
Abstract
Studies of the genetics of Alzheimer’s disease (AD) have largely focused on single nucleotide variants and short insertions/deletions. However, most of the disease heritability has yet to be uncovered, suggesting that there is substantial genetic risk conferred by other forms of genetic variation. There are over one million short tandem repeats (STRs) in the genome, and their link to AD risk has not been assessed. As pathogenic expansions of STR cause over 30 neurologic diseases, it is important to ascertain whether STRs may also be implicated in AD risk. Here, we genotype 312,731 polymorphic STR tracts genome-wide using PCR-free whole genome sequencing data from 2981 individuals (1489 AD case and 1492 control individuals). We implement an approach to identify STR expansions as STRs with tract lengths that are outliers from the population. We then test for differences in aggregate burden of expansions in case versus control individuals. AD patients harbor a 1.19-fold increase of STR expansions compared to healthy elderly controls (p = 8.27×10-3, two-sided Mann-Whitney test). Individuals carrying >30 STR expansions have a 3.69-fold higher odds of having AD and have more severe AD neuropathology. AD STR expansions are highly enriched within active promoters in post-mortem hippocampal brain tissues and particularly within SINE-VNTR-Alu (SVA) retrotransposons. Together, these results demonstrate that expanded STRs within active promoter regions of the genome associate with risk of AD.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder in the United States and has a growing prevalence in our aging population, yet there is a lack of effective treatments1. Delineating the genetic basis of AD is crucial to uncovering the underlying genes and molecular mechanisms and can spur development of more targeted therapies. Despite studies in hundreds of thousands of individuals, most of the genetic risk for AD has yet to be identified2. One source of genetic variation that has not been explored in AD is the approximately one million short tandem repeats (STRs) tracts in the human genome, which are classically defined as DNA sequences composed of repeated units of 2–6 bp motifs. STR tract length (i.e., number of repeat units) is highly polymorphic in the population and linked to widespread gene expression changes, thus representing a potential source of functional genetic variation3,4,5,6,7,8. Moreover, pathogenic expansions in the tract lengths of STRs cause over 30 monogenic neurological disorders such as Huntington’s disease9,10. Thus, it is important to assess whether STRs may also contribute to the risk of AD.
Pathogenic expansions of specific STR tracts cause >30 monogenic disorders such as fragile X syndrome, myotonic dystrophy, and Huntington’s disease9,10. A single pathogenic STR expansion confers the majority of the genetic risk for these disorders. However, the role of STRs in promoting genetic risk for polygenic disorders such as AD is not well understood. Our ability to understand the relationship between STRs and polygenic disorders has been limited by the need for genome-wide measures of STR lengths in large sample sizes. Recent studies in large cohorts have revealed that patients with autism spectrum disorder and schizophrenia carry a higher burden of germline STR expansions11,12,13. These studies suggest that, in contrast to our traditional view of a single STR conferring disease risk for monogenic disorders, many STRs distributed throughout the genome in aggregate can contribute to neuropsychiatric disease risk in a polygenic fashion.
The majority of STRs in the genome are not within genes and the role of these intergenic STRs with disease is less well-understood. While known disease-associated STR tracts are within gene bodies (exons, introns, or untranslated regions)9, pathogenic-length expansions of disease-associated STRs has been linked to alterations beyond protein-coding functions, including severe disruption to histone modifications, DNA methylation, and genome folding14. Polymorphic changes in intergenic STRs are also known to correlate with expression of nearby genes often in a tissue-specific manner5,15,16. Moreover, STRs correlated with gene expression are enriched at transcriptional start sites and colocalize with putative enhancers5. Of note, prior studies have also found widespread alterations in the epigenome profiled in post-mortem brain tissue from patients with AD17,18,19. Whether the lengths of intergenic STRs are altered in AD and interplay with epigenetic changes has yet to be explored.
Measuring the lengths of STRs is challenging, especially when interrogating across the entire genome in many individuals. The repetitive nature of STRs makes sequencing and downstream processing prone to errors, and their tract lengths often exceed the lengths of traditional short-read next-generation sequencing reads. However, computational tools have been developed to overcome these challenges to infer the lengths of STRs from short-read sequencing data20,21,22,23, which have enabled genome-wide genotyping of STRs in cohorts of individuals11,12,13. Consequently, they provide a valuable opportunity to test the association of numerous STRs with disease risk across large cohorts of patients and uncover novel links between STRs and disease risk.
In this work, we sought to understand whether STRs may promote genetic risk of AD. We applied cutting-edge computational tools ExpansionHunter22 and gangSTR20 to genotype STRs genome-wide using PCR-free WGS data from 2981 individuals with and without AD from the Alzheimer’s Disease Sequencing Project (ADSP). We implemented a rigorous approach to identify STR expansions based on having extreme STR tract lengths. Across the genome, we identified 9641 unique STR tracts that had STR expansions in at least one individual. Strikingly, individuals carrying a high burden of expanded STRs had 3.69 higher odds of having AD and had worse AD neuropathology, thus representing one of the strongest genetic effects on AD risk. STR expansions seen in individuals with AD are enriched in promoter regions active in post-mortem hippocampal tissue. These results suggest a model whereby the cumulative effect of multiple STR expansions across the genome promotes genetic risk of AD and generates important insights into the genetic architecture of AD.
Results
Genome-wide profiling of STR tracts in an AD cohort
In this study, we utilized a discovery cohort of 2393 samples (1213 AD cases and 1180 controls) from the National Institute of Aging Alzheimer’s Disease Centers (ADC) cohort of the ADSP WGS dataset (see Supplementary Table 1 for participant characteristics). For replication of our single STR associations, we utilized a replication cohort of 588 samples from the Religious Orders Study/Memory Aging Project (ROSMAP; n = 309 AD cases, 279 controls). These samples have been sequenced on peripheral blood-derived genomic DNA using a PCR-free Illumina whole genome sequencing (WGS) strategy to >30× coverage using 150 bp paired end sequencing reads. We restricted our analyses to White individuals of European ancestry (based on both self-reported non-Hispanic White race/ethnicity and based on genetic ancestry as determined by principal component analysis [PCA] coordinates) (Supplementary Fig. 1a,b). There were no apparent differences between AD cases and controls based on PCA coordinates (Supplementary Fig. 1c), sequencing centre (Supplementary Fig. 1d), or sequencing cohort (Supplementary Fig. 1e). To limit potential technical artifacts that could lead to spurious results, we restricted our analysis to samples sequenced to between 30× and 50× coverage genome-wide. There was no statistically significant difference in sequencing coverage between case and control samples (p = 0.055, two-sided Student’s t-test) (Supplementary Fig. 1f). Together, these samples represent a deeply phenotyped case/control cohort that has been carefully filtered to mitigate the potential for technical artifacts and confounders.
To understand the role of STRs in AD, we sought to perform an unbiased genome-wide assessment of the relationship between STRs and AD risk (Fig. 1a). There are approximately ~1 million STR tracts in the genome, which is computationally infeasible to apply across thousands of samples using existing methods. To limit the search space for our analyses and reduce computational costs, we focused on STRs that are polymorphic (i.e., vary in tract length in the population). To identify polymorphic STRs, we first applied the gangSTR algorithm on a catalogue of 895,826 STRs across the genome to a subset of 495 individuals of European ancestry from the ADSP (n = 246 AD cases and 249 controls). We identified 237,197 STRs that were polymorphic in these 495 samples. We merged this set of 237,197 STRs with 174,262 STRs previously identified to be polymorphic22 to result in a union set of 312,731 unique polymorphic STRs (Supplementary Data 1). We used this panel of 312,731 STRs throughout this study to provide genome-wide assessment of polymorphic STRs.

a Schematic of study design. Middle section shows six example STRs (STR1-6), with rows representing STRs, columns representing individuals, and numbers representing non-reference STR tract lengths. Long expansions are bolded. STR1 is an example of a rare STR expansion seen only in individuals with AD. At the bottom are three models for testing for associations with AD case/control status. In model 1 (left), STR tract lengths are treated as a continuous variable. In model 2 (middle), STR expansions are defined as those longer than a given STR tract length threshold, and we compare the number of individuals with a given STR expansion between AD cases and controls. In model 3 (right), we identify rare, long STR expansions, and we compare the number of such STR expansions per individual across the genome between cases and controls. b Genomic distribution of STR panel. c Histogram of repeat unit lengths (number of base pairs [bp] in STR motif) for STR panel. Inset shows the subset of STRs with repeat units 7 bp or longer. d Histogram of STR tract lengths (number of repeat units) in the GRCh38 reference genome for each STR in the panel. e Histogram of median STR tract lengths relative to the GRCh38 reference genome as genotyped by ExpansionHunter. Negative values indicate a shorter median STR tract length relative to reference genome and positive values indicate longer median tract lengths relative to reference genome.
As genotyping STR tract lengths is challenging using short-read sequencing data, we performed comparisons with long-read sequencing using PacBio technology. We utilized four samples from the 1000 Genomes Project that were not used in this study24. We called STR tract lengths from PacBio sequencing data using the TRGT software25. We compared STR genotypes from PacBio data to STR genotypes identified using ExpansionHunter and gangSTR from short-read Illumina data on these same four samples. Overall, we observed strong correlations in STR tract lengths as estimated by ExpansionHunter and gangSTR when compared with PacBio (Spearman r2 ~ 0.96–0.97) (Supplementary Fig. 2a-h). Focusing on STR tracts length ≥10 repeat units longer than the reference genome identified by PacBio long-read sequencing, we found that ExpansionHunter achieved 70.0–73.3% sensitivity, while gangSTR achieved 58.0–64.7% sensitivity (Supplementary Fig. 2i). In contrast, ExpansionHunter demonstrated 63.0–67.7% specificity compared with 81.7–85.0% specificity for gangSTR for STR tracts length ≥10 repeat units longer than the reference genome as identified in the Illumina short read data, (Supplementary Fig. 2j). Together, these results suggest that ExpansionHunter is more sensitive for STR expansions, while gangSTR has higher specificity, similar to as observed previously26.
The STRs in the polymorphic STR set were largely not within protein-coding regions of the genome: 47.4% of STRs were within introns, 36.7% in distal noncoding regions (defined as being outside of gene bodies and at least 3 kb from the nearest transcriptional start site [TSS]), and 14.1% in promoter regions (defined as ≤3 kb upstream from a TSS) (Fig. 1b). Only 0.76% of STRs were in coding exons. The high number of polymorphic STRs in gene promoters is consistent with prior studies showing that STRs are enriched in gene promoters and have important gene regulatory roles27,28. The majority (97.7%) of STRs had a repeat unit size between 2–6 bp, which is the classic definition of an STR (Fig. 1c). Only 0.39% of STRs had a repeat unit size larger than 15 bp. In the GRCh38 reference genome, STRs in the panel generally had short tract lengths, with a median of 6 repeat units per tract and 99.8% of STR tract lengths ≤30 repeat units (Fig. 1d).
We next used the ExpansionHunter and gangSTR algorithms to genotype our panel of 312,731 polymorphic STRs. We did find overall high concordance between ExpansionHunter and gangSTR genotypes. For 61.2% of STRs, gangSTR and ExpansionHunter genotypes were at least 90% concordant across individuals (Supplementary Fig. 3a). The tract lengths were strongly correlated across STRs between gangSTR and ExpansionHunter (Pearson r2 = 0.83) (Supplementary Fig. 3b). We do note that tract lengths in ExpansionHunter tended to be longer than gangSTR, which is consistent with our validation using long-read sequencing and the work of others26. Nonetheless, we found that the median STR tract length measured by ExpansionHunter were largely concordant with the tract lengths in the GRCh38 reference genome (Fig. 1e). For the remainder of the paper, we performed our primary association analyses using ExpansionHunter as this algorithm has been shown to be more sensitive for detecting longer STR tract lengths26.
Testing for single STR associations with AD risk
We first tested the lengths of each STR for association with AD risk, treating STR lengths as a continuous variable (Fig. 1a, model 1). We applied a logistic regression model to test the association of the tract length of each STR with AD risk, accounting for sample covariates (age, sex, and the first 3 PCs) and technical covariates (sample sequencing coverage and local sequencing coverage). Throughout, we applied a dominant model, such that only the longer of the two alleles for a given STR in an individual was considered. As the tract lengths of many STRs are not normally distributed, we performed a rank-based inverse normal transformation of STR tract lengths prior to association testing. We filtered out STRs within segmental duplicated regions of the genome from our results as these regions resulted in a high number of artifactual genotype calls (see Methods). This resulted in a final set of 293,752 genome-wide STRs that we report. Our statistical association test provided well-calibrated results (λ genomic control=0.997), suggesting minimal evidence of systemic technical artifacts or population stratification (Fig. 2a; Supplementary Data 2). We identified one STR associated with a clinical diagnosis of AD at a Bonferroni-corrected p-value threshold of 1.70 × 10−7 (Supplementary Fig. 4a). This STR was a TTTA repeat at chr19:44921097–44921125 (p-value 4.29 × 10−9, logistic regression test) and is located approximately 11.7 kb downstream from the APOE gene, which has the strongest known genetic association with AD risk29.

a Quantile-quantile plot of single STR association statistics. X-axis shows the expected distribution of −log10(p-values) under a uniform p-value distribution. Y-axis shows observed −log10(p-values). Each point represents a separate STR. b Volcano plot for STR tract lengths under a single STR association test. Each point represents a single STR, with colours reflecting statistical significance of association. X-axis reflects standard mean difference, which is the difference in mean STR lengths between cases and controls divided by the standard deviation of STR lengths across the whole cohort. Positive values reflect longer mean STR tract lengths in cases as compared with controls. λ genomic control (λGC) values are listed in figure legend. c Ratio of number of STRs with tract lengths that are longer in cases relative to the number of STRs with tract lengths longer in controls at different statistical significance thresholds. d Quantile–quantile plot of p-values from hypergeometric tests comparing number of STR expansions in cases versus controls for each STR. X-axis shows the expected distribution of −log10(p-values) under a uniform p-value distribution. Y-axis shows observed −log10(p-values) from a two-sided Fisher’s exact test. Each point represents a separate STR, and points are coloured by the STR tract length threshold. e Relative burden of STR expansions in case versus control individuals at different STR tract length and frequency thresholds. Values above 1.0 reflect higher burden of expansions observed in cases than controls. For a–c observed p-values are from single STR test performed using a logistic regression model of the tract length of each STR with AD risk, accounting for sample and technical covariates.
We performed several validation steps for our association analysis. We first repeated the association analysis using two alternative statistical models. To ensure that associations do not result from the inverse normal transformation of STR lengths, we repeated the logistic regression test on untransformed genotypes using the same model and covariates. We also applied a non-parametric test (Mann–Whitney test) without covariates. Application of either the untransformed STR lengths in a logistic regression model or a non-parametric Mann–Whitney test led to highly similar results (correlation Pearson r2 = 0.91 and 0.94 respectively) (Supplementary Fig. 4b,c). We further performed the association analyses using genotypes generated by a second STR genotyping software programme called gangSTR20. The STR association near APOE remained statistically significant using either of these alternative statistical models or using genotypes from gangSTR.
We replicated our association test in the ROSMAP replication cohort (n = 309 cases, 279 controls). The expansion at chr19:44921097–44921125:TTTAN repeat near the APOE gene robustly replicated across these additional cohorts (meta-analysis p-value 1.43 × 10–12 Supplementary Data 2). Since this STR is near the APOE gene that is known to be associated with AD risk, we next assessed whether this STR represents an independent association. When re-performing the STR association test with APOE genotype as a covariate, the statistical association of this STR was fully attenuated (association p-value = 0.079 after adjusting for APOE genotype), suggesting that this STR is in linkage disequilibrium with the APOE genotype and does not represent an independent genetic association. Together, these results suggest that there is not a predominant STR that independently drives genetic risk for AD. However, based on power calculations, we note that given our current sample sizes, we have 80% power to identify STR associations for STR allelic odds ratio >1.20 (Supplementary Fig. 4d). Thus, we currently lack statistical power to detect STR associations with more modest effect sizes (see Discussion).
Additionally, there were 11,511 STRs nominally associated with AD risk at a p-value threshold of p < 0.05. Among these nominally-associated STRs, we noticed that there was a clear bias toward more STRs having longer mean STR tract lengths in individuals with AD cases as compared to STRs having longer mean tract lengths in controls (Fig. 2b). This contrasts with the null hypothesis, in which we would expect an equal number of STRs with longer mean tract lengths in AD cases as there are STRs with longer mean tract length in controls. This skew toward more STRs with longer tract lengths in cases versus longer in controls was more marked at increasingly stringent p-value cutoffs. For example, at an association p-value threshold of <0.001, there were 1.15-fold as many STRs where the mean tract length was longer in cases than in controls (Fig. 2c). Together, these results show that while no single STR drove genetic risk of AD, longer STR tract lengths appeared to be systematically associated with higher AD risk.
In the above analysis, we treated STR tract lengths as a continuous variable for association testing. However, in known STR-associated diseases, there is usually a threshold at which a given STR tract length becomes pathogenic9, suggesting that large expansions of STR tract lengths rather than population variation confer disease status. Thus, we performed association analyses to test whether the number of individuals with a given STR expansion differs between AD cases and controls (Fig. 1a, model 2). The challenge with testing this model is that the tract length at which an STR becomes pathogenic is not known a priori. We thus performed our analyses at different pre-specified STR tract length thresholds of ≥1, ≥5, ≥10, or ≥20 repeat units longer than the GRCh38 reference genome.
After excluding 20,653 STRs with low genotyping rates, we tested 273,099 STRs for associations between expansions and AD case/control status in 2393 samples (1213 AD cases and 1180 controls) from the discovery cohort from ADC. For each STR, we counted the number of case and control individuals with and without an STR expansion as defined by a given STR tract length threshold and performed a hypergeometric test to assess for differences in number of expansions between case and control individuals. We found that association results were well-calibrated across all STR tract length thresholds tested (Fig. 2d; Supplementary Data 3). However, besides the chr19:44921097–44921125:TTTAN STR near APOE at a STR tract length threshold of ≥1 repeat unit (p-value = 7.74 × 10−11, two-sided Fisher’s exact test), there were no other STRs that were statistically significant after correcting for multiple hypothesis testing.
While no single STR showed a statistically significant difference in the number of expansions between case and control individuals, we next tested whether there may be a difference in the cumulative burden of expansions across the genome in cases versus controls. We identified STR expansions at different STR length thresholds (≥1, ≥5, ≥10, or ≥20 repeat units longer than reference) and at different STR expansion frequency cutoffs (seen once, ≤5, ≤10, ≤100, or no cutoff in n = 2393 individuals) and counted the total number of STR expansions per individual. Strikingly, we found that individuals with AD carried a higher burden of longer and rare STR expansions. For example, individuals with AD carried a 3.28-fold higher number of STRs ≥20 repeat units longer than the reference and seen only once in the cohort (Fig. 2e). This observed increased burden of STRs in individuals with AD was attenuated when examining either shorter STR alleles or more common alleles. For example, there was only a 1.08-fold increased burden in AD individuals for STRs ≥1 repeat unit longer than reference and seen once in the cohort. Similarly, there was only a 1.03-fold increased burden in AD individuals for STRs ≥20 repeat units longer than reference but when no frequency cutoff was placed. These results suggest that a high burden of rare expanded STR alleles associates with risk of AD.
Identification of an increased burden of rare STRs expansions in AD
Given our finding of an increased burden of STR expansions in individuals with AD, we next sought to systematically identify rare, long STR expansions without pre-specifying an STR length threshold since the tract length threshold is not known a priori for any given STR. To increase statistical power to identify rare STR expansions, we combined samples from across the ADC and ROSMAP cohorts (total n = 2981; n = 1492 controls and 1489 cases). Since we are focused on rare expansions, we lack statistical power to test for the association between rare expansions in AD case/control status. Thus, for remaining analyses, we aggregate the number of expansions per individual and test whether there is a difference in the burden of rare STR expansions between case and control individuals (Fig. 1a, model 3).
To detect STR expansions, we implemented an approach using density-based spatial clustering of applications with noise (DBSCAN), which we extended from the work of Trost et al.13 This approach obviates the need for selecting an arbitrary expansion length threshold and frequency cutoff, but instead identifies individuals carrying outlier STR tract lengths for each STR. In our implementation of DBSCAN, we additionally accounted for the effects of sample and technical covariates (sex, age, first three principal components of ancestry, genomic sequencing coverage for each sample, local sequencing coverage for each sample at the STR). We excluded 15 individuals who had >148 STR expansions in their genome (see Methods), resulting in 2966 individuals (n = 1481 control individuals and n = 1485 AD cases). Applying DBSCAN, we identified expansions in 9641 unique STRs across the genome in these 2966 individuals. We classified expansions to those seen only in AD cases (n = 4412 STRs), expansions seen only in controls (n = 3365 STRs), or seen in both cases and controls (n = 1864 STRs) (Fig. 3a; Supplementary Data 4–5). There were 6276 unique STR expansions seen in individuals with AD (hereafter referred to as “AD STR expansions”) and 5229 unique STR expansions seen in control individuals (hereafter referred to as “control STR expansions”). AD STR expansions were slightly but statistically significantly longer than control STR expansions (mean tract length of 39.3 and 37.3 repeat units longer than the GRCh38 reference genome tract length, respectively; p < 1.1 × 10−3, two-sided Mann–Whitney test) (Fig. 3b). 50.1% of AD STR expansions were dinucleotide repeats and just 2.1% of AD STR expansions had a repeat unit of 6 bp or longer (Fig. 3c). Many of the STR expansions were observed in more than one individual (Supplementary Fig. 5a). 1497 STR expansions were present in more than one AD case individual, including 213 STR expansions present in five or more AD case individuals.

a Three separate STRs are shown as examples of AD-only STR expansions, control-only STR expansions, or shared STR expansions as identified by DBSCAN. For each STR, y-axis shows tract length for the STR, where the STR tract length for each individual is represented by one point and ranked by their tract length. Orange points represent AD cases and green points represent controls. b STR tract lengths in number of repeat units longer than GRCh38 reference genome for control STR expansions (green; n = 7823 expansions) and AD STR expansions (orange; n = 9276 expansions). c Repeat unit lengths for STR expansions seen in controls (green) and AD cases (orange). Histogram shows values as proportion of STR expansions seen in controls and cases respectively. d Number of STR expansions identified by DBSCAN per individual in controls (green; n = 1485 individuals) versus cases (orange; n = 1481 individuals). Each point represents one individual. Right panel is zoomed in for individuals with fewer than 50 STRs. e Odds ratio for AD case/control status for individuals carrying varying numbers of STR expansions. Odds ratios > 1.0 represent higher odds of having AD. p-values are calculated using a two-sided Fisher’s exact. *p-value < 0.05. ***p-value < 0.001. f Stacked bar chart of Braak stages for individuals with ≤ 10 STR expansions (top) or > 30 expansions (bottom). Braak stages are from 0–6, with higher values reflecting more severe neuropathology. For b and d box plots elements: centre line, median; box limits, upper and lower quartiles; and whiskers, 1.5× interquartile range.
We also compared STR tract lengths of these expansions in the ADSP cohort to 476 individuals of European ancestry from the 1000 Genomes Project30. We found that for STRs in which an STR expansion was observed, the median STR tract length in the ADSP cohort was similar to samples from the 1000 Genomes Project (Spearman r2 = 0.99) (Supplementary Fig. 5b). However, for 96.5% of STRs in which an STR expansion was observed, the maximum STR tract length in the ADSP cohort was much longer when compared to samples from the 1000 Genomes Project, suggesting that most STR expansions are rare polymorphisms in the population and/or unique to our dataset (Supplementary Fig. 5c).
We next tested for differences in the burden of STRs in AD cases versus controls. There was a mean of 6.27 and 5.27 STR expansions in AD cases versus controls, representing a 1.19-fold higher burden of STR expansions in cases (p = 8.27 × 10−3, two-sided Mann–Whitney test) (Fig. 3d). The median number of STR expansions was 4.0 for both AD cases and controls. Strikingly, we found that individuals who carried > 30 STR expansions had an odds ratio of 3.69 for having AD (Fig. 3e, p-value 4.30 × 10−4, two-sided Fisher’s exact test; 95% confidence interval for odds ratio 1.83–8.06). In contrast, individuals with ≤10 expansions had a odds ratio of 0.77 for having AD. Importantly, we found that at higher STR expansion burden thresholds, we observed higher odds ratio for AD and increasing statistical significance (Fig. 3e).
We also rigorously examined whether our results may be due to potential confounders. In our above analyses, we had included local and genome-wide sequencing coverage as covariates in the DBSCAN analysis, as we had observed a modest but statistically significant correlation between the sequencing coverage and the number of STR expansions identified (Spearman r2 = 0.17; p = 1.79 × 10−12; linear regression F-test) (Supplementary Fig. 6a). However, even when sequencing coverage was not included as a covariate in the DBSCAN analysis, we continued to find an increased burden of STR expansions in individuals with AD (p = 4.45 × 10−3; two-sided Mann–Whitney test) and that individuals carrying >30 STR expansions had an increased odds for having AD (p-value 4.30 × 10−4, two-sided Fisher’s exact test; odds ratio 3.69 [95% CI 1.83–8.06]) (Supplementary Fig. 6b,c). We also did not observe any clear association of the STR expansion burden and the first two principal components of ancestry (Supplementary Fig. 7a). Moreover, even when correcting up to 10 principal components in the DBSCAN, we continued to observe an increased number of STR expansions in individuals with AD (p = 4.43 × 10−3; two-sided Mann–Whitney test) and that individuals carrying >30 STR expansions had an increased odds for having AD (p-value 4.30 × 10−4, two-sided Fisher’s exact test; odds ratio 3.69; [95% CI 1.83–8.06]) (Supplementary Fig. 7b-c).
We next ascertained if our observations were reproducible using gangSTR20, a separate STR genotyping algorithm. We note that there were fewer STR expansions identified by gangSTR (mean 1.028, median 1.00 STR expansions per individual) as compared to ExpansionHunter (mean 5.77, median 4.00 STR expansions per individual). This is consistent with our analyses and prior work demonstrating higher sensitivity of ExpansionHunter for identifying STR expansions as compared to gangSTR (Supplementary Fig. 2i)26. Using gangSTR, we again found a higher burden of STR expansions in cases (p = 2.93 × 10−4, two-sided Mann–Whitney test) (Supplementary Fig. 8a). We also found that individuals who carry >10 STR expansions as identified by gangSTR have a 3.14 odds ratio for having AD, though this was not statistically significant (95% CI odds ratio 0.73–21.0; p-value 0.19, two-sided Fisher’s exact test) (Supplementary Fig. 8b).
We next assessed whether individuals with a high burden of STR expansions also had differences in neuropathology as measured by Braak staging which reflects the degree of spread of tau pathology31. Braak staging data was available for 1188 individuals of the 2981 individuals in the cohort (n = 365 controls and 823 AD cases). Braak stages are scored from 0 to 6, with 0 representing absence of AD neuropathology and 6 being the most severe spread of AD neuropathology31. Using the same expansion burden thresholds as we used for AD risk, we found that individuals with >30 expansions had worse AD neuropathology compared to individuals with ≤10 expansions (p = 0.01, Kruskal–Wallis rank sum test) (Fig. 3f).
Since APOE is the strongest genetic effect on AD risk in the genome29, we next assessed whether there was any relationship between APOE genotype and STR expansion burden. We did not observe any relationship between the number of ε4 alleles and burden of STR expansions in the genome (p = 0.096, Kruskal–Wallis rank sum test) (Supplementary Fig. 9a). Moreover, the increased burden of STR expansions observed in individuals with AD remained when adjusting for APOE genotype in the DBSCAN analysis (p = 4.57 × 10−3, two-sided Mann–Whitney test) and that individuals carrying >30 STR expansions had an increased odds for having AD (95% CI odds ratio 1.83–8.06; p-value 4.30 × 10−4, two-sided Fisher’s exact test; odds ratio 3.69) (Supplementary Fig. 9b,c). Despite correcting for participant age in our DBSCAN analysis, we did identify a modest correlation between participant age and number of STR expansions (Spearman r2 = 0.0017; p = 0.025, linear regression F-test) (Supplementary Fig. 9d).
STR expansions are enriched in active regulatory regions of the genome
The majority of STRs tested in our study are not within coding portions of genes (Fig. 1b), precluding any simple interpretations of the mechanisms by which they may promote disease pathogenesis. We first annotated the genomic distributions of the 6276 AD STR expansions (those seen in individuals with AD) and found that they had similar distributions to the background of 293,752 STRs we tested. Specifically, the majority of AD STR expansions were in distal intergenic regions (38.1%; defined as being >3 kb from the nearest TSS) or in promoter regions (14.7%; ≤3 kb upstream from the nearest TSS) (Fig. 4a). However, AD STR expansions (median 24.7 kb) were further from the nearest TSS than background STRs (median 22.2 kb) (p-value = 6.10 × 10−10, two-sided Student’s t-test), which appeared to be driven by a slightly larger subset of STR expansions that were >100 kb upstream of the nearest TSS (Fig. 4b).

a Genomic distribution of all (background) STRs tested (top) compared to AD STRs expansions (bottom). b Distribution of distances to nearest TSS for all STRs tested (top) or AD STR expansions (bottom). c Enrichment of AD STR expansions in different chromatin states from the 18-state chromHMM model for adult hippocampus. d Enrichment of AD STR expansions using a permutation-based test. Dotted vertical line shows the number of AD STR expansions observed to overlap “flanking TSS” (chromatin state 2 from chromHMM) in adult hippocampus using a permutation-based test. e Enrichment of AD expanded STRs in peaks from different histone marks based on ChIP-seq in bulk adult hippocampal tissue. f Enrichment of AD STR expansions in different TE classes. g Enrichment of AD expanded STRs in SVA elements using a permutation-based test. h Proportion of STRs at given genomic distances from the nearest SVA. For c,e,f, height of points along the x-axis represents the statistical significance of enrichment, as measured by two-sided Fisher’s exact test. Size of points represents the magnitude of enrichment as represented by the log2(fold enrichment), and points are ordered by statistical significance for enrichment. For d,g dotted line reflects the observed number of AD STR expansions overlapping the genomic annotation, and histogram represents the number of AD STR expansions overlapping each of 1000 randomly permuted genomic regions. For d,g p-values represent empiric p-values calculated as the number of permutations exceeding observed STR overlap divided by total number of permutations.
Given that the majority of AD STR expansions were not within protein-coding regions, we next explored whether these AD STRs may be enriched in any chromatin features. We utilized all AD STR expansions (including those also seen in control individuals) since the STR expansions we identified in our study are likely not penetrant for AD. We tested whether AD STR expansions were enriched in chromatin states based on bulk post-mortem hippocampus ChIP-seq data from ENCODE32, using the 18 chromatin state partitions in chromHMM33. We found that “flanking TSS” (chromatin state 2) and “active TSS” (chromatin state 1) reflecting active promoter regions were the most enriched chromatin states for AD STR expansions (p-value = 2.82 × 10−18 and 1.66 × 10−14 respectively, two-sided Fisher’s exact test) (Fig. 4c). We validated the enrichment using a permutation-based approach by randomly drawing genomic regions and testing for their overlap with AD STR expansions. This permutation-based approach recapitulated the enrichment of AD STR expansions in “flanking TSS” (empirical enrichment p-value < 0.001) (Fig. 4d). The enrichment of AD STR expansions in “flanking TSS” was stronger in brain tissues compared to tissues from other parts of the body (Supplementary Fig. 10). We also examined STRs present in individuals with AD only and found enrichment p-values of 8.80 × 10−12 and 3.45 × 10−8 for chromatin states 1 and 2 respectively. We also validated the chromHMM enrichment results using ChIP-seq data from post-mortem hippocampal tissue in ENCODE32. Across ChIP-seq peaks for seven different histone marks in hippocampal tissue, we found that AD STR expansions were most highly enriched in H3K4me3 (p = 3.54 × 10−11, two-sided Fisher’s exact test) and H3K27ac peaks (p = 1.59 × 10−7). H3K4me3 marks active promoters, and H3K27ac marks active promoters and/or enhancers. Chromatin marked by both H3K4me3 and H3K27ac had even stronger enrichment (p = 2.84 × 10−11) (Fig. 4e). These findings demonstrate that AD STR expansions are enriched in active promoter regions in the brain.
Given that many transposable elements (TE) contain or are in proximity to STRs34, we next assessed whether AD STR expansions are enriched for any specific TE classes. We tested for enrichment across five broad categories of TEs: Alu, Human Endogenous Retroviruses (HERVs), long interspersed nuclear elements (LINE)-1 and -2, and SINE-VNTR-Alus (SVA). AD STR expansions were highly enriched for SVA elements (p-value 1.90 × 10−8, two-sided Fisher’s exact test) (Fig. 4f), which we also validated using a permutation-based approach (Fig. 4g). We also examined STRs present only in individuals with AD and found enrichment p-values within SVA elements of 7.58 × 10−8. 0.49% of AD STRs were inside the boundaries of an SVA element, compared with 0.15% of all STRs tested (p-value 5.7 × 10−11; two-sided two-proportion z-test), 0.55% of AD STR expansions were within 500 base pairs of an SVA element as compared to 0.38% of all STRs tested (p-value 0.035; two-sided two-proportion z-test) (Fig. 4h). Our observation of a strong enrichment of STR expansions within active promoter regions in the brain is consistent with prior work suggesting that SVA elements having been co-opted during evolution for enhancers and promoters in neurons35,36,37.
AD STR expansions are enriched in disease pathways with relevance to AD
While the majority of the 6276 AD STR expansions were not within protein-coding regions (Fig. 4a), we sought to understand whether they may be enriched near genes with certain biological or molecular functions. For each STR expansion, we identified the gene with the closest TSS within ± 250 kb. We then performed a gene ontology enrichment analysis for genes near AD STR expansions (n = 6276 STRs associated with 2373 unique genes) compared to all STRs tested (n = 293,752 STRs associated with 14,126 unique genes). We found that many of the most strongly associated gene sets were related to neuron biology, such as “neuron projection morphogenesis”, and “axon development” (Fig. 5a). Thus, while most AD STR expansions were not protein-coding, they are highly enriched near genes implicated in biological processes with known relevance to AD pathophysiology.

a Gene set enrichment for AD STR expansions relative to all STRs tested. The top 10 most highly associated gene ontology terms are shown. Height of points along the x-axis represents the statistical significance of enrichment, as measured by a two-sided Fisher’s exact test. b Same as a except only for STRs expansions within gene bodies. c Number of STR expansions within gene bodies identified by DBSCAN per individual in controls (green; n = 1485 individuals) versus cases (orange; n = 1481 individuals). d Odds ratio for AD case/control status for individuals carrying varying numbers of STR expansions within gene bodies. Odds ratios > 1.0 represent higher odds of having AD. p-values are calculated using a two-sided Fisher’s exact test. *p-value < 0.05. Box plot elements: centre line, median; box limits, upper and lower quartiles; and whiskers, 1.5x interquartile range. e GRID2 gene is shown with thick vertical lines representing exons and horizontal lines representing introns. Locations of AD STR expansions (orange) and control individual STR expansions (green) are shown. Each circle represents an STR expansion seen in one individual.
Since assigning intergenic STR expansions to a cognate gene is challenging, we next examined AD STR expansions in gene bodies (i.e., those occurring in exons, untranslated regions, or introns of genes). We note that both protein coding and non-protein coding genes were used in this analysis. We compared AD STR expansions found within gene bodies (n = 1327) to all STRs tested within gene bodies (n = 13,594) and identified strong enrichments in many gene sets related to synaptic function (Fig. 5b). These enrichments are notable given the pathologic role of synaptic dysfunction in AD38. We also found a higher burden of STR expansions in gene bodies between AD cases and control individuals (p-value 0.021, two-sided Mann–Whitney test) (Fig. 5c). Moreover, individuals with more than 10 STR expansions in gene bodies had a 2.02 odds ratio for having AD (95% CI odds ratio 1.18–3.58; p-value 0.02, two-sided Fisher’s exact test) (Fig. 5d). There was no difference in the number of STR expansions in protein-coding sequences only between cases and controls (p-value 0.89, two-sided Mann–Whitney test), though the number of such expansions was low (median 0.0 and mean 0.20 protein-coding STR expansions per individual).
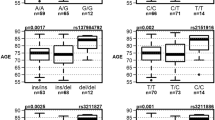
As an example, we highlight the GRID2 gene, in which we identified seven STR expansions across nine individuals with AD compared to just one STR expansion seen in a single control individual (Table 1; Fig. 5e; two-sided Fisher’s exact p-value 0.039). GRID2 encodes a subunit of the glutamate receptor and has recognized roles in synaptic transmission39. We also provide per-gene counts of STR expansions including hypergeometric statistical testing results across all genes (Supplementary Data 6). Together, these results demonstrate that AD STR expansions are highly enriched in and near genes implicated in biological processes with known relevance to AD pathophysiology and thus the AD STR expansions may represent molecular drivers of disease. Moreover, these results show that while these STR expansions were detected in blood-derived DNA, they appear to function in the brain, where much of the pathophysiology of AD is presumed to occur.
Discussion
In this work, we perform genome-wide profiling of STRs in 2981 individuals with and without AD to understand whether STRs associate with risk of AD. In contrast to the known STR-associated disorders, we do not find that a single STR individually drives genetic risk for AD. Instead, we identify thousands of STR expansions distributed throughout the genome with a higher burden of STR expansions in cases as compared to controls. Moreover, expanded STRs in AD cases are enriched in active promoters in the brain and in SVA TEs. These results suggest a model in which a polygenic burden of STR expansions distributed throughout the genome promotes risk of AD.
Remarkably, we found that individuals with AD carried an excess burden of expanded STRs. Each individual with AD had a mean of 6.27 STR expansions, though we note this estimate is likely an underestimate given the lower sensitivity of STR genotyping software on long expansions and our stringent quality filters. Nonetheless, this suggests that a polygenic burden of expanded STRs rather than a single STR promotes risk of AD. This polygenic effect is similar to what has been observed for STRs in autism spectrum disorder11,13 and in schizophrenia12. The distributed nature of these AD-associated STR expansions throughout the genome rather than within one or a few genes suggests that general genomic instability is a pathologic hallmark of the genomes in AD. This concept of an increased burden of STRs in individuals with AD is consistent with prior findings that individuals with AD have a higher burden of rare coding single nucleotide variants40 and structural variants41.
This observation of an increased burden of STR expansions in AD suggests one of two mechanistic models, which are not mutually exclusive. First, STR expansions may be an epiphenomenon of a disease process that promotes genomic instability. For example, AD pathology or biological aging may promote STR instability to result in an increased number of STR expansions in AD. The second possibility is that inherited and/or somatically unstable STR expansions promote disease pathogenesis. In support of this second model, we find that STR expansions in cases are preferentially localized in active promoters of genes with important neuronal functions. We note that in our gene set enrichment analyses, we assigned noncoding STRs to the nearest gene, though noncoding elements are known to often act at long genomic distances. Expression quantitative trait loci (eQTL)-based approaches may offer a better approach for assigning noncoding STRs to a cognate gene, but given our focus on rare STR expansions further limited by the relative paucity of available gene expression data in these samples, we did not have the statistical power to apply eQTL approaches. Nonetheless, these results suggest that STR expansions may have a functional role in AD onset and progression, though mechanistic studies will be needed to dissect the exact causal relationships between STRs and AD.
Strikingly, we found that AD-associated STR expansions were colocalized within active promoters in brain tissues. The AD STR expansions were particularly enriched within SVA elements, which have been proposed to have been co-opted for enhancers and promoters during human evolution35,36,42. SVA elements have also been shown to be important sources of tandem repeat variation and evolution in the human genome and particularly near neuronal genes37. Thus, our findings suggest that STRs at these SVA elements in active gene regulatory elements are prone to instability, particularly near genes with roles in AD pathophysiology. These findings in the context of existing literature underscore an important relationship between TEs, genome instability, and disease.
There are several important limitations to our study. First, while we expect molecular drivers of AD to act in the brain, we only have access to peripheral blood-derived DNA. We did however find that AD STR expansions were enriched in active promoters in the brain and near genes involved in neuronal function, suggesting that even though STR lengths were measured in blood-derived cells, they may manifest their effect on AD risk in the brain. Similarly, without paired blood and brain-derived DNA, we were unable to test for somatic instability of STRs in brain tissues. Second, we used short-read WGS where the accuracy of genotyping STRs is limited, though our comparisons with long-read sequencing identified a sensitivity of 66.4–68.3% for detecting STR expansions using ExpansionHunter (Supplementary Fig. 2i)26. We mitigated these concerns by using two different software programmes to replicate our results. We also compared our STR lengths to control individuals well-matched on ancestry and sequencing coverage (Supplementary Fig. 1c, e; Supplementary Table 1) and as such, we expect that any STR genotyping artifacts would be randomly allocated between AD and control individuals. Future work may also apply long-read sequencing approaches such as PacBio or Nanopore sequencing that can improve accuracy of STR genotyping, but are currently limited by cost. Analyses of long-read data will become an important future direction as these technologies become more feasible to apply across thousands of samples. Third, our study focuses on a cohort of 3000 individuals of European ancestry, limiting our statistical power and applicability of our results across diverse populations. With more computationally efficient approaches in the future, we will be able to explore larger sample sizes to increase our statistical power to identify STR associations, especially as our single STR associations, out sample sizes only provide statistical power to detect STRs with larger effect sizes (~1.2 allelic odds ratio; see Supplementary Fig. 4d). Studying diverse populations will allow us to test whether our results are applicable across other groups, especially as STRs are known to be more highly polymorphic in other populations43. Finally, our study design uses observational data in humans and so we cannot directly test causality. Future studies performing in vitro mechanistic dissection of these STRs will be required to establish a causal role of STRs in AD.
Together, our work identifies novel links between STR expansions and regulatory elements in AD. These results underscore the importance of uncovering the role of STRs in a broad range of diseases and understanding the mechanisms by which these STRs may promote disease risk.
Methods
Our research complies with all ethical regulations and has received Institutional Review Board exemption from the University of Pennsylvania.
Cohort Description
The Alzheimer’s Disease Sequencing Project (ADSP) is a collaborative project aimed at identifying new variants, genes, and therapeutic targets in AD44. In the R3 release of ADSP, genetic data from 16,905 individuals were aggregated across 24 cohorts and whole genome sequencing was performed by Illumina HiSeqX, HiSeq2000, HiSeq2500, and NovaSeq platforms.
In this work, we used individuals from the ADC and ROSMAP cohorts within the ADSP. For single STR analyses, we used ADC as the discovery cohort (total n = 2393; 1213 AD cases and 1180 controls) and ROSMAP as the replication cohort (total n = 588; n = 309 AD cases and 279 controls). We utilized AD case/control status as adjudicated and provided by the ADSP. Age of participants refers to the age at disease onset for cases and age at latest assessment for control participants. We restricted our analyses to individuals with self-reported non-Hispanic White race/ethnicity. We further restricted to individuals with European genetic ancestry based on PCA coordinates, which were provided by the ADSP. Based on manual inspection of PCA plots, we used samples with PC1 < −0.0037 and PC2 <0.02 to represent individuals of European ancestry. To minimize the impact of sequencing coverage on STR calls, we removed samples with <30× or >50× sequencing coverage across the genome. We also restricted our analyses to samples sequenced on Illumina HiSeqX machines. All samples meeting these criteria were included in our analyses. We note that have focused on this subset of individuals rather than the full ADSP release given the computational costs of our analyses. All individuals have been previously consented for AD genetic research by their local Institutional Review Boards.
Generation of polymorphic STR panel
To reduce computational burden, rather than testing all STRs genome-wide, we generated a custom panel of polymorphic STRs. To generate this panel, we first ran gangSTR20. using the hg38_ver13 catalogue provided by the gangSTR authors (https://s3.amazonaws.com/gangstr/hg38/genomewide/hg38_ver13.bed.gz) on 246 AD cases and 249 AD controls from the ADC cohort in ADSP. We used gangSTR v2.4.0 on this hg38_ver13 reference panel using default parameters, except --max-proc-read 100000 and –rescue-count 10. STRs were determined to be polymorphic if at least one individual in the cohort had a non-reference STR length and the genotyping rate across the 495 samples was ≥ 90%. In total, there were 237,197 STRs that met these criteria. We next merged these 237,197 STRs with a set 174,262 STRs previously identified to be polymorphic22. This resulted in a union set of 312,731 unique polymorphic STRs.
Genotyping STR tract lengths using Illumina short-read data
We genotyped STR tract lengths using ExpansionHunter v522 on our panel of 312,731 polymorphic STRs using default parameters. Prior to running ExpansionHunter, the polymorphic STR panel was converted to json format required for ExpansionHunter.
We also genotyped STR tract lengths using gangSTR20 on our panel of 312,731 polymorphic STRs. To increase sensitivity of gangSTR, we added in offtarget alignment locations for any STR that had a p-value <0.05 in the ExpansionHunter single STR association analysis. To determine offtarget alignment locations, we used wgsim v1.11 to simulate 10000 sequencing reads for each STR (https://github.com/lh3/wgsim). We used the following parameters in wgsim: -e 0.005 -d 500 -s 100 -N 10000 -1 150 -2 150 -r 0 -R 0 -X 0. Simulated reads were then realigned back to the GRCh38 reference genome using bwa mem version 0.7.17 with default parameters45. The offtarget alignment locations of simulated reads in the GRCh38 reference genome were then extracted using scripts provided by the gangSTR authors, and we used the top 5 offtarget locations for each STR. gangSTR v2.4.2 was run on this custom reference panel with default parameters except --max-proc-read 100000.
Assessment of genotyping accuracy using PacBio long-read data
To assess the accuracy of gangSTR and ExpansionHunter applied to Illumina short-read sequencing data, we also performed STR genotyping on long-read data. We used PacBio Revio long-read data generated on n = 4 individuals (HG00096, HG00268, HG00358, and NA20509) from the 1000 Genomes Project24. We downloaded PacBio data from https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC3/working/20220831_JAX_HiFi/. We aligned PacBio data using Minimap2 v2.2446 with option -ax map-hifi and all default parameters. We then called STR tract lengths from PacBio data using TRGT v0.8.025 with default parameters on the set of 312,731 polymorphic STRs.
For comparison, we also analysed Illumina short-read sequencing data for the same four samples from the 1000 Genomes Project. We downloaded aligned Illumina short-read sequencing data from https://www.internationalgenome.org/data/. We genotyped the n = 312,731 polymorphic STRs using gangSTR v2.4.2 with no offtarget locations and all default parameters except --max-proc-read 100000. We genotyped the same set of 312,731 polymorphic STRs using ExpansionHunter v5 with all default parameters. To measure concordance, we only examined STRs were the longer allele had a tract length ≥10 repeat units longer than the reference STR tract length in GRCh38. For concordance calculations, we required the genotyped tract length to be within ± 20% between PacBio and the corresponding genotypes from Illumina data.
Single STR association analysis
We performed association analyses of STR genotype with AD risk under a dominant model by testing the longer of two alleles for each STR. Given that many of the STR genotypes were not normally distributed, we first performed rank-based inverse normal transformation of STR genotypes. To test for the association of each STR with AD case/control status, we applied a logistic regression model, controlling for sample covariates (sex, age, and the first three PCs) and technical covariates (genomic sequencing coverage for each sample and local sequencing coverage at each STR for each sample). We used ADC as the discovery cohort (total n = 2393; 1213 AD cases and 1180 controls) and ROSMAP as the replication cohort (total n = 588; n = 309 AD cases and 279 controls).
We report the logistic regression p-value of the rank-based inverse normal transformed STR genotype on AD risk. We separately report the regression p-value in a logistic regression model without inverse normal transformation to derive absolute effect sizes on AD case/control status. Finally, to ensure results were not due to the non-normal distribution of the STR genotypes, association analyses were also performed using a non-parametric association test (two-sided Mann-Whitney test) without covariates.
For reporting of STR association analyses, we removed all STRs that were in segmentally duplicated regions (http://hgdownload.cse.ucsc.edu/goldenpath/hg38/database/genomicSuperDups.txt.gz) (n = 18,979), resulting in a final list of 293,752 STRs that were reported in our association analyses. Multiple hypothesis testing correction was performed using Bonferroni correction, with a p-value threshold of 1.70×10-7 (0.05/293752). P-values between the discovery and replication cohorts were meta-analysed using Fisher’s method.
Power analyses for single STR associations
To estimate power for single STR associations, we simulated STRs with five alleles (i.e., five different STR tract lengths). We simulated a sample size of 3000 individuals, where 600 individuals had each of the five alleles. We tested power at different allelic odds ratios (i.e., the odds ratio per unit increase in STR tract length) ranging from 1.01 to 1.25 in step sizes of 0.01. For each allelic odds ratio, we performed 10,000 permutations of case/control status by randomly permuting each individual to a case or a control with equal probability. For each permutation, we then performed a logistic regression of case/control status against the simulated STR tract length. For each allelic odds ratio, we then counted the number of permutations where the logistic regression p-value was less than the Bonferroni-corrected p-value threshold of 1.70 × 10−7 (0.05/293752). Power was calculated as the number of simulations reaching statistical significance divided by the number of permutations (n = 10,000).
Hypergeometric test for STR expansions
We performed a burden test for the number of STR expansions in case versus control individuals for each STR. We performed this analysis using ExpansionHunter genotyping data on the ADC discovery cohort (total n = 2393; 1213 AD cases and 1180 controls)
In addition to excluding STRs in segmentally duplicated regions as described above, we also excluded 20,653 STRs that had genotyping rates <90% in either the cases or the controls, resulting in a set of 273,099 STRs. We first identified STR expansions for each of the 273,099 STRs in our custom STR panel as STR tract lengths that were ≥ 1, 5, 10, or 20 repeat units longer than the GRCh38 reference STR tract length. For each STR, we then constructed a 2 × 2 contingency table of the number of individuals with and without an STR expansion as defined by these thresholds. We applied a two-sided Fisher’s exact test to each STR to test whether there is a difference in burden of STR expansions in cases versus controls. Multiple hypothesis testing correction was performed using Bonferroni correction, with a p-value threshold of 1.83 × 10−7 (0.05/273099).
We also performed analyses restricting to STR expansions observed in only one individual, ≤5, ≤10, ≤100, or with no frequency cutoff in the combined n = 2981 individuals. We then compared the total number of STR expansions in AD case versus control individuals using a two-sided Mann-Whitney test.
Identifying STR expansions using DBSCAN
We applied the DBSCAN outlier detection method to identify STR expansions, which we extended from the work of Trost et al.13 Briefly, DBSCAN is an unsupervised density-based clustering method that can be used to identify outlier groups47, here representing extreme STR tract lengths for each STR. DBSCAN defines a cluster based on the minimum number of data points (μ) reachable to each other by a maximum distance (ε). Data points not reachable by the clusters are classified outliers if they have an STR tract length that is higher than those of cluster members. Specifically, ε was set as the maximum of twice the mode of STR lengths, and μ was set as the log2 of the number of samples.
To increase statistical power to identify rare STR expansions, we combined samples from across the ADC and ROSMAP cohorts (total n = 2981; n = 1492 controls and 1489 cases). We ran DBSCAN on each of the 293,752 STRs in our panel (after removing STRs in segmentally duplicated regions as described above). For each STR, the longer of the two alleles for each individual was used as input for DBSCAN. To account for potential confounders, we first performed linear regression for the tract lengths of each STR to regress out the effects of sample covariates (sex, age, and the first three PCAs) and technical covariates (genomic sequencing coverage for each sample and local sequencing coverage at each STR for each sample). We input the residuals from the linear regression into DBSCAN, with ε and μ as determined above. We ran DBSCAN on these residuals to identify outlier STR lengths. We applied DBSCAN separately for STR genotyping results from ExpansionHunter and gangSTR. We excluded individuals from downstream analyses if the number of STR expansions identified was more than 2 standard deviations away from the mean STR expansion count for the cohort; 15 individuals were excluded by this filter. DBSCAN was implemented using the dbscan package v1.1-11 (https://github.com/cran/dbscan) in R v3.6.348,49.
Testing for enrichment of STRs in chromHMM and ChIP-seq annotations
We downloaded chromHMM partitions under the 18 state model for the adult human hippocampus (ENCODE tissue ID E071) from the Roadmap Epigenomics Consortium (https://egg2.wustl.edu/roadmap/web_portal/chr_state_learning.html)32,33. We then tested whether AD STR expansions were enriched in each chromatin state, relative to a background of all STRs in our custom panel of polymorphic STRs. We performed statistical testing using a two-sided Fisher’s exact test. Prior to statistical testing, we removed all regions located within segmental duplications from the analysis. We considered an STR to be located within a chromHMM partition if at least one bp of the STR was within the partition.
We also tested whether AD STR expansions were enriched in certain histone marks as assayed by ChIP-seq. We downloaded ChIP-seq peak data from ENCODE for the adult hippocampus for H3K4me1, H3K4me3, H3K9ac, H3K27ac, H3K27me3, and H3K36me3 (https://www.encodeproject.org/matrix/). For each biological sample, we used IDR-replicated peaks from ENCODE. If there were multiple biological samples for each histone mark, we merged peaks across samples using BEDTools v2.2650. We then performed the enrichment analysis as above.
For both the chromHMM data and ENCODE ChIP-seq data, we also performed enrichment analyses using a permutation-based approach. We calculated how many of the n = 6276 AD STR expansions directly overlapped the peak set. We then generated 1000 random sets of 6276 peaks using regioneR v1.30.0 using default parameters except per.chromsome=F51. Random peak sets were generated against the GRCh38 reference genome, with masking of segmental duplications. We counted how many AD STR expansions directly overlapped each random peak set. We derived an empirical p-value by counting the number of random permutations with equal or more overlaps than the observed number of overlaps and dividing by the total number of permutations (n = 1000).
Testing for STR enrichments in transposable elements
To identify locations of TE in the genome, we obtained the RepeatMasker file from the UCSC Table Browser (https://genome.ucsc.edu/cgi-bin/hgTables)52. We tested for enrichment of AD STR expansions across 5 broad classes of TEs: Alu, HERVs, LINE1, LINE2, and SVA elements. For each TE class, we tested whether AD STR expansions were enriched in the TE, relative to a background of all STRs in our custom STR panel. We performed statistical testing using a two-sided Fisher’s exact test. Prior to statistical testing, we removed all genomic regions within segmental duplications. We considered an STR to be located within a TE if at least one bp of the STR was within the TE coordinates from the RepeatMasker file.
Gene Ontology Enrichment Analysis
We performed gene set enrichment analysis using the clusterProfiler v4.2.2 package in R v3.6.353. For all STRs, we compared AD STR expansions (n = 6276) to all STRs in the reference STR panel (n = 293,752). For this analysis, we assigned each STR to the gene with the closest TSS within ± 250 kb. We assigned genes using transcriptional start sites annotated in Ensembl release 112 in human reference genome GRCh38.14 https://useast.ensembl.org/index.html, which was loaded into R using the biomaRt package v2.58.054,55. All genes in the Ensembl gene annotation were used, including non-protein coding genes. We also performed separate analyses for STRs within the gene body (either introns, exons, or untranslated regions), where we compared genes with AD STR expansions within the gene body (n = 1327) to all genes with containing an STR from the reference STR panel (n = 13,594). We then tested the enrichment of genes assigned to AD STR expansions compared to genes assigned to background STRs using the enrichGO function in clusterProfiler, with the following parameters: keyType = “ENTREZID”, ont = ”ALL”, p-value Cutoff = 0.05, q-value Cutoff = 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The individual level genotype and phenotype data analysed in this study are accessible from NIAGADS Data Sharing Service (DSS) via qualified access (accession number NG00067). The individual level genotype and phenotype data are not provided with this manuscript due to data use restrictions in place to protect participant privacy. Formal requests to access individual level genotype and phenotype datasets can be submitted to NIAGADS DSS https://dss.niagads.org/. The source data underlying main figures are provided with this paper and available at https://doi.org/10.5281/zenodo.13917278. The gangSTR STR panel file used in this study is openly available at https://s3.amazonaws.com/gangstr/hg38/genomewide/hg38_ver13.bed.gz. PacBio long-read sequencing data used in this study are openly available at https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC3/working/20220831_JAX_HiFi/. Illumina short-read sequencing data used in this study are openly available at https://www.internationalgenome.org/data/. The segmental duplications file from UCSC Genome Browser is openly available at http://hgdownload.cse.ucsc.edu/goldenpath/hg38/database/genomicSuperDups.txt.gz. chromHMM data is openly available at https://egg2.wustl.edu/roadmap/web_portal/chr_state_learning.html. ChIP-seq data from ENCODE Project used in this study are openly available at https://www.encodeproject.org/matrix/. The RepeatMasker file of TE annotations is openly available at https://genome.ucsc.edu/cgi-bin/hgTables. The Ensembl gene annotation file is openly available at https://useast.ensembl.org/index.html. All other data supporting the findings described in this manuscript are available in the article and associated Supplementary Information files. Source data are provided with this paper.
Code availability
All code used in this manuscript are available at https://github.com/mhguo1/AD_STR56.
References
2023 Alzheimer’s disease facts and figures. Alzheimers. Dement. 19, 1598–1695 (2023).
Escott-Price, V. & Hardy, J. Genome-wide association studies for Alzheimer’s disease: bigger is not always better. Brain Commun 4, fcac125 (2022).
Willems, T., Gymrek, M., Highnam, G., Mittelman, D. & Erlich, Y. The landscape of human STR variation. Genome Res. 24, 1894–1904 (2014).
Dib, C. et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380, 152–154 (1996).
Gymrek, M. et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet. 48, 22–29 (2016).
Olsson, T., Barcellos, L. F. & Alfredsson, L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 13, 25–36 (2017).
Shi, Y. et al. Characterization of genome-wide STR variation in 6487 human genomes. Nat. Commun. 14, 2092 (2023).
Fazal, S. et al. Large scale in silico characterization of repeat expansion variation in human genomes. Sci. data 7, 294 (2020).
Hannan, A. J. Tandem repeats mediating genetic plasticity in health and disease. Nat. Rev. Genet. 19, 286–298 (2018).
Depienne, C. & Mandel, J.-L. 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am. J. Hum. Genet. 108, 764–785 (2021).
Mitra, I. et al. Patterns of de novo tandem repeat mutations and their role in autism. Nature 589, 246–250 (2021).
Wen, J. et al. Rare tandem repeat expansions associate with genes involved in synaptic and neuronal signaling functions in schizophrenia. Mol. Psychiatry 28, 475–482 (2023).
Trost, B. et al. Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature 586, 80–86 (2020).
Haws, S. A., Simandi, Z., Barnett, R. J. & Phillips-Cremins, J. E. 3D genome, on repeat: Higher-order folding principles of the heterochromatinized repetitive genome. Cell 185, 2690–2707 (2022).
Jakubosky, D. et al. Properties of structural variants and short tandem repeats associated with gene expression and complex traits. Nat. Commun. 11, 2927 (2020).
Fotsing, S. F. et al. The impact of short tandem repeat variation on gene expression. Nat. Genet. 51, 1652–1659 (2019).
Nativio, R. et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 21, 497–505 (2018).
Nativio, R. et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat. Genet. 52, 1024–1035 (2020).
Klein, H.-U. et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 22, 37–46 (2019).
Mousavi, N., Shleizer-Burko, S., Yanicky, R. & Gymrek, M. Profiling the genome-wide landscape of tandem repeat expansions. Nucleic Acids Res. 47, e90 (2019).
Dashnow, H. et al. STRling: a k-mer counting approach that detects short tandem repeat expansions at known and novel loci. Genome Biol 23, 257 (2022).
Dolzhenko, E. et al. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35, 4754–4756 (2019).
Dolzhenko, E. et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol 21, 102 (2020).
Ebert, P. et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372, eabf7117 (2021).
Dolzhenko, E. et al. Characterization and visualization of tandem repeats at genome scale. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-02057-3 (2024).
Weisburd, B., Tiao, G. & Rehm, H. L. Insights from a genome-wide truth set of tandem repeat variation. BioRxiv. https://www.biorxiv.org/content/10.1101/2023.05.05.539588v1 (2023).
Quilez, J. et al. Polymorphic tandem repeats within gene promoters act as modifiers of gene expression and DNA methylation in humans. Nucleic Acids Res. 44, 3750–3762 (2016).
Sawaya, S. et al. Microsatellite tandem repeats are abundant in human promoters and are associated with regulatory elements. PLoS One 8, e54710 (2013).
Corder, E. H. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993).
Byrska-Bishop, M. et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185, 3426–3440.e19 (2022).
Braak, H. & Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259 (1991).
Bernstein, B. E. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Ernst, J. & Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 12, 2478–2492 (2017).
Ahmed, M. & Liang, P. Transposable elements are a significant contributor to tandem repeats in the human genome. Comp. Funct. Genomics 2012, 947089 (2012).
Trizzino, M., Kapusta, A. & Brown, C. D. Transposable elements generate regulatory novelty in a tissue-specific fashion. BMC Genomics 19, 468 (2018).
Quinn, J. P. & Bubb, V. J. SVA retrotransposons as modulators of gene expression. Mob. Genet. Elements 4, e32102 (2014).
Sulovari, A. et al. Human-specific tandem repeat expansion and differential gene expression during primate evolution. Proc. Natl. Acad. Sci. USA. 116, 23243–23253 (2019).
Long, J. M. & Holtzman, D. M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 179, 312–339 (2019).
Kashiwabuchi, N. et al. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluR delta 2 mutant mice. Cell 81, 245–252 (1995).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Wang, H. et al. Structural Variation Detection and Association Analysis of Whole-Genome-Sequence Data from 16,905 Alzheimer’s Diseases Sequencing Project Subjects. medRxiv https://doi.org/10.1101/2023.09.13.23295505 (2023).
Barnada, S. M. et al. Genomic features underlie the co-option of SVA transposons as cis-regulatory elements in human pluripotent stem cells. PLoS Genet 18, e1010225 (2022).
Ziaei Jam, H. et al. A deep population reference panel of tandem repeat variation. Nat. Commun. 14, 6711 (2023).
Beecham, G. W. et al. The Alzheimer’s Disease Sequencing Project: Study design and sample selection. Neurol. Genet. 3, e194 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows – Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Ester, M., Kriegel, H.-P., Sander, J., Xu, X. & others. A density-based algorithm for discovering clusters in large spatial databases with noise. in Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (KDD-96). 226–231 (1996).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing (2019).
Hahsler, M., Piekenbrock, M. & Doran, D. dbscan: fast density-based clustering with R. J. Stat. Softw. 91, 1–30 (2019).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Gel, B. et al. regioneR: an R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics 32, 289–291 (2016).
Rosenbloom, K. R. et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 43, D670–D681 (2015).
Xu, S. et al. Using clusterProfiler to characterize multiomics data. Nat. Protoc. 19, 3292–3320 (2024).
Durinck, S., Spellman, P. T., Birney, E. & Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191 (2009).
Durinck, S. et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440 (2005).
Guo, M. H., Lee, W.-P., Vardarajan, B., Schellenberg, G. D. & Phillips-Cremins, J. E. Polygenic burden of short tandem repeat expansions promotes risk for Alzheimer’s disease. https://doi.org/10.5281/zenodo.13917273 (2024).
Acknowledgements
MHG, GS, and JEPC wish to dedicate this work in honour of our late colleague Dr. Christopher D. Brown, Department of Genetics, University of Pennsylvania. Dr. Brown inspired the initial concept for this study, and his generous feedback was invaluable for the early stages of this work.
We thank members of the Cremins lab for helpful discussions. This work is supported by NIH National Institute of Neurologic Disorders and Stroke (R01-NS11422 to JEPC; R25-NS065745 to MHG), NIH National Institute on Aging (RF1-AG074328, P30-AG072979, U54-AG052427, and U24-AG041689 to WPL; P30-AG072979 to MHG), NIH National Institute of Mental Health (1R01MH120269 to JEPC), NSF CAREER Award (CBE-1943945 to JEPC), NSF EFRI Award (EFMA1933400 to JEPC), and Chan Zuckerberg Initiative Neurodegenerative Disease Pairs Awards (2020-221479-5022 and DAF2022-250430 to JEPC).
Data for this study were prepared, archived, and distributed by the NIAGADS DSS at the University of Pennsylvania (U24-AG041689), funded by the National Institute on Aging.
Alzheimer’s Disease Sequencing Project
The ADSP is comprised of two AD genetics consortia and three National Human Genome Research Institute (NHGRI) funded Large Scale Sequencing and Analysis Centers (LSAC). The two AD genetics consortia are the Alzheimer’s Disease Genetics Consortium (ADGC) funded by NIA (U01 AG032984), and the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) funded by NIA (R01 AG033193), the National Heart, Lung, and Blood Institute (NHLBI), other National Institute of Health (NIH) institutes and other foreign governmental and non-governmental organizations. The Discovery Phase analysis of sequence data is supported through UF1AG047133 (to Drs. Schellenberg, Farrer, Pericak-Vance, Mayeux, and Haines); U01AG049505 to Dr. Seshadri; U01AG049506 to Dr. Boerwinkle; U01AG049507 to Dr. Wijsman; and U01AG049508 to Dr. Goate and the Discovery Extension Phase analysis is supported through U01AG052411 to Dr. Goate, U01AG052410 to Dr. Pericak-Vance and U01 AG052409 to Drs. Seshadri and Fornage.
Sequencing for the Follow Up Study (FUS) is supported through U01AG057659 (to Drs. PericakVance, Mayeux, and Vardarajan) and U01AG062943 (to Drs. Pericak-Vance and Mayeux). Data generation and harmonization in the Follow-up Phase is supported by U54AG052427 (to Drs. Schellenberg and Wang). The FUS Phase analysis of sequence data is supported through U01AG058589 (to Drs. Destefano, Boerwinkle, De Jager, Fornage, Seshadri, and Wijsman), U01AG058654 (to Drs. Haines, Bush, Farrer, Martin, and Pericak-Vance), U01AG058635 (to Dr. Goate), RF1AG058066 (to Drs. Haines, Pericak-Vance, and Scott), RF1AG057519 (to Drs. Farrer and Jun), R01AG048927 (to Dr. Farrer), and RF1AG054074 (to Drs. Pericak-Vance and Beecham).
The ADGC cohorts include: Adult Changes in Thought (ACT) (U01 AG006781, U19 AG066567), the Alzheimer’s Disease Research Centers (ADRC) (P30 AG062429, P30 AG066468, P30 AG062421, P30 AG066509, P30 AG066514, P30 AG066530, P30 AG066507, P30 AG066444, P30 AG066518, P30 AG066512, P30 AG066462, P30 AG072979, P30 AG072972, P30 AG072976, P30 AG072975, P30 AG072978, P30 AG072977, P30 AG066519, P30 AG062677, P30 AG079280, P30 AG062422, P30 AG066511, P30 AG072946, P30 AG062715, P30 AG072973, P30 AG066506, P30 AG066508, P30 AG066515, P30 AG072947, P30 AG072931, P30 AG066546, P20 AG068024, P20 AG068053, P20 AG068077, P20 AG068082, P30 AG072958, P30 AG072959), the Chicago Health and Aging Project (CHAP) (R01 AG11101, RC4 AG039085, K23 AG030944), Indiana Memory and Aging Study (IMAS) (R01 AG019771), Indianapolis Ibadan (R01 AG009956, P30 AG010133), the Memory and Aging Project (MAP) (R01 AG17917), Mayo Clinic (MAYO) (R01 AG032990, U01 AG046139, R01 NS080820, RF1 AG051504, P50 AG016574), Mayo Parkinson’s Disease controls (NS039764, NS071674, 5RC2HG005605), University of Miami (R01 AG027944, R01 AG028786, R01 AG019085, IIRG09133827, A2011048), the Multi-Institutional Research in Alzheimer’s Genetic Epidemiology Study (MIRAGE) (R01 AG09029, R01 AG025259), the National Centralized Repository for Alzheimer’s Disease and Related Dementias (NCRAD) (U24 AG021886), the National Institute on Aging Late Onset Alzheimer’s Disease Family Study (NIA- LOAD) (U24 AG056270), the Religious Orders Study (ROS) (P30 AG10161, R01 AG15819), the Texas Alzheimer’s Research and Care Consortium (TARCC) (funded by the Darrell K Royal Texas Alzheimer’s Initiative), Vanderbilt University/Case Western Reserve University (VAN/CWRU) (R01 AG019757, R01 AG021547, R01 AG027944, R01 AG028786, P01 NS026630, and Alzheimer’s Association), the Washington Heights-Inwood Columbia Aging Project (WHICAP) (RF1 AG054023), the University of Washington Families (VA Research Merit Grant, NIA: P50AG005136, R01AG041797, NINDS: R01NS069719), the Columbia University Hispanic Estudio Familiar de Influencia Genetica de Alzheimer (EFIGA) (RF1 AG015473), the University of Toronto (UT) (funded by Wellcome Trust, Medical Research Council, Canadian Institutes of Health Research), and Genetic Differences (GD) (R01 AG007584). The CHARGE cohorts are supported in part by National Heart, Lung, and Blood Institute (NHLBI) infrastructure grant HL105756 (Psaty), RC2HL102419 (Boerwinkle) and the neurology working group is supported by the National Institute on Aging (NIA) R01 grant AG033193.
The CHARGE cohorts participating in the ADSP include the following: Austrian Stroke Prevention Study (ASPS), ASPS-Family study, and the Prospective Dementia Registry-Austria (ASPS/PRODEM-Aus), the Atherosclerosis Risk in Communities (ARIC) Study, the Cardiovascular Health Study (CHS), the Erasmus Rucphen Family Study (ERF), the Framingham Heart Study (FHS), and the Rotterdam Study (RS). ASPS is funded by the Austrian Science Fond (FWF) grant number P20545-P05 and P13180 and the Medical University of Graz. The ASPS-Fam is funded by the Austrian Science Fund (FWF) project I904), the EU Joint Programme – Neurodegenerative Disease Research (JPND) in frame of the BRIDGET project (Austria, Ministry of Science) and the Medical University of Graz and the Steiermärkische Krankenanstalten Gesellschaft. PRODEM-Austria is supported by the Austrian Research Promotion agency (FFG) (Project No. 827462) and by the Austrian National Bank (Anniversary Fund, project 15435. ARIC research is carried out as a collaborative study supported by NHLBI contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C). Neurocognitive data in ARIC is collected by U01 2U01HL096812, 2U01HL096814, 2U01HL096899, 2U01HL096902, 2U01HL096917 from the NIH (NHLBI, NINDS, NIA and NIDCD), and with previous brain MRI examinations funded by R01-HL70825 from the NHLBI. CHS research was supported by contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and grants U01HL080295 and U01HL130114 from the NHLBI with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629, R01AG15928, and R01AG20098 from the NIA. FHS research is supported by NHLBI contracts N01-HC-25195 and HHSN268201500001I. This study was also supported by additional grants from the NIA (R01s AG054076, AG049607 and AG033040 and NINDS (R01 NS017950). The ERF study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP grant number 018947 (LSHG-CT-2006-01947) and also received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013)/grant agreement HEALTH-F4- 2007-201413 by the European Commission under the programme “Quality of Life and Management of the Living Resources” of 5th Framework Programme (no. QLG2-CT-2002- 01254). High-throughput analysis of the ERF data was supported by a joint grant from the Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043). The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, the Netherlands Organization for Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the municipality of Rotterdam. Genetic data sets are also supported by the Netherlands Organization of Scientific Research NWO Investments (175.010.2005.011, 911-03-012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), and the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA), project 050-060-810. All studies are grateful to their participants, faculty and staff. The content of these manuscripts is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the U.S. Department of Health and Human Services.
The FUS cohorts include: the Alzheimer’s Disease Research Centers (ADRC) (P30 AG062429, P30 AG066468, P30 AG062421, P30 AG066509, P30 AG066514, P30 AG066530, P30 AG066507, P30 AG066444, P30 AG066518, P30 AG066512, P30 AG066462, P30 AG072979, P30 AG072972, P30 AG072976, P30 AG072975, P30 AG072978, P30 AG072977, P30 AG066519, P30 AG062677, P30 AG079280, P30 AG062422, P30 AG066511, P30 AG072946, P30 AG062715, P30 AG072973, P30 AG066506, P30 AG066508, P30 AG066515, P30 AG072947, P30 AG072931, P30 AG066546, P20 AG068024, P20 AG068053, P20 AG068077, P20 AG068082, P30 AG072958, P30 AG072959), Alzheimer’s Disease Neuroimaging Initiative (ADNI) (U19AG024904), Amish Protective Variant Study (RF1AG058066), Cache County Study (R01AG11380, R01AG031272, R01AG21136, RF1AG054052), Case Western Reserve University Brain Bank (CWRUBB) (P50AG008012), Case Western Reserve University Rapid Decline (CWRURD) (RF1AG058267, NU38CK000480), CubanAmerican Alzheimer’s Disease Initiative (CuAADI) (3U01AG052410), Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA) (5R37AG015473, RF1AG015473, R56AG051876), Genetic and Environmental Risk Factors for Alzheimer Disease Among African Americans Study (GenerAAtions) (2R01AG09029, R01AG025259, 2R01AG048927), Gwangju Alzheimer and Related Dementias Study (GARD) (U01AG062602), Hillblom Aging Network (2014-A-004-NET, R01AG032289, R01AG048234), Hussman Institute for Human Genomics Brain Bank (HIHGBB) (R01AG027944, Alzheimer’s Association “Identification of Rare Variants in Alzheimer Disease”), Ibadan Study of Aging (IBADAN) (5R01AG009956), Longevity Genes Project (LGP) and LonGenity (R01AG042188, R01AG044829, R01AG046949, R01AG057909, R01AG061155, P30AG038072), Mexican Health and Aging Study (MHAS) (R01AG018016), Multi-Institutional Research in Alzheimer’s Genetic Epidemiology (MIRAGE) (2R01AG09029, R01AG025259, 2R01AG048927), Northern Manhattan Study (NOMAS) (R01NS29993), Peru Alzheimer’s Disease Initiative (PeADI) (RF1AG054074), Puerto Rican 1066 (PR1066) (Wellcome Trust (GR066133/GR080002), European Research Council (340755)), Puerto Rican Alzheimer Disease Initiative (PRADI) (RF1AG054074), Reasons for Geographic and Racial Differences in Stroke (REGARDS) (U01NS041588), Research in African American Alzheimer Disease Initiative (REAAADI) (U01AG052410), the Religious Orders Study (ROS) (P30 AG10161, P30 AG72975, R01 AG15819, R01 AG42210), the RUSH Memory and Aging Project (MAP) (R01 AG017917, R01 AG42210Stanford Extreme Phenotypes in AD (R01AG060747), University of Miami Brain Endowment Bank (MBB), University of Miami/Case Western/North Carolina A&T African American (UM/CASE/NCAT) (U01AG052410, R01AG028786), and Wisconsin Registry for Alzheimer’s Prevention (WRAP) (R01AG027161 and R01AG054047).
The four LSACs are: the Human Genome Sequencing Center at the Baylor College of Medicine (U54 HG003273), the Broad Institute Genome Center (U54HG003067), The American Genome Center at the Uniformed Services University of the Health Sciences (U01AG057659), and the Washington University Genome Institute (U54HG003079). Genotyping and sequencing for the ADSP FUS is also conducted at John P. Hussman Institute for Human Genomics (HIHG) Center for Genome Technology (CGT).
Biological samples and associated phenotypic data used in primary data analyses were stored at Study Investigators institutions, and at the National Centralized Repository for Alzheimer’s Disease and Related Dementias (NCRAD, U24AG021886) at Indiana University funded by NIA. Associated Phenotypic Data used in primary and secondary data analyses were provided by Study Investigators, the NIA funded Alzheimer’s Disease Centers (ADCs), and the National Alzheimer’s Coordinating Center (NACC, U24AG072122) and the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS, U24AG041689) at the University of Pennsylvania, funded by NIA. Harmonized phenotypes were provided by the ADSP Phenotype Harmonization Consortium (ADSP-PHC), funded by NIA (U24 AG074855, U01 AG068057 and R01 AG059716) and Ultrascale Machine Learning to Empower Discovery in Alzheimer’s Disease Biobanks (AI4AD, U01 AG068057). This research was supported in part by the Intramural Research Program of the National Institutes of health, National Library of Medicine. Contributors to the Genetic Analysis Data included Study Investigators on projects that were individually funded by NIA, and other NIH institutes, and by private U.S. organizations, or foreign governmental or nongovernmental organizations.
ROSMAP
We are grateful to the participants in ROSMAP. This work is supported by the US National Institutes of Health [U01 AG046152, R01 AG043617, R01 AG042210, R01 AG036042, R01 AG036836, R01 AG032990, R01 AG18023, RC2 AG036547, P50 AG016574, U01 ES017155, KL2 RR024151, K25 AG041906-01, R01 AG30146, P30 AG10161, R01 AG17917, R01 AG15819, K08 AG034290, P30 AG10161 and R01 AG11101.
Author information
Authors and Affiliations
Contributions
Study conception and design: M.H.G. and J.E.P-C. Acquisition of study data: W-P.L. and G.D.S. Genotyping of STRs: M.H.G., B.V., and W-P.L. Analyses of STR associations: M.H.G. and J.E.P-C. Writing and figure preparation: led by M.H.G. and J.E.P-C., and reviewed by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kishore Kumar and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, M.H., Lee, WP., Vardarajan, B. et al. Polygenic burden of short tandem repeat expansions promotes risk for Alzheimer’s disease. Nat Commun 16, 1126 (2025). https://doi.org/10.1038/s41467-025-56400-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56400-0
This article is cited by
-
Whole-genome sequencing analyses suggest novel genetic factors associated with Alzheimer’s disease and a cumulative effects model for risk liability
Nature Communications (2025)
-
PolyGR-containing aggregates link with pathology and clinical features of Alzheimer’s disease
Acta Neuropathologica (2025)
