
Abstract
Protein O-GlcNAcylation is a post-translational modification coupled to cellular metabolic plasticity. Aberrant O-GlcNAcylation has been observed in many cancers including endometrial cancer (EC), a common malignancy in women. However, clinical characterization of dysregulated O-GlcNAcylation homeostasis in EC and interrogating its molecular mechanism remain incomplete. Here we report that O-GlcNAcylation level is positively correlated with EC histologic grade in a Chinese cohort containing 219 tumors, validated in The Cancer Genome Atlas dataset. Increasing O-GlcNAcylation in patient-derived endometrial epithelial organoids promotes proliferation and stem-like cell properties, whereas decreasing O-GlcNAcylation limits the growth of endometrial cancer organoids. CRISPR screen and biochemical characterization reveal that tumor suppressor F-box only protein 31 (FBXO31) regulates O-GlcNAcylation homeostasis in EC by ubiquitinating the O-GlcNAc transferase OGT. Downregulation of O-GlcNAcylation impedes EC tumor formation in mouse models. Collectively, our study highlights O-GlcNAcylation as a useful stratification marker and a therapeutic vulnerability for the advanced, poorly differentiated EC cases.
Similar content being viewed by others


Introduction
Post-translational modifications (PTMs) endow the proteome with functional plasticity to cope with intrinsic and extrinsic perturbations under various developmental and disease conditions. Protein O-GlcNAcylation, catalyzed by a pair of evolutionarily conserved enzymes O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), is a PTM involving the covalent addition of single O-linked N-acetylglucosamine (O-GlcNAc) modification to serine or threonine residue of intracellular proteins1. The GlcNAc moieties are supplied by a metabolite uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), whose synthesis via the hexosamine biosynthesis pathway (HBP) requires fructose-6-P, acetyl-CoA, glutamine, and UTP, substrates from all major cellular metabolic processes. As a result, O-GlcNAcylation is sensitive to nutrient availability and intrinsic metabolic reprogramming. Meanwhile, O-GlcNAcylation is highly responsive to a wide range of extrinsic stimuli, including osmotic, oxidative, hyperthermic, and genotoxic stresses1,2,3, making it an important cellular stress sensing mechanism. O-GlcNAcylation is required for the maintenance of pluripotency of embryonic stem cells (ESCs). Its level declines as ESCs differentiate, alongside the cellular metabolic switch from glycolysis to oxidative phosphorylation4. Cancer cells often hijack embryonic programs to support their cell fate transition and abnormal proliferation, adopting a metabolic lifestyle relying on aerobic glycolysis (Warburg effect). Altered O-GlcNAcylation has been observed in cell lines of many cancers5,6,7,8, probably as a result of their increased nutrient consumption, or imbalanced enzymatic activity of OGT and OGA due to somatic mutations or altered protein stability9,10,11,12,13. To date, systemic assessment of O-GlcNAcylation level in major cancer cohorts and functional dissection of its homeostasis in patient-derived organoids haven’t been conducted.
Endometrial cancer (EC), the incidence of which has increased over 50% during the past two decades, is the most common cancers within the female reproductive system in developed countries14. In China, as of 2022, there were ~77,700 newly diagnosed EC cases and 13,500 estimated EC cancer deaths15. EC comprises a panel of tumors that are clinically and biologically heterogeneous, with obesity and conditions associated with metabolic syndrome such as diabetes being its risk factors16. It can be grouped into type I or type II tumors according to the clinical and endocrine features17, or classified as endometrioid carcinoma, serous carcinoma, carcinosarcoma, or clear-cell carcinoma based on its histopathological characteristics18. The Cancer Genome Atlas Research Network (TCGA) study of uterine corpus endometrial carcinoma (UCEC) has established a more precise genomic classification including four molecular subtypes: POLE-mutated, microsatellite instable (MSI), copy-number low, and copy-number high tumors19. More recently, integration of proteomic analysis to the genomic classification has accelerated the identification of clinically actionable molecular targets in EC20,21. Yet, PTMs, which add tremendous functional complexity to the proteome, remain to be comprehensively characterized in EC samples and complemented into the current classification system. O-GlcNAcylation as an important PTM responsive to cellular metabolism and stress has been linked to the molecular etiology of EC. Both OGT and OGA manifested highest alterations, mainly gene mutation and amplification, in EC among female reproductive cancer types22. The mRNA levels of both OGT and OGA were increased in EC samples of higher histologic grade23. More recently, elevated O-GlcNAcylation level in EC tissues was observed using a small tissue microarray24. O-GlcNAcylation was reported to promote proliferation, migration, and epithelial-mesenchymal transition (EMT) in cultured EC cell lines by regulating Wnt/β-catenin and Hippo-YAP signaling pathways22,24,25,26. These observations suggest that altered O-GlcNAcylation may contribute to EC progression, and it is worthy of thorough interrogation in large EC cohorts to determine whether O-GlcNAcylation can be utilized both as a stratification factor and a potential druggable target.
In this study, utilizing a Chinese EC cohort containing 219 tumors and the TCGA UCEC dataset, we uncover that O-GlcNAcylation level correlates with histologic grade, International Federation of Gynecology and Obstetrics (FIGO) stage, and patients’ prognosis. Moreover, we experimentally demonstrate that upregulation of O-GlcNAcylation promotes proliferation and stem-like cell properties in non-cancerous endometrial epithelial organoids (EE-Os), whereas downregulation of O-GlcNAcylation impedes the proliferation of endometrial cancer organoids (EC-Os). Furthermore, we identify FBXO31 as a key regulator of O-GlcNAcylation homeostasis, by controlling the ubiquitin-dependent protein degradation of OGT. Using subcutaneous xenograft mouse models, we show that treatment with small molecular inhibitor targeting OGT restrains tumor formation of EC cells. Our findings highlight that O-GlcNAcylation is a useful factor complementary to the current classification system to better stratify EC patients, and targeting the dysregulated O-GlcNAcylation homeostasis is a promising differentiation therapeutic strategy worthy of clinical exploitation for high grade EC patients.
Results
Elevated O-GlcNAcylation level is positively correlated with histologic grade and poor prognosis in endometrial cancer
To get a glimpse of global O-GlcNAcylation level in EC tissues, we obtained an EC tissue array from Xinchao Biotech (Shanghai, China) containing 23 peritumoral and 31 tumoral endometrial specimens, and performed immunohistochemistry (IHC) analyses with the anti-O-GlcNAc monoclonal antibody RL2, which was raised against the nuclear pore complex-lamina fraction of rat liver and is widely used to detect O-GlcNAcylation in a broad range of species in different applications24,27,28,29,30,31,32,33,34, as well as the antibodies of OGT and OGA (Fig. 1a). The amount of O-GlcNAcylation and OGT expression were significantly higher in epithelial cells of EC tissues relative to the control (Fig. 1b–e), consistent with a previous report24. The expression of OGA however showed no significant difference between peritumoral and tumoral endometrial tissues (Fig. s1a, b).

a A flowchart illustrating the process of clinical sample selection, data collection, and analysis. All samples were derived from patients receiving their initial treatment, and none of the patients had concurrent or previous tumors. Paraffin-embedded (FFPE); Immunohistochemistry (IHC). b, c Representative images depicting IHC staining of O-GlcNAcylation (RL2) and OGT in EC tumoral and peritumoral tissues in the FFPE tissue array. O-GlcNAcylation (RL2) and OGT immunostaining were intense in the glandular epithelium of the tumor. Scale bars: 50 µm. d, e Quantitative analysis of the levels of O-GlcNAcylation (RL2) and OGT in the EC tissue arrays. The levels of O-GlcNAcylation and OGT were assessed semi-quantitatively based on both the intensity and area of the staining. The product of proportion and intensity score was used as the final IHC score (0–12). Tumoral tissue (n = 31); peritumoral tissue (n = 23). The results are presented as mean ± SD. Statistical significance was calculated using unpaired two-tailed Student’s t-test. f Representative images of IHC staining showing varying levels of O-GlcNAcylation in serial sections of EC tissues of different histologic grades (well differentiated G1, moderately differentiated G2, and poorly differentiated G3. G1, n = 71; G2, n = 106; G3, n = 42). Scale bar: 50 µm. g Percentage of samples with high or low level of O-GlcNAcylation in different histologic grade groups. High and low categories were determined using a scoring system (high score: 8–12; low score: 0–6). (G1, n = 71; G2, n = 106; G3, n = 42). Statistical significance between groups was calculated using two-sided Fisher’s exact test. h, i Kaplan–Meier survival curves of PFS and OS of the EC patients stratified by the level of O-GlcNAcylation derived from their IHC scores. (Patients in high-RL2 group, n = 89; Patients in low-RL2 group, n = 115). Statistical significance was determined by the log-rank test. The source data for (d-e, g, h, i) are provided in the Source Data file.
To elaborate the relationship between the O-GlcNAcylation level and clinical characteristics of EC, we expanded the analyses to an EC cohort containing 219 tumor patients who received hysterectomy in the Department of Gynecology, Xiangya Hospital, Central South University (Fig. 1a). The paraffin-embedded EC tissue sections were subjected to IHC analysis, and the O-GlcNAcylation level revealed by the RL2 antibody staining for each specimen was semi-quantified to categorize the patients. Specifically, the IHC results were quantified by two independent assessors, and confirmed by a pathologist, based on both the proportion of positively stained tumor cells which was assessed by a value of 0–4 (0: negative; 1: 1–25%; 2: 26–50%; 3: 51–75%; 4: 76–100%), and the intensity of the staining which was scored using a value of 0 to 3 (0: negative; 1: weak; 2: medium; 3: strong). The product of the proportion and the intensity values was used as the final IHC score for each sample35,36,37. The patients were then divided into high O-GlcNAcylation (High-RL2) and low O-GlcNAcylation (Low-RL2) groups according to their IHC scores (High-RL2: 8–12; Low-RL2: 0–6). This high- and low-O-GlcNAcylation status exhibited significant association with histologic grade, FIGO stage, and distant metastasis of EC (Supplementary table 1). Consistently, the level of O-GlcNAcylation manifested a marked increase in EC tissues from patients with more advanced histologic grade (Fig. 1f), and the high O-GlcNAcylation cases were significantly enriched in the histologic grade 3 (G3) patients’ group (Fig. 1g). Further statistical analysis established a positive correlation between the O-GlcNAcylation level and tumor histologic grade (Goodman-Kruskal gamma statistic p ≤ 0.0001; 2-sided gamma-knife gamma = 0.473), as well as distant metastasis (Goodman-Kruskal gamma statistic p = 0.003; 2-sided gamma-knife gamma = 1). Kaplan-Meier analysis indicated that patients in the high O-GlcNAcylation group exhibited significantly shorter progression-free survival (PFS) and overall survival (OS) than that in the low O-GlcNAcylation group (Fig. 1h, i). Univariate analysis revealed that O-GlcNAcylation level, alongside age, FIGO stage, and myometrial invasion, was significantly associated with PFS. Subsequent multivariate Cox regression analysis using all the statistically significant variables (p < 0.05) identified O-GlcNAcylation level and age as independent predictors of the clinical outcome of EC patients (Supplementary table 2).
Calculated virtual O-GlcNAc index is correlated with tumor histologic grade and survival in TCGA endometrial cancer dataset
We wanted to validate the correlations observed in our EC cohort using the TCGA UCEC dataset. Kaplan-Meier analysis of the OS based on either OGT or OGA expression showed no statistical difference (Fig. s1c, d), suggesting that the mRNA abundance of OGT or OGA alone is insufficient to reflect the O-GlcNAcylation level and the amount of OGT protein in EC might be regulated translationally or post-translationally. To better estimate the O-GlcNAcylation level, we sent 40 high O-GlcNAcylation and 15 low O-GlcNAcylation frozen EC samples according to their corresponding RL2 IHC scores for RNA-seq (Fig. s1e). Gene set enrichment analysis (GSEA) revealed that the high O-GlcNAcylation group was enriched for expression of genes involved in EMT and angiogenesis (Fig. s1f, g, Supplementary data 1). Next, we calculated the Pearson’s correlation coefficient (r) of the transcripts level of each gene with the O-GlcNAcylation IHC scores, and included the top 1000 genes with r > 0.3 in the O-GlcNAcylation correlated gene set (Supplementary data 2). Gene ontology (GO) analysis revealed that they were highly enriched in biological processes related to cilium (Fig. s1h, Supplementary data 3). This result was in agreement with the observed correlation between O-GlcNAcylation level and the histologic grade of EC, because multiciliogenesis is a marker of differentiation of endometrial epithelial cells38.
We subsequently constructed a mathematical model based on the expression matrix of the O-GlcNAcylation correlated gene set using machine learning algorithms in R to calculate a virtual O-GlcNAc index for each sample in the TCGA UCEC cohort (Fig. s1e, Supplementary data 4). The calculated O-GlcNAc index in the TCGA dataset exhibited a significant association with histologic grade and FIGO stage (Fig. 2a, Supplementary data 5), consolidating the observations made in our EC cohort. Patients in the advanced histologic grade G3 group had a higher O-GlcNAc index in comparison to that in the grade 1 (G1) or grade 2 (G2) group (Fig. 2b). Similarly, EC patients at FIGO stages II, III, or IV demonstrated an increased O-GlcNAc index than that at stage I (Fig. 2c). Of note, analysis of the relationship between the O-GlcNAc index and EC molecular subtypes revealed that patients of the copy-number high molecular subtype, which had the worst clinical outcome among all EC cases39, exhibited a significantly higher O-GlcNAc index than that of other molecular subtypes (Fig. 2d). The O-GlcNAc index also increased with age, an independent predictor of the clinical outcome of EC patients (Fig. 2e). We further stratified the EC patients in the TCGA cohort into high O-GlcNAcylation and low O-GlcNAcylation groups using the median O-GlcNAc index as the cutoff. Patients from the high O-GlcNAcylation group experienced significantly shorter progression-free interval (PFI) and OS than that from the low O-GlcNAcylation group (Fig. 2f, g).

a Heatmap displaying the expression profiles of the 1000 O-GlcNAcylation correlated genes in the TCGA UCEC RNA-seq dataset (n = 589). The EC samples are annotated by clinical parameters, including Body Mass Index (BMI), menopause status, diabetes, histologic grades, molecular subtypes (integrative cluster), International Federation of Gynecology and Obstetrics (FIGO) stage, age, and primary diagnosis. Patients were categorized into O-GlcNAcylation high or O-GlcNAcylation low group using the median of the calculated O-GlcNAc index as the threshold. The symbol (*) indicates a statistically significant difference of the calculated O-GlcNAc index among the patients’ groups according to the indicated clinical parameter. Statistical significance was determined by two-sided Wilcoxon test, **p < 0.01, ****p < 0.0001. b–e The O-GlcNAc index in different EC groups stratified by histologic grade, FIGO stage, integrative cluster, or age in the TCGA UCEC dataset. For histologic grade (b): G1 (n = 99), G2 (n = 119), and G3 (n = 324). For FIGO stage (c): Stage I (n = 335), Stage II (n = 51), Stage III (n = 127), and Stage IV (n = 29). For integrative clusters (d): POLE (n = 17), copy number low (n = 90), microsatellite unstable (n = 65), and copy number high (n = 61). For age (e), n = 177 and 362. The box bounds the interquartile range divided by the median, with the whiskers extending to a maximum of 1.5 times the interquartile range beyond the box. Outliers are shown as dots. Statistical significance was determined by two-sided Wilcoxon test. f, g Kaplan–Meier survival curves for Progression-free interval (PFI) and OS of EC groups with high or low O-GlcNAc index in the TCGA UCEC dataset. Statistical significance was determined by the log-rank test.
In summary, the elevated O-GlcNAcylation level in EC tissues is correlated with more advanced histologic grade and poorer clinical outcome of the patients, both in our EC cohort and the TCGA UCEC dataset.
Generation of endometrial organoids that recapitulate molecular properties of non-cancerous and cancerous endometrial epithelia
Endometrial organoids mirror many molecular and functional traits of the in vivo endometrial tissues, manifesting glandular self-organization, apicobasal polarity, mucus production, and responsiveness to sex hormones40,41. To dissect the functional impact of altered O-GlcNAcylation level on endometrial tissues, we generated endometrial organoids from biopsy or surgical samples, including three eutopic endometrial epithelial organoids (EE-Os) from patients with endometriosis as non-cancerous controls and three endometrial cancer organoids (EC-Os) (Supplementary data 6).
The EE-Os were usually monocystic, with well-polarized epithelial cells forming hollow spheres in the three-dimensional (3D) extracellular matrix (Fig. s2a). IHC analysis showed that the EE-Os retained many characteristics of endometrial epithelium, including production of mucins, expressions of estrogen receptor (ER) and progesterone receptor (PR) (Fig. s2b). The EC-Os however manifested more irregular cell organizations, often with dense and polycystic phenotypes (Fig. s2a). The EC-Os faithfully reflected the histopathological characteristics of their primary EC tissues, such as the presence or absence of P53 and FOXA2, expressions of ER and PR (Fig. s2c). Additionally, the EC-Os demonstrated substantial proliferative activity as indicated by the Ki-67 staining (Fig. s2c). We performed IHC analysis with RL2 antibody on the paraffin-embedded endometrial organoids and the matched primary tissues, and confirmed that the O-GlcNAcylation status remained unchanged (Fig. s2d). In accordance with their primary tissues, the EC-Os manifested significantly higher O-GlcNAcylation level than the EE-Os (Fig. s2e).
Increase of O-GlcNAcylation by inhibition of OGA promotes proliferation and stemness in non-cancerous endometrial epithelial organoids
Small molecular inhibitors have been developed to modulate the activities of O-GlcNAc cycling enzymes OGT and OGA, and are widely used to dissect the functions of O-GlcNAcylation in vitro and in vivo42,43,44,45,46,47,48,49,50,51,52. We treated the EE-Os with the OGA inhibitor Thiamet-G (TMG) to increase the cellular O-GlcNAcylation level (Fig. 3a, b). The addition of TMG resulted in enhanced colony formation and organoid growth of EE-Os (Fig. 3c, d), along with a rise in the number of mitotic cells within each EE-O (Fig. 3e, f). Acetylated alpha-tubulin (Ac-tubulin) and PAEP are differentiation markers for multiciliated epithelial cells and secretory cells respectively in the endometrium41. TMG treatment reduced the number of both PAEP positive cells and Ac-tubulin labeled multiciliated cells (Fig. 3g, h), suggesting that the elevated O-GlcNAcylation level caused de-differentiation of the endometrial cells in the EE-Os. We further examined the expression levels of a panel of stemness markers of the endometrium, including SSEA-1, SOX9, ALDH1, OCT4, CD133, and SOX2. In contrast to PAEP whose mRNA level was decreased upon TMG treatment, all the examined stemness markers showed upregulated expressions (Fig. 3i). Given that the GSEA results suggested that high O-GlcNAcylation could promote EMT and angiogenesis (Fig. s1f, g), we also examined the expression of genes involved in these two processes in the TMG-treated EE-Os. While the expression of two epithelial markers E-cadherin and ZO-1 showed no difference, four out of six of the mesenchymal markers analyzed, namely, FN-1, Snail1, TWIST2, and MMP1, exhibited elevated expression after TMG treatment (Fig. s3a). Among the analyzed angiogenesis-related genes, increase of O-GlcNAcylation in EE-Os by TMG treatment upregulated the expression of PDGFA and VEGFC. Yet, the expression of the rest of the angiogenesis genes was unaltered or downregulated (Fig. s3b). These findings suggest that elevated O-GlcNAcylation can promote de-differentiation of endometrial epithelial cells, as well as their EMT and angiogenesis capacities to varying degrees.

a Bright-field images of endometrial epithelial organoids (EE-Os) depicting responses to Thiamet G (TMG) at day 1 and day 3. Representative images from control dimethyl sulfoxide (DMSO) and 10 µM TMG treated EE-O groups are presented. Scale bar: 50 µm. b Immunoblot with RL2 antibody assessing O-GlcNAcylation level in EE-Os treated with DMSO, 5 µM, or 10 µM TMG for 48 h. Actin was used as the loading control. c Comparison of the EE-O numbers at day 3 of culture after 10 µM TMG or DMSO treatment. d Measurement of cross-sectional area of EE-Os at day 3 after treatment with 10 µM TMG versus DMSO. (Organoids derived from 6 biological replicates, n = 94 and 165 organoids). e Representative immunofluorescence images of control and TMG-treated EE-Os stained with PH3 (red), Tubulin (green), DAPI (blue), and F-actin labeled by Phalloidin (magenta). Scale bar: 5 µm. f Quantification of phospho-histone H3 (PH3) positive cells in each EE-O. (n = 31 and 22 organoids). g Representative immunofluorescence images of control and TMG-treated EE-Os. Ciliated epithelium is labeled by acetylated alpha-tubulin (Ac-tubulin, green), secretory cells by PAEP (red), DAPI (blue), and F-actin (magenta). Scale bar: 50 µm. Insets show magnification of the area in the white box, scale bar: 5 µm. h Quantification of the number of ciliated cells (Ac-tubulin + ) in each EE-O (n = 23 and 25 organoids). i qPCR analysis of stemness markers’ expression in EE-Os treated with TMG or DMSO, normalized to actin mRNA level. j Minimum-Distortion Embedding (MDE) projection of scRNA-seq data of DMSO and TMG treated EE-Os. k Subclustered epithelial populations of EE-Os (left), and the proportion of each subcluster in control and TMG-treated groups (right). Results in (a, b) show a representative example from n = 3 independent experiments. Results in (c, d) were derived from n = 6 biologically independent experiments, and results in (f–i) were derived from n = 3 biologically independent experiments, with p-values calculated by unpaired two-tailed Student’s t-test and data presented as mean ± SD. The source data for (b–d, f, h, i, k) are provided in the Source Data file.
To further characterize the influence of TMG treatment on different cell subtypes in the EE-Os, the control and TMG treated EE-Os were subject to single-cell RNA-seq analysis (Fig. 3j). The cells were clustered and classified into six major subtypes according to the specific expression of known markers38,53: pre-ciliated, ciliated, stem, proliferative, O-GlcNAc-related stem-like, and inflammatory (Fig. s3c-f, Supplementary data 7). Of note, we identified an O-GlcNAc-related stem-like subtype in which the cells displayed activated signaling pathways regulating the pluripotency of stem cells, as well as the O-glycan biosynthesis (Fig. s3d). The TMG treatment of EE-Os resulted in a substantial decrease of cells in the ciliated and pre-ciliated subtypes, and a concurrent increase of cells in the proliferative and O-GlcNAc-related stem-like subtypes (Fig. 3k). This result supports that upregulation of O-GlcNAcylation level promotes proliferation and stemness of endometrial epithelial cells.
For comparison, we treated the EE-Os with the OGT inhibitor OSMI-1 to analyze the effects of downregulation of O-GlcNAcylation on the non-cancerous endometrial epithelial cells. OSMI-1 treatment only mildly impacted the growth of EE-Os, and the number of organoids formed was unaffected (Fig. s4a–c). TUNEL staining revealed no significant apoptosis in the OSMI-1 treated EE-Os, and the number of differentiated cells with multiple cilia remained comparable (Fig. s4d, e). We also examined the expression of the marker genes for stemness, EMT, and angiogenesis in the EE-Os treated with OSMI-1, and no concordant changes were observed (Fig. s4f–h). These results suggest that inhibition of OGT only has minimal influence on the cells in EE-Os.
Decrease of O-GlcNAcylation by inhibition of OGT impedes proliferation and induces differentiation and cell death in endometrial cancer organoids
To investigate whether a decrease of O-GlcNAcylation can inhibit the growth of tumor cells in EC-Os, we treated the EC-Os with OSMI-1, a chemical inhibitor of OGT (Fig. 4a, b). The addition of OSMI-1 impeded the formation and growth of EC-Os. A significant fraction of the EC-Os displayed darkening and cell lysing in the presence of OSMI-1, resulting in reductions in both the number and size of the EC-Os compared to time-matched control (Fig. 4a–d). TUNEL staining revealed that many cells in the OSMI-1 treated EC-Os underwent apoptosis (Fig. 4e). We performed immunofluorescence on the remaining EC-Os with relatively normal size and morphology. Mitotic cells as visualized by phospho-histone H3 (PH3) staining became barely detectable in EC-Os after OSMI-1 treatment (Fig. 4f, g). Meanwhile, the population of both the PAEP positive secretory cells and Ac-tubulin labeled multiciliated cells increased in these EC-Os (Fig. 4h, i), suggesting that OSMI-1 treatment promoted differentiation. Consistently, the expression of stemness markers, including SSEA-1, SOX9, ALDH1, OCT4, CD133, and SOX2, was significantly downregulated in the OSMI-1 treated EC-Os, accompanying the upregulation of the differentiation marker PAEP (Fig. 4j). The expression of all the mesenchymal markers, including FN-1, Vimentin, Snail1, TWIST2, TGFB1, and MMP1, was downregulated (Fig. 4k). Many of the angiogenesis markers also manifested reduced expression in the EC-Os treated with OSMI-1 (Fig. 4l).

a Representative bright-field images of endometrial cancer organoids (EC-Os) treated with 50 µM OSMI-1 at day 1 and day 3. Scale bar: 50 µm. b Immunoblot assessing O-GlcNAcylation level in EC-Os treated with 25 µM or 50 µM OSMI-1 for 48 h. Actin was used as the loading control. c Comparison of the numbers of EC-Os at day 3 after treatment with OSMI-1 versus the control DMSO. d Cross-sectional area of EC-Os at day 3 after OSMI-1 treatment compared to control (DMSO). (n = 101 and 57 organoids). e TUNEL staining showing apoptotic cells in EC-Os after 50 µM OSMI-1 treatment. Nuclei are visualized with DAPI (blue). Scale bar: 50 µm. f Representative immunofluorescence images of control and OSMI-1 treated EC-Os. Mitotic cells (PH3, red), tubulin (green), DAPI-labeled nuclei (blue), and Phalloidin labeled F-actin (magenta). Scale bar: 10 µm. g Quantification of the number of PH3+ cells. (n = 42 and 46 organoids). h Representative immunofluorescence images of control and OSMI-1 treated EC-Os showing ciliated epithelial cells (Ac-tubulin, green), secretory cells (PAEP, red), DAPI (blue), and F-actin (magenta). Scale bar: 50 µm. Insets show magnification of the area in the white box, scale bar: 5 µm. i Quantification of the number of Ac-tubulin+ cells. (n = 40 and 28 organoids). (j–l) qPCR analyses of stemness (j), EMT (k), and angiogenesis (l) markers in EC-Os treated with OSMI-1 or DMSO, normalized to actin mRNA. m Measurement of cell viability in patients-derived endometrial organoids with the indicated inhibitors. RLU represents relative light units. 5 replicates per each patient-derived organoid. Results in (a, b) show a representative example from n = 3 independent experiments. Results in (c, d) represent n = 6 biologically independent experiments, and results in (g, i-l) represent n = 3 biologically independent experiments, with p-values calculated by unpaired two-tailed Student’s t-test and data presented as mean ± SD. The source data for (b–d, g, i–m) are provided in the Source Data file.
To ascertain the effects observed after OSMI-1 treatment in the EC-Os was on-target, we directly knocked down the expression of OGT using short hairpin RNAs (shRNA) and repeated the analyses. Similar to the observations made with OSMI-1, knockdown of OGT by shRNA also downregulated O-GlcNAcylation level and significantly inhibited the formation and growth of EC-Os (Fig. s5a–d). The cells in EC-Os after OGT knockdown exhibited increased apoptosis (Fig. s5e), and the remaining ones were often multiciliated (Fig. s5f, g), with upregulated expression of the differentiation marker PAEP and downregulated expression of the stemness genes (Fig. s5h). These results suggest that a decrease of O-GlcNAcylation level induced by different means invariably leads to growth limitation and enhanced differentiation and apoptosis of tumor cells in EC-Os.
We also treated the EC-Os with the OGA inhibitor TMG to check if there was a tumor-promoting effect. TMG treatment led to a marked increase in the number and the size of EC-Os (Fig. s5i–k). The expression of stemness genes such as SOX9, ALDH1, CD133, and SOX2 was also upregulated, while the differentiation marker PAEP was downregulated (Fig. s5l). Additionally, all the mesenchymal markers and many of the angiogenesis genes manifested increased expression in the TMG treated EC-Os (Fig. s5m, n), suggesting that inhibition of OGA further enhanced malignancy.
The experiments with OGT and OGA inhibitors in the endometrial organoids showed that the EE-Os were less sensitive to the downregulation of O-GlcNAcylation than the EC-Os. To validate this, we conducted a 3D cell viability assay using EC-Os from the two EC patients with relatively high O-GlcNAcylation level, as well as two EE-Os from non-EC patients. In addition to OSMI-1, we also included its derivative, OSMI-454, in the assay. While treatment with TMG promoted the proliferation of both EE-Os and EC-Os, inhibition of OGT by OSMI-1 or OSMI-4 demonstrated a reduction of viability in EC-Os than the control EE-Os (Fig. 4m). These findings indicate that OGT inhibitors can effectively restrain the expansion of tumor organoids in vitro, particularly that with an inherently high O-GlcNAcylation level.
Genome-wide screen for tumor suppressors that maintain O-GlcNAcylation homeostasis
To identify crucial factors regulating O-GlcNAcylation homeostasis in EC, we conducted a comprehensive genome-wide CRISPR-Cas9 knockout screen. A lentiviral single guide RNA (sgRNA) library targeting 19,050 genes (6 sgRNAs/gene) was transduced into 293T cells, along with 1000 nontargeting control sgRNAs, at a multiplicity of infection (MOI) of 0.3 to ensure each cell expressed only one sgRNA. Following cell staining with the anti-O-GlcNAc antibody RL2, we isolated the top 5% RL2-positive cells via fluorescence-activated cell sorting (FACS) and conducted deep sequencing of the sgRNAs from this cell population (Fig. 5a, b and Fig. s6a). The sgRNA abundance was then used to calculate a robust rank aggregation (RRA) score for each gene using MAGeCK55, and the genes were ranked accordingly, with a smaller RRA score indicated greater essentiality (Supplementary Data 8). We reviewed the literatures and collected known regulators whose inactivation could impact cellular O-GlcNAcylation homeostasis56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71. Genes that negatively regulate O-GlcNAcylation, such as TSC2, SIRT1, and TP53, had smaller RRA scores and were enriched in the first half of the gene list, comparing to the known positive regulators of O-GlcNAcylation (Fig. 5c). We performed Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis on the 1038 high-confidence genes (p < 0.05) from the genome-wide screen. These genes were enriched in pathways including ECM-receptor interaction, thermogenesis, histidine metabolism, proteoglycan in cancer, and maturity onset diabetes of the young (Fig. 5d, Supplementary Data 9).

a Schematic representation of the FACS-based genome-wide CRISPR-Cas9 screen for putative regulators of O-GlcNAcylation homeostasis. Fluorescence-Activated Cell Sorting (FACS); Immunofluorescence (IF); CRISPR: Clustered Regularly Interspaced Short Palindromic Repeats; Human genome-wide CRISPR/Cas9 knockout (GeCKO). The elements in this figure were created using BioGDP.com (https://BioGDP.com). b Validation of the sensitivity of RL2 staining (red) with 293T cells transfected with OGT (green). Nuclei are labeled with DAPI (blue). Scale bar: 5 µm. c Genes plotted according to their relative ranking analysis (RRA) enrichment scores, with known O-GlcNAcylation regulators highlighted in red and blue. Knockdown (KD). d Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showing enrichment of putative O-GlcNAcylation regulators in the indicated pathways (statistical analysis was performed using a hypergeometric test to calculate p-values). Analysis was performed on the 1038 high-confidence genes (p < 0.05). e Venn diagram showing the overlap between the 526 human UCEC Tumor Suppressor Genes (TSGs) and the 1038 high-confidence genes from the O-GlcNAcylation screen. Source data are provided in the Source Data file. f Immunofluorescent detection of O-GlcNAcylation level by RL2 (red) in WT (Wild Type) and FBXO31-KO (FBXO31-Knockout) 293T cells. Nuclei were stained with DAPI (blue). Scale bar: 5 µm. g Kaplan-Meier analysis of the OS of the EC patients stratified by the expression level of FBXO31 (http://kmplot.com/analysis/). EC cases were stratified using the median cut-off, and statistical significance was determined using the log-rank test. Results in (b, f) show a representative example from n = 3 independent experiments.
To further pinpoint key regulators that impact O-GlcNAcylation level in EC tissues, we cross-referenced the 1038 positive hits in the screen with 526 putative tumor suppressor genes of EC72,73. As a result, 18 overlapping genes were identified, including ACVR1C, AGTR1, CADM2, PRKAA1, CDKN1C, CMTM3, DIRAS3, SIK1, EPHB4, GATA5, ITGAV, KLF10, MAP3K8, PLA2G2A, PTPN11, RNASEL, SPARCL1, and FBXO31 (Fig. 5e). We conducted Kaplan-Meier analysis using the TCGA UCEC dataset for each gene, and found that only FBXO31 showed downregulated expression in EC that was associated with poor survival (Figs. s6b and 5g). Therefore, we generated FBXO31 knockout (FBXO31-KO) 293T cells using CRISPR (Fig. s6c). Immunostaining with RL2 antibody confirmed that the O-GlcNAcylation level was significantly increased in the FBXO31-KO cells (Fig. 5f).
FBXO31 interacts with and ubiquitinates OGT to limit the O-GlcNAcylation level
FBXO31 functions as a substrate recognition component in the SCF ubiquitin E3 ligase complex to control the degradation of many proteins74,75,76,77,78,79. Accordingly, FBXO31 might regulate O-GlcNAcylation level by directly binding and ubiquitinating the O-GlcNAc transferase OGT. To confirm the interaction between FBXO31 and OGT, we performed a pull-down assay using bacterially purified GST-OGT to incubate with lysates of 293T cells expressing GFP-FBXO31. Western blot showed that GST-OGT pulled down a significant amount of GFP-FBXO31 relative to GST control (Fig. 6a). We further validated the interaction using co-immunoprecipitation in 293T cells overexpressing Flag-OGT and GFP-FBXO31. GFP-FBXO31 was co-immunoprecipitated with Flag-OGT, and both the amounts of Flag-OGT and GFP-FBXO31 in the immunoprecipitant were increased in the presence of the proteasome inhibitor MG132 (Fig. 6b).

a Immobilized recombinant GST-OGT protein but not GST control absorbed GFP-FBXO31 from 293T cell lysates. GST and GST-OGT were detected by Coomassie brilliant blue (CBB) staining, and FBXO31 was detected by western blotting with FBXO31 antibody. b Co-immunoprecipitation of GFP-FBXO31 with Flag-OGT in 293T cell lysates. The presence of MG132 enhanced the interaction between Flag-OGT and GFP-FBXO31. c Western blotting assessing the protein level of OGT as well as the global O-GlcNAcylation (RL2) level in 293T cells transfected with increasing amount of GFP-FBXO31. d Western blotting quantification of the protein level of endogenous OGT in 293T cells transfected with GFP-FBXO31. MG132 was added to inhibit the ubiquitination-mediated proteasome degradation. e Western blotting detecting the protein level of endogenous OGT and its ubiquitination in 293T cells transfected with different amount of HA-Ub and GFP-FBXO31. f In vitro ubiquitination of His-OGT by the SCF complex together with FBXO31. HA-tagged SCF components (Skp1, Cul1, and Roc1) and HA-FBXO31 were affinity-purified using anti-HA-conjugated magnetic beads from 293T cell lysates. The purified protein complex was incubated with E1 (UBA1), E2 (UBE2D1), Ub, and His-OGT in ubiquitination buffer. The reaction was halted by the addition of SDS sample buffer, and the samples were subjected to western blotting using the indicated antibodies. g In vivo ubiquitination assay was performed to evaluate the ubiquitination levels of exogenous Flag-OGT in 293T cells transfected with HA-tagged Ub and GFP-FBXO31 or its F-box domain deletion mutant GFP-FBXO31ΔF. h Western blotting detecting the O-GlcNAcylation (RL2) and OGT levels in WT and FBXO31-KO 293T cells. i Western blotting quantitation of OGT protein level following cycloheximide (CHX) treatment in WT and FBXO31-KO 293T cells. Results in (d, i) represent n = 3 independent experiments, with p-values calculated by unpaired two-tailed Student’s t-test and data presented as mean ± SD. Samples derive from the same experiment and gels were processed in parallel. (a–c, e–h) show representative examples from n = 3 independent experiments. The source data for results in (a–i) are provided in the Source Data file.
To assess whether the interaction with FBXO31 controlled the protein homeostasis of OGT, we transfected 293T cells with increasing amounts of GFP-FBXO31 and detected the levels of OGT as well as O-GlcNAcylation by western blot. Both the OGT protein and cellular O-GlcNAcylation levels demonstrated a negative correlation with the amount of GFP-FBXO31 (Fig. 6c). Additionally, the downregulation of OGT induced by GFP-FBXO31 overexpression was significantly reversed by MG132, suggesting that FBXO31 controlled the OGT level via the ubiquitin-dependent proteasome degradation process (Fig. 6d). To ascertain that FBXO31 could induce ubiquitination of OGT, we immunoprecipitated OGT from 293T cell lysates overexpressing GFP-FBXO31 and HA-ubiquitin. Western blot detected strong polyubiquitination of OGT in the presence of GFP-FBXO31 (Fig. 6e). We further tested whether FBXO31 could ubiquitinate OGT in vitro. The SCF complex was affinity-purified with anti-HA magnetic beads from 293T cells expressing HA-tagged Skp1, Cul1, and Roc1 with or without FBXO31, and then incubated with bacterially purified E1, E2, ubiquitin, and His-OGT. Polyubiquitination signals of His-OGT were detected, suggesting that the FBXO31-containing SCF complex could directly ubiquitinate OGT (Fig. 6f). Skp1 in the SCF complex recruits F-box proteins via their F-box motif. We mutated the F-box of FBXO31 (FBXO31∆F) and assessed its ability to induce polyubiquitination of OGT in 293T cells. Overexpression of HA-ubiquitin and GFP-FBXO31 resulted in strong polyubiquitination of the immunoprecipitated Flag-OGT, which was significantly reduced when GFP-FBXO31 was replaced with the GFP-FBXO31∆F mutant (Fig. 6g). These results confirmed that FBXO31, together with other components of SCF complex, possessed a ubiquitin E3 ligase activity toward OGT. We further evaluated the impact of FBXO31 in controlling the cellular OGT homeostasis using FBXO31-KO 293T cells. Both the OGT and O-GlcNAcylation levels were increased in FBXO31-KO cells (Fig. 6h). Cycloheximide (CHX) treatment, which blocked new protein synthesis, uncovered that the half-life of OGT was significantly extended in FBXO31-KO cells relative to control (Fig. 6i), indicating that FBXO31 is indispensable for limiting the cellular OGT level.
We wanted to confirm if the observed regulation of OGT by FBXO31 also held true in endometrial cancer cells. To this end, we overexpressed GFP-FBXO31 in the Ishikawa cells. Immunofluorescent staining showed that both OGT and the O-GlcNAcylation level visualized by RL2 were markedly decreased in the GFP-FBXO31 positive cells (Fig. s7a). We subsequently generated FBXO31 knockout (FBXO31-KO) Ishikawa cells using CRISPR, and western blot indicated that OGT and O-GlcNAcylation were upregulated in these cells (Fig. s7b). Co-immunoprecipitation assay with endogenous FBXO31 and OGT in Ishikawa cells detected only weak interaction (Fig. s7c). However, this interaction became evident with exogenous GFP-FBXO31 and Flag-OGT that were overexpressed in Ishikawa cells (Fig. s7d). To test if FBXO31 could similarly mediate ubiquitination of OGT in Ishikawa cells, we immunoprecipitated OGT from the Ishikawa cell lysates overexpressing GFP-FBXO31 and HA-ubiquitin. Substantial polyubiquitination signal of OGT was detected in the presence of GFP-FBXO31 (Fig. s7e), confirming that FBXO31 could also control OGT protein level via the ubiquitin-dependent proteasome degradation process in endometrial cancer cells.
O-GlcNAcylation was reported to increase the stability of several target proteins, such as YAP, β-catenin, and c-Myc, therefore promoting tumor progression25,29,80. To test if the FBXO31-OGT regulatory axis impacted the homeostasis of these proteins in EC, we examined their protein levels in WT and FBXO31-KO Ishikawa cells. While YAP and β-catenin showed minimum changes, c-Myc was markedly increased in FBXO31-KO cells, and this increase was largely dependent on OGT, as knockdown of OGT could restore its level to that seen in WT (Fig. s7f). We further investigated whether c-Myc was a bona fide O-GlcNAcylated target in EC. To this end, c-Myc was immunoprecipitated from Ishikawa cell lysate, and its O-GlcNAc modification was detected by western blot with RL2 (Fig. s7g). The amount of O-GlcNAcylated c-Myc was significantly higher in the FBXO31-KO Ishikawa cells than in the WT (Fig. s7h). These results indicate that c-Myc is an important cellular target downstream of FBXO31 and OGT to promote EC progression.
Loss of FBXO31 promotes endometrial organoids growth by increasing O-GlcNAcylation
We next investigated the clinical relevance of the regulation of O-GlcNAcylation homeostasis by FBXO31 using the endometrial specimens in our EC cohort. IHC staining revealed that the protein level of FBXO31 was significantly downregulated in EC relative to normal endometrial tissues, often manifesting an anti-correlation pattern to that of O-GlcNAcylation (Fig. 7a, b). We semi-quantified the expression level of FBXO31 based on the IHC signals, and found that the FBXO31 protein level in the low O-GlcNAcylation EC group was markedly higher than that in the high O-GlcNAcylation group (Fig. 7c). We further analyzed the relationship between the calculated virtual O-GlcNAc index and the expression level of FBXO31 in the TCGA UCEC cohort, and observed a significant negative correlation (Fig. 7d). Western blot uncovered that the FBXO31 protein level was decreased, accompanying the increase of OGT level, in EC-Os comparing to the EE-Os (Fig. 7e), suggesting that the elevated O-GlcNAcylation in EC tissues was due to upregulation of OGT. We categorized the cases in our EC cohort into FBXO31-low and FBXO31-high groups. The FBXO31 expression exhibited significant association with the O-GlcNAcylation status and histologic grade (Supplementary Table 3).

a Representative IHC images of FBXO31 in EC and peritumoral tissues from an FFPE tissue array. Scale bar: 50 µm. b Quantitative analysis of FBXO31 levels in the EC tissue array. FBXO31 expression was semi-quantified based on staining intensity and area. Tumoral tissue (n = 31); peritumoral tissue (n = 23). Results are presented as mean ± SD. Statistical significance was calculated using unpaired two-tailed Student’s t-test. c Percentage of samples with high or low FBXO31 level by IHC in the two different O-GlcNAcylation level groups (Patients in high-RL2 group, n = 83; Low-RL2 group, n = 38). Statistical significance was calculated using two-sided Fisher’s exact test. d Spearman two-sided correlation analysis between the calculated virtual O-GlcNAc index and the expression of FBXO31 (Transcripts Per Million, TPM), n = 542. e Protein levels of OGT and FBXO31 were assessed by western blotting in EC-Os and EE-Os derived from different patients. f Immunofluorescence detection of O-GlcNAcylation (RL2, green) and FBXO31 (red) in control and shFBXO31 infected EE-Os. The nuclei were stained with DAPI (blue) and F-actin with Phalloidin (magenta). Scale bar: 50 µm. g qPCR analysis of stemness markers’ expression in control shNT and shFBXO31 infected EE-Os, normalized to actin mRNA level. h Quantification of organoid numbers of the control and shFBXO31 infected EE-Os after 3D culture. Representative bright-field images are provided on the left. Scale bar: 300 µm. i Quantification of organoid numbers in shFBXO31 treated EE-Os, with OSMI-1 or DMSO treatment at day 3. Representative bright-field images are shown on the left. Scale bar: 150 µm. j Quantification of organoid numbers of EC-Os overexpressing GFP or GFP-FBXO31. Bright-field and fluorescent images of the treated EC-Os are shown on the left. Scale bar: 50 µm. Results in (g) represent n = 3 biologically independent experiments, and results in (h–j) represent n = 6 biologically independent experiments, with p-values calculated by unpaired two-tailed Student’s t-test and data presented as mean ± SD. e, f show a representative example from n = 3 independent experiments. The source data for results in (b–e, g–j) are provided in the Source Data file.
To elucidate the functional impact of FBXO31 alterations in endometrial tissues, we knocked down the expression of FBXO31 using lentivirus-mediated expression of shRNAs in EE-Os. Downregulation of FBXO31 resulted in an increased amount of O-GlcNAcylation in EE-Os (Figs. 7f and s8a). Particularly, the growth of EE-Os was significantly enhanced by FBXO31 knockdown, in alignment with upregulated expression of the stemness markers SSEA-1, SOX9, ALDH1, OCT4, CD133, and SOX2 (Fig. 7g, h). This enhanced growth of EE-Os after FBXO31 knockdown could be inhibited by OSMI-1 treatment, indicating that it was a result of elevated O-GlcNAcylation (Fig. 7i). Reciprocally, given that FBXO31 was downregulated in EC-Os, we supplemented the EC-Os with GFP-FBXO31 or GFP control using lentivirus-mediated transduction. Overexpression of GFP-FBXO31 downregulated O-GlcNAcylation and significantly impeded the formation of EC-Os (Figs. 7j and s8b).
In summary, our results identify FBXO31 as one of the key rheostats that control the O-GlcNAcylation homeostasis by ubiquitinating OGT. FBXO31 is frequently downregulated in EC, resulting in stabilization of OGT and elevation of cellular O-GlcNAcylation level that advance endometrial malignancy.
Chemical inhibition of OGT limits endometrial tumor growth in mouse models
The in vitro characterization of the impact of O-GlcNAcylation on EC cells suggested that targeting OGT to decrease O-GlcNAcylation level is a promising therapeutic strategy. We therefore investigated the antitumor effects of the OGT inhibitor OSMI-1 in a xenograft mouse model using Ishikawa cells. Ten days after the subcutaneous transplantation of Ishikawa cells, tumor-bearing mice were randomly divided into three groups and administered either DMSO (vehicle solvent), TMG (20 mg/kg/day), or OSMI-1 (10 mg/kg/day) via intraperitoneal injection (Fig. 8a). The TMG treatment resulted in increased tumor growth and shortened lifespan, while OSMI-1 treatment reduced tumor volume compared to the control, and the mice exhibited increased survival (Fig. 8b, c). IHC staining of the dissected tumor tissues confirmed that OSMI-1 treatment decreased O-GlcNAcylation level compared to TMG or the control DMSO (Fig. 8d). Consistently, the expression of the differentiation marker PAEP in the tumor tissues was increased, and many of the stemness genes were mildly downregulated after OSMI-1 treatment (Fig. s8c). In contrast, TMG treatment resulted in downregulation of PAEP expression and upregulation of the stemness markers in the dissected tumor tissues (Fig. s8d).

a Schematic representation of the treatment schedule in the Ishikawa cells xenograft model. On day 10 after subcutaneous injection of EC cells, mice were treated daily with DMSO, TMG, or OSMI-1 for 15 days. Tumor growth and survival were monitored till the endpoint. The mouse elements in this figure were created using BioGDP.com (https://BioGDP.com). b Tumor growth curves of Ishikawa xenografts in different treatment groups as indicated (DMSO group, n = 8 mice; TMG group, n = 7 mice; OSMI−1 group, n = 8 mice). The results are presented as mean ± SEM. c Survival curves for mice bearing Ishikawa xenografts across different treatment groups. (DMSO group, n = 8 mice; TMG group, n = 7 mice; OSMI-1 group, n = 8 mice). Statistical significance was determined by log-rank test. d Representative Hematoxylin and Eosin (HE) and IHC staining of mouse tumor tissues from different treatment groups. Scale bar: 50 µm. e Schematic representation of the treatment schedule in the WT and FBXO31-KO Ishikawa cells xenograft model. On day 10 of tumor growth, mice with WT or FBXO31-KO cells xenografts received daily treatment with DMSO or OSMI-1 for 15 days. Tumor growth was assessed till the endpoint. The mouse elements in this figure were created using BioGDP.com (https://BioGDP.com). f Tumor growth curves of WT and FBXO31-KO Ishikawa cells xenografts in different treatment groups as indicated (WT DMSO control group, n = 10 mice; WT OSMI-1 treatment group, n = 9; FBXO31-KO DMSO group, n = 10 mice; FBXO31-KO OSMI-1 treatment group, n = 9 mice). The results are presented as mean ± SEM. Statistical significance was calculated using unpaired two-tailed Student’s t-test. g Photograph of the excised tumors from different treatment groups as indicated. h Immunofluorescence detection of CD31 (red) in tumor tissues from the indicated treatment groups. Nuclei were stained with DAPI (blue). Scale bar: 50 µm. i Quantitative analysis of CD31-positive blood vessel areas. The results are presented as mean ± SD. Statistical significance was calculated using unpaired two-tailed Student’s t-test, n = 6 mice. The source data for results in (b–i) are provided in the Source Data file.
Next, we investigated whether the deletion of FBXO31 promoted EC tumor formation in the mouse model and sensitized the tumors to OSMI-1 treatment. We subcutaneously injected WT and FBXO31-KO Ishikawa cells into nude mice and monitored tumor growth. Ten days after the transplantation, tumor-bearing mice were randomly divided into groups and administered either DMSO (vehicle solvent) or OSMI-1 (10 mg/kg/day) via intraperitoneal injection (Fig. 8e). The tumors formed by the FBXO31-KO Ishikawa cells grew much faster than that of the WT cells (Fig. 8f), and they upregulated the expression of many stemness genes such as SOX9, ALDH, OCT4, CD133, and SOX2 (Fig. s8f). OSMI-1 treatment decreased the O-GlcNAcylation level in these tumor tissues (Fig. s8e), downregulated the expression of the stemness genes (Fig. s8g), and significantly limited the tumors’ growth in both the WT and FBXO31-KO groups (Figs. 8f and s8e). Moreover, compared to the WT, the FBXO31-KO tumors showed increased sensitivity to the OSMI-1 treatment (Fig. 8f, g). In addition, we observed that the tumors formed by the FBXO31-KO Ishikawa cells harbored richer vasculature as indicated by the CD31 immunofluorescent staining (Fig. 8h). The OSMI-1 treatment significantly blocked angiogenesis and the formation of blood vessels in the FBXO31-KO tumors (Fig. 8i).
Together, these findings confirm that loss of FBXO31 promotes EC tumor formation in vivo by enhancing the stemness as well as angiogenesis, and downregulation of O-GlcNAcylation by inhibiting OGT can suppress these tumors, indicating that targeting the dysregulated O-GlcNAcylation homeostasis in EC is a promising therapeutic strategy worthy of further clinical exploitation.
Discussion
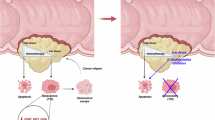
This study delves deeply into the intricate relationship between O-GlcNAcylation homeostasis and the progression of EC, elucidating the clinical significance of abnormal O-GlcNAcylation level and unveiling an important regulatory module controlling its homeostasis in endometrial tissues (Fig. 9).

FBXO31-mediated ubiquitination of OGT maintains a relatively low level of O-GlcNAcylation in the non-cancerous endometrium. Inactivation of FBXO31 in endometrial cancer tissues results in accumulation of OGT and concurrent increase of O-GlcNAcylation that promote endometrial malignancy. The uterus elements in this figure were created using BioGDP.com (https://BioGDP.com).
Integrative analysis of O-GlcNAcylation in a large clinical EC cohort to assess its relationships with current histomorphologic and molecular subtypes of EC had not been conducted till this study. A previous report using 76 EC samples revealed that the two executing enzymes of O-GlcNAcylation, OGT and OGA, manifested increased mRNA levels in ECs of higher histologic grade relative to the well-differentiated tumors23. A more recent IHC analysis on a tissue microarray containing 28 EC specimens showed that both the OGT and O-GlcNAcylation levels were increased in EC tissues than the adjacent normal endometrial tissues24. This pilot study indicated that increased O-GlcNAcylation was associated with histologic grade, clinical stage, and lymph node metastasis. However, when repeating the IHC analysis using the same tissue microarray, we only observed increased OGT and O-GlcNAcylation levels in ECs but failed to associate O-GlcNAcylation level with any of the clinical parameters, probably due to the differences in inclusion and exclusion criteria and the limited sample size. Nonetheless, when we expanded the analysis to our EC cohort containing 219 patients, the clinical significance of O-GlcNAcylation became invariable. The O-GlcNAcylation level shows strong association with histologic grade, FIGO stage, and poor prognosis. The caveat of our EC cohort is that it only contains endometrioid carcinoma cases and lacks information of molecular subtypes. Therefore, we further included the TCGA UCEC dataset in our analysis, by building a mathematical model to calculate an estimated O-GlcNAc index for each TCGA UCEC case. This computational analysis not only confirmed the observations made with our EC cohort, but also revealed that EC patients belonging to the copy-number high molecular subtype group have significantly higher O-GlcNAcylation level than that in the other groups. Agreeingly, one feature of the copy-number high molecular subtype is TP53 mutations18, and p53 is a known negative regulator of O-GlcNAcylation level not only identified in our genome-wide screen but also reported in a previous study56. Loss-of-function of p53 in tumor cells increases glucose uptake, aerobic glycolysis, and pentose phosphate pathway (PPP) flux, thereby promoting O-GlcNAcylation level56. These findings suggest that O-GlcNAcylation is a useful factor complementary to the current classification system to better identify EC patients with poor clinical outcomes.
Our understanding on the molecular circuitry controlling the cellular O-GlcNAcylation homeostasis is incomplete. Given that O-GlcNAcylation is dependent on nutrient availability, metabolic factors such as GFPT1, POLDIP2, and PPM1K have been reported to influence O-GlcNAcylation level by modulating the metabolic flux of the HBP pathway56,59,64,70,71. However, emerging evidence indicates that O-GlcNAcylation may also be regulated by non-nutrient dependent mechanisms, particularly at the protein level of OGT57,58,62,66,67,68. OGT is regulated by the balance of ubiquitination and deubiquitination9,10,12,13. The E3 ligases XIAP and E6AP have been reported to promote the ubiquitin-dependent proteasome degradation of OGT10,11. The histone demethylase LSD2 displays an atypical ubiquitin E3 ligase activity toward OGT in the A549 cells9. However, we found that the expression of these reported E3 ligases of OGT has no clinical relevance in ECs. Instead, our unbiased screen uncovered that FBXO31, together with other components in the SCF complex, functions as an E3 ligase for OGT. Loss of FBXO31 stabilizes OGT and increases cellular O-GlcNAcylation level, thereby promoting the progression of endometrial malignancy. Consistently, EC patients with low FBXO31 expression exhibited more advanced histologic grade and poor survival. FBXO31 is a tumor suppressor gene located in the 16q24.3 region, with frequently observed loss of heterozygosity in several cancers, including breast, ovarian, hepatocellular, and prostate cancers81,82. The FBXO31-OGT regulatory axis reported in this study is worthy of investigation in these cancers as well.
How the aberrant O-GlcNAcylation downstream of the FBXO31-OGT regulatory module promotes EC progression is not fully understood and can be complex. Loss of FBXO31 leads to accumulation of OGT that can increase O-GlcNAcylation on thousands of nuclear and cytoplasmic proteins, including many EC-related oncogenes and tumor suppressors, such as PI3K, PTEN, ARID1A83, p5384, c-Myc85, YAP24,29,37, and β-catenin86. Our study revealed that c-Myc protein level was increased in FBXO31-KO EC cells in an OGT-dependent manner, suggesting that c-Myc can be one of the key tumor-promoting factors controlled by the FBXO31-OGT axis. Additionally, proteins involved in the regulation of pluripotency, such as Oct487, Sox288, and Sox989, are able to be modified by O-GlcNAcylation, which may contribute to the observation that increased O-GlcNAcylation promotes stemness of EC cells. It is noteworthy that in addition to OGT, the SCFFBXO31 complex can also ubiquitinate other protein substrates, including cell cycle regulators, such as cyclin D174, Cdt175, MDM277, and cyclin A90; signaling molecules, such as c-Myc91, β-catenin92, and MKK676; as well as EMT factors, Snail193 and Slug94. Considering how FBXO31 recognizes its substrates remains unclear and FBXO31 can physically interact with OGT, it is compelling to speculate that O-GlcNAcylation can modulate the substrates recognition of the SCFFBXO31 complex. This hypothesis warrants future molecular and structural characterizations.
Last but not least, our pilot experiments in mouse xenograft models validated that the OGT inhibitor OSMI-1 has anti-tumor activity in vivo. The administration of OSMI-1 reduced O-GlcNAcylation level in tumors formed by the subcutaneously injected Ishikawa cells, downregulated the expression of many genes involved in stemness, EMT, and angiogenesis, inhibited the formation of blood vessels, and significantly limited the tumors’ growth. These results suggest that hyper O-GlcNAcylation is a shared vulnerability for the FBXO31 mutated as well as many high histologic grade EC cases, and targeting this dysregulated O-GlcNAcylation homeostasis holds promising therapeutic significance. Future elaboration of the spatiotemporal dynamics of the O-GlcNAcylation landscapes during the progression of ECs with high-throughput, tissue-specific proteomic profiling methods will further consolidate the foundation of targeting O-GlcNAcylation to develop new therapeutic strategies in clinical settings.
Methods
Ethics
This research complies with all relevant ethical regulations approved by the Ethics Committee of Central South University, including the use of human tissues and mouse experiments.
Human tissues
All fresh tissues and paraffin-embedded (FFPE) tissues were prospectively obtained from patients with endometrial diseases at Xiangya Hospital, Central South University. Clinical data and histopathological characteristics were retrieved from patient records and routine pathology reports. The study was approved by the Medical Ethics Committee of Central South University (No. 202103076, No. 201910255), and all participating patients provided informed written consent. The study was registered with and approved by the Human Genetics Resource (HGR) office of the Minister of Science and Technology of China (No. 2024BAT00742). Surgical tumor tissue from consented patients with a confirmed diagnosis of EC was included in this study. The inclusion criteria for patient enrollment were as follows: no prior anticancer therapies, no diagnosis or history of other concurrent malignancies, and availability of follow-up data. For this study, all pathology reports were reviewed by two pathologists. Patients whose original biopsies did not indicate endometrial cancer or whose histology was insufficiently informative were excluded. The endometrial tissues for generating the EE-Os were obtained from endometriosis patients who underwent hysteroscopic biopsy for endometrial polyps without prior drug treatment. Samples were selected based on tissue availability without bias toward any specific parameters.
Organoids culturing from endometrial surgical samples
The endometrial organoids were generated as previously described95. Tumor tissues and non-cancerous control tissues were isolated and stored in ice-cold serum-free DMEM medium supplemented with 1% penicillin-streptomycin. The tissues were then washed in ice-cold DPBS (Biological Industries) supplemented with penicillin-streptomycin and minced into small pieces. The tissues were digested by collagenase IV (1-2 mg/mL; 17104019, Thermo Fisher Scientific) in the presence of ROCK inhibitor (10 µM; SCM075, Merck Millipore) and penicillin-streptomycin for 1 h on a shaker at 37 °C, then incubated for 15 min in TrypLE (1 ×; 12604013, Thermo Fisher Scientific) supplemented with ROCK inhibitor and penicillin-streptomycin. Subsequently, the tissue digests were stopped by ice-cold serum-free DMEM/F12 and after centrifugation, a 100 μm cell strainer was used to obtain cell pellets. The strainers were inverted over a Petri dish, and the glandular elements were backwashed, transferred to a centrifuge tube, and pelleted by centrifugation. Larger undigested tissue fragments retained on the strainer were collected for further digestion. Finally, the cell pellets were resuspended in 70% Matrigel mixed with 30% DMEM/F12 (356231, Corning and 11039021, Gibco, respectively) and seeded in 50 μL droplets in non-treated 24-well plates. After incubation at 37 °C and 5% CO2 in a cell culture incubator for 20–30 min, the pre-warmed organoid complete medium (DMEM/F12 supplemented with 1% penicillin-streptomycin, 2% B27 supplement minus vitamin A (12587010, Gibco), 5% R-spondin-1 conditioned medium, 1% chemically defined lipid concentrate (11905031, Gibco), recombinant human Noggin 100 ng/mL (HY-P7051A, MCE), 1% N2 (17502048, Gibco), N-acetyl-L-cysteine 1.25 mM (A7250, Sigma Aldrich), Nicotinamide 10 µM (73240, Sigma Aldrich), recombinant human EGF 50 ng/mL (236-EG-01M, R&D Systems), Y-27632 10 µM (SCM075, Sigma Aldrich), 17-β estradiol 10 nM (E8872, Sigma Aldrich), SB202190 0.1 µM (S7067, Sigma Aldrich), A83-01 0.25 µM (SML0788, Sigma Aldrich), recombinant human IGF 40 ng/mL (100-11, Peprotech), recombinant human HGF 20 ng/mL (100-39, Peprotech), IL-6 5 ng/mL (200-06, Peprotech)) was added. The organoid medium was changed every 2 days, and the organoids were passaged after 7-10 days of culture. Organoids of low passage number (P3-P6) were used for the experiments described. To assess clonogenic capacity, organoids were dissociated into single cells with TrypLE supplemented with Y-27632, filtered through a 40-µm cell strainer and resuspended in 70% Matrigel / 30% DMEM/F12 supplemented with Y-27632 at 1000 cells per well in 96-well plates. The organoids formed were counted after 10–15 days. A randomly selected field of view at 10 × magnification in each well was used to count organoids and measure their cross-sectional area.
Immunohistochemistry (IHC) and Immunofluorescence (paraffin sections)
An endometrial cancer tissue array was purchased from Xinchao Biotechnology Company (HUteA060CS01, Shanghai, China), consisting of 26 pairs of tumoral and peritumoral tissue specimens, along with an additional 8 cases of cancerous tissue without paired peritumoral tissue. After removing the incomplete tissue spots, 31 cases of cancer tissue and 23 cases of peritumoral tissue were included in IHC analysis. IHC was performed as previously described96, with primary antibody incubation overnight after antigen retrieval and endogenous peroxidase activity blocking on paraffin sections. The IHC staining signal levels were blindly scored by two independent assessors without knowledge of clinical parameters, and confirmed by a pathologist. Based on the proportion of positive stained-tumor cells which was assessed on a value of 0–4: 0 (negative), 1 (1–25%), 2 (26–50%), 3 (51–75%), or 4 (76–100%) and the intensity of staining which was scored on a value of 0-3: 0 (negative), 1 (weak), 2 (medium), or 3 (strong). The final IHC score was calculated by multiplying the staining intensity score (0–3) and the proportion of positively stained tumor cells score (0–4), resulting in a staining index (SI) ranging from 0 to 12. Low and high expression were defined as SI 0–6 and SI 8–12, respectively36. Immunofluorescence was performed for CD31 staining, a marker of endothelial cells. Briefly, the slides were de-paraffinized, and avidin and biotin were added with the blocking agents. The blocking agent was blotted off, and the first antibody was added to incubate overnight at 4 °C. The slides were washed and incubated with the second antibody at room temperature for 30 min. DAPI was applied to the slides and coverslip was placed, and the slides were kept in dark97. All the antibodies used in this study were listed in Supplementary Data 10.
Survival analysis
Progression-free survival (PFS) was calculated as the time between the surgery that procured the sample and the date of disease progression or a new metastatic event in a different location. Overall survival (OS) was defined as the interval between the date of surgery and the date of death or last follow-up. Progression-free interval (PFI) was defined as the duration from surgery to the first occurrence of disease progression or death after treatment. The curves were stratified based on the O-GlcNAcylation level. Log-rank test was used to compare the two groups over a follow-up time of 61 months. Kaplan-Meier survival curves were generated and compared using GraphPad Prism (version 8.0.2).
Generation of O-GlcNAc index prediction model
The RNA-seq data of 15 low O-GlcNAcylation level (RL2 by IHC) tumor tissues and 40 high O-GlcNAcylation level tumor tissues were processed to identify the O-GlcNAcylation correlated genes. The gene expression matrix of these 55 EC samples was correlated with the O-GlcNAcylation IHC staining index using the Pearson correlation method in the mlr3.filters package within the mlr3 framework in R. The top 1000 genes with a correlation coefficient >0.3 were included in the O-GlcNAc correlated gene set. Subsequently, mlr3 learners including six regression model-based approaches (regr.lm, regr.glmnet, regr.kknn, regr.ranger, regr.rpart, regr.svm) were applied to the expression matrix of the 1000 O-GlcNAcylation correlated genes. The O-GlcNAc indices for the 55 EC tissues were calculated, subjecting to 5-fold cross-validations of training and ranking based on predefined performance metrics. The reliability of the prediction model was assessed by comparing the calculated O-GlcNAc indices with actual IHC SI scores. The regr.glmnet demonstrated the lowest mean squared error (MSE) and was selected for the establishment of the final prediction model. The O-GlcNAc indices were then calculated using the prediction model for the 589 EC samples in TCGA. The patients were categorized into high and low O-GlcNAcylation groups using the median of the calculated O-GlcNAc indices. Wilcoxon Mann-Whitney tests were used to assess differences between the two groups in terms of histologic grade, FIGO stage, molecular subtype, age, and diabetes. Log-rank tests were employed to compare the OS and PFI differences between the high and low O-GlcNAcylation groups, and Kaplan-Meier survival curves were generated and compared using R (version 4.03).
Immunofluorescence of organoids
Immunofluorescence staining experiments were performed on organoids as previously described98. When the organoids reached a size of ~100 μm, they were selected for staining. After washing twice with pre-cooled DPBS, 500 μL of cell recovery solution (354253, Corning) was added to each well, and the Matrigel was dissolved on ice to ensure that the morphology of the organoids was not disrupted. After 30 min, all the organoids were collected into a 15 mL centrifuge tube, fixed with 4% paraformaldehyde for 30 min, and then centrifuged to remove the supernatant. Next, 10 mL of 1% PBST was added to stop the tissue fixation. After blocking with Organoid Washing Buffer (OWB, 0.1% Triton X-100, 0.2% BSA in DPBS), the primary antibody was added and incubated overnight at 4 °C with shaking at 60 rpm. On the following day, the organoids were washed three times with OWB for 2 h each time, and then the corresponding fluorescent secondary antibody was added. The organoids were incubated overnight on a shaker in the dark. On the third day, 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI, D9542, Sigma) at 10 mg/mL was added for 30 min. After washing with OWB, the samples were spun down at 70 × g for 5 min at 4 °C. Finally, the organoids were resuspended with fructose-glycerol clearing solution (60% glycerol and 2.5 M fructose in ddH2O) and imaged using an LSM880 confocal microscope (Zeiss) and a CSU-W1 spinning disk field scanning confocal system (Nikon). A cell death detection (TUNEL) kit (Roche) was used to identify dead cells in accordance with the company’s description. All the antibodies used in this study were listed in Supplementary Data 10.
Lentiviral transduction of organoids
For organoid lentiviral transduction, pLKO.1-puro vectors and TK-PCDH-copGFP-T2A-Puro vectors were used. The organoids were washed twice with pre-cooled DPBS, and 500 μL of TrypLE (12604013, Thermo Fisher Scientific) was added to each well for 10 min at 37 °C. The Matrigel was disrupted by pipetting the mixture up and down repeatedly during digestion. TrypLE was inactivated by adding 10 mL of ice-cold serum-free DMEM/F12, and the mixture was centrifuged for 5 min at 200 × g. After digestion, the organoids were made into single cells or cell mass and resuspended in virus infection solution containing ROCK inhibitor, polybrene, and concentrated lentivirus in organoid culture media. The cell suspension was added to a 6-well plate, spun at 2000 rpm for 1 h, and then incubated at 37 °C for 5-6 h. The cells were then transferred to a 15 mL centrifuge tube, washed twice with serum-free DMEM/F12, and seeded in a prewarmed 24-well plate with 70% Matrigel. Then, 500 µL of organoid medium was added to each well, followed by incubation at 37 °C with 5% CO2 for 20 min. The medium was changed every 2 days. Puromycin selection (1 μg/mL) in organoid culture was conducted for 3–4 days to establish stably infected organoids. The stable organoids were validated by western blot or quantitative RT-PCR.
Cell Titer-Glo® 3D viability assay
Tumor organoids were recovered from the Matrigel and dissociated. 2000 cells were seeded in 96-well plates and allowed to form organoids for 7 days. Then, TMG (10 µM, Selleck; S7213), OSMI-1 (50 µM, Selleck; S9835), OSMI-4 (20 µM, MCE; HY-11436), or DMSO (0.1%, Sigma Aldrich, D2650) was added and viability was measured after 72 h using Cell Titer Glo 3D cell (Promega, Cat# G9681) following the manufacturer’s instructions95. The Cell Titer-Glo® 3D Cell reagent was thawed and equilibrated at room temperature for 30 min. The reagent was mixed 1:1 with organoid complete medium and added to the plate. After a 30 min incubation at 37°C, the luminescence was measured on the PerkinElmer Envision.
Tumor xenograft implantation in nude mice
In this in vivo study, 4-week-old female nude mice without a thymus (BALB/C) were purchased from Hunan SJA Laboratory Animal Co., Ltd (Changsha, China). All animal experiments were conducted in accordance with the Animal Welfare Law and were approved by the Ethics Committee of Central South University (No. 202103076, No. 202411200). The mice were housed at a maximum of five per cage under a 12 h light/dark cycle at 22–25°C and 50–70% humidity. The animals were provided with standard growth maintenance chow (GMCF, purchased from Beijing Keao Xieli Feed Limited, Beijing, China; Product ID: 24083213) and allowed free access to water. Ishikawa or Ishikawa FBXO31-KO endometrial cancer cells were suspended in 100 µL DPBS and injected subcutaneously into the left flank (5 × 106 cells per mouse). For the tumor TMG or OSMI-1 treatment study, after 10 days, upon tumor onset, mice were randomly divided into three groups: the DMSO group (vehicle solvent, n = 8), the TMG group (20 mg/kg/day, n = 7), and the OSMI-1 group (10 mg/kg/day, n = 8). The vehicle solvent, comprising 5% DMSO, 40% PEG300 (Selleck), and 5% TWEEN80 (Selleck), was prepared according to the manufacturer’s instructions to improve the solubility of OSMI-1 and TMG for intraperitoneal injections in mice99. The mice received daily intraperitoneal injections over 15 days. Tumor growth, body weight, and survival of the animals were monitored twice a week. The maximal tumor volume allowed by the Ethical Committee for Animal Experiment of the Central South University is 2000 mm3, which was not exceeded in this study. Experimental endpoints were reached when tumors exceeded 20 mm in diameter or ruptured, or when mice became moribund, showing signs of lateral recumbency, cachexia, lack of response to noxious stimuli, or observable weight loss (≥20% of body weight)100. For the FBXO31-KO tumor study, upon tumor onset, nude mice were randomly divided into four groups: the WT DMSO control group (vehicle solvent, 5% DMSO + 40% PEG300 + 5% TWEEN80, Selleck, n = 10), the WT OSMI-1 treatment group (10 mg/kg/day, n = 9), the FBXO31-KO DMSO group (vehicle solvent, 5% DMSO + 40% PEG300 + 5% TWEEN80, Selleck, n = 10), and the FBXO31-KO OSMI-1 treatment group (10 mg/kg/day, n = 9). Tumor growth and body weight of the animals were monitored three times a week. Tumor volume was calculated by measuring the short (l) and long (L) diameters (volume = l2 × L/2). Mice were sacrificed for examination 28 days after tumor inoculation. At the end of the study, mice were euthanized, and the tumors were processed for further analyses.
Quantitative RT-PCR
RNA extraction was performed using TRIzol (87804, Life Technologies) according to the manufacturer’s protocol for all samples, including cells, organoids, and primary tissues. The extracted RNA was then reverse transcribed to cDNA using the PrimeScript RT Reagent Kit (RR037A, Takara). The cDNA was used as a template for qPCR, which was performed using the SYBR Green qPCR Master Mix (QST-100, SolomonBio) on the QuantStudio 3 Real-Time PCR system (Applied Biosystems). All the primers were listed in Supplementary Data 11.
Western blot and immunoprecipitation
Cells were lysed in sample buffer (2% SDS, 10% glycerol, and 62.5 mM Tris-HCl, pH 6.8) supplemented with 1 × protease inhibitor cocktail (P8340, Sigma). The protein concentration was measured using a BCA kit (P0009, Beyotime). Cell lysates were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. The membrane was then blocked with 5% non-fat dry milk for 1 h at room temperature and probed with the indicated primary antibodies overnight at 4°C. Antigen-antibody complexes were detected by incubating with horseradish peroxidase secondary antibodies followed by ECL substrates (WBKLS0500, Millipore). For immunoprecipitation experiments, cells were washed twice with ice-cold PBS and then lysed in lysis buffer (20 mM Tris-HCl (pH 8.0), 137 mM NaCl, 1% NP-40, 2 mM EDTA) on ice for 30 min. Cell lysates were gently mixed with specific antibodies overnight at 4°C under gentle rotation, then incubated with protein A/G beads (SC-2003, Santa Cruz) for 1-2 h at 4°C Immunoprecipitants were washed three times with lysis buffer. After the final wash, the supernatant was aspirated and discarded, and the pellet was resuspended in 2 × SDS sample buffer (0.125 M Tris HCl (pH 6.8), 4% SDS, 20% glycerol, 2% β-mercaptoethanol, 0.02% bromophenol blue). The sample was then subjected to reducing SDS-PAGE and western blot. All the antibodies used in this study were listed in Supplementary Data 10.
Cell culture and generation of cell lines
HA-R-Spondin1-Fc 293T cell line (3710-001-01, R&D Systems) was used to produce R-spondin-1 conditional media. HEK293T (ATCC, CRL-3216) and Ishikawa cells (Sigma, 99040201) were maintained in DMEM (06-1055-57-1 ACS, Vivocell) supplemented with 10% FBS. All cells were cultured at 37 °C in a humidified incubator with 5% CO2 and periodically screened for Mycoplasma contamination. Human FBXO31 knockout cell lines were generated according to previously published protocol79. To generate 293T and Ishikawa FBXO31-KO cell lines, the cells were transfected with LentiCRISPR-V2 plasmid carrying sgFBXO31 (Supplementary Data 11) and further selected with 1 μg/mL puromycin (s7417, Selleck) for 3 days. The cells were then plated at single-cell density in 100 mm Petri dishes, and the individual clones that emerged were picked and replated into 24-well plates. The loss of FBXO31 expression was confirmed by western blot and Sanger sequencing. Genomic DNA was extracted using QuickExtract (Epicenter). Genotyping PCRs were performed with KOD FX DNA Polymerase (KFX-101, Toyobo) using primers flanking the genomic target site. The resulting PCR products were purified and sequenced to confirm the presence of indel events. To further validate the mutational status of candidate clones, the PCR products underwent TA cloning (Invitrogen) and were sequenced to distinguish the amplified products of distinct alleles. Clones with confirmed insertion or deletion events were also validated by western blot analysis.
In vivo and in vitro ubiquitination assay
For detection of ubiquitinated proteins in vivo, 293T and Ishikawa cells were co-transfected with expression vectors for HA-ubiquitin and the indicated proteins. Polyubiquitinated OGT was detected by immunoprecipitation of OGT with ANTI-FLAG® M2 Affinity Gel (A2220, Merck Millipore) or OGT antibody under denaturing conditions followed by western blot with an anti-HA antibody. In vitro ubiquitination was performed as previously described101. The SCF-FBXO31 (E3) complexes were immunopurified from the cell lysate using Pierce™ Anti-HA Magnetic Beads (88836, Thermo Fisher Scientific) and incubated with His-OGT fusion protein expressed and purified from E. coli as previously reported102 in the presence of recombinant purified E1 (UBA1; 11990-H20B, sinobiological), E2 (UBE2D1; 11432-H07E, sinobiological), recombinant human ubiquitin protein (U-100H, Boston Biochem), and ubiquitination buffer (20 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 0.5 mM DTT, 2 mM ATP). The reaction was stopped by adding 2 × SDS sample buffer and boiling for 10 min.
CRISPR-Cas9 screen and data analysis
The human genome-scale CRISPR knockout library (GeCKO v2, Addgene #1000000048) in the lentiCRISPR v2 vector (Addgene, #52961) consists of 123,411 sgRNAs that target 19,050 protein-coding genes (6 sgRNAs per gene) and 1000 nontargeting control sgRNAs was used103,104. The human GeCKO v2 library was transduced into 293T cells by lentivirus at a multiplicity of infection of 0.3. Cells were selected with puromycin for 7 days followed by fluorescence-activated cell sorting (FACS) based on their O-GlcNAcylation staining intensities. An unsorted sample was used to assess sgRNA library coverage, and the sorted RL2 high population was subjected to genomic DNA extraction. The inserted sgRNA library was amplified by two steps of PCR for next-generation sequencing. Each screen was performed twice. For data analysis, reads from the fastq files generated by sequencing were tallied for each guide by taking the first 20 bp from each read and mapping to the identical short gRNA sequence. For each screen, a table of reads per guide that includes the counts from the RL2 high population of both replicates was generated and loaded into MAGeCK55. Top genes were determined based on their mean log2 fold change, FDR, and robust ranking aggregation (RRA) score.
Data download
The TCGA UCEC dataset used in this study, including the gene raw count data (htseq-count files), and the annotated somatic simple nucleotide variation files (MuTect2 VCF), were downloaded using the gdc-client v1.6.0. The clinical OS and PFI information were obtained from Liu et al.105.
RNA-seq and bioinformatic analysis
Total RNA was isolated from EC tissues, and libraries were generated using the NEBNext UltraTM RNA Library Prep Kit (New England Biolabs) for the Illumina system. Sequencing was conducted using the Illumina Novaseq 6000 platform (Novogene). Trim Galore v0.6.4 was employed to eliminate adapter sequences and remove reads of poor quality. Subsequently, the reads from each RNA-seq data were aligned to the hg38 genome assembly using STAR v2.7.2 d. The key alignment parameters were set as follows: ‘--outFilterMismatchNoverLmax 0.04 --outSAMtype BAM SortedByCoordinate --outFilterMultimapNmax 500 --outMultimapperOrder Random --outSAMmultNmax 1’. Gene expression was quantified using featureCounts v2.0.0. Heatmaps were created using R package heatmap v1.0.12. Differential expression analysis was conducted using the negative binomial distribution with the ‘DESeq’ and ‘results’ functions from DESeq2, applying cut-off values of adjusted p-value < 0.05 and |log2 FC | > 1. All GSEA analyses presented in this study were based on hallmark gene sets and performed using the R package ‘clusterProfiler’. The GO enrichment analysis was performed using the function ‘enrichGO’ from the R package clusterProfiler v3.10.1106.
Organoid single-cell analysis
EE-Os were treated with 10 mM TMG or vehicle control (0.1% DMSO). Following treatment, the organoids were dissociated into single cells using TrypLE digestion, and the mixture was passed through a 40-μm cell strainer. The cells were then counted and viability assessed. Single-cell suspensions (2 × 105 cells/mL) in PBS (HyClone) were loaded onto microwell chip using the Singleron Matrix® Single Cell Processing System. Barcoding beads were subsequently collected from the microwell chip, followed by reverse transcription of the mRNA captured to obtain the cDNA. After PCR amplification, the amplified cDNA was then fragmented and ligated with sequencing adapters. The scRNA-seq libraries were constructed according to the protocol of the GEXSCOPE® Single Cell RNA Library Kits (Singleron)107. Individual libraries were diluted to 4 nM, pooled, and sequenced on Illumina Novaseq 6000 with 150 bp paired end reads. Raw reads were processed to generate gene expression profiles using CeleScope v2.0.7 (Singleron) with default parameters. Briefly, barcodes and UMIs were extracted from R1 reads and corrected. Adapter sequences and polyA tails were trimmed from R2 reads and the trimmed R2 reads were aligned to the hg38 transcriptome using STAR (v2.6.1b). Uniquely mapped reads were then assigned to exons with featureCounts (v2.0.1). Successfully assigned reads with the same cell barcode, UMI and gene were grouped to generate the gene expression matrix. Omicverse V1.5.4 was used for quality control, dimensionality reduction and clustering under Python 3.8. The following criteria were used to filter the expression matrix: (1) cells with gene count <500 were excluded; (2) cells detected genes <250 were excluded; (3) cells with mitochondrial content >15% were excluded; (4) genes expressed in <3 cells were excluded. After filtering, 20,736 cells were retained for the downstream analyses. The raw count matrix was normalized by total counts per cell and logarithmically transformed into normalized data matrix. The top 3000 highly variable genes were selected by setting flavor = ‘seurat’. Principal Component Analysis (PCA) was performed on the scaled variable gene matrix, and 50 principal components were used for clustering and dimensional reduction. ‘Harmony’ was employed to integrate samples. Cells were separated into 6 clusters using Leiden algorithm with the resolution parameter at 0.25. Subsequently, omicverse was used to calculate the ranking of highly differential genes in each cluster to identify marker genes. Cell clusters were visualized using Minimum-Distortion Embedding (mde). Cell types were annotated based on the cell type auto-annotation tool SCSA, and the known cellular markers from the literature38,53,108,109,110: epithelial cells (EPCAM, KRT8, KRT18), stem cells (LGR5, SOX9, POU5F1, PROM1, AXIN2), proliferative cells (MMP7, TOP2A, MKI67), ciliated cells (PIFO, FOXJ1, TPPP3), pre-ciliated cells (CDC20B, DYDC2, CCNO), and inflammatory cells (IL4I1, IL32, S100A9, CD14, IL1RN). The O-GlcNAc-related stem-like cells annotation was mainly based on the results of KEGG pathway enrichment. The expression of markers used to identify each cell type was visualized using a violin plot.
AUCell geneset enrichment analysis
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were collected and used as functional genesets for AUCell scoring. AUCell scores of gene sets were visualized using sc.pl.embedding. p-values from t-tests were used for estimating the statistical significance between cell types and groups.
Statistical analysis
The experiments were conducted in at least three independent biological replicates, and the data were presented as mean ± SD. If not specified, the Student’s t-test was used to perform a statistical significance test between different groups, and p < 0.05 was considered significant. Overall survival curves were estimated by the Kaplan-Meier method and Cox proportional hazards model. All statistical and correlation analyses were performed using the GraphPad Prism 8.0 software (GraphPad Software) and SPSS 26.0 (SPSS Software).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing data generated in this study, including both RNA-seq and scRNA-seq raw data, have been deposited in the Genome Sequence Archive for Human (GSA) under the accession number HRA007070. These raw data files can be accessed at https://download.cncb.ac.cn/gsa-human/HRA007070. These data have been registered with the Human Genetics Resource Office in China, with the registration number: 2024BAT00742. The sequencing raw data of the genome-scale CRISPR knockout screen generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under the accession code GSE287788. These data are publicly available. Source data are provided with this paper. All the other data are available within the article and its Supplementary Information. Source data are provided with this paper.
Code availability
The original code for the mathematical model of the virtual O-GlcNAc index has been deposited in Zenodo (https://doi.org/10.5281/zenodo.14292333).
References
Yang, X. & Qian, K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 18, 452–465 (2017).
Zachara, N. E. & Hart, G. W. O-GlcNAc a sensor of cellular state: the role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim. Biophys. Acta. 1673, 13–28 (2004).
Chatham, J. C., Zhang, J. & Wende, A. R. Role of O-linked N-acetylglucosamine protein modification in cellular (Patho) physiology. Physiol. Rev. 101, 427–493 (2021).
Sheikh, M. A., Emerald, B. S. & Ansari, S. A. Stem cell fate determination through protein O-GlcNAcylation. J. Biol. Chem. 296, 100035 (2021).
Slawson, C. & Hart, G. W. O-GlcNAc signalling: implications for cancer cell biology. Nat. Rev. Cancer 11, 678–684 (2011).
de Queiroz, R. M., Carvalho, E. & Dias, W. B. O-GlcNAcylation: The sweet side of the cancer. Front. Oncol. 4, 132 (2014).
Trinca, G. M. & Hagan, C. R. O-GlcNAcylation in women’s cancers: breast, endometrial and ovarian. J. Bioenerg. Biomembr. 50, 199–204 (2018).
Le Minh, G. & Reginato, M. J. Role of O-GlcNAcylation on cancer stem cells: connecting nutrient sensing to cell plasticity. Adv. Cancer Res. 157, 195–228 (2023).
Yang, Y., Yin, X., Yang, H. & Xu, Y. Histone demethylase LSD2 acts as an E3 ubiquitin ligase and inhibits cancer cell growth through promoting proteasomal degradation of OGT. Mol. Cell 58, 47–59 (2015).
Seo, H. G. et al. Mutual regulation between OGT and XIAP to control colon cancer cell growth and invasion. Cell Death Dis. 11, 815 (2020).
Peng, K. et al. Regulation of O-linked N-acetyl glucosamine transferase (OGT) through E6 stimulation of the ubiquitin ligase activity of E6AP. Int. J. Mol. Sci. 22, 10286 (2021).
Tang, J. et al. Targeting USP8 inhibits O-GlcNAcylation of SLC7A11 to promote ferroptosis of hepatocellular carcinoma via stabilization of OGT. Adv. Sci. 10, e2302953 (2023).
Tang, J. et al. The deubiquitinase EIF3H promotes hepatocellular carcinoma progression by stabilizing OGT and inhibiting ferroptosis. Cell Commun. Signal. 21, 198 (2023).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca. Cancer J. Clin. 71, 209–249 (2021).
Han, B. et al. Cancer incidence and mortality in China, 2022. J. Natl Cancer Cent. 4, 47–53 (2024).
Lu, K. H. & Broaddus, R. R. Endometrial acncer. N. Engl. J. Med. 383, 2053–2064 (2020).
Bokhman, J. V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 15, 10–17 (1983).
Murali, R., Soslow, R. A. & Weigelt, B. Classification of endometrial carcinoma: more than two types. Lancet Oncol. 15, e268–e278 (2014).
The Cancer Genome Atlas Research, N. et al. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013).
Jamaluddin, M. F. B. et al. Proteomic and functional characterization of intra-tumor heterogeneity in human endometrial cancer. Cell Rep. Med. 3, 100738 (2022).
Dou, Y. et al. Proteogenomic insights suggest druggable pathways in endometrial carcinoma. Cancer Cell 41, 1586–1605 (2023).
Jaskiewicz, N. M. & Townson, D. H. Hyper-O-GlcNAcylation promotes epithelial-mesenchymal transition in endometrial cancer cells. Oncotarget 10, 2899–2910 (2019).
Krzeslak, A., Wojcik-Krowiranda, K., Forma, E., Bienkiewicz, A. & Brys, M. Expression of genes encoding for enzymes associated with O-GlcNAcylation in endometrial carcinomas: clinicopathologic correlations. Ginekol. Pol. 83, 22–26 (2012).
Zhai, L. et al. O‑GlcNAcylation mediates endometrial cancer progression by regulating the HippoYAP pathway. Int. J. Oncol. 63, 90 (2023).
Zhou, F. et al. Elevated glucose levels impair the WNT/beta-catenin pathway via the activation of the hexosamine biosynthesis pathway in endometrial cancer. J. Steroid. Biochem. Mol. Biol. 159, 19–25 (2016).
Ciesielski, P., Jozwiak, P., Forma, E. & Krzeslak, A. TET3- and OGT-dependent expression of genes involved in epithelial-mesenchymal transition in endometrial cancer. Int. J. Mol. Sci. 22, 13239 (2021).
Gomes, L. C., Odedra, D., Dikic, I. & Pohl, C. Autophagy and modular restructuring of metabolism control germline tumor differentiation and proliferation in C. elegans. Autophagy 12, 529–546 (2016).
Chen, L. et al. Direct stimulation of de novo nucleotide synthesis by O-GlcNAcylation. Nat. Chem. Biol. 20, 19–29 (2024).
Zhang, X. et al. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat. Commun. 8, 15280 (2017).
Federici, M. et al. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation 106, 466–472 (2002).
Yu, S. B. et al. Neuronal activity-driven O-GlcNAcylation promotes mitochondrial plasticity. Dev. Cell 59, 2143–2157 (2024).
Guo, B. et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat. Cell Biol. 16, 1215–1226 (2014).
Liu, Y. Y. et al. O-GlcNAcylation of MORC2 at threonine 556 by OGT couples TGF-beta signaling to breast cancer progression. Cell Death Differ. 29, 861–873 (2022).
Encarnacion-Rosado, J. et al. Targeting pancreatic cancer metabolic dependencies through glutamine antagonism. Nat. Cancer 5, 85–99 (2024).
Jiang, M. et al. O-GlcNAcylation promotes colorectal cancer metastasis via the miR-101-O-GlcNAc/EZH2 regulatory feedback circuit. Oncogene 38, 301–316 (2019).
Wu, N. et al. O-GlcNAcylation promotes colorectal cancer progression by regulating protein stability and potential catcinogenic function of DDX5. J. Cell Mol. Med. 23, 1354–1362 (2019).
Li, X. et al. OGT regulated O-GlcNAcylation promotes papillary thyroid cancer malignancy via activating YAP. Oncogene 40, 4859–4871 (2021).
Garcia-Alonso, L. et al. Mapping the temporal and spatial dynamics of the human endometrium in vivo and in vitro. Nat. Genet. 53, 1698–1711 (2021).
Brooks, R. A. et al. Current recommendations and recent progress in endometrial cancer. Ca. Cancer J. Clin. 69, 258–279 (2019).
Boretto, M. et al. Development of organoids from mouse and human endometrium showing endometrial epithelium physiology and long-term expandability. Development 144, 1775–1786 (2017).
Turco, M. Y. et al. Long-term, hormone-responsive organoid cultures of human endometrium in a chemically defined medium. Nat. Cell Biol. 19, 568–577 (2017).
Zhao, M. et al. Protein O-GlcNAc modification links dietary and gut microbial cues to the differentiation of enteroendocrine L cells. Cell Rep. 32, 108013 (2020).
Zhao, M. et al. Epithelial STAT6 O-GlcNAcylation drives a concerted anti-helminth alarmin response dependent on tuft cell hyperplasia and Gasdermin C. Immunity 55, 623–638 (2022).
Li, X. et al. Bladder cancer-derived small extracellular vesicles promote tumor angiogenesis by inducing HBP-related metabolic reprogramming and SerRS O-GlcNAcylation in endothelial cells. Adv. Sci. 9, e2202993 (2022).
Zhao, M. et al. Deficiency in intestinal epithelial O-GlcNAcylation predisposes to gut inflammation. Embo Mol. Med. 10, e8736 (2018).
Li, Y. N. et al. Dynamic changes in O-GlcNAcylation regulate osteoclast differentiation and bone loss via nucleoporin 153. Bone Res. 10, 51 (2022).
Shan, X. et al. Identification of a diketopiperazine-based O-GlcNAc transferase inhibitor sensitizing hepatocellular carcinoma to CDK9 inhibition. Febs J. 290, 4543–4561 (2023).
Andres-Bergos, J. et al. The increase in O-linked N-acetylglucosamine protein modification stimulates chondrogenic differentiation both in vitro and in vivo. J. Biol. Chem. 287, 33615–33628 (2012).
Chen, J. et al. Inhibition of O-GlcNAc transferase activates type I interferon-dependent antitumor immunity by bridging cGAS-STING pathway. Elife 13, RP94849 (2024).
McGreal, S. R. et al. Modulation of O-GlcNAc levels in the liver impacts acetaminophen-induced liver injury by affecting protein adduct formation and glutathione synthesis. Toxicol Sci 162, 599–610 (2018).
Lee, S. J. & Kwon, O. S. O-GlcNAc transferase inhibitor synergistically enhances doxorubicin-induced apoptosis in HepG2 cells. Cancers 12, 3154 (2020).
Taylor, E. W. et al. O-GlcNAcylation of AMPA receptor GluA2 is associated with a novel form of long-term depression at hippocampal synapses. J. Neurosci. 34, 10–21 (2014).
Fitzgerald, H. C., Dhakal, P., Behura, S. K., Schust, D. J. & Spencer, T. E. Self-renewing endometrial epithelial organoids of the human uterus. Proc. Natl. Acad. Sci. USA. 116, 23132–23142 (2019).
Wu, X. et al. Discovery of a novel OGT inhibitor through high-throughput screening based on homogeneous time-resolved fluorescence (HTRF). Bioorganic Chem. 139, 106726 (2023).
Li, W. et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014).
Kawauchi, K., Araki, K., Tobiume, K. & Tanaka, N. Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-N-acetyl glucosamine modification. Proc. Natl. Acad. Sci. USA. 106, 3431–3436 (2009).
Zhang, F., Snead, C. M. & Catravas, J. D. Hsp90 regulates O-linked beta-N-acetylglucosamine transferase: a novel mechanism of modulation of protein O-linked beta-N-acetylglucosamine modification in endothelial cells. Am. J. Physiol. Cell Physiol. 302, C1786–C1796 (2012).
Deplus, R. et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. Embo. J. 32, 645–655 (2013).
Ledee, D. et al. c-Myc alters substrate utilization and O-GlcNAc protein posttranslational modifications without altering cardiac function during early aortic constriction. Plos One 10, e135262 (2015).
Li, Y. N., Hu, J. A. & Wang, H. M. Inhibition of HIF-1alpha affects autophagy mediated glycosylation in oral squamous cell carcinoma cells. Dis. Markers 2015, 239479 (2015).
Muthusamy, S., Hong, K. U., Dassanayaka, S., Hamid, T. & Jones, S. P. E2F1 transcription factor regulates O-linked N-acetylglucosamine (O-GlcNAc) transferase and O-GlcNAcase expression. J. Biol. Chem. 290, 31013–31024 (2015).
Sodi, V. L. et al. mTOR/MYC axis regulates O-GlcNAc transferase expression and O-GlcNAcylation in breast cancer. Mol. Cancer Res. 13, 923–933 (2015).
Zhang, X. et al. MAPK/ERK signaling pathway-induced hyper-O-GlcNAcylation enhances cancer malignancy. Mol. Cell Biochem. 410, 101–110 (2015).
Li, T. et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab. 25, 374–385 (2017).
Zibrova, D. et al. GFAT1 phosphorylation by AMPK promotes VEGF-induced angiogenesis. Biochem. J. 474, 983–1001 (2017).
Berthier, A. et al. Combinatorial regulation of hepatic cytoplasmic signaling and nuclear transcriptional events by the OGT/REV-ERBalpha complex. Proc. Natl. Acad. Sci. USA 115, E11033–E11042 (2018).
Lai, C. Y. et al. Identification of UAP1L1 as a critical factor for protein O-GlcNAcylation and cell proliferation in human hepatoma cells. Oncogene 38, 317–331 (2019).
Deng, X. et al. ROCK2 mediates osteosarcoma progression and TRAIL resistance by modulating O-GlcNAc transferase degradation. Am. J. Cancer Res. 10, 781–798 (2020).
Lu, S. et al. SIRT1 regulates O-GlcNAcylation of tau through OGT. Aging (Albany Ny) 12, 7042–7055 (2020).
Paredes, F., Williams, H. C., Quintana, R. A. & San Martin, A. Mitochondrial protein Poldip2 (Polymerase delta interacting protein 2) controls vascular smooth muscle differentiated phenotype by O-linked GlcNAc (N-Acetylglucosamine) transferase-dependent inhibition of a ubiquitin proteasome system. Circ. Res. 126, 41–56 (2020).
Walter, L. A. et al. Inhibiting the hexosamine biosynthetic pathway lowers O-GlcNAcylation levels and sensitizes cancer to environmental stress. Biochemistry 59, 3169–3179 (2020).
Zhao, M., Sun, J. & Zhao, Z. TSGene: a web resource for tumor suppressor genes. Nucleic Acids Res. 41, D970–D976 (2013).
Zhao, M., Kim, P., Mitra, R., Zhao, J. & Zhao, Z. TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 44, D1023–D1031 (2016).
Santra, M. K., Wajapeyee, N. & Green, M. R. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature 459, 722–725 (2009).
Johansson, P. et al. SCF-FBXO31 E3 ligase targets DNA replication factor Cdt1 for proteolysis in the G2 phase of cell cycle to prevent re-replication. J. Biol. Chem. 289, 18514–18525 (2014).
Liu, J. et al. F-box only protein 31 (FBXO31) negatively regulates p38 mitogen-activated protein kinase (MAPK) signaling by mediating lysine 48-linked ubiquitination and degradation of mitogen-activated protein kinase kinase 6 (MKK6). J. Biol. Chem. 289, 21508–21518 (2014).
Malonia, S. K., Dutta, P., Santra, M. K. & Green, M. R. F-box protein FBXO31 directs degradation of MDM2 to facilitate p53-mediated growth arrest following genotoxic stress. Proc. Natl. Acad. Sci. USA. 112, 8632–8637 (2015).
Jeffery, J. M. et al. FBXO31 protects against genomic instability by capping FOXM1 levels at the G2/M transition. Oncogene 36, 1012–1022 (2017).
Duan, S. et al. Loss of FBXO31-mediated degradation of DUSP6 dysregulates ERK and PI3K-AKT signaling and promotes prostate tumorigenesis. Cell Rep. 37, 109870 (2021).
Itkonen, H. M. et al. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res. 73, 5277–5287 (2013).
Tan, Y., Liu, D., Gong, J., Liu, J. & Huo, J. The role of F-box only protein 31 in cancer. Oncol. Lett. 15, 4047–4052 (2018).
Tekcham, D. S. et al. F-box proteins and cancer: an update from functional and regulatory mechanism to therapeutic clinical prospects. Theranostics 10, 4150–4167 (2020).
Wulff-Fuentes, E. et al. The human O-GlcNAcome database and meta-analysis. Sci. Data. 8, 25 (2021).
Yang, W. H. et al. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat. Cell Biol. 8, 1074–1083 (2006).
Chou, T. Y., Hart, G. W. & Dang, C. V. c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J. Biol. Chem. 270, 18961–18965 (1995).
Zhu, Y. & Hart, G. W. Dual-specificity RNA aptamers enable manipulation of target-specific O-GlcNAcylation and unveil functions of O-GlcNAc on beta-catenin. Cell 186, 428–445 (2023).
Jang, H. et al. O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network. Cell Stem Cell 11, 62–74 (2012).
Kim, D. K. et al. O-GlcNAcylation of Sox2 at threonine 258 regulates the self-renewal and early cell fate of embryonic stem cells. Exp. Mol. Med. 53, 1759–1768 (2021).
Sun, C. et al. Glucose regulates tissue-specific chondro-osteogenic differentiation of human cartilage endplate stem cells via O-GlcNAcylation of Sox9 and Runx2. Stem Cell Res. Ther. 10, 357 (2019).
Dutta, P. et al. The tumor suppressor FBXO31 preserves genomic integrity by regulating DNA replication and segregation through precise control of cyclin A levels. J. Biol. Chem. 294, 14879–14895 (2019).
Islam, S. et al. Feedback-regulated transcriptional repression of FBXO31 by c-Myc triggers ovarian cancer tumorigenesis. Int. J. Cancer 150, 1512–1524 (2022).
Baek, D. et al. Ubiquitin-specific protease 53 promotes osteogenic differentiation of human bone marrow-derived mesenchymal stem cells. Cell Death Dis. 12, 238 (2021).
Zou, S. et al. FBXO31 suppresses gastric cancer EMT by targeting Snail1 for proteasomal degradation. Mol. Cancer Res. 16, 286–295 (2018).
Manne, R. K. et al. A MicroRNA/Ubiquitin ligase feedback loop regulates slug-mediated invasion in breast cancer. Neoplasia 19, 483–495 (2017).
Boretto, M. et al. Patient-derived organoids from endometrial disease capture clinical heterogeneity and are amenable to drug screening. Nat. Cell Biol. 21, 1041–1051 (2019).
Liu, H. et al. Transcriptional pausing induced by ionizing radiation enables the acquisition of radioresistance in nasopharyngeal carcinoma. J. Mol. Cell Biol. 15, mjad044 (2023).
Wu, J. K. et al. A switch in Notch gene expression parallels stem cell to endothelial transition in infantile hemangioma. Angiogenesis 13, 15–23 (2010).
Dekkers, J. F. et al. High-resolution 3D imaging of fixed and cleared organoids. Nat. Protoc. 14, 1756–1771 (2019).
Hsu, Y. S., Wu, P. J., Jeng, Y. M., Hu, C. M. & Lee, W. H. Differential effects of glucose and N-acetylglucosamine on genome instability. Am. J. Cancer Res. 12, 1556–1576 (2022).
Oba, T. et al. Overcoming primary and acquired resistance to anti-PD-L1 therapy by induction and activation of tumor-residing cDC1s. Nat. Commun. 11, 5415 (2020).
Choo, Y. S. & Zhang, Z. Detection of protein ubiquitination. J. Vis. Exp. 19, 1293 (2009).
Gundogdu, M. et al. The O-GlcNAc transferase intellectual disability mutation L254F distorts the TPR helix. Cell Chem. Biol. 25, 513–518 (2018).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014).
Shalem, O. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014).
Liu, J. et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400–416 (2018).
Yu, C. et al. ARID1A loss derepresses a group of human endogenous retrovirus-H loci to modulate BRD4-dependent transcription. Nat. Commun. 13, 3501 (2022).
Dura, B. et al. scFTD-seq: freeze-thaw lysis based, portable approach toward highly distributed single-cell 3’ mRNA profiling. Nucleic Acids Res. 47, e16 (2019).
Lai, Z. Z. et al. Single-cell transcriptome profiling of the human endometrium of patients with recurrent implantation failure. Theranostics 12, 6527–6547 (2022).
Tan, Y. et al. Single-cell analysis of endometriosis reveals a coordinated transcriptional programme driving immunotolerance and angiogenesis across eutopic and ectopic tissues. Nat. Cell Biol. 24, 1306–1318 (2022).
Fonseca, M. A. S. et al. Single-cell transcriptomic analysis of endometriosis. Nat. Genet. 55, 255–267 (2023).
Acknowledgements
We gratefully acknowledge Drs. Daan van Aalten, Kum Kum Khanna, Xiaowei Yang, Timothy Mitchison, Xuebiao Yao, Chao Xu, Cuiting Yong, Lisha Wu, Wenqing Yang, and Hongqiang Qin for reagents or inspiring discussions. This project has been supported by the National Natural Science Foundation of China (grants 92153301, 32170821, and 32370821 to K.Y, 32101034 to F.C), National Key Research and Development Program of China (2021YFC2701200), Department of Science & Technology of Hunan Province (grants 2023RC1028, 2023SK2091, and 2021JJ10054 to K.Y).
Author information
Authors and Affiliations
Contributions
Conceptualization: K.Y.; Methodology: N.Z., Y.M., H.N., C.H., L.S., L.L., C.Y., P.M., C.K., S.M., F.C., Y.Z., K.Y.; Validation: H.N., C.H., L.S., K.F.; Software: N.Z., Y.M., S.M.; Formal Analysis: N.Z., Y.M., K.Y.; Investigation: N.Z., Y.M., H.N., S.M., K.Y.; Resources: Y.Z., K.Y.; Data Curation: N.Z., Y.M., H.N.; Writing-Original Draft: N.Z., K.Y.; Writing-Review & Editing: K.Y.; Visualization: N.Z., Y.M., S.M., K.Y.; Supervision: K.Y.; Project Administration: L.L., K.Y.; Funding Acquisition: Y.Z., K.Y.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, N., Meng, Y., Mao, S. et al. FBXO31-mediated ubiquitination of OGT maintains O-GlcNAcylation homeostasis to restrain endometrial malignancy. Nat Commun 16, 1274 (2025). https://doi.org/10.1038/s41467-025-56633-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56633-z
This article is cited by
-
HIF1A regulates follicular atresia through O-GlcNAcylation-mediated VEZF1/ET-1/FOXO1/BAX signaling in porcine granulosa cells
Journal of Animal Science and Biotechnology (2025)
