Version 73). The 198 RiBP genes can be divided into 2 different subclasses based on their detailed function (85 RPs, 113 AMFs) from Reactome and previous study63. The remaining 102 AMFs were obtained through literature review, primarily including the articles by Dörner et al. 16 and Singh et al. 17. TFs was downloaded from AnimalTFDB v3.064. Meanwhile, the list of known oncogenes and tumor suppressors was downloaded from the COSMIC database (version 94)65.
Differential expression analysis
We obtained The Cancer Genome Atlas (TCGA) datasets encompassing raw counts, gene expression profiles, and associated clinical information from Xena Browser. Our analysis focused on 15 cancer types, specifically Bladder Urothelial Carcinoma (BLCA), BC, Colon Adenocarcinoma (COAD), Head and Neck Squamous (HNSC), Kidney Chromophobe (KICH), Kidney Renal Clear Cell Carcinoma (KIRC), Kidney Renal Papillary Cell Carcinoma (KIRP), Liver Hepatocellular Carcinoma (LIHC), Lung adenocarcinoma (LUAD), Lung Squamous Cell Carcinoma (LUSC), Prostate Adenocarcinoma (PRAD), Rectum Adenocarcinoma (READ), Stomach Adenocarcinoma (STAD), Thyroid Carcinoma (THCA), and Uterine Corpus Endometrial Carcinoma (UCEC). Differential expression analysis of 300 RiBP genes across tumor and adjacent normal tissues for each cancer type was conducted using the ‘edgeR’ package in R. Genes were considered significantly dysregulated if they exhibited a p-value ≤ 1E-3. Heatmaps illustrating the log2-fold changes of these 300 RiBPs across the 15 cancer types were generated using the ‘pheatmap’ and ‘ggplot2’ packages in R. Statistical significance was assessed using the ‘ggsignif’ package in R.
Microarray data analysis
Microarray data pertinent to breast cancer were sourced from the Gene Expression Omnibus (GEO) database, specifically from the datasets GSE70947 and GSE76250.
Copy number alteration analysis
TCGA copy number data were obtained from the Broad Institute and analyzed using GISTIC2 software66 to identify recurrent regions of amplification or deletion. In each cancer type, genes were annotated with their respective copy number variations (CNVs), defining a CNV value > 0.3 as gain and < -0.3 as loss. Correlations between DCAF13 mRNA expression levels and CNV were assessed through Pearson and Spearman analyses using R (version 3.6.1).
Somatic mutation analysis
TCGA mutation data were sourced from the Broad Institute and subjected to statistical analysis using our custom Python scripts across 15 distinct cancer types. We selectively retained mutations classified as missense, nonsense, or frameshift (either insertions or deletions). The same analytical approach was applied to transcription factors (TFs), known oncogenes and tumor suppressors.
Cell proliferation index analysis
Data based on shRNA screens targeting 14,222 genes across 216 cancer cell lines were obtained from the DepMap67 (https://depmap.org/portal/download/). Of these, 139 RiBPs with available RNA silencing data were identified. In our analysis, core-AMFs were considered to significantly influence cell proliferation if it displayed a log fold-change score (quantile normalized) of ≥ 2 across five shRNAs. Finally, we compared the distributions of log2-transformed ratios of shRNA concentrations between the terminal cell populations and the initial DNA pools across known oncogenes, core-AMFs, and non-cancer genes using the two-sided Wilcoxon rank-sum test.
Statistical analysis and reproducibility
Data analysis was performed using GraphPad Prism version 7. Each experimental group underwent triplicate analyses, and all experiments intended for statistical evaluation were replicated a minimum of three times. Results are expressed as the mean ± standard error of the mean (SEM). Two-tailed Student’s t test and Two-sided Wilcoxon rank-sum test were used for the statistical comparison between the two groups, and One-way analysis of variance (ANOVA) was used among multiple groups, respectively. Survival study was analyzed through Two-sided log-rank (Mantel-Cox) test. Similarity analysis was performed using Two-tailed Pearson and Spearman correlation test. A significant difference was considered when the p value was less than 0.05. Dots overlaid on the bar graphs are the value measured for biological replicates. The micrographs are representative images (n = 3 biological replicates). The immunoblotting results shown in the plot are representative, based on at least two biological replicates. No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were randomized.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The ChIP-seq data generated in this study has been deposited in the Gene Expression Omnibus (GEO) repository under the accession number GSE242318. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX partner repository68,69 with the dataset identifier PXD044918. TFs list was downloaded from AnimalTFDB v3.0. RBP list in Supplementary Fig. was obtained from EuRBPDB. Source data are provided with this paper.
Code availability
The analysis code used in this study is available at https://zenodo.org/records/14334730.
References
Domostegui, A. et al. Impaired ribosome biogenesis checkpoint activation induces p53-dependent MCL-1 degradation and MYC-driven lymphoma death. Blood 137, 3351–3364 (2021).
Sulima, S. O., Hofman, I. J. F., De Keersmaecker, K. & Dinman, J. D. How ribosomes translate cancer. Cancer Discov. 7, 1069–1087 (2017).
Izumikawa, K. et al. LYAR potentiates rRNA synthesis by recruiting BRD2/4 and the MYST-type acetyltransferase KAT7 to rDNA. Nucleic Acids Res 47, 10357–10372 (2019).
Yang, X. M. et al. Nucleolar HEAT repeat containing 1 up-regulated by the mechanistic target of rapamycin complex 1 signaling promotes hepatocellular carcinoma growth by dominating ribosome biogenesis and proteome homeostasis. Gastroenterology 165, 629–646 (2023).
Bohnsack, K. E. & Bohnsack, M. T. Uncovering the assembly pathway of human ribosomes and its emerging links to disease. EMBO J. 38, e100278 (2019).
Li, Z. et al. Nuclear export of pre-60S particles through the nuclear pore complex. Nature 618, 411–418 (2023).
Zisi, A. et al. Small molecule-mediated disruption of ribosome biogenesis synergizes with FGFR inhibitors to suppress glioma cell growth. Neuro Oncol. 25, 1058–1072 (2023).
Barna, M. et al. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456, 971–975 (2008).
Grummt, I. Life on a planet of its own: regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 17, 1691–1702 (2003).
Nguyen le, X. T. & Mitchell, B. S. Akt activation enhances ribosomal RNA synthesis through casein kinase II and TIF-IA. Proc. Natl Acad. Sci. USA 110, 20681–20686 (2013).
Liebhaber, S. A., Wolf, S. & Schlessinger, D. Differences in rRNA metabolism of primary and SV40-transformed human fibroblasts. Cell 13, 121–127 (1978).
Peltonen, K. et al. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer cell 25, 77–90 (2014).
Khot, A. et al. First-in-human RNA polymerase i transcription inhibitor CX-5461 in Patients with Advanced Hematologic Cancers: Results of a Phase I Dose-Escalation Study. Cancer Discov. 9, 1036–1049 (2019).
Pataki, J., Dombradi, V., Sarvary, A. & Szollosi, G. J. Breast cancer screening and its associating factors among hungarian women aged 45-65: a cross-sectional study based on the European health interview surveys from 2009 to 2019. BMC Public Health 23, 1679 (2023).
Moudry, P., Chroma, K., Bursac, S., Volarevic, S. & Bartek, J. RNA-interference screen for p53 regulators unveils a role of WDR75 in ribosome biogenesis. Cell Death Differ. 29, 687–696 (2022).
Dörner, K., Ruggeri, C., Zemp, I. & Kutay, U. Ribosome biogenesis factors-from names to functions. Embo j. 42, e112699 (2023).
Singh, S., Vanden Broeck, A., Miller, L., Chaker-Margot, M. & Klinge, S. Nucleolar maturation of the human small subunit processome. Science 373, eabj5338 (2021).
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic Acids Res 48, D498–D503 (2020).
Koboldt, D. C. et al. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Ciriello, G. et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163, 506–519 (2015).
Pelletier, J., Thomas, G. & Volarević, S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat. Rev. Cancer 18, 51–63 (2018).
Bursać, S., Prodan, Y., Pullen, N., Bartek, J. & Volarević, S. Dysregulated Ribosome Biogenesis Reveals Therapeutic Liabilities in Cancer. Trends cancer 7, 57–76 (2021).
Cowley, G. S. et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data 1, 140035 (2014).
Cheng, J. et al. In vitro structural maturation of an early stage pre-40S particle coupled with U3 snoRNA release and central pseudoknot formation. Nucleic Acids Res 50, 11916–11923 (2022).
Bax, R., Vos, H. R., Raue, H. A. & Vos, J. C. Saccharomyces cerevisiae Sof1p associates with 35S Pre-rRNA independent from U3 snoRNA and Rrp5p. Eukaryot. Cell 5, 427–434 (2006).
Angrisani, A., Vicidomini, R., Turano, M. & Furia, M. Human dyskerin: beyond telomeres. Biol. Chem. 395, 593–610 (2014).
Welting, T. J., Kikkert, B. J., van Venrooij, W. J. & Pruijn, G. J. Differential association of protein subunits with the human RNase MRP and RNase P complexes. RNA (N. Y., N. Y.) 12, 1373–1382 (2006).
Schmidt, E. K., Clavarino, G., Ceppi, M. & Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. methods 6, 275–277 (2009).
Zhou, L. et al. T cell proliferation requires ribosomal maturation in nucleolar condensates dependent on DCAF13. The Journal of cell biology 222 (2023).
Chu, C. et al. Systematic discovery of Xist RNA binding proteins. Cell 161, 404–416 (2015).
Zhao, J., Yuan, X., Frodin, M. & Grummt, I. ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Mol. Cell 11, 405–413 (2003).
Liao, J. Y. et al. EuRBPDB: a comprehensive resource for annotation, functional and oncological investigation of eukaryotic RNA binding proteins (RBPs). Nucleic Acids Res 48, D307–D313 (2020).
Dvorackova, M. & Fajkus, J. Visualization of the Nucleolus Using 5’ Ethynyl Uridine. Methods Mol. Biol. 2672, 377–385 (2023).
Nabet, B. et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431–441 (2018).
Shin, Y. & Brangwynne, C. P. Liquid phase condensation in cell physiology and disease. Science 357 (2017).
Ide, S., Imai, R., Ochi, H. & Maeshima, K. Transcriptional suppression of ribosomal DNA with phase separation. Science advances 6 (2020).
Sobell, H. M. Actinomycin and DNA transcription. Proc. Natl Acad. Sci. USA 82, 5328–5331 (1985).
Sun, Y. et al. Targeting neddylation sensitizes colorectal cancer to topoisomerase I inhibitors by inactivating the DCAF13-CRL4 ubiquitin ligase complex. Nat. Commun. 14, 3762 (2023).
Jin, J., Arias, E. E., Chen, J., Harper, J. W. & Walter, J. C. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23, 709–721 (2006).
Radko-Juettner, S. et al. Targeting DCAF5 suppresses SMARCB1-mutant cancer by stabilizing SWI/SNF. Nature 628, 442–449 (2024).
Ray, S. et al. Topoisomerase IIα promotes activation of RNA polymerase I transcription by facilitating pre-initiation complex formation. Nat. Commun. 4, 1598 (2013).
Snyder, N. A. & Silva, G. M. Deubiquitinating enzymes (DUBs): Regulation, homeostasis, and oxidative stress response. J. Biol. Chem. 297, 101077 (2021).
Thul, P. J. et al. A subcellular map of the human proteome. Science 356 (2017).
Kim, J. O. et al. Deubiquitinating enzyme USP37 regulating oncogenic function of 14-3-3γ. Oncotarget 6, 36551–36576 (2015).
Pan, T. et al. USP49 potently stabilizes APOBEC3G protein by removing ubiquitin and inhibits HIV-1 replication. eLife 8, e48318 (2019).
Kiran, S., Dar, A., Singh, S. K., Lee, K. Y. & Dutta, A. The Deubiquitinase USP46 Is Essential for Proliferation and Tumor Growth of HPV-Transformed Cancers. Mol. Cell 72, 823–835.e825 (2018).
Cai, J. et al. Induction of deubiquitinating enzyme USP50 during erythropoiesis and its potential role in the regulation of Ku70 stability. J. investigative Med.: Off. Publ. Am. Federation Clin. Res. 66, 1–6 (2018).
Yang, S. et al. USP52 acts as a deubiquitinase and promotes histone chaperone ASF1A stabilization. Nat. Commun. 9, 1285 (2018).
Bragado, L. et al. SUMO conjugation regulates the activity of the Integrator complex. Nucleic Acids Res 50, 12444–12461 (2022).
Chen, Y. et al. The ubiquitin-specific protease USP36 SUMOylates EXOSC10 and promotes the nucleolar RNA exosome function in rRNA processing. Nucleic Acids Res 51, 3934–3949 (2023).
Kim, W. et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 (2011).
Cao, L., Liu, X., Zheng, B., Xing, C. & Liu, J. Role of K63-linked ubiquitination in cancer. Cell death Discov. 8, 410 (2022).
Bierhoff, H., Dundr, M., Michels, A. A. & Grummt, I. Phosphorylation by casein kinase 2 facilitates rRNA gene transcription by promoting dissociation of TIF-IA from elongating RNA polymerase I. Mol. Cell Biol. 28, 4988–4998 (2008).
Bruserud, O. & Reikvam, H. Casein Kinase 2 (CK2): A Possible Therapeutic Target in Acute Myeloid Leukemia. Cancers (Basel) 15 (2023).
Zhang, Y. et al. Visualizing the nucleoplasmic maturation of human pre-60S ribosomal particles. Cell Res33, 867–878 (2023).
Mendillo, M. L. et al. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150, 549–562 (2012).
Marcel, V. et al. p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell 24, 318–330 (2013).
Peng, H. et al. N(6)-methyladenosine (m(6)A) in 18S rRNA promotes fatty acid metabolism and oncogenic transformation. Nat. Metab. 4, 1041–1054 (2022).
Gallagher, J. E. et al. RNA polymerase I transcription and pre-rRNA processing are linked by specific SSU processome components. Genes Dev. 18, 2506–2517 (2004).
Vannini, A. & Cramer, P. Conservation between the RNA polymerase I, II, and III transcription initiation machineries. Mol. Cell 45, 439–446 (2012).
Caron, P. et al. WWP2 ubiquitylates RNA polymerase II for DNA-PK-dependent transcription arrest and repair at DNA breaks. Genes Dev. 33, 684–704 (2019).
Alfeghaly, C., Behm-Ansmant, I. & Maenner, S. Study of genome-wide occupancy of long non-coding RNAs Using chromatin isolation by RNA Purification (ChIRP). Methods Mol. Biol. 2300, 107–117 (2021).
Guimaraes, J. C. & Zavolan, M. Patterns of ribosomal protein expression specify normal and malignant human cells. Genome Biol. 17, 236 (2016).
Hu, H. et al. AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res 47, D33–d38 (2019).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res 47, D941–d947 (2019).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
Dwane, L. et al. Project Score database: a resource for investigating cancer cell dependencies and prioritizing therapeutic targets. Nucleic Acids Res 49, D1365–d1372 (2021).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res 47, D1211–D1217 (2019).
Chen, T. et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res 50, D1522–D1527 (2022).
Li, Y. et al. Proteogenomic data and resources for pan-cancer analysis. Cancer cell 41, 1397–1406 (2023).
Acknowledgements
This study was supported by grants from the National Key Research and Development Program of China (2021YFA0909300, D. Y); the Natural Science Foundation of China (82273033, J-Y. L; 82072924, J-Y. L; 82073067, D. Y; 81872140, D. Y); Guangdong Science and Technology Department (2022B1515020100, J-Y. L; 2023B1212060013; 2020B1212030004); Guangzhou Bureau of Science and Information Technology (202201020575, J-Y. L; 2024A04J6554, J-Y. L).
Author information
Authors and Affiliations
Contributions
J-Y.L., D.Y., and YQ.Z. conceived and designed the research. Z-Z.Y. and B.Y. performed most of the experiments with assistance from HY.Y., XY.M., B.T., BQ.Z., Y-X.C., Y-M.D., JS.D., ZL.Z., YM.S., JY.Z., DN.L., J-H.H., Y.Z., KS.H., S.Z., KD.Z., and Y-C.Z. J-Y.L. and Z-Z.Y. wrote the manuscript. All the authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, ZZ., Yang, B., Yan, H. et al. DCAF13-mediated K63-linked ubiquitination of RNA polymerase I promotes uncontrolled proliferation in Breast Cancer. Nat Commun 16, 557 (2025). https://doi.org/10.1038/s41467-025-55851-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-55851-9
This article is cited by
-
MDM2 promotes CELF6 ubiquitination-dependent degradation to promote neuroblastoma cell proliferation
Cell Death & Disease (2025)
-
Targeting ubiquitination in disease and therapy
Signal Transduction and Targeted Therapy (2025)
