
Abstract
Non-coding 7SL RNA is an ancestor to mammalian Alu and B1 SINE RNAs and is thought to function exclusively within the Signal Recognition Particle (SRP), aiding in the translocation of secretory proteins into the endoplasmic reticulum for export. Here, we discover a function of 7SL/SRP unrelated to protein secretion. Under acute heat shock, 7SL and SRP together selectively arrest cellular transcription and translation machineries during early response to stress. Under thermal stress, 7SL is upregulated, accumulates in the nucleus, and binds to target genes repressed by heat shock. Concurrently, in the cytosol, SRP binds to ribosomes and inhibits new protein synthesis. Translational suppression occurs independently of the signal peptide and is abrogated by depleting SRP. Translation inhibition extends to the mitochondria, as nuclear-encoded genes with mitochondrial functions are enriched among SRP targets. Thus, apart from its role in protein export, 7SL/SRP orchestrates a global response to acute stress that encompasses the nucleus, cytosol, and mitochondria across transcription and translation.
Similar content being viewed by others


Introduction
Short Interspersed Nuclear Elements (SINEs) are a prominent group of repetitive elements that constitute over 10% of the mammalian genome1,2, the best characterized of which are human Alu’s and mouse B1 and B2 elements3. These elements play crucial regulatory roles under conditions of perturbed homeostasis, such as during viral infection or heat shock4,5,6,7. Following exposure to stress, SINEs are transcribed by RNA polymerase III (POL-III), giving rise to noncoding transcripts that regulate the transcription and translation in response to stress5,8,9,10,11,12,13,14,15. Upon heat shock, human Alu RNA binds to promoters and negatively affects RNA Polymerase II (POL-II)-mediated transcription13,15. Similarly, mouse B2 RNA plays a crucial role in the heat-dependent regulation of POL-II activity. B2 RNA serves two purposes with respect to transcription: It negatively regulates POL-II transcription by generally inhibiting the formation of the preinitiation complex9,11 and controlling POL-II pause-release at stress-response genes14. Dynamic regulation of B2 RNA during acute stress thereby enables the selective expression of stress-response genes while effectively impeding pervasive POL-II transcription. Beyond their nuclear role, SINE transcripts also give rise to shorter RNA species known as small cytosolic Alu (scAlu) RNA in human16,17,18 and small cytosolic B1(scB1) RNA in mouse17,19. Together with the signal recognition particle proteins 9 and 14 (SRP9 and SRP14), scAlu RNA and scB1 RNA form species-specific ribonucleoprotein particles (RNPs) that interact with ribosomes and inhibit translation in a stress-dependent manner10,12.
Mouse B1 and human Alu SINEs have a common evolutionary origin in a noncoding element known as 7SL (Fig. 1a). Mammalian 7SL is a ~300 nt noncoding RNA transcribed by POL-III20. Its central 7SL-specific domain is flanked by Alu-like sequences (Fig. 1a and Supplementary Fig. 1a). Over time, 7SL gave rise to FAM (fossil Alu monomer), which then gave rise to FRAM (free right Alu monomer) and FLAM (free left Alu monomer)—the precursors of primate Alu—as well as protoB1, the precursor to rodent B1 (Fig. 1a, left panel)21,22. Primate Alu evolved from 7SL through the deletion of the 7SL-specific domain and fusion of the 5’ and 3’ regions23, followed by a duplication of the Alu-like sequences (Fig. 1a, right panel). Unlike Alu and B1, however, 7SL is produced only from 2 known loci, Rn7s1 and Rn7s224, in mammals and is therefore not considered a repetitive SINE element. 7SL is recognized primarily as the RNA component of the Signal Recognition Particle (SRP)25—an RNP comprising six interacting proteins SRP9, SRP14, SRP19, SRP54, SRP68, and SRP72 which are together required for co-translational targeting of secretory proteins across the endoplasmic reticulum (ER) membrane26,27,28,29. When the secretory signal peptide (SP) of newly synthetized proteins is exposed, the SRP binds to translating ribosomes and guides them to the ER30.

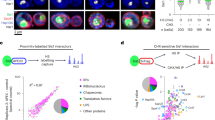
a Schematic illustration of the evolutionary connection between 7SL and 7SL-related ncRNA (left panel)21. 7SL gives rise to mammalian Alu and B1 SINEs. Human Alu repeats originate from 7SL sequences that have lost their central, 7SL-specific portion followed by Alu domain duplication (right panel)23. Created in BioRender. Bujisic, B. (2025) https://BioRender.com/h93t574. b PRO-Seq analysis40 of nascent 7SL transcription after exposure to 42 °C heat shock for indicated periods of time in mouse embryonic fibroblasts (MEFs). Source data are provided as a Source Data file. c Cell fractionation experiment: 7SL Northern Blot of the cytosolic, nuclear, or total NIH3T3 cells incubated at 37 °C or after 15 min exposure to 45 °C (HS) (upper panel). SYBR gold staining of the gel was used as a loading control (lower panel). d 7SL RNA FISH at 37 °C (pre-HS) or after 1 h exposure to 42 °C (post-HS) in mouse NIH3T3 cells. DAPI staining depicts nuclei. e Total (input), cytosolic, membrane, and nuclear fractions were isolated from NIH3T3 cells incubated at 37 °C or after 15 min exposure to 42, 43, 44, or 45 °C. Immunoblots were performed for SRP72, SRP54, Tubulin (cytosolic marker), and H3K27me3 (nuclear marker). f 7SL CHART-Seq tracks over Rn7s1 and Rn7s2 loci on chromosome 12. Tracks represent the Antisense (AS) signal subtracted for Sense (S) signal for two biological replicates in pre- and post-HS conditions. g Venn diagram demonstrating the number of 7SL CHART-Seq peaks in pre- and post-HS conditions. Peaks were called using MACS91 only if appeared in both biological replicates. h Proportion of 7SL CHART peaks with associated genomic features. Analysis performed using ChIPSeeker92.
Beyond this well-established role in ER translocation, we began to suspect additional functions, given 7SL’s close evolutionary relationship to the stress-response element, Alu. Interestingly, early in vitro studies31,32,33 showed that the interaction between the SRP9-SRP14 subunits and ribosomes can cause inhibition of translation34,35,36. However, the in vivo impact on translation has remained uncertain because studies performed in yeast37,38 and mammalian cells39 have provided opposing conclusions. 7SL has also not been previously implicated in the stress response, and unlike Alu’s and B2’s, its total expression levels are also thought to be stable during heat stress6. Here, we explore the idea that the ancestral 7SL RNA also functions in stress and provide pioneering evidence that 7SL and SRP together orchestrate transcriptional and translational responses during heat shock as part of a global mechanism to divert resources to manage stress.
Results
Heat shock induces de novo 7SL expression, nuclear accumulation, and binding to chromatin
To test whether 7SL expression is upregulated in mouse fibroblasts exposed to 42 °C for up to 60 min, we examined nascent synthesis of 7SL RNA using a previously published PRO-Seq dataset40. We observed increased de novo transcription within 2.5 min of heat shock exposure and lasting at least 60 min (Fig. 1b). Thus, like B2 and Alu RNAs, heat stress acutely induces de novo 7SL RNA synthesis. Unlike B2 and Alu, however, 7SL is thought to be predominantly cytosolic, in keeping with its function within the SRP. To investigate whether subcellular distribution of 7SL RNA is affected during stress, we fractionated cytosolic and nuclear compartments in cells before and after exposure to heat shock and performed a Northern Blot analysis of each fraction to examine changes in 7SL levels (Fig. 1c). These experiments revealed a significant increase in 7SL in the nuclear fraction paralleled by a reduction in the cytosolic fraction after heat shock (Fig. 1c). To determine if new transcription is required for the nuclear accumulation, we treated cells prior to heat exposure with a potent transcription inhibitor, Actinomycin D, at concentrations known to inhibit POL-III. Interestingly, 7SL still accumulated in the nucleus despite transcription inhibition (Supplementary Fig. 1b, right panel). In contrast, transcription inhibition prevented heat-induced nuclear tRNA accumulation (Supplementary Fig. 1b, left panel)—a phenomenon previously described for tRNA during thermal stress41. To further investigate 7SL nuclear localization, we performed RNA fluorescence in situ hybridization (FISH) (Fig. 1d, upper panels). Heat shock for 1 h at 42 °C resulted in a strong increase in nuclear 7SL RNA signals as compared to the control non-stressed condition (Fig. 1d, lower panels). To understand whether 7SL translocates from the cytosol alone, or together with interacting SRP proteins, we fractionated cells following exposure to temperatures ranging from 37 °C to 45 °C (Fig. 1e), and performed immunoblots for SRP72 and SRP54—two major protein constituents of the signal recognition particle. We observed a dose-dependent nuclear accumulation of both tested 7SL-binding proteins. Together, these experiments demonstrate that, in addition to de novo transcription induction of 7SL, there is a significant translocation of the 7SL and SRP protein pool from the cytosol to the nucleus during heat shock.
Given this, we investigated whether 7SL associates with chromatin like other SINE RNA during heat shock (HS)14,15. To examine chromatin binding in an unbiased manner and capture both, free 7SL and 7SL within the ribonucleoprotein particle, we performed capture hybridization analysis of RNA targets sequencing (CHART-Seq; Supplementary Fig. 1c)42,43,44 of pre-HS cells (37 °C) and post-HS cells (1 h at 42 °C). To avoid contamination with SINE RNA, biotinylated antisense capture probes were designed to 7SL-specific sequences (Supplementary Fig. 1d). The antisense probe was highly specific to 7SL RNA as demonstrated by a prominent ~300 nt single band by Northern Blot analysis (Supplementary Fig. 1e). Control experiments were performed in parallel using a 7SL sense probe to rule out hybridization to the DNA locus. As expected, the 7SL sense probe failed to produce a Northern Blot signal (Supplementary Fig. 1e). 7SL RNA-target DNA hybrids eluted with RNAseH digestion and DNA libraries of two independent replicates were prepared for sequencing. Statistically significant 7SL binding peaks were then called using MACS Software. Results from two independent replicates were similar. Sense probes failed to demonstrate enrichment shown for the antisense probe (Supplementary Fig. 1f).
As expected, CHART-seq detected a major RNA-binding signal at Rn7s1 and Rn7s2, the sites of 7SL expression proximal to the Nemf and Rps29 loci (Fig. 1f and Supplementary Fig. 1f). RNA signals were observed both before and after HS. Intriguingly, in addition to these loci, 7SL peaks were observed at 316 other chromatin sites in pre-HS conditions (Fig. 1g, blue). In post-HS cells, 556 binding sites became apparent—of which 246 sites were shared with pre-HS cells (Fig. 1g, red and Supplementary Data 1). These data demonstrate that a total of 310 new 7SL peaks were acquired during heat shock stress. We note that the genomic binding profiles of 7SL were different from those of two other noncoding RNAs, TERRA42 and JPX24 (Supplementary Fig. 2a). Importantly, 7SL binding sites were distinct from sites detected by JPX and TERRA CHART probes. Reciprocally, JPX and TERRA sites were distinct from sites detected by 7SL CHART probes. These exclusive findings argue for the specificity of 7SL CHART pulldowns. In both pre- and post-HS cells, 7SL peak annotation analysis using ChIPSeeker showed a major preference for promoters, specifically at a distance of <1 kb from the promoter region (Fig. 1h). De novo motif search revealed two motifs, one A-rich and one G-rich (Supplementary Fig. 2b). Altogether, these data demonstrate that, during acute heat shock, 7SL is upregulated, accumulates in the nucleus together with SRP proteins, and binds chromatin specifically to promoter regions.
7SL-SRP bound genes are suppressed by heat shock
To understand the functional effects of 7SL-SRP binding to chromatin, we asked if 7SL target genes respond to heat stress. Most POL-II-transcribed genes are downregulated by heat shock as part of a global response to conserve resources for stress management14,40. Here we compared 7SL peak intensities pre- vs post-HS and found significantly stronger signals in post-HS at 7SL-bound genes (Fig. 2a). Augmentation of 7SL RNA binding was notable in both independent replicates as there was a significant shift to the right in the distribution frequencies in both cases (Supplementary Fig. 3a). Gene Ontology analysis45 showed that 7SL predominantly bound genes involved in transcriptional regulation (Fig. 2b), such as the POL-II transcription factor Btf3, (Fig. 2c and Supplementary Fig. 3b) and the mediator complex subunit Med4 (Fig. 2d and Supplementary Fig. 3c). Notably, binding occurred specifically to the promoter region of 7SL target genes.

a Heatmap shows the intensity of 7SL CHART-seq signal over the binding site pre- and post-HS. The indicated region represents the peak center −/+ 3 kb. b Gene Ontology analysis (Biological process)45 was performed for 7SL CHART-Seq peaks detected in both replicates in pre- or post-HS conditions. −Log10 Benjamini P value is indicated for each term. Benjamini P value is adjusted p value for the multiple testing corrections. Source data are provided as a Source Data file. c Genome browser (IGV) view of 7SL CHART-Seq signal covering a region of the Btf3 gene. Tracks represent the Antisense (AS) signal subtracted for Sense (S) signal for two biological replicates pre- and post-HS. d Genome browser (IGV) view of 7SL CHART-Seq signal covering a region of the Med4 gene. Tracks represent the Antisense (AS) signal subtracted for Sense (S) signal for two biological replicates pre- and post-HS. e Genome browser (IGV) view of 7SL CHART-Seq signal covering a region of the pre-ribosomal Rn45s gene. Tracks represent the Antisense (AS) signal subtracted for Sense (S) signal for two biological replicates pre- and post-HS. f RT-qPCR analysis of the DNA eluate following 7SL CHART pre- and post-HS. For each indicated genomic sequence the enrichment ratio was calculated between the Antisense (AS) and Sense (S) 7SL probe. Data were presented as mean values ± SD. Source data are provided as a Source Data file. g Analysis of PRO-Seq expression patterns of 7SL target genes 1 h after exposure to heat shock (42 °C). Distribution of the entire PRO-Seq data set (blue) was compared to distribution of 7SL RNA-target genes (orange). Four different gene groups are shown based on the type of transcriptional regulation: downregulated (DownReg), unexpressed (UnExp), unregulated (UnReg), or upregulated (UpReg) genes. Source data are provided as a Source Data file. h Cumulative distribution function (CDF) of the Log2 fold change following exposure to heat shock for the entire PRO-Seq dataset (black line) and for 7SL RNA-target genes (red line). P value is calculated using Kolmogorov–Smirnov (KS), a two-sided test. Source data are provided as a Source Data file.
Interestingly, POL-I-transcribed rRNA precursor locus Rn45s was also targeted by 7SL (Supplementary Data 1). PRO-seq analysis showed that, after 1 h of heat shock40, there was a significant downregulation in the synthesis of 28S and 18S rRNAs (Supplementary Fig. 3d). The reduction coincided with an increase in 7SL expression (Fig. 1b) and 7SL binding over the corresponding rDNA locus Rn45s, as demonstrated by increased CHART signals in post-HS cells (Fig. 2e and Supplementary Fig. 3e). CHART RT-qPCR also demonstrated increased 7SL binding over 18S, 28S and External Transcribed Spacer 1 (ETS1) rDNA loci (Fig. 2f). Thus, heat shock results in a substantial increase of 7SL binding to both POL-I and POL-II genes.
Next, we investigated the effect of increased 7SL binding on the target genes during heat shock. We tried to delete the two 7SL loci or knockdown nuclear 7SL using ASOs, but all attempts were unsuccessful, likely due to the abundance and essential nature of the 7SL transcript for SRP assembly and function. To circumvent this issue, we compared expression patterns of 7SL target genes relative to the overall expression landscape after exposure to heat shock. As previously noted40,46, heat shock globally altered transcription profiles after 1 h. Overall, 46.5 and 18.8% of genes were downregulated and upregulated, respectively (Fig. 2g and Supplementary Data 2). However, among 7SL target genes, 66.1% were downregulated, while only 11.9% were upregulated. Thus, 7SL binding correlates with transcriptional downregulation during heat shock. We then compared heat shock-induced fold changes in expression levels for 7SL target genes versus all other genes. Cumulative distribution function (CDF) analysis demonstrated a significant shift of 7SL target genes to the left, indicating a substantial downregulation of gene expression (Fig. 2h). Overall, our data indicate that, during heat shock, 7SL increasingly binds to both POL-I and POL-II genes, and this augmented interaction correlates with downregulation of target genes. These data suggest that 7SL-SRP may play a role in transcriptional suppression during acute thermal stress.
SRP selectively arrests transcription during heat shock
Given that 7SL targets rDNA loci and other POL-I-transcribed genes, we next investigated whether 7SL might localize within the nucleolus during heat shock. Notably, RNA FISH indicated a diffuse presence of 7SL throughout the nucleus, including the nucleolus, prior to heat shock (Fig. 1d). Upon heat shock, however, we also observed a major upregulation and relocalization of 7SL in the nucleus, and its enrichment in intranuclear foci resembling nucleoli (Fig. 1d). To determine if these foci corresponded to nucleoli, we performed an RNA FISH coupled with immunofluorescence to detect 7SL simultaneously with nucleophosmin, a nucleolar marker. In pre-HS cells, a very modest enrichment in nucleophosmin-stained nucleoli could be observed (Supplementary Fig. 4), indicating that 7SL is indeed already present in nucleoli prior to heat shock, consistent with its previously described nucleolar localization and the role in SRP biogenesis47. Within 15 min of 45 °C heat shock, the degree of 7SL enrichment within the nucleolus became notable as demonstrated by considerable colocalization with nucleophosmin (Fig. 3a), thus confirming the 7SL shift into the nucleolus during acute heat shock. These data indicate that displacement of 7SL into the nucleolus may aid 7SL’s suppressive effect on POL-I genes, including rDNA.

a 7SL RNA FISH coupled with nucleophosmin immunostaining after 15 min exposure to heat shock (45 °C) in NIH3T3 cells. Nucleophosmin staining shows nucleoli. DAPI staining depicts nuclei. b DESeq2 analysis of transcriptional changes following exposure to heat shock (10 min, 45 °C) in siScr-transfected NIH3T3 cells (upper panel). 4125 DEGs detected. P adjusted value cutoff = 0.01. DESeq2 analysis of transcriptional changes following exposure to heat shock (10 min, 45 °C) in siSrp72 transfected cells (lower panel). 1633 DEGs detected. P adjusted value cutoff = 0.01. P adjusted values were derived from DESeq2 analysis and are based on Wald test with the adjustment by Benjamini and Hochberg method for multiple testing corrections. c Venn diagrams represent the overlap of heat shock-induced downregulated (left) or upregulated (right) differentially expressed genes (DEGs) in siScr and siSrp72 transfected NIH3T3 cells.
This 7SL displacement was also intriguing, as the nucleolus is the site of SRP biogenesis and where 7SL is assembled into the RNP28,47,48. This raised the intriguing possibility that the SRP itself may play a role in the response to heat stress. To investigate this, we examined the effects of depleting an essential SRP subunit, SRP72, using siRNAs and compared effects to that of a control, scrambled siRNA (siScr). Following 48 h knockdown, we subjected cells to acute and severe (10 min, 45 °C) heat shock and conducted RNA-Seq analysis of pre- and post-HS cells. In control cells treated with scrambled siRNA (siScr), there were no changes in SRP72 levels (Supplementary Fig. 4b). In these cells, 10 min of heat shock resulted in 2294 downregulated DEG’s and 1831 upregulated DEG’s (Fig. 3b, c and Supplementary Data 3), with more genes becoming downregulated, as expected. Intriguingly, these transcriptomic changes were significantly blunted by SRP72 depletion (Supplementary Fig. 4b): 10 min of heat shock resulted in only 1265 downregulated DEG’s and 368 upregulated DEG’s as compared to pre-HS SRP-depleted cells (Fig. 3b, c and Supplementary Data 4). Thus, both 7SL RNA and the SRP critically contribute to transcriptional reprogramming during acute heat shock.
Heat shock induces the binding of 7SL-SRP to ribosomes
Because 7SL and SRP are predominantly cytosolic, we next considered the possibility that these factors—which are classically associated with the translocation of secretory proteins49—might also play a role in translation regulation during heat shock. This idea was particularly attractive to us, as previous studies found that SRP9-SRP14 subunits can cause inhibition of translation31,32,33,34,35,36, though the in vivo impact has been controversial37,38,39. Furthermore, if 7SL regulated both transcription and translation during stress, this ancestral noncoding RNA might have evolved to shut down gene expression on a global scale as a way to divert resources during acute stress.
Here, to determine if the SRP complex associates with ribosomes during stress, we performed sucrose density gradient centrifugation of the cytosol before and after heat shock. In line with previous results50, RT-qPCR of 18S rRNA demonstrated depletion of ribosomes from heavy gradient fractions (f10–f14) under heat shock, with a corresponding accumulation in lighter fractions (f6–f9), indicative of a reduction in mRNA translation rates under heat stress (Supplementary Fig. 5a). Interestingly, heat shock also induced a significant association of 7SL with ribosomes (Fig. 4a, b and Supplementary Fig. 5b), whereas it did not induce a strong association for the related, similarly sized noncoding RNA, 7SK51 (Fig. 4a, b and Supplementary Fig. 5b). Notably, for 7SL, there was a shift from the soluble pool to the ribosome-associated pool. We observed that protein components of the SRP behaved similarly. For instance, SRP54 and SRP72 showed a similar pattern of distribution across sucrose gradients as 7SL (Fig. 4c). Indeed, heat stress redistributed SRP54 and SRP72 towards heavier, ribosome-associated fractions marked by ribosomal subunits RPL23a and RPS6.

a RT-qPCR analysis of 7SL (top) and 7SK (bottom) RNAs in 14 sucrose density gradient fractions pre- and post-HS (15 min, 45 °C) in NIH3T3 cells (left). Statistical analysis of the sum of 7SL RNA in ribosome-free and ribosome-associated fractions (right). Data were presented as mean values ± SD. P values were calculated using an unpaired, two-sided t-test. Source data are provided as a Source Data file. b Fold change of RNA levels in pre- and post-HS conditions calculated per sucrose gradient fraction. P, calculated by unpaired, two-sided t-test, comparing 7SL to 7SK RNA. Data were presented as mean values ± SD. Source data are provided as a Source Data file. c TCA-precipitated proteins derived from 14 sucrose density gradient fractions pre- and post-HS were analyzed by immunoblot for SRP54, SRP72, RPL23a, and RPS6. d RT-qPCR enrichment analysis for 7SL and 7SK RNA following RPL23a UV-RIP in pre- and post-HS conditions (15 min, 45 °C) in NIH3T3 cells. To correct for putative 7SL and 7SK RNA interactions with free L23a, enrichments were normalized by the percentage of 28S rRNA pulled down by L23a (L23a interacting with 28S rRNA is within the large ribosomal subunit). Data were presented as mean values ± SD. P values were calculated using an unpaired, two-sided t-test. Source data are provided as a Source Data file. e SRP54-RPL23a Proximity Ligation Analysis (PLA) pre- and post-HS (15 min, 45 °C) in NIH3T3 cells. Images were analyzed by confocal fluorescent microscopy (left) and quantified (right). The boxplot displays the minimum to maximum values, with the boxes showing the interquartile range (25th to 75th percentiles), and the center line indicating the median. P values were calculated using a two-sided Welch’s t-test. Source data are provided as a Source Data file.
Furthermore, UV-crosslink RNA immunoprecipitations (UV-RIP) with RT-qPCR showed that 7SL’s interaction with RPL23a increased significantly following heat shock, whereas no such increase was observed for 7SK (Fig. 4d), suggesting that the 7SL-SRP complex directly associates with ribosomes. These observations were supported by proximity ligation assay (PLA) for RPL23a and SRP54, which demonstrated a significant increase in association between the two proteins during heat stress (Fig. 4e and Supplementary Fig. 5c). Together, these data demonstrate that the 7SL-SRP complex shifts from the free cytosolic pool to become ribosome-bound during acute thermal stress.
7SL-SRP acutely suppresses translation during heat shock
The apparent increase in SRP-ribosome association during heat shock prompted us to investigate whether the SRP may contribute to known heat-mediated inhibition of translation. To study this, we depleted SRP14 and SRP72 by siRNA and performed Click-iT labeling of nascent proteins in the presence or absence of heat shock (Fig. 5a, left panel). Without heat shock, depleting SRP14 or SRP72 had no effect on translation (Fig. 5a, right panel). In contrast, exposure to heat shock strongly decreased protein synthesis (Fig. 5a, right panel, lane 4), as expected. However, depleting SRP14 or SRP72 blunted the ability of cells to suppress translation in response to heat shock—indicating that the SRP is indeed required for the stress-induced block in translation (Fig. 5a, right panel, lanes 5, 6).

a Schematic representation of Click-iT™ AHA labeling of nascent proteins (left panel). Following SRP14 and SRP72 knock down for 48 h, Click-iT™ AHA labeling was performed in human HCT8 cells in absence (10 min at 37 °C pretreatment, followed by 25 min of Click-iT™ AHA labeling at 37 °C) or presence of heat shock (10 min at 45 °C pretreatment, followed by 25 min of Click-iT™ AHA labeling at 45 °C). Immunoblots were done for Biotin, SRP72, SRP14, and Tubulin (right panel). b Puromycilation of nascent proteins was analyzed in mouse NIH3T3 cells following 48 h SRP72 knockdown pre- (10 min at 37 °C pretreatment, followed by 25 min of 10 µg/ml Puromycin labeling at 37 °C) and post-HS (10 min at 45 °C pretreatment, followed by 25 min of 10 µg/ml Puromycin labeling at 45 °C). Immunoblots for Puromycin, SRP72, and total-eIF2α are shown (see Material and Methods for experimental details). c DESeq2 analysis of transcriptional changes following siScr or siSrp72 transfection for 48 h in NIH3T3 cells at basal conditions (37 °C). Thirty-three significantly differentially expressed genes (DEGs) were detected. P adjusted value cutoff = 0.01. P adjusted values were derived from DESeq2 analysis and are based on the Wald test with the adjustment by Benjamini and Hochberg method for multiple testing corrections. d Venn diagram representing the intersection between 33 siSrp72-driven DEGs and 97 genes directly regulated by ATF4/CHOP56. Atf3 and Ero1l genes were detected in both datasets. e Following SRP72 knockdown for 48 h, nascent translation was assessed in mouse NIH3T3 cells in the presence or absence of an integrated stress response inhibitor (ISRIB). After 1 h pretreatment with 500 nM ISRIB at 37 °C, puromycilation was performed pre- (10 min at 37 °C pretreatment, followed by 25 min of 10 µg/ml Puromycin labeling at 37 °C) and post-HS (10 min at 45 °C pretreatment, followed by 25 min of 10 µg/ml Puromycin labeling at 45 °C). Where indicated, ISRIB at 500 nM was maintained until cell harvest. Immunoblots were performed for Puromycin, SRP72, phospho-eIF2α, and total-eIF2α. f Cell viability was assessed in heat shock-treated HCT8 cells 48 h after siRNA-mediated SRP54 or SRP72 knock-down. Heat shock was performed at 45 °C for indicated times. Cell viability was assessed following 24 h recovery at 37 °C. Cell Counting Kit 8 (WST-8/CCK8) reagent was added for 3 h, and viability was calculated as a percent change in 460 nm absorbance relative to pre-HS conditions. P values were calculated using an unpaired, two-sided t-test. Data were presented as mean values ± SD. Source data are provided as a Source Data file.
Click-iT labeling requires methionine starvation (see Fig. 5a, left panel, and Material and Methods) that could confound result interpretation by triggering an additional layer of stress response. We thus turned to an orthogonal method of labeling nascent peptides with puromycin, an antibiotic mimic of tyrosyl-tRNA52. The effects of SRP72 depletion and heat shock on protein synthesis monitored by puromycylation and Click-iT labeling were comparable (Fig. 5b). To exclude the possibility that the observed effects on attenuation of translational repression during heat shock may be mediated by SRP72’s function outside of SRP, we assessed SRP72 interaction with other SRP subunits (SRP14-9) pre- and post-HS (Supplementary Fig. 6a). Pull-down experiments did not show conspicuous alterations in SRP72-SRP14-9 interaction before and after exposure to heat, suggesting that the effects observed by SRP72 knock-down reflect the function of whole SRP. This was further confirmed using downregulation of SRP54, the most evolutionarily conserved SRP subunit (Supplementary Fig. 6b). Collectively, these data indicate that SRP is required for acute translation inhibition during heat shock.
We next sought to exclude the possibility that the effects of SRP54 and SRP72 depletion are mediated through SRP9/14-scAlu, an RNP that contains small cytoplasmic Alu (scAlu) and shares two of the SRP subunits, SRP9 and SRP1410,12,53. We depleted SRP14, SRP54 or SRP72 (Supplementary Fig. 6c) and performed Northern Blot analysis for full-length Alu RNA and scAlu (Supplementary Fig. 6d). As expected, we observed a heat shock-dependent increase in Alu RNA. On the other hand, scAlu levels did not change except when siSrp14 was depleted (Supplementary Fig. 6d), suggesting that the blunting of translation inhibition in SRP54 or SRP72 depleted cells was not due to loss of scAlu.
Yet another potential confounding factor is that, even in the absence of heat shock, depletion of SRP54 can induce transcription changes in heat shock-related genes54,55. To investigate whether the depletion of SRP72 provoked similar perturbations, we monitored the effects of SRP72 depletion on the transcriptome of NIH3T3 cells using RNA-Seq. Depletion of SRP72 resulted only in minor perturbations in the transcriptome as compared to siScr-transfected cells (Fig. 5c). Only 32 DEG’s were observed between siSrp72 and siScr cells, apart from Srp72 itself, and none were known heat shock-related genes (Supplementary Data 5) Additionally, SRP72 downregulation did not appear to induce other types of stress responses (Fig. 5d and Supplementary Fig. 6e). For instance, there was only a marginal overlap between SRP72-depleted cells transcriptomic changes and those caused by Activating Transcription Factor 4 (ATF4) and C/EBP homologous protein (CHOP)56, two master transcriptional regulators induced during the Integrated Stress Response (ISR)57. This overlap comprised only two genes- Ero1l and Atf3 (Fig. 5d). Moreover, after intersection of 33 siSrp72 DEGs with 575 genes targeted by ATF4 or CHOP within 3 kb from their transcription start site, only two additional genes overlapped—Car6 and Rab39b (Supplementary Fig. 6e). These data indicate that SRP72 depletion does not cause a major stress response and that pre-exposure to stress and tolerance acquisition58 are unlikely reasons for loss of translation inhibition in siSrp72 cells during heat shock.
Cellular response to a variety of stressors (e.g., heat shock, ER dysfunction, amino acid deprivation, viral infection, toxins, heme deficiency) triggers ISR, which is centered on stress-sensing kinases (PERK, PKR, HRI, or GCN2)-dependent phosphorylation of the α subunit of the eukaryotic translation initiation factor eIF259. Phosphorylation of eIF2α suppresses the GEF activity of eIF2B, thus limiting the recycling of the ternary complex, which delivers initiator tRNA to the ribosome59. This leads to a reduction of global protein synthesis, and concomitant induction of proteins encoded by upstream open reading frame (uORF) containing mRNAs including ATF4 and CHOP59. Notwithstanding that the transcriptional profiles of SRP72 depletion and ATF4- and CHOP-driven ISR transcriptional responses were clearly distinct (Fig. 5d and Supplementary Fig. 6e), we further sought to establish the relationship between SRP and ISR. Depletion of SRP72 led to slight increase in eIF2α phosphorylation at baseline as compared to control (Fig. 5e). Heat shock however, led to a substantial increase in eIF2α phosphorylation which appeared to be SRP72-independent (Fig. 5e). Notwithstanding that eIF2α phosphorylation levels were comparable between control and SRP72-depleted cells, the inhibitory effects of heat shock on protein synthesis were alleviated only in SRP72-depleted cells (Fig. 5e, compare lanes 5 and 6). Finally, adding an ISR inhibitor, ISRIB60 1 h prior to heat shock partially attenuated translation inhibition under heat shock to a comparable level as SRP72 depletion (Fig. 5e). When ISRIB and SRP72 depletion were combined, the two treatments additively attenuated translation inhibition under the heat shock, resulting in a nearly complete recovery of global protein synthesis (Fig. 5e, compare lanes 6 and 7 with lane 8).
Collectively, these data suggest that SRP and ISR operate in parallel to shut down protein synthesis during the heat shock response. In light of this, we were curious to learn if SRPs affect cell survival under heat stress (Fig. 5f). Following heat exposure, the viability of cells transfected with siSrp54 or siSrp72 was compared to siScr-treated cells. Intriguingly, our data demonstrate that SRP54 or SRP72 deficiency promotes survival under heat shock. Thus, while SRPs help cells cope with thermal stress, we suggest that they may either exact a survival toll on cells with an overly aggressive heat shock response and/or promote an adaptive response by inducing apoptosis in cells that have been damaged by heat shock.
Ribo-Seq analysis reveals selective translational suppression
Of interest was whether SRP inhibition affected all mRNAs or only a select group of translating mRNAs during acute thermal stress. To address this, we performed Ribo-Seq61, a next-generation sequencing-based method of detecting and actively translating mRNAs by quantifying ribosome-protected fragments (RPFs). Prior to heat shock, we observed comparable translatomes (i.e., collection of actively translated mRNAs on a transcriptome-wide scale) in both control (siScr) cells and cells depleted of SRP72 (siSrp72) (Fig. 6a, left panels). This was consistent with experiments showing that depletion of SRP72 in the absence of stress does not exert a major effect on global protein synthesis (Fig. 5). Meta-ORF analysis mapping reads between the TIS (translation initiation site) and TTS (translation termination site) showed that exposing cells to 10 min of heat shock resulted in a marked decrease in ribosome-protected mRNA footprints under control conditions (siScr), whereas depleting SRP72 blunted this effect (Fig. 6a, right panels)—again consistent with above experiments addressing nascent protein synthesis (Fig. 5). These data indicated a loss of RPFs (active translation) under heat shock and their restoration upon SRP72 depletion.

a Heatmap represents a meta-ORF analysis of indicated Ribo-Seq datasets. After 48 h of siScr or siSrp72 transfection, NIH3T3 cells were subjected to for 10 min to 37 °C (pre-HS) or 45 °C (post-HS). k-means clustering was used to group samples into two separate clusters. Results represent the average of two biological replicates. TIS signifies translation initiation site. TTS represents the translation termination site. b Metaplots derived from Ribo-Seq Cluster1 and Cluster2 for indicated experimental conditions. In the metaplot 0 represents translation initiation site (TIS) and 100 indicates translation termination site (TTS). Source data are provided as a Source Data file. c Graphs of elongation index (Elg idx) derived from Cluster1 and Cluster2 metaplots. The elongation index was calculated as an RPF signal ratio between 10 and 90% of the meta-ORF and the first 10% of the meta-ORF. The whiskers represent the 10th and 90th percentiles, the boxes show the interquartile range (25th to 75th percentiles), and the center line indicates the median. P values were calculated using an unpaired, two-sided t-test. Source data are provided as a Source Data file. d Graph of translation efficiency (TE) calculated as the number of ribosome-protected fragments divided by the number of RNA-Seq reads for each transcript. The whiskers represent the 10th and 90th percentiles, the boxes show the interquartile range (25th to 75th percentiles), and the center line indicates the median. P values were calculated using an unpaired, two-sided t-test. Source data are provided as a Source Data file.
Using k-means clustering, we grouped mRNAs into two clusters based on their RPF patterns with Cluster2 accounting for >90% of mRNAs (Fig. 6a, b and Supplementary Data 6). Both clusters exhibited a heat-induced drop in the RPF signal around the TIS and across the entire meta-ORF, consistent with inhibition of translation initiation. Notably, Cluster2 was distinguished from Cluster1 by a sensitivity to SRP72 depletion (siSrp72) specifically during heat shock (Fig. 6b, red versus black lines, bottom panel). Of note, in line with previous observations37, under basal conditions, SRP72 knock-down did not cause a noticeable change in translation. SRP, therefore, serves as a heat shock-specific inhibitor of translation.
Given that 7SL regulates transcription (Figs. 1–3), we wished to rule out the possibility that the translational decrease was secondary to transcription suppression. We calculated the translation efficiency (TE), a measure of translation that normalizes for changes in mRNA levels by computing the number of RPFs per copy of a messenger RNA62. Heat shock induced a substantial decrease in TE across a subset of genes, which was in part alleviated by SRP72 depletion (Fig. 6d). Importantly, SRP-dependent changes in TE were specific to heat stress. Overall, our data indicate that, during heat shock, SRP negatively regulates protein synthesis not only by reducing transcriptional outputs, but also by reducing translation efficiency of a subset of mRNAs.
SRP preferentially suppresses the translation of nuclear-encoded mitochondrial factors under heat stress
Next, to identify cohorts of mRNAs whose TEs were specifically affected by SRP72 depletion under heat shock, we performed a detailed, transcriptome-wide analysis of changes in TEs in control vs. SRP72-depleted NIH3T3 cells by calculating normalized RPF/RNA-seq read log2 ratios for individual genes before and after heat shock. We defined 4 clusters based on their SRP72 dependency (Fig. 7a, Supplementary Fig. 7a, b, and Supplementary Data 7). Clusters 1 and 2 genes were more difficult to disentangle for the effect of SRP72 on transcription and translation. Cluster1 represents mRNAs whose translation was not regulated by SRP, as SRP72 knock-down did not have a major effect on TEs in pre- or post-HS conditions—i.e., translation remained proportional to mRNA levels. Cluster2 mRNAs showed a small increase in translation efficiency upon SRP72 knock-down in both pre- and post-HS cells, indicating that their translation is negatively regulated by SRP under both conditions. On the other hand, Cluster3 mRNAs showed transcriptional upregulation after SRP72 knock-down, but greater mRNA abundance did not translate to more protein synthesis—consistent with the idea of “translational buffering” wherein mRNA translation offsets the effects of alterations in transcription to maintain the levels of corresponding proteins constant63. In contrast to the other three clusters, Cluster4 comprised genes prominently regulated by SRP solely at the level of translation and under thermal stress. Intriguingly, the Ribo-Seq signal of Cluster4 mRNAs was elevated in siSrp72 cells under heat shock, without underlying changes in mRNA levels (Fig. 7a). TE calculation further reinforced this observation by demonstrating a significant, heat shock specific “recovery” in translation of this subset of mRNAs after SRP72 depletion (Fig. 7b).

a Heatmap represents a parallel view of RNA-Seq and Ribo-Seq Log2 fold changes of siScr/siSrp72 in pre- and post-HS conditions. Datasets were divided into four clusters using k-means clustering. TSS stands for transcription start site. TES represents the transcription end site. TIS signifies translation initiation site. TTS represents the translation termination site. b Translation efficiency graph for Cluster4 for indicated conditions. Cluster4 comprises 19% of all genes. The whiskers represent the 10th and 90th percentiles, the boxes show the interquartile range (25th to 75th percentiles), and the center line indicates the median. P values were determined using an unpaired, two-sided t-test. Source data are provided as a Source Data file. c Functional annotation clustering for Cluster4 genes45. Enrichment scores and −Log10 Benjamini P values are represented for each of the top three represented terms. Benjamini P value is the adjusted p value for the multiple testing correction. Source data are provided as a Source Data file. d Graph of mitochondrial (left) and signal peptide-containing genes (right) representation per TE cluster. Values above 1 indicate enrichment. Values below 1 indicate depletion. Source data are provided as a Source Data file. e Genome browser (IGV) view of RNA-Seq and Ribo-Seq signal covering Cyc1 and Apex1 genes in pre-HS (black) or post-HS (red) conditions under siScr or siSrp72. Replicate 1. f Model: 7SL and SRP orchestrate transcriptional and translational responses during heat shock. See Discussion for description.
We next analyzed Cluster4 genes by performing functional annotation clustering45. Intriguingly, this analysis revealed that “mitochondria” was the most highly enriched functional category while the “isopeptide bond” and “endoplasmic reticulum” categories scored significantly lower (Fig. 7c). Consistent with this observation, when Cluster4 was evaluated by KEGG pathway analysis45, the “metabolic pathway” category was identified as the most significantly enriched (Supplementary Fig. 7c). We further investigated the distribution of nuclear-encoded mitochondrial genes (as defined by MitoCarta3.064) across all four clusters by calculating the representation factor—a measure of the likelihood of the observed number of mitochondrial genes overlapping with each cluster, divided by the expected number of overlapping mitochondrial genes if randomly drawn. We observed a significant enrichment of mitochondrial genes within Cluster4 and confirmed their dependence on SRP for translational suppression during heat stress (Fig. 7d and Supplementary Fig. 7d).
We then performed the same analysis for proteins containing signal peptides (SPs; as defined by http://www.signalpeptide.de65,66,67). Intriguingly, these factors were enriched in Cluster3—the group of transcriptionally upregulated genes (Cluster3) in siSrp72 cells—and were under-represented in Cluster4, the group that underwent exclusive translational regulation (Cluster4) (Fig. 7d and Supplementary Fig. 7e). Accordingly, when compared to no-SP genes, the SP-genes did not exhibit a significant change in TE after SRP72 depletion at basal or during heat shock (Supplementary Fig. 7f). This indicated that SRP-mediated translation suppression shows no preference for SP-containing genes and is a separate phenomenon from SRP’s function in protein secretion.
SRP-mediated regulation of nuclear-encoded mitochondrial gene translation was confirmed by evaluating genome browser tracks of representative genes coding for mitochondrial proteins- Cyc1 and Apex1. Exposure to heat shock led to a loss of the RPF signal over the Cyc1 and Apex1 ORF’s and these effects were attenuated in SRP72-depleted cells (Fig. 7e and Supplementary Fig. 7g). On the other hand, steady-state levels of these mRNAs did not show major alterations (Fig. 7e and Supplementary Fig. 7g). Together, these data additionally implicate the SRP in selective translational suppression of nuclear-encoded mitochondrial mRNAs during heat shock. Thus, the SRP integrates the heat shock response across the nucleus, cytosol, and mitochondria and plays a maladaptive role under heat-induced homeostasis alterations.
Discussion
Herein, we have identified 7SL RNA and SRP as regulators of transcriptional and translational changes during heat shock. Once thought to only control the translocation of the signal peptide-containing proteins across the ER membrane, our study unveils 7SL and the SRP in the global regulation of transcription and translation across the nucleus, cytosol, and mitochondria. 7SL/SRP is, therefore, part of a cell-wide coordinated response to heat stress. On the basis of data presented here, we propose that 7SL and SRP concurrently act in the nucleus, cytosol, as well as mitochondria (via inhibiting translation of nuclear-encoded mitochondrial proteins), during heat shock (Fig. 7f). By suppressing transcription and translation of housekeeping genes, cells in acute stress can conserve resources in order to mobilize essential stress-response factors.
In the nucleus, heat shock induces both, transcription-dependent and transcription-independent 7SL RNA accumulation, leading to nucleolar enrichment as well as global nuclear levels. The data we have shown demonstrate that cytosolic 7SL and SRP proteins re-enter the nucleus and significantly contribute to the overall nuclear increase. Our CHART-Seq results indicate that 7SL increasingly binds the DNA under stress, however, further studies will be needed to clarify whether this interaction is mediated through free 7SL or 7SL within fully/partially assembled SRP (therefore labeled as Ribonucleoprotein particle (RNP) in Fig. 7f). The possibility that 7SL interacts with the genome as an independent RNA could be explained by an imbalance between the increasing 7SL levels and the non-changing amounts of the SRP proteins during heat exposure. This could result in a free pool of 7SL that is not tethered to SRP. The increased pool of 7SL could then drive binding to specific POL-I and POL-II target genes. Interestingly, various SRP proteins have been proposed to interact with chromatin independently of fully assembled SRP and regulate transcriptional outcomes68,69. Therefore, further studies will be needed to clarify the exact form of 7SL or SRP in which they interact with the genome in the context of heat shock.
As the target genes are generally downregulated by heat shock, we suggest that 7SL binding, leads to transcription inhibition of these target genes. Notably, in addition to rRNA transcripts, genes that are preferentially targeted by 7SL are enriched in regulators of transcription. This suggests that by modifying the expression of transcriptional regulators, 7SL and SRP may also profoundly alter transcriptional wiring in response to heat shock. Intriguingly, these chromatin-related functions of 7SL RNA are akin to those described for other POL-III transcripts that have been reported to interact with the genome and regulate gene expression. For instance, short noncoding RNA species transcribed by POL-III, such as Alu8,15, B29,14, and 7SK70 have been found to negatively affect POL-II transcription. Interestingly, while global POL-II and POL-I activity undergoes inhibition in heat stress40, POL-III transcription of SINE-related sequences is elevated2,6. In conjunction with these reports, our observations that heat shock increases 7SL expression, nuclear accumulation, and its binding to POL-II and POL-I genes suggests that POL-III and its transcription products may be key drivers of the stress response, with the capacity to regulate the two other RNA polymerases and their genetic outputs.
Our data implicate SRP protein subunits in the regulation of mRNA levels during heat stress, as there was a significant nuclear accumulation (Fig. 1e) and blunting of transcription downregulation when SRP72 is depleted (Fig. 3c). While this study does not address the underlying mechanism of SRP-mediated downregulation of mRNA levels during heat shock, it is plausible that the SRP-driven inhibition of translation may lead to degradation of the underlying mRNA as suggested by multiple studies that investigated mechanisms whereby translation rates directly impact on half-life of mRNA71,72,73. Moreover, 7SL-mediated transcription downregulation and SRP-mediated mRNA metabolism may represent a synchronous molecular mechanism by which the 7SL/SRP components fine-tune transcriptional outputs in a coordinated fashion during heat shock and further orchestrate them with translational programs to shape the proteome.
Importantly, the SRP has already been implicated in the regulation of the stability of mRNAs coding for SP-containing proteins through Regulation of Aberrant Protein Production (RAPP)74—a protein quality control mechanism where an adequate interaction between the nascent SP-containing polypeptides and the SRP is needed to prevent the destruction of the corresponding, actively translating mRNA. While, through RAPP, depletion of SRP54 leads to failure in SP recognition and degradation of the corresponding mRNA74, depletion of SRP72 did not result in major global changes in mRNA abundances. Specifically, SRP72 depletion altered levels of only 33 mRNAs (Fig. 5c). Also, in contrast to SRP54 knock-down, SRP72 silencing failed to induce a stress response (Fig. 5c, d). Notably, SRP54 is the major and the most evolutionary conserved component of the signal recognition process, as it recognizes and interacts with SP-containing proteins either as a free polypeptide or as a part of the SRP75,76. On the other hand, the SRP72 protein forms a structural bridge between the signal recognition domain (SRP54) and the elongation arrest domain (SRP9/14) of the Signal Recognition Particle36. Therefore, we speculate that upon SRP72 knock-down, a significant level of SRP54-mediated signal recognition is still maintained either through the remaining SRP or through the free SRP54.
Our study also demonstrates significant effects of 7SL/SRP in the cytosol. Heat shock shifts the function of 7SL/SRP from translocation of secretory proteins to inhibition of peptide synthesis. 7SL/SRP binds ribosomes and suppresses translational outputs. This observation sheds new light on the physiological importance of SRP-ribosome interaction and addresses a long-standing question37,38 of whether the SRP binding to ribosomes can affect protein synthesis in vivo. In agreement with a recent study37, under basal conditions, we do not notice significant changes in translational activity upon SRP54 or SRP72 depletion. However, under heat stress, SRP54 or SRP72 depletion resulted in partial recovery of protein synthesis, demonstrating that SRP regulates translation under stress conditions. Moreover, our study suggests that SRP-mediated regulation of translation is likely to be independent of eIF2α phosphorylation and ISR.
We find that SRP preferentially regulates de novo synthesis of nuclear-encoded mitochondrial proteins. Given the importance of mitochondria in cell metabolism and energy production, we predict that, in conditions of heat stress, the SRP inhibits de novo synthesis of mitochondrial proteins and, therefore, impairs mitochondrial function77. Interestingly, we also found that SRPs reduce the viability of cells under heat stress, potentially in part due to mitochondrial dysfunction. When SRP54 and SRP72 were depleted, cell survival was paradoxically enhanced under thermal shock. However, the improved survival may be adaptive, as apoptosis of cells that have been damaged by heat shock may aid the overall survival of the organism. Finally, we demonstrate that, under heat shock, the SRP-dependent regulation of translation does not show a preference for the SP-containing proteins. We speculate that exposure to heat shock may reduce the specificity of the SRP-mediated recognition of secretory and membrane proteins’ signal peptides, leading to a more promiscuous SRP binding to ribosomes and consequent inhibition of translation. Finally, it is plausible that the dissociation of factors that bind to nascent peptide chains (e.g., METAP2, chaperones, and NAC) may facilitate the binding of SRP to ribosomes under heat shock78.
In summary, our study uncovers aspects of 7SL RNA and SRP biology that go beyond signal peptide recognition and protein translocation to include a global response to acute stress. At the present time, it is not known how the 7SL/SRP mechanism fits into the overall molecular circuitry during the response to thermal stress79,80. Further studies will be required to understand how 7SL/SRP interdigitates with other major stress-response regulators such as stress granules81,82,83 for translation and HSF140,84,85,86 and NELF87,88 pathways for transcription regulation. The fact that 7SL is an ancestor to other stress-related SINEs (Alu, B1) could suggest that 7SL ancestors may have initially evolved to handle stress in an RNA world and that their function in protein translocation was later usurped by SRP proteins. Notably, a past study performed in barley suggested that, under heat shock, the SRP inhibits the production of secretory proteins and leads to the degradation of secretory messenger RNA89. While mechanisms remained elusive, this research in plants suggests that the heat stress specific regulation of protein synthesis by SRP could represent a phenomenon with cross-kingdom conservation.
Methods
Cell culture and siRNA transfection
NIH3T3 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum, 1% Penicilin-Streptomycin-Neomycin (PSN) antibiotic mixture, and 1% non-essential amino acids from GibcoTM. HCT8 cells were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum, 1% Penicilin-Streptomycin-Neomycin (PSN) antibiotic mixture and 1% non-essential amino acids from GibcoTM. All the siRNA (listed in Supplementary Data 8) were ordered through DharmaconTM and used at 20 nM final concentration. siRNAs were delivered to cells by two rounds of transfection using Lipofectamine® RNAiMAX Transfection Reagent.
SRP pull down
About 100 × 103 HCT8 cells were seeded into six-well plates. The next day, cells were transfected with 2.5 µg P14-9VN plasmid for overexpressing human SRP14-9 heterodimer or pcDNA3.1 empty vector as control using Lipofectamine™ 3000 (Thermo Fisher). p14-9VN was created by Katharina Strub (Addgene plasmid # 50930). Twenty-four hours later, cells were exposed to heat shock (45 °C for 15 min), washed with cold 1 × PBS, and collected by cell scraper in 1 × PBS, 1 × cOmplete™, EDTA-free Protease Inhibitor Cocktail (Sigma-Aldrich). Cells were lysed with sonication (5S ON, 30S OFF, 20% amplitude for two times). Cell lysate was bound to Anti-FLAG® M2 Magnetic Beads (Sigma-Aldrich), washed with 1 × PBS, 0.1% NP-40 and eluted with 1 × PBS, 0.1% NP-40, 0.8 mg/mL FLAG peptide (Thermo Fisher).
Western blot
Protein lysates were prepared with NP-40 lysis buffer (1% NP-40, 137 mM NaCl, 20 mM Tris pH8, 2 mM EDTA pH8) or, in the case of Click-IT metabolic labeling, with SDS lysis buffer (1% SDS in 50 mM Tris-HCl, pH 7.5). Buffers were supplemented with cOmplete™, an EDTA-free Protease Inhibitor Cocktail. Gel electrophoresis was performed in 1X TBE using Bio-Rad Mini-PROTEAN® TGX™ Precast Protein Gels and proteins were transferred to Immobilon-P membranes using Hoefer Te77xp semi-dry apparatus. Membranes were blocked in 5% non-fat dry milk for 30 min and incubated with primary antibodies overnight at 4 °C. Next day, membranes were washed three times in 1x PBS, 0.05% Tween-20 and incubated with adequate HRP-containing secondary antibodies for 1 h at room temperature. Membranes were developed using Western Lightning Plus-ECL, Enhanced Chemiluminescence Substrate (PerkinElmer). Uncropped Western Blot images have been provided as a Source Data file. Antibodies used in this study are listed in Supplementary Data 9.
RNA FISH
About 50 × 103 NIH3T3 cells were plated in 500ul of DMEM medium on top of isopropanol-sterilized and gelatin-coated coverslips. Two days later, cells were exposed to heat shock (42 °C for 1 h, or 45 °C for 15 min), washed with ice-cold 1x PBS, and pre-extracted for 5 min using CSKT buffer (100 mM NaCl, 300 mM sucrose, 10 mM PIPES, 3 mM MgCl2, 0.5% Triton X-100, pH 6.8). Next, cells were fixed for 10 min at room temperature with 4% paraformaldehyde in 1x PBS, rinsed once with 1x PBS, and dehydrated in 70, 80, 90, and 100% EtOH for 2 min per step and air-dried. Coverslips were, then, incubated in humidified chambers overnight at room temperature with 3′ FAM6—labeled 7SL antisense (5′-AGTTTTGACCTGCTCCGTTTCCGAC-3′) or 7SL sense (5′-GTCGGAAACGGAGCAGGTCAAAACT-3′) probe. The next day, samples were washed 3 × 5 min in 25% Formamide, 2x SSC at room temperature, and mounted using Vectashield mounting medium with Dapi.
RT-qPCR
Cells were lysed with TRIzolTM reagent and total RNA was isolated by phenol/chloroform extraction. Random Primers (Promega, C1181) and SuperScript™ III Reverse Transcriptase kit (Life Technologies, 18080085) were used for cDNA generation. Quantitative PCR was performed using iTaq™ Universal SYBR® Green Supermix (BIO-RAD, 1725125). Oligo sequences utilized for RT-qPCR are shown in Supplementary Data 10.
7SL RNA CHART-Seq
7SL RNA CHART-Seq was done as previously described90 with adaptations. Briefly, 26 × 106 NIH3T3 cells were used per condition. Cells were resuspended in 2 ml of DMEM prewarmed to 37 °C and incubated for 1 h at 37 °C (pre-HS) or 42 °C (post-HS). Following formaldehyde crosslinking and sonication steps, overnight hybridization was done at room temperature with biotinylated 7SL sense/control probe (5′-GTCGGAAACGGAGCAGGTCAAAACT/3BioTEG/-3′) or 7SL antisense probe (5′-AGTTTTGACCTGCTCCGTTTCCGAC/3BioTEG/-3′). For CHART enrichment, 120 µl of DEPC H2O washed MyOne™ Streptavidin C1 beads (Life Technologies, 65-002), without previous blocking, were incubated with hybridization reactions for 1 h at 37 °C with rotation. Beads containing RNA-DNA hybrids were washed and eluted for RNA (to demonstrate 7SL RNA pull-down specificity by RT-qPCR) and for DNA in the presence or absence of RNaseH (NEB, M0297L). Eluted DNA was further analyzed by RT-qPCR to validate the pulldown. To calibrate the library preparation steps, 2 pg of drosophila DNA was spiked-in to each sample. Then, we prepared sequencing libraries using the NEBNext Ultra DNA Library Prep Kit for Illumina (E7370S, NEB) and sequenced on Illumina Novaseq, creating 150 bp paired-end sequencing reads.
CHART-Seq analysis
CHART reads were aligned to mouse mm10 reference genome with Bowtie2, followed by removal of reads that aligned to more than one position in the genome, duplicated reads, or reads with low mapping quality score. Reads were also aligned to the drosophila reference genome, and the number of aligned reads was obtained. Heatmaps and BigWig files were created with Deeptools and tracks were normalized according to the number of reads aligned to the drosophila genome. Then, Sense track was subtracted from the Antisense track, indicated as AS-S. Peak calling was performed with MACS291 with a p value cutoff of 05e-04. Peaks were called significant if appeared in both replicates of the same condition. Annotation of peaks was performed with ChIPseeker92.
De novo motif analysis
For de novo motif analysis within the 7SL RNA CHART-Seq peaks, MEME Suite software93 https://meme-suite.org/meme/tools/meme was used with following settings: Motif Site Distribution - ANR: Any number of sites per sequence, Maximum Number of Motifs- 3, Motif E-value Threshold - no limit, Minimum Motif Width – 6, Maximum Motif Width – 50, Minimum Sites per Motif – 2, Maximum Sites per Motif – 1000, Bias on Number of Sites - 0.8. The top two most significantly enriched motifs were shown.
Proximity ligation assay
The cells were cultured on coverslips and subjected to a 15-min heat shock at 45 °C. Subsequently, they were washed and fixed using 4% Paraformaldehyde, followed by three washes with 1X PBS. To prevent nuclear interference, cytoplasmic-specific permeabilization was performed by treating the cells with 0.2% Saponin in 1X PBS at room temperature for 7 minutes. Antibodies against SRP54 and RPL23a were used at a dilution of 1:500 for primary protein targeting prior to the proximity ligation assay (PLA). The PLA was conducted according to the manufacturer’s instructions using the Duolink® In Situ Starter Kit Mouse/Rabbit (Sigma, DUO92101). However, certain modifications were made: antibody incubations were carried out at room temperature for 2 h, ligation reactions were performed at 37 °C for 1 h, and polymerization reactions were conducted at 37 °C for 2 h. Image acquisition was performed using a Nikon 90i Microscope equipped with a 603/1.4 NA VC Objective lens, Orca ER CCD Camera (Hamamatsu), and Volocity Software (PerkinElmer). The acquisition time was adjusted using the same parameters for all conditions and replicates. Polymerized ligated spots were quantified for each field, normalized by the total number of cells in the field, and expressed as a PLA score or interactions per cell. The results are presented as violin plots, where the median is depicted as a continuous line and the quartiles as dotted lines.
UV-RIP
About 20 × 106 cells were UV-crosslinked (200 µJ/cm2) in cold 1x PBS. Subsequently, cells were spun for 5 min at 450×g, resuspended in 800 µl of lysis buffer (1x PBS, 1% NP-40, 0.5% Sodium Deoxycholate, 400 U/ml Protector RNase Inhibitor (Sigma-Aldrich Inc., 3335402001), 1x cOmplete™, EDTA-free Protease Inhibitor Cocktail) and rotated for 30 min at 4 °C. 30U of TURBO™ DNase (2 U/µl) (Life Technologies, AM2239) was added to the lysates and tubes were placed for 15 min at 37 °C. Samples were, then, cleared for 15 min at 12,500×g, supernatants were collected, and protein concentration was measured. About 300 µg of proteins was combined with 5 µg of RPL23a or Normal rabbit IgG antibody conjugated to 35 µl of Protein G Dynabeads (Life Technologies, 10003D), and reactions were rotated overnight at 4 °C. About 30 µg of the lysates was saved for 10% input. The next day, beads were washed six times by pipetting 20x up and down in the first wash buffer (1x PBS, 1% NP-40, 0.5% sodium deoxycholate, 150 mM NaCl) followed by three washes by pipetting 20x up and down in the second wash buffer (50 mM Tris pH 7.5, 50 mM NaCl, 10 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate). Samples and inputs were, then, incubated for 30 min at 55 °C with 100 µg ProteinaseK in 100 µl of ProteinaseK buffer (100 mM Tris pH 7.5, 50 mM NaCl, 10 mM EDTA, 0.5% SDS). Subsequently, 900 µl of TRIzolTM reagent was added to each sample and RNA was extracted in 100 µl of DEPC H2O using Direct-zol™ RNA Miniprep Plus (Zymo Research, R2072). AboutT 10 µl of the RNA extract was used for cDNA synthesis. Oligo sequences used for RT-qPCR are shown in the Supplementary Data 10. Enrichments were calculated relative to input and normalized to 28S rRNA enrichments in RPL23a immunoprecipitations.
Sucrose density gradient fractionation
Sucrose gradients were prepared according to the previously described94. Briefly, filtered 60% sucrose stock solution was combined with H2O and 10x sucrose gradient buffer (200 mM Hepes pH 7.6, 1 M KCl, 50 mM MgCl2, 100 µg/ml Cycloheximide, 1x cOmplete™, EDTA-free Protease Inhibitor Cocktail, 100U/ml SUPERase-In (Life Technologies, AM2696) to achieve 5% and 50% sucrose in 1x sucrose gradient buffer. Linear 5 to 50% sucrose gradients were formed in 14 × 89 mm ultracentrifuge tubes (Seton Scientific, 7030) using a Biocomp gradient station. Cell pellets obtained from six confluent 15 cm tissue culture plates were incubated for 15 min at 37 °C or 45 °C in 2 ml of cell medium prewarmed to 37 °C. After two washes in cold 1x PBS, cell pellets were lysed in 1 ml of lysis buffer (100 mM Tris pH 7.5, 50 mM KCl, 25 mM MgCl2, 1 mM DTT, 100 µg Cycloheximide, 1x cOmplete™, EDTA-free Protease Inhibitor Cocktail, 1 U/µl SUPERase-In) and rotated for 10 min at 4 °C. Lysates were cleared for 20 min at 13,800×g and 1 ml of the supernatant was loaded on top of the sucrose gradients. Samples were subjected to 5 h ultracentrifugation at 107,000×g, 4 °C using SW41i rotor and slow brake. Consequently, each gradient was divided into 14 fractions using a Biocomp gradient station. From each fraction, 100 µl was used for RNA extraction, and 500 µl was subjected to TCA protein precipitation.
Click-iT™ AHA labeling of nascent proteins
HCT8 cells were washed three times in 1x PBS and incubated for 1 h at 37 °C in a methionine-free medium (RPMI, Thermo Fischer, A1451701). Subsequently, cells were kept at 37 °C (pre-HS) or exposed to heat shock for 10 min at 45 °C in a methionine-free medium prewarmed to 45 °C. Next, Click-iT™ AHA (Life Technologies, C10102) labeling was added to cells at 40 µM final concentration for an additional 25 min at 37 °C or 45 °C. After Click-iT™ AHA labeling, cells were harvested in SDS lysis buffer (1% SDS in 50 mM Tris-HCl, pH 7.5) and protein biotinylation was performed using Click-iT™ Protein Reaction Buffer Kit (Life Technologies, C10276). Protein samples were separated using Bio-Rad Mini-PROTEAN® TGX™ Precast Protein Gels, transferred to Immobilon-P membrane, and developed with Streptavidin-HRP.
Northern blot
Total RNA was isolated according to manufacturer’s instruction using TRIzolTM reagent and 2–5 µg of the RNA was loaded on 8 M Urea/6% PAGE gel. To evaluate RNA loading and integrity samples were stained in 1x TBE 1x SybrGold (Life Technologies, S-11494) for 5 min. Gel pictures were acquired using Amersham Imager 680. Following an overnight transfer to Amersham Hybond N+ membrane (RPN303B), samples were UV-crosslinked and incubated with a radiolabeled probe overnight at 58 °C. For 7SL Northern Blot detection, the following radiolabeled probes were used: antisense (5′- AGTTTTGACCTGCTCCGTTTCCGAC-3′) and control/sense (5′- GTCGGAAACGGAGCAGGTCAAAACT-3′). tRNA Lys was detected using the following radiolabeled probe 5′- CCCTGAGATTAAGAGTCTCATGCTC-3′.
For Alu RNA Northern blot, the experimental design was identical to Click-iT™ AHA labeling of nascent proteins (see above). Briefly, human HCT8 cells were starved in an amino acid-depleted medium for 1 h. Then, cells were exposed to 10 min heat shock pretreatment (10 min, 45 °C) followed by 25 min at 45 °C in the presence of 40 µM Click-iT™ AHA labeling. For Alu and scAlu RNA detection, we used the following radiolabeled oligo probe 5′-GGATGGTCTCGATCTCCTGACCTC-3′. After an overnight probe hybridization at 58 °C, the membrane was washed three times for 10 min in 2X SSC, 0.1% SDS at 58 °C, exposed and analyzed using an Amersham Typhoon Phosphoimager.
Puromycilation of nascent proteins
Cells were incubated at 37 °C (pre-HS) or exposed to heat shock for 10 min at 45 °C in a corresponding cell medium prewarmed to 45 °C. Next, Puromycin Dihydrochloride (Life Technologies, A1113803) labeling was added to cells at 10 µg/ml final concentration for an additional 25 min at 37 or 45 °C. After Puromycin labeling, cells were lysed and samples were separated using Bio-Rad Mini-PROTEAN® TGX™ Precast Protein Gels, transferred to Immobilon-P membrane and incubated overnight at 4 °C with 1:1000 dilution of anti-Puromycin primary antibody (Sigma-Aldrich Inc., MABE343). In experiments with ISRIB (Sigma-Aldrich Inc., SML0843-5MG), the compound was administered 1 h before heat shock exposure and during the heat shock treatment. Blots were developed using secondary antibody Anti-Mouse IgG (H + L), HRP Conjugate (Promega, PAW4021).
PRO-seq repeat enrichment analysis
Repeat enrichment analysis for 7SL RNA and 18S and 28S ribosomal RNAs was performed by utilizing Repeat Enrichment Estimator95 and Repeat Masker Database release 2015080796. Match counts for repeat types were normalized by the total number of reads of PRO-Seq pre-HS and heat shock conditions40.
Ribo-seq
Ribo-seq was performed according to Ingolia et al.61 with several adaptations. Briefly, 800 × 103 NIH3T3 cells were plated per well in a six-well plate and transfected either with siScr or siSrp72. 24 h later medium was replaced, and transfection was repeated. Twenty-four hours later medium was removed, and cells were exposed to heat shock (45 °C, 10 min) or not (37 °C, 10 min) in 1 ml medium prewarmed to 37 °C or 45 °C. After the treatment, plates were placed on ice, the medium was removed, and the cells were washed in 2 ml per well in cold 1X PBS without cycloheximide (CHX). PBS was removed, and cells were lysed in 450 µl of lysis buffer. (*Polysome buffer: 20 mM Tris pH 7.5, 150 mM NaCl, 100 µg/ml CHX, 5 mM MgCl2, 1 mM DTT. *Lysis buffer: Polysome buffer + 25 U/ml TURBO™ DNase + 1% Triton X-100. Cells were spun for 10 min at 20,000×g, and the supernatant was collected. About 300 µl of the lysate was used for ribosome footprinting. After RNAseI treatment, samples were layered on top of 900 µl of 1 M sucrose in polysome buffer in Beckman polycarbonate tubes (No. 343778) and ultracentrifugation was performed for 30 min at 120,000×g using TLA120.2 rotor @ 4 °C. Subsequently, supernatant was removed, and pellet was resuspended in 700 µl of Qiazol. RNA from ribosomal pellet was extracted using miRneasy kit and eluted in 50 µl of RNAse-free water. Forward library DNA primer was used as a lower marker for footprint excision. Following each precipitation step, pellets were additionally washed with 500 µl of 70% EtOH to remove residual salts. Circularized cDNA libraries were amplified eight cycles and three reactions were pooled together for sequencing.
Ribo-seq analysis
Ribo-seq data analysis was conducted by following the protocol from ref. 61 with minor modifications. Briefly, raw sequencing reads were pre-processed by Trim-galore software (https://github.com/FelixKrueger/TrimGalore), and trimmed sequences were aligned to rRNA sequence to filter out reads from rRNA. Collected non-rRNA sequence reads were aligned to the mouse genome (mm10) by TopHat297 with the parameters “—no-novel-juncs” and “—b2-very-sensitive”. Meta-analysis for coding-sequence regions was conducted by utilizing deepTools298. The lengths of CDS regions for all genes were normalized to 100 for the meta-analysis. Normalization by total number of reads (RPKM) was used for clustering in Fig. 6a (and Supplementary Fig. 7a).
Elongation index (Elg idx) was calculated by taking the ratio of the average signal intensity of gene body regions in 10–90% range of CDS regions to the average signal intensity from the first 10% of 5′ CDS regions.
Translation efficiency (TE) was defined by the ratio between average Ribo-seq signal from CDS regions and average RNA-Seq signal for each gene.
Representation factor analysis
Statistical significance of the overlap between two groups of genes was calculated based on hypergeometric distribution. The representation factors were defined by the observed number of overlapping genes divided by the expected number of overlapping genes if randomly drawn. List of Signal Peptide-containing proteins was derived from http://www.signalpeptide.de65,66,67.
RNA-seq
RNA used for RNA-Seq was obtained from a fraction of cell lysates used for the Ribosome footprinting experiment. After cell lysis in 450 µl of the lysis buffer, 70 µl of the lysate was mixed with 630 µl Qiazol. Total RNA was extracted using a Direct-zol™ RNA Miniprep Plus kit. mRNA was isolated from 100 to 200 ng of total RNA with NEBNext® Poly(A) mRNA Magnetic Isolation Module (NEB, E7490S). cDNA libraries were prepared according to the protocol for NEBNext® Ultra™ II Directional RNA Library Prep Kit for Illumina (NEB, E7760S). Libraries were amplified 12 cycles and sent for Illumina sequencing. The experiment was performed using two biological replicates.
DESeq2 analysis
Short-read sequencing by the NovaSeq 6000 system was performed by Novogene. Trim-galore (https://github.com/FelixKrueger/TrimGalore) was used for preprocessing of the raw reads, and TopHat297 was used to align the RNA-Seq reads to mm10 genome with default parameters. Generated bam files were further utilized by DESeq299, featureCounts100, and enhancedVolcano (https://github.com/kevinblighe/EnhancedVolcano) for analyzing the differentially expressed genes. Normalization was done by a total number of reads (RPKM). P adjusted values were derived from the Wald test using the Benjamini and Hochberg method for multiple testing corrections.
CCK8 viability assay
10K HCT8 cells was plated per well in a 96-well plate on top of a transfection master mix containing siRNAs siSrp54, siSrp72, or a non-targeting control (siScr). Twenty-four hours later, the medium was replaced, and transfection was repeated. Twenty-four hours later, plates were placed at 45 °C incubator for indicated time durations (0–240 min) and placed back in a 37 °C incubator after the treatment. The next day, the medium was replaced with 100 µl of fresh RPMI medium containing 10% Cell Counting Kit 8 (WST-8/CCK8) reagent (ab228554). Three hours later, absorbance was measured at 460 nM.
Cell fractionation
About 30 × 106 was used for each fractionation. After 15 min exposure to 45 °C, cells were harvested and resuspended in 1 ml of lysis buffer (10 mM Tris pH8, 140 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 1 mM DTT, and RNAse Inhibitor) by slow pipetting (50 “ups and downs”) using 1 ml pipette. After spinning for 3 min at 1000×g at 4 °C, supernatants (cytoplasmic fractions) were harvested for RNA or protein extraction and stored. Pelleted nuclear fractions were resuspended in 1 ml of the lysis buffer supplemented with 10% detergent stock (3.3% (w/v) Sodium Deoxycholate, 6.6% (v/v) NP-40) and incubated for 5 min under slow vortexing. Samples were spun for 3 min at 1000×g at 4 °C, superanatants (post-nuclear/membrane fractions) were also tested in Western Blots. Pellets were rinsed in 1 ml of lysis buffer (10 mM Tris pH8, 140 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 1 mM DTT, and RNAse Inhibitor) and spun again for 3 min at 1000×g at 4 °C. Supernatants were discarded and pellets were lysed in 1 ml of TRIzolTM reagent for RNA extractions or resuspended in lysis buffer (10 mM Tris pH8, 140 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 1 mM DTT, and RNAse Inhibitor) and sonicated for protein extraction. About 250 µl of the cytosolic fraction was mixed with 750 µl TRIzolTM LS reagent and subjected to phenol-chloroform RNA extraction. About 250 µl of the nuclear fraction TRIzolTM reagent lysate was mixed with 750 µl TRIzolTM LS reagent and subjected to phenol-chloroform RNA extraction.
Statistics and reproducibility
All experiments have been successfully repeated, and statistical analysis was performed using biologically independent replicates. The Western Blot, Northern Blot, and microscopy images presented in the manuscript are representative examples from three independent experiments, all yielding consistent results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-Seq and Ribo-seq data generated in this study have been deposited at GEO under accession code GSE243286. Pro-seq data referenced in this study is available under code GSE71708. Source data are provided with this paper.
References
Vassetzky, N. S. & Kramerov, D. A. SINEBase: a database and tool for SINE analysis. Nucleic Acids Res. 41, D83–D89 (2013).
Deininger, P. Alu elements: know the SINEs. Genome Biol. 12, 236 (2011).
Walters, R. D., Kugel, J. F. & Goodrich, J. A. InvAluable junk: the cellular impact and function of Alu and B2 RNAs. IUBMB Life 61, 831–837 (2009).
Dunker, W., Zhao, Y., Song, Y. & Karijolich, J. Recognizing the SINEs of infection: regulation of retrotransposon expression and modulation of host cell processes. Viruses 9, 386 (2017).
Hernandez, A. J. et al. B2 and ALU retrotransposons are self-cleaving ribozymes whose activity is enhanced by EZH2. Proc. Natl Acad. Sci. USA 117, 415–425 (2020).
Liu, W. M., Chu, W. M., Choudary, P. V. & Schmid, C. W. Cell stress and translational inhibitors transiently increase the abundance of mammalian SINE transcripts. Nucleic Acids Res. 23, 1758–1765 (1995).
Tucker, J. M. & Glaunsinger, B. A. Host noncoding retrotransposons induced by DNA viruses: a SINE of infection? J. Virol. 91, e00982–17 (2017).
Hasler, J. & Strub, K. Alu elements as regulators of gene expression. Nucleic Acids Res. 34, 5491–5497 (2006).
Allen, T. A., Von Kaenel, S., Goodrich, J. A. & Kugel, J. F. The SINE-encoded mouse B2 RNA represses mRNA transcription in response to heat shock. Nat. Struct. Mol. Biol. 11, 816–821 (2004).
Berger, A. et al. Direct binding of the Alu binding protein dimer SRP9/14 to 40S ribosomal subunits promotes stress granule formation and is regulated by Alu RNA. Nucleic Acids Res. 42, 11203–11217 (2014).
Espinoza, C. A., Allen, T. A., Hieb, A. R., Kugel, J. F. & Goodrich, J. A. B2 RNA binds directly to RNA polymerase II to repress transcript synthesis. Nat. Struct. Mol. Biol. 11, 822–829 (2004).
Ivanova, E., Berger, A., Scherrer, A., Alkalaeva, E. & Strub, K. Alu RNA regulates the cellular pool of active ribosomes by targeted delivery of SRP9/14 to 40S subunits. Nucleic Acids Res. 43, 2874–2887 (2015).
Yakovchuk, P., Goodrich, J. A. & Kugel, J. F. B2 RNA and Alu RNA repress transcription by disrupting contacts between RNA polymerase II and promoter DNA within assembled complexes. Proc. Natl Acad. Sci. USA 106, 5569–5574 (2009).
Zovoilis, A., Cifuentes-Rojas, C., Chu, H. P., Hernandez, A. J. & Lee, J. T. Destabilization of B2 RNA by EZH2 activates the stress response. Cell 167, 1788–1802 e13 (2016).
Mariner, P. D. et al. Human Alu RNA is a modular transacting repressor of mRNA transcription during heat shock. Mol. Cell 29, 499–509 (2008).
Bovia, F., Fornallaz, M., Leffers, H. & Strub, K. The SRP9/14 subunit of the signal recognition particle (SRP) is present in more than 20-fold excess over SRP in primate cells and exists primarily free but also in complex with small cytoplasmic Alu RNAs. Mol. Biol. Cell 6, 471–484 (1995).
Chang, D. Y. & Maraia, R. J. A cellular protein binds B1 and Alu small cytoplasmic RNAs in vitro. J. Biol. Chem. 268, 6423–6428 (1993).
Chang, D. Y. et al. A human Alu RNA-binding protein whose expression is associated with accumulation of small cytoplasmic Alu RNA. Mol. Cell Biol. 14, 3949–3959 (1994).
Hsu, K., Chang, D. Y. & Maraia, R. J. Human signal recognition particle (SRP) Alu-associated protein also binds Alu interspersed repeat sequence RNAs. Characterization of human SRP9. J. Biol. Chem. 270, 10179–10186 (1995).
Walter, P. & Blobel, G. Disassembly and reconstitution of signal recognition particle. Cell 34, 525–533 (1983).
Bovia, F. & Strub, K. The signal recognition particle and related small cytoplasmic ribonucleoprotein particles. J. Cell Sci. 109, 2601–2608 (1996).
Labuda, D. & Zietkiewicz, E. Evolution of secondary structure in the family of 7SL-like RNAs. J. Mol. Evol. 39, 506–518 (1994).
Ullu, E. & Tschudi, C. Alu sequences are processed 7SL RNA genes. Nature 312, 171–172 (1984).
Oh, H. J. et al. Jpx RNA regulates CTCF anchor site selection and formation of chromosome loops. Cell 184, 6157–6173 e24 (2021).
Walter, P. & Blobel, G. Purification of a membrane-associated protein complex required for protein translocation across the endoplasmic reticulum. Proc. Natl Acad. Sci. USA 77, 7112–7116 (1980).
Akopian, D., Shen, K., Zhang, X. & Shan, S. O. Signal recognition particle: an essential protein-targeting machine. Annu. Rev. Biochem. 82, 693–721 (2013).
Keenan, R. J., Freymann, D. M., Stroud, R. M. & Walter, P. The signal recognition particle. Annu. Rev. Biochem. 70, 755–775 (2001).
Kellogg, M. K., Miller, S. C., Tikhonova, E. B. & Karamyshev, A. L. SRPassing co-translational targeting: the role of the signal recognition particle in protein targeting and mRNA protection. Int. J. Mol. Sci. 22, 6284 (2021).
Walter, P., Ibrahimi, I. & Blobel, G. Translocation of proteins across the endoplasmic reticulum. I. Signal recognition protein (SRP) binds to in-vitro-assembled polysomes synthesizing secretory protein. J. Cell Biol. 91, 545–550 (1981).
Koch, H. G., Moser, M. & Muller, M. Signal recognition particle-dependent protein targeting, universal to all kingdoms of life. Rev. Physiol. Biochem. Pharm. 146, 55–94 (2003).
Walter, P. & Blobel, G. Translocation of proteins across the endoplasmic reticulum III. Signal recognition protein (SRP) causes signal sequence-dependent and site-specific arrest of chain elongation that is released by microsomal membranes. J. Cell Biol. 91, 557–561 (1981).
Lipp, J., Dobberstein, B. & Haeuptle, M. T. Signal recognition particle arrests elongation of nascent secretory and membrane proteins at multiple sites in a transient manner. J. Biol. Chem. 262, 1680–1684 (1987).
Wolin, S. L. & Walter, P. Signal recognition particle mediates a transient elongation arrest of preprolactin in reticulocyte lysate. J. Cell Biol. 109, 2617–2622 (1989).
Strub, K. & Walter, P. Assembly of the Alu domain of the signal recognition particle (SRP): dimerization of the two protein components is required for efficient binding to SRP RNA. Mol. Cell Biol. 10, 777–784 (1990).
Siegel, V. & Walter, P. Elongation arrest is not a prerequisite for secretory protein translocation across the microsomal membrane. J. Cell Biol. 100, 1913–1921 (1985).
Halic, M. et al. Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature 427, 808–814 (2004).
Chartron, J. W., Hunt, K. C. & Frydman, J. Cotranslational signal-independent SRP preloading during membrane targeting. Nature 536, 224–228 (2016).
Mason, N., Ciufo, L. F. & Brown, J. D. Elongation arrest is a physiologically important function of signal recognition particle. EMBO J. 19, 4164–4174 (2000).
Lakkaraju, A. K., Mary, C., Scherrer, A., Johnson, A. E. & Strub, K. SRP keeps polypeptides translocation-competent by slowing translation to match limiting ER-targeting sites. Cell 133, 440–451 (2008).
Mahat, D. B., Salamanca, H. H., Duarte, F. M., Danko, C. G. & Lis, J. T. Mammalian heat shock response and mechanisms underlying its genome-wide transcriptional regulation. Mol. Cell 62, 63–78 (2016).
Torrent, M., Chalancon, G., de Groot, N. S., Wuster, A. & Madan Babu, M. Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Sci. Signal 11, eaat6409 (2018).
Chu, H. P. et al. TERRA RNA antagonizes ATRX and protects telomeres. Cell 170, 86–101 e16 (2017).
Simon, M. D. et al. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature 504, 465–469 (2013).
Simon, M. D. et al. The genomic binding sites of a noncoding RNA. Proc. Natl Acad. Sci. USA 108, 20497–20502 (2011).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Vihervaara, A., Duarte, F. M. & Lis, J. T. Molecular mechanisms driving transcriptional stress responses. Nat. Rev. Genet. 19, 385–397 (2018).
Politz, J. C. et al. Signal recognition particle components in the nucleolus. Proc. Natl Acad. Sci. USA 97, 55–60 (2000).
Massenet, S. In vivo assembly of eukaryotic signal recognition particle: a still enigmatic process involving the SMN complex. Biochimie 164, 99–104 (2019).
Wild, K., Halic, M., Sinning, I. & Beckmann, R. SRP meets the ribosome. Nat. Struct. Mol. Biol. 11, 1049–1053 (2004).
Shalgi, R. et al. Widespread regulation of translation by elongation pausing in heat shock. Mol. Cell 49, 439–452 (2013).
Ji, C. et al. Interaction of 7SK with the Smn complex modulates snRNP production. Nat. Commun. 12, 1278 (2021).
Yarmolinsky, M. B. & Haba, G. L. Inhibition by puromycin of amino acid incorporation into protein. Proc. Natl Acad. Sci. USA 45, 1721–1729 (1959).
Brown, J. D. et al. Subunits of the Saccharomyces cerevisiae signal recognition particle required for its functional expression. EMBO J. 13, 4390–4400 (1994).
Mutka, S. C. & Walter, P. Multifaceted physiological response allows yeast to adapt to the loss of the signal recognition particle-dependent protein-targeting pathway. Mol. Biol. Cell 12, 577–588 (2001).
Tikhonova, E. B. et al. Defective human SRP induces protein quality control and triggers stress response. J. Mol. Biol. 434, 167832 (2022).
Han, J. et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490 (2013).
Pakos-Zebrucka, K. et al. The integrated stress response. EMBO Rep. 17, 1374–1395 (2016).
Zakrzewska, A. et al. Genome-wide analysis of yeast stress survival and tolerance acquisition to analyze the central trade-off between growth rate and cellular robustness. Mol. Biol. Cell 22, 4435–4446 (2011).
Costa-Mattioli, M. & Walter, P. The integrated stress response: from mechanism to disease. Science 368, eaat5314 (2020).
Sidrauski, C., McGeachy, A. M., Ingolia, N. T. & Walter, P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife 4, e05033 (2015).
Ingolia, N. T., Brar, G. A., Rouskin, S., McGeachy, A. M. & Weissman, J. S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 7, 1534–1550 (2012).
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. & Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009).
Kusnadi, E. P., Timpone, C., Topisirovic, I., Larsson, O. & Furic, L. Regulation of gene expression via translational buffering. Biochim. Biophys. Acta Mol. Cell Res. 1869, 119140 (2022).
Rath, S. et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547 (2021).
Hegde, R. S. & Bernstein, H. D. The surprising complexity of signal sequences. Trends Biochem. Sci. 31, 563–571 (2006).
Martoglio, B. & Dobberstein, B. Signal sequences: more than just greasy peptides. Trends Cell Biol. 8, 410–415 (1998).
von Heijne, G. Patterns of amino acids near signal-sequence cleavage sites. Eur. J. Biochem. 133, 17–21 (1983).
Gussakovsky, D., Booy, E. P., Brown, M. J. F. & McKenna, S. A. Nuclear SRP9/SRP14 heterodimer transcriptionally regulates 7SL and BC200 RNA expression. RNA 29, 1185–1200 (2023).
Li, J. et al. A novel histone H4 arginine 3 methylation-sensitive histone H4 binding activity and transcriptional regulatory function for signal recognition particle subunits SRP68 and SRP72. J. Biol. Chem. 287, 40641–40651 (2012).
Flynn, R. A. et al. 7SK-BAF axis controls pervasive transcription at enhancers. Nat. Struct. Mol. Biol. 23, 231–238 (2016).
Ingolia, N. T., Hussmann, J. A. & Weissman, J. S. Ribosome profiling: global views of translation. Cold Spring Harb. Perspect. Biol. 11, a032698 (2019).
Peltz, S. W., Brown, A. H. & Jacobson, A. mRNA destabilization triggered by premature translational termination depends on at least three cis-acting sequence elements and one trans-acting factor. Genes Dev. 7, 1737–1754 (1993).
Presnyak, V. et al. Codon optimality is a major determinant of mRNA stability. Cell 160, 1111–1124 (2015).
Karamyshev, A. L. et al. Inefficient SRP interaction with a nascent chain triggers a mRNA quality control pathway. Cell 156, 146–157 (2014).
Lutcke, H., High, S., Romisch, K., Ashford, A. J. & Dobberstein, B. The methionine-rich domain of the 54 kDa subunit of signal recognition particle is sufficient for the interaction with signal sequences. EMBO J. 11, 1543–1551 (1992).
Zopf, D., Bernstein, H. D. & Walter, P. GTPase domain of the 54-kD subunit of the mammalian signal recognition particle is required for protein translocation but not for signal sequence binding. J. Cell Biol. 120, 1113–1121 (1993).
Cortes Sanchon, A. et al. ER-misfolded proteins become sequestered with mitochondria and impair mitochondrial function. Commun. Biol. 4, 1350 (2021).
Gamerdinger, M., Hanebuth, M. A., Frickey, T. & Deuerling, E. The principle of antagonism ensures protein targeting specificity at the endoplasmic reticulum. Science 348, 201–207 (2015).
Lindquist, S. The heat-shock response. Annu. Rev. Biochem. 55, 1151–1191 (1986).
Richter, K., Haslbeck, M. & Buchner, J. The heat shock response: life on the verge of death. Mol. Cell 40, 253–266 (2010).
Buchan, J. R. & Parker, R. Eukaryotic stress granules: the ins and outs of translation. Mol. Cell 36, 932–941 (2009).
Marcelo, A., Koppenol, R., de Almeida, L. P., Matos, C. A. & Nobrega, C. Stress granules, RNA-binding proteins and polyglutamine diseases: too much aggregation? Cell Death Dis. 12, 592 (2021).
Protter, D. S. W. & Parker, R. Principles and properties of stress granules. Trends Cell Biol. 26, 668–679 (2016).
Jolly, C., Usson, Y. & Morimoto, R. I. Rapid and reversible relocalization of heat shock factor 1 within seconds to nuclear stress granules. Proc. Natl Acad. Sci. USA 96, 6769–6774 (1999).
Chowdhary, S., Kainth, A. S., Paracha, S., Gross, D. S. & Pincus, D. Inducible transcriptional condensates drive 3D genome reorganization in the heat shock response. Mol. Cell 82, 4386–4399 e7 (2022).
Pincus, D. Regulation of Hsf1 and the heat shock response. Adv. Exp. Med. Biol. 1243, 41–50 (2020).
Rawat, P. et al. Stress-induced nuclear condensation of NELF drives transcriptional downregulation. Mol. Cell 81, 1013–1026 e11 (2021).
Aoi, Y. et al. NELF regulates a promoter-proximal step distinct from RNA Pol II pause-release. Mol. Cell 78, 261–274 e5 (2020).
Chu, B., Brodl, M. R. & Belanger, F. C. Heat shock inhibits release of the signal recognition particle from the endoplasmic reticulum in barley aleurone layers. J. Biol. Chem. 272, 7306–7313 (1997).
Wang, C. Y., Jegu, T., Chu, H. P., Oh, H. J. & Lee, J. T. SMCHD1 merges chromosome compartments and assists formation of super-structures on the inactive X. Cell 174, 406–421 e25 (2018).
Zhang, Y. et al. Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137 (2008).
Yu, G., Wang, L. G. & He, Q. Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015).
Machanick, P. & Bailey, T. L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011).
Gandin, V. et al. Polysome fractionation and analysis of mammalian translatomes on a genome-wide scale. J. Vis. Exp. 17, 51455 (2014).
Day, D. S., Luquette, L. J., Park, P. J. & Kharchenko, P. V. Estimating enrichment of repetitive elements from high-throughput sequence data. Genome Biol. 11, R69 (2010).
Bao, W., Kojima, K. K. & Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Acknowledgements
We thank all the Lee lab members for their critical input during discussions. We thank Christopher Ball for the critical reading of the manuscript. We thank Niklas Grimm for his help with microscopy setups. We thank Dig B. Mahat and John T. Lis for their help with the PRO-Seq analysis. We thank Predrag Jovanovic for helpful discussions regarding sucrose gradient ultracentrifugation. This work was supported by Swiss National Science Foundation Early Postdoc Mobility Fellowship P2LAP3_174617 (to B.B.), The Pew Biomedical Sciences Fellowship 2021-2024 (to C.R.), and NIH Grant R01-HD097665 to J.T.L. I.T. is supported by a Fonds de recherche du Québec (FRQS) Senior Investigator award and the Terry Fox Foundation (TFF) Oncometabolism Team Grants TFF-242122.
Author information
Authors and Affiliations
Contributions
B.B. and J.T.L. designed the study, analyzed data, and wrote the manuscript. I.T. helped to design and interpret translation studies. L.X. performed SRP pull-downs and evaluated translation changes after SRP54 downregulation. CHART-Seq was done by U.W. and PLA by C.R. B.B. performed all the remaining experiments. B.B., U.W., and H.-G.L. conducted bioinformatic analyses.
Corresponding author
Ethics declarations
Competing interests
J.T.L. is a cofounder of Fulcrum Therapeutics, an advisor to Skyhawk Therapeutics, and a non-executive Director of GSK. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bujisic, B., Lee, HG., Xu, L. et al. 7SL RNA and signal recognition particle orchestrate a global cellular response to acute thermal stress. Nat Commun 16, 1630 (2025). https://doi.org/10.1038/s41467-025-56351-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56351-6
