
Abstract
In an era where swift and precise diagnostic capabilities are paramount, we introduce NAPTUNE (Nucleic acids and Protein Biomarkers Testing via Ultra-sensitive Nucleases Escalation), an innovative platform for the amplification-free detection of nucleic acids and protein biomarkers in less than 45 minutes. Using a tandem cascade of endonucleases, NAPTUNE employs apurinic/apyrimidinic endonuclease 1 (APE1) to generate DNA guides, enabling the detection of target nucleic acids at femtomolar levels. The sensitivity is elevated to attomolar levels through the action of Pyrococcus furiosus Argonaute (PfAgo), which intensifies probe cleavage, thereby boosting both sensitivity and specificity within an innovative in-situ cascade circuit. This technology not only streamlines rapid, onsite diagnostics without pre-amplification but also demonstrates exceptional accuracy in identifying a broad spectrum of nucleic acids and crucial cancer-related protein biomarkers directly from clinical samples. The development of a portable device for point-of-care testing further underscores NAPTUNE’s potential to transform diagnostic processes, especially in resource-limited environments, marking a significant diversity forward in medical diagnostics and patient care.
Similar content being viewed by others

Introduction
The early and reliable detection of nucleic acids and proteins biomarkers is critical in disease control, precise treatment, and surveillance monitoring1,2. Gold standard methods such as polymerase chain reaction (PCR) and quantitative real-time polymerase chain reaction (qRT-PCR) have revolutionized diagnostics by enabling the detection of nucleic acids, and immunological methods are widely used for detecting protein markers. However, these methods often involve operational complexity, time consumption (from 4 to 6 h), operational complexity, and reliance on specialized equipment, leading to a growing demand for faster, more accurate, and user-friendly diagnostic tools3,4,5,6. These challenges underscore the urgent need to explore other diagnostic strategies that are not only rapid but also exhibit high specificity and sensitivity.
Point-of-care testing (POCT) is essential for environmental monitoring, food safety, and disease prevention. It can be performed at the point of need or in environments with limited resources.7,8,9,10. Recent developments in programmable nucleases with sequence-specific catalytic properties, particularly within the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas) systems, have garnered attention for their potential in molecular diagnostics. Certain Cas proteins exhibit collateral cleavage on non-specific targets11,12,13,14,15,16,17. This unique feature has enabled the development of CRISPR-based POCT platforms such as SHERLOCK and DETECTR, which demonstrate high specificity and sensitivity18,19,20,21. Despite the promise, challenges, including the reliance on the protospacer adjacent motif (PAM) and increased cost and instability associated with guide RNA may impede the widespread use of CRISPR/Cas systems in diagnostics.
Argonaute proteins are categorized into two primary groups, including eukaryotic Argonautes (eAgos) and prokaryotic Argonautes (pAgos)22,23,24. The eAgos play a pivotal role in RNA interference pathways, engaging in various cellular processes such as transcriptional and posttranscriptional gene silencing, host defense mechanisms, and others25,26. The pAgos can be subdivided into short-length and full-length Ago23. Most pAgos utilize short DNA guides (gDNA) and operate without the need for a PAM, cleaving the complementary strand between the 10th and 11th bases from the 5′-end of the gDNA27,28,29. However, mesophilic pAgos such as Clostridium perfringens Argonaute (CpAgo), Intestinibacter bartlettii Argonaute (IbAgo), and Clostridium butyricum Argonaute (CbAgo), have been reported to lack the ability to digest dsDNA targets with high GC content due to their inability to unwind dsDNA, potentially limiting their applications in nucleic acids detection30,31. In contrast, recent advancements in thermophilic pAgos, such as Pyrococcus furiosus Argonaute (pfAgo) and Thermus thermophilus Argonaute (TtAgo), have positioned them prominently in nucleic acids testing and as focal points in biosensor research. Leveraging their high-temperature tolerant characteristics, thermophilic pAgos overcome the challenge of opening double-stranded DNA (dsDNA)32,33. Nonetheless, their current application is confined to nucleic acids detection with amplification, particularly RNA detection. We suppose the possibility of changing the existing behavior of pAgos to utilize successive cleavage of single-stranded DNA (ssDNA) as a robust signal transduction mechanism, aiming to extend the applications of pAgos beyond nucleic acids diagnostics to include non-nucleic acids targets, such as cancer-related protein biomarkers diagnostics.
In this study, we present a comprehensive solution through the development of a new concept termed Nucleic acids and Protein Biomarkers Testing via Ultra-sensitive Nucleases Escalation (NAPTUNE) for the detection of nucleic acids without pre-amplification and protein biomarkers within 45 min using tandem endonuclease. The engineered NAPTUNE employs the APE1 and PfAgo endonucleases in a synergistic manner. First, the APE1 itself can establish a positive feedback loop for the target nucleic acids detection with high specificity and exponential signal amplification. Meanwhile, the APE1-triggered ceaseless cleavage products with 5′-phosphate ends can then be regarded as a gDNA guide to activate the PfAgo-mediated cis-cleavage system on the secondary and tertiary probes, leading to the exciting potential for converting APE1 ability toward target into the “APE1-Argonaut” in situ cascade amplified fluorescence signal mode. This amplification-free model eliminates the need for complex amplification steps, making the detection process faster, more cost-effective, and suitable for POCT applications. This proposed approach capitalizes on the cooperative enzymatic activities of APE1 and PfAgo, paving the way for an enhanced and efficient signal amplification process to achieve the hypersensitive, amplification-free, rapid, highly specific, single-tube and on-site detection of nucleic acids, especially non-coding RNAs (ncRNAs).
We validate the NAPTUNE platform using RNA samples isolated from both human breast adenocarcinoma and human cervical cancer cell lines, as well as neuroblastoma (NB) patient tissue. Employing straightforwardly synthesized probes, we further showcase the adaptability of the platform by deploying it to detect the protein biomarkers such as APE1 and FEN1, whose expression levels are typically elevated in different cancer cells (e.g., HepG2, MCF-7, HeLa) and various types of cancers (e.g., liver, breast, lung, colorectal, gastric), utilizing the in-situ cascade strategy integral to the NAPTUNE framework. Additionally, a portable device has been engineered to augment the platform’s accessibility and ease of use. The NAPTUNE platform is expected to substantially enhance the capability for onsite profiling of nucleic acids and protein biomarkers, enabling even individuals with minimal training to operate it effectively. This advancement is poised to markedly improve diagnostic precision and therapeutic outcomes in clinical settings.
Results
APE1 detects target and generates DNA guide for PfAgo
PfAgo has been thoroughly explored for its structural mechanisms and applications in nucleic acid detection. This protein typically forms a complex with a short (~16 nt) DNA fragment that serves as a guide (Supplementary Fig. 1a). Although these DNA guides are not sequence-specific, PfAgo recognition requires the presence of a 5′ phosphate group, a process mediated by Mg²⁺ (Fig. 1a, b). Importantly, the first base of the guide DNA does not participate in target recognition as it is buried within PfAgo, resulting in the guide DNA complementarity with the target DNA starting from the second base (Fig. 1a). Moreover, since all detection methods rely on the cis-cleavage of PfAgo on the probe or target, the resultant signal readout is typically not strong. Furthermore, the reprogramming of the guide DNA can only be achieved in vitro, which restricts the broader application potential of PfAgo.

a Detailed view of the PfAgo-gDNA-tDNA structure, illustrating how PfAgo bypasses the first base of the guide DNA. b The diagram depicts PfAgo’s interaction with guide RNA in the presence of magnesium, requiring a phosphate group. c Schematic diagram of APE1 for cleavage of AP site. Blue strand showing target and designed strand with AP site showing red. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/j94m460]. d Detailed hypothesized mechanism where APE1-generated DNA fragments with a 5′ phosphate are loaded into PfAgo as guide DNA. This panel shows APE1 cleavage, guide DNA loading, and targeting by PfAgo in a DNA-guided manner. e Denaturing PAGE (polyacrylamide gel electrophoresis) displaying the APE1 activity for DNA-RNA structure. This experiment was repeated three times. f Denaturing PAGE showing the PfAgo nuclease activity for cleavage of P2 within the context of APE1 digestion reaction. This experiment was repeated three times. g Schematic depiction of dephosphorylation of 5′ terminal labeled phosphate group by ALP unable to trigger PfAgo activity. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/j94m460]. h Denaturing PAGE showing the inactivity of PfAgo under the introduction ALP into the APE1 digestion reaction. This experiment was repeated three times. i Real-time profiles of signal response using fluorophore-quencher labeled P2 enable monitoring of the cleavage of P2 by PfAgo-gDNA. All the experiments were conducted in triplicate and graphs were represented by mean (bold line) ± standard deviation (SD).
Meanwhile, APE1, a key enzyme in the BER pathway, has the unique ability to recognize apurinic/apyrimidinic (AP) sites and mediate Mg2+ nucleophilic attacks on the phosphate site at the 5′ end of these sites (Fig. 1c, Supplementary Fig. 1b). This action results in the generation of a DNA fragment with a 5′ phosphate group, with the first nucleotide being baseless due to the AP site (Fig. 1c). Based on these properties, we hypothesized that this DNA fragment generated by APE1 could effectively serve as a guide DNA for PfAgo to target the secondary probe (P2) (Fig. 1d). To validate our hypothesis, we first confirmed APE1’s ability to generate an effective 5′ phosphate group through experiments involving APE1-mediated cleavage and T4 ligase-mediated ligation (Supplementary Fig. 1c, d).
Additionally, we introduced DNA probe 1 (P1), containing an AP site, to assess APE1 activity. This probe binds to a target RNA through 22 complementary bases. We selected microRNA 21 (miR-21), a well-known RNA biomarker frequently associated with diseases such as cancer and heart disease, as the target for detection34,35,36. The results showed clear DNA cleavage at the AP site on P1 in the presence of just 0.1 pM target RNA, demonstrating the sensitivity of the system (Fig. 1e). We extended this assay to determine the minimal RNA concentration detectable by APE1, with results showing detection capabilities down to 1 fM (Supplementary Fig. 1e, f), indicating robust activity and the potential for APE1 to continuously generate guide DNA.
To further confirm the functionality of the DNA fragment as a guide, we introduced a P2 designed to complement the sequence of the APE1-generated fragment. Figure 1f clearly demonstrates that PfAgo exhibited strong DNA cleavage on P2 when combined with APE1, P1, and the target RNA. In contrast, there was no cleavage on P2 in the absence of APE1, indicating that PfAgo’s activity was dependent on the product of P1 cleavage by APE1 (Fig. 1f). Additionally, to underscore the importance of the 5′ phosphate, we introduced alkaline phosphatase (ALP) into the reaction system, which hydrolyzes the 5′ phosphate, thereby preventing the proper loading of guide DNA and subsequently shutting down PfAgo cleavage activity on P2 (Fig. 1g). Real-time tracking of this process using fluorophore-quencher (FQ) labeled P2 confirmed that cleavage action was evident when APE1, P1, target RNA, and PfAgo were present, whereas minimal activity was observed upon the introduction of ALP (Fig. 1h, i).
In conclusion, this study proposes a diagnostic platform in which APE1 not only detects minimal concentrations of nucleic acids like RNA but also continuously generates short DNA guides for PfAgo, enhancing secondary signal generation on the cleavage of P2. This integration not only opens a new method for generating DNA guides for Ago proteins but also advances the combination of nucleic acid detection technologies.
NAPTUNE development based on in situ cascade
Leveraging this distinctive feature, we adeptly devised an innovative in situ cascade signal amplification system by harnessing the synergistic action of tandem APE1 and PfAgo endonucleases, named NAPTUNE. As shown in Fig. 2a, we introduced three FQ-labeled ssDNA probes, denoted as P1, P2, and probe 3 (P3). In the presence of the target RNA, the formation of DNA P1-RNA duplex structure is promptly subjected to cleavage by APE1, resulting in the release of the quencher-labeled fragment and manifesting a distinct and discernible signal output. Subsequently, in a remarkable feedback loop, the target can be turned over once again to recognize P1, reigniting a ceaseless cycle of signal amplification. This dynamic and self-sustaining process ensures robust and continuous signal amplification (Fig. 2a). Simultaneously, the 5′-phosphate ssDNA produced by APE1 serves as an in-situ gDNA, initiating PfAgo’s cleavage of DNA P2 and thereby enhancing signal intensity. In addition, the regenerated gDNA from the truncated P2, in conjunction with guide-free PfAgo, facilitates a specific subsequent cleavage of P3, resulting in the generation of a significantly robust signal output (Fig. 2a). In theory, this amplification could be infinite, depending on the number of fissile DNA probes introduced into the system. This intricately orchestrated in situ cascade circuit-generated amplification process not only enhances the sensitivity of the NAPTUNE platform but also enables ultra-sensitive, amplification-free detection, establishing it as a promising and efficient tool for precise target identification.

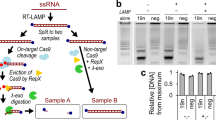
a Schematic for the working mechanism of NAPTUNE for recognition of nucleic acids. Briefly, probe 1 binds to RNA target and APE1 cuts the AP site of DNA-RNA structure, leading to target cycling for amplified signal output. Meanwhile, the APE1-cleaved product can be regarded as a gDNA for PfAgo system, resulting in two consecutive cutting with the additional P2 and P3, which leads to resulting in gradual signal enhancement by in situ cascade circuit. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/j94m460]. b Denaturing PAGE showing the PfAgo nuclease activity for cleavage of P1, P2, and P3 within the context of APE1 digestion reaction. This experiment was repeated three times. c Real-time monitoring of signal changes due to cleavage on FAM-BHQ1-labeled probes. The reaction begins with APE1-P1, followed by the addition of PfAgo-P2 at 45 min and P3 at 70 min, respectively. d Real-time analysis of pre-mixed APE1-PfAgo cascade signal amplification, the reaction of the control group is conducted only with probes and without target RNA, APE1 in the reaction are overlaid for comparison, all the probes are labeled with FAM-BHQ1 fluorophore. e Quantification and comparison of the sensitivity of APE1-P1, APE1-P1-PfAgo-P2 (NAPTUNE without P3), and APE1-P1-PfAgo-P2-P3 (NAPTUNE) demonstrate that NAPTUNE offers a 1000-fold improvement in detection sensitivity compared to using APE1 alone. NC, negative control assay with buffer to substitute miR-21 target. Each group consists of 12 parallel assays in a microplate reader (Tecan Spark GmbH, Austria). Statistical analysis was conducted using a two-tailed t-test. Statistical significance was determined as follows: ns (not significant) for p > 0.05, * for p ≤ 0.05, ** for p ≤ 0.01, *** for p ≤ 0.001, and **** for p ≤ 0.0001.
The experimental outcome substantiates the hypothesis that APE1-generated DNA fragments effectively guide, trigger, and augment the subsequent enzymatic activity of PfAgo, unveiling a synergistic interplay between these enzymatic components within our innovative NAPTUNE platform. As anticipated, the P3 probe also exhibited susceptibility to cleavage by the PfAgo protein within the APE1-mediated cyclic signal amplification reaction (Fig. 2b). To investigate whether this synergistic interplay is conserved across different pAgo families, we selected a mesophilic pAgo, CbAgo, which is distantly related to PfAgo (Supplementary Fig. 2a). Structural alignment revealed a Cα-RMSD (alpha carbon-root-mean-square deviation) of 5.7 (Supplementary Fig. 2b). Notably, our studies showed that the CbAgo protein exhibited behavior analogous to that of PfAgo (Supplementary Fig. 2c, d), highlighting the universality and versatility of this amplification strategy. Overall, this versatility opens the door to potential applications of various pAgos in diagnostic settings, further expanding the scope and adaptability of our NAPTUNE platform.
Next, we performed step-by-step fluorescence tracking within the APE1-PfAgo cascade by supplementing FQ-labeled probes (Fig. 2c). Our investigations unveiled that APE1 initiated an initial burst of fluorescence upon the introduction of P1, followed by the APE1-P1 reaction induced rapid surge in fluorescence, which was attributed to the distinct cleavage behavior of P2 by PfAgo. Notably, the introduction of P3 further augmented the fluorescence signal, highlighting the cumulative effect of the APE1-PfAgo cascade in enhancing the signal (Fig. 2c). Subsequently, we combined P1, P2, P3, APE1, and PfAgo in a single pot reaction and conducted a detailed assessment of the cascade behavior (Fig. 2d). Remarkably, the setup containing all components achieved the highest signal, while the group lacking P3 showed higher intensity than the group with only P1, demonstrating a similar activation pattern driven solely by APE1’s cleavage action. Importantly, in the absence of the miR-21 target or APE1, no discernible effects were observed (Fig. 2d). Furthermore, we aimed to test the limit of detection (LOD) using NAPTUNE. Notably, NAPTUNE with P2 and P3 can detect RNA at concentrations as low as 1 aM, significantly improving on the 1 fM detection limit of APE1-P1 alone, representing a 1000-fold enhancement (Fig. 2e).
Collectively, these findings strongly endorse the feasibility and effectiveness of our proposed NAPTUNE platform, emphasizing its robust signal amplification capabilities and its potential for sensitive and reliable target detection.
Engineering optimization of NAPUNE assay
In pursuit of enhancing the simplicity, convenience, and avoiding contamination of the NAPTUNE platform, our focus shifted towards streamlining the procedure through the implementation of a one-pot reaction involving APE1 and PfAgo cascade reaction. Although there is little difference in fluorescence signal intensity, the one-pot efficiency of APE1-activated fluorescence takes a longer time to reach the plateau than the two-step assay (Fig. 3a, b). This discrepancy may arise from the inherent complexity of the molecular interactions within the system. When P1 engages with the target, its efficacy may encounter interference from P2 and P3 due to the simultaneous presence of all assay reagents within the reaction tube. Alternatively, by designing a routine 0.2 mL affiliated with a mini tube in which the PfAgo reaction system is placed to separate PfAgo and APE1 reaction system, this one-tube design not only eliminates the need for a lid-opening operation but also minimizes the risk of potential cross-contamination and mutual interference between the reaction systems (Supplementary Fig. 3a). As shown in Fig. 3c, it became evident that the specific design outperformed the conventional arrangement of the PfAgo reaction system on the tube wall or in the tube cap. Notably, the APE1 buffer demonstrated commendable compatibility with the PfAgo cleavage reaction (Supplementary Fig. 3b–f), contributing to the design’s superior performance in comparison.

a Real-time analysis of APE1-PfAgo cascade in two-step assay. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/p72c816]. b Real-time analysis of APE1-PfAgo cascade in one-pot assay. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/p72c816]. c Study of the APE1-PfAgo one-tube reaction method involved inner tube, tube cap, and tube wall placement of PfAgo reaction mixture droplet to achieve the best performance. 1 pM of target were used in this study. All the experiments were conducted in three technical replicates and error bars represent mean value ± SD (n = 3). [Created in BioRender. Dbs, D. (2025) https://BioRender.com/p72c816]. d Schematic diagram of the designed AP sites from position 1 to 22 covering the miR-21 sequence of P1. 1 pM of target were used in this study. e Cleavage efficiency of APE1 activity for different AP site positions. The structure model (PDB code: 1DE8) highlights the substrate DNA fragment length besides the AP site. Error bars represent the mean ± SD, where n = 3 technical replicates. f Denaturing PAGE displaying the APEI activity for different AP site positions. g Denaturing PAGE evaluation of the best activity of APE1 in different temperatures. h Denaturing PAGE study of the best temperature for PfAgo-mediated cleavage. i Fluorescence profiling of APE1 cleavage reaction as a function of time in the presence of different miR-21 concentrations (NC, negative control assay with buffer to substitute miR-21 target, 0.1 pM, 1.0 pM, and 10.0 pM). j Real-time fluorescence changes of PfAgo cleavage reaction as a function of time in the presence of different miR-21 concentrations (NC, negative control assay with buffer to substitute miR-21 target, 0.1 pM, 1.0 pM, and 10.0 pM). All Denaturing PAGE experiments were repeated three times.
Extensive research has highlighted the intricate relationship between APE1 activity and the neighboring bases surrounding the AP site. In our quest to refine the NAPTUNE platform, we focused our efforts on pinpointing the optimal position for the AP site on P1. We strategically devised 22 extended variants of P1, each spanning 40 bases, meticulously ensuring that the “X” position could encompass every base of the miR-21 target (Fig. 3d). Notably, our observations unveiled a noteworthy trend: the proficiency of APE1 in recognizing the AP site within the middle region of enzyme cleavage significantly surpassed its efficacy in regions proximal to the 5′-end or 3′-end of the DNA-RNA structure (Fig. 3e). This intriguing phenomenon can be attributed to the inadequacy of shorter DNA fragments to provide the requisite stability and structure necessary to support APE1 activity (Structural model shown inside of Fig. 3e). Moreover, our findings elucidated that the optimal position for the AP site within the designed P1 was position 11 (P1@11) (Fig. 3e). This discernment offers invaluable insights for guiding the design of biosensors, highlighting the critical importance of strategic positioning within the probe structure.
Although the previous study showed that the APE1 cleavage of dsDNA was set at 37 °C, we observed that the best activity of APE1 for incision of AP site of the annealed DNA-RNA structure formed by P1@11 and miR-21 was 42 °C (Fig. 3g). Meanwhile, we further evaluated the length of gDNA (Supplementary Fig. 3g) and PfAgo-mediated cleavage of substrate at different temperature from 50 °C to 90 °C, showing high cleavage efficiency at 90 °C (Fig. 3h). Next, we focused on testing the other optimal conditions for the NAPTUNE platform, as illustrated in Supplementary Fig. 3h, i. We discerned that the reaction sustaining a pH of 8.0 with 250 nM PfAgo, 100 mU/mL APE1, 100 nM P1, and 200 nM for both P2 and P3, yielded the best performance of NAPTUNE. After this comprehensive optimization, we meticulously evaluated the turnaround time of the NAPTUNE platform. The real-time fluorescence results unequivocally indicated that the signal from APE1 cleavage reached saturation within a mere 20 min (Fig. 3i). Concurrently, the optimal reaction time for the PfAgo system was meticulously determined to be 25 min (Fig. 3j). This efficient orchestration of reaction times underscored the effectiveness of the streamlined one-tube NAPTUNE platform, significantly simplifying the assay workflow. As a result, the turnaround time was notably condensed to a mere 45 min.
NAPTUNE demonstrates multiplexing efficiency and detection fidelity
Given that numerous ncRNAs, including short microRNAs (miRNAs), long ncRNAs (lncRNAs), and circular RNAs (circRNAs), often exhibit elevated expression levels in various cancers37,38,39,40,41,42, their potential suitability for monitoring cancer progression and recurrence is compelling. Significantly, our developed assay presents a promising avenue for showcasing this potential. Designing P1 to target different ncRNAs is exceptionally convenient and straightforward (Fig. 4a). This exemplifies the remarkable programmability inherent in our NAPTUNE platform, emphasizing its versatility and adaptability for diverse applications in the realm of cancer research and diagnostics. We selected circRNA1141 (circRNA hsa_circ_0001141), HOTAIR (lncRNA HOX antisense intergenic RNA), and miR-21as models to demonstrate their feasibility, and their targeted sequences were shown in Fig. 4b. Notably, NAPTUNE platform displayed good performance for the three types of ncRNAs. The sensitivity was determined to be as low as 1 aM for miR-21, HOTAIR, and circRNA1141 (Fig. 4c, Supplementary Fig. 4a). The sensitivity was slightly higher or comparable to that of the standard qRT-PCR (0.1 aM of miR-21, 1 aM of HOTAIR and 500 aM of circRNA1141) assays (Fig. 4d, Supplementary Fig. 4b–d). Our assays were also shown to afford highly specific detection with minimal cross-reactivity from the ncRNAs targets (Fig. 4e). To further evaluate the fidelity of our assay for detecting RNA, we investigated the effect of mismatches with single-point, two bases, and three bases mutations from position 7 to 17 of miR-21 target (Supplementary Fig. 5a). We found that one base mismatches near the AP site (positions 11, 12, 13, and 14) slightly affected APE1 cleavage activity, whereas mutations further away from the AP site had a more pronounced impact on APE1 cleavage efficiency (Supplementary Fig. 5b, upper panel). For double and triple base-pair mismatches, only mutations involving AP site (M11-12 and M10-12) showed a slight decrease in APE1 activity (Supplementary Fig. 5b, lower panel). Together, those results revealed that mutations closer to the AP site have a lower effect on the activity of APE1 as compared to mutations away from the AP site.

a Schematic representation of NAPTUNE for detection of circRNAs, lncRNAs, and miRNAs. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/p72c816]. b Detailed P1 designed for the three types of ncRNAs detection, circRNA1141, HOTAIR, and miR-21. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/p72c816]. c Quantifications of NAPTUNE for the detection of different concentrations of circRNA1141, HOTAIR, and miR-21. Dotted lines are plotted to show the background fluorescence level. All the experiments were conducted in three technical replicates and error bars represent mean value ± SD (n = 3). d Comparing the limit of detections (LODs) for the NAPTUNE and qRT-PCR analyses of the three types of ncRNAs targets (1 pM each). e Evaluation of specificity across three types of ncRNAs targets (1 pM each) by NAPTUNE. The color intensity represents the average signal level of three technical replicates for each target. NC, negative control assay with buffer to substitute RNA target. f The diagram illustrates the detection of multiple targets in a single-pot assay. This method employs various types of fluorophore-quencher labeled probes to monitor different target RNAs. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/j94m460]. g Representative results of simultaneous HOTAIR and miR-21 detection using NAPTUNE. In this assay, the HEX signal (red) indicates miR-21, while FAM (green) reflects HOTAIR. CircRNA1141 was used as the non-target RNA. h Quantification of multiple target (1 Pm each) detection results from 12 parallel assays in a microplate reader. CircRNA1141 was used as the non-target RNA.
Based on these findings, the NAPTUNE assay demonstrates robust capabilities in detecting a diverse array of RNAs, exhibiting performance that is either superior to or on par with that of qRT-PCR. Moving forward, we aimed to assess the NAPTUNE’s capacity for simultaneous detection of multiple target RNAs within a one-tube setup. To this end, we combined APE1 and PfAgo enzymes with two distinct sets of probes, each targeting different RNA sequences: HOTAIR and miR-21, respectively (Fig. 4f). Each probe set was uniquely labeled with a specific FQ pair to facilitate differential detection. Probes targeting HOTAIR were tagged with FAM, which emits at 518 nm, while those targeting miR-21 were labeled with HEX, which has an emission peak at 553 nm. This labeling strategy leverages the distinct excitation and emission spectra of FAM and HEX, enabling the use of a fluorescence scanner to separately identify and quantify the signals corresponding to each RNA target (Fig. 4f). The fluorescence detection thus reflects the specific cleavage of each probe, indicating the presence or absence of the respective RNA molecules.
Our experimental results demonstrated the simultaneous detection capability of the NAPTUNE assay. When the tested sample contained only HOTAIR, the fluorescence emission was predominantly green, whereas samples containing only miR-21 emitted red fluorescence. In samples where both target RNAs were present, the resultant emission was a distinct yellow, indicating the simultaneous detection of both targets (Fig. 4g). Quantitative analysis of these results revealed that the NAPTUNE assay is exceptionally sensitive and precise in detecting multiplexed RNA targets within a single assay format (Fig. 4h). This level of multiplexing efficiency and detection fidelity underscores the potential of the NAPTUNE assay for complex biological analyses and diagnostic applications.
NAPTUNE displays robust activity in handling clinical samples and cancer cells
The versatility and applicability of the NAPTUNE platform were explored through the detection of miR-21 in a range of human tumor cell lines, including human breast adenocarcinoma cells (MCF-7) and the human cervical cancer cells (Hela), and using the human normal hepatocytes LO2 as the control group. To ensure the robustness and accuracy of our platform, we employed qRT-PCR as the gold standard for validation (Fig. 5a, b), the NAPTUNE platform presents a shorter assay time and easier-to-interpret signals. The comprehensive assessment of miR-21 expression levels across MCF-7 and Hela showcased varying degrees of elevation compared to LO2, consistently mirroring the findings of the qRT-PCR analysis (Fig. 5c and Supplementary Fig. 6a). This validation process underscores the reliability and precision of the NAPTUNE platform in accurately detecting miR-21 expression levels across diverse cancer cell types.

a Schematic representation of NAPTUNE distinguishes RNA biomarkers from different tissues and cell lines. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/j94m460]. b Comparison of representative results from real-time signal profiles using NAPTUNE and qRT-PCR. c Quantification of the detection results of NAPTUNE and qRT-PCR for the relative expression of miR-21 in Hela and MCF-7 cells (two-tailed t-test, ** for p ≤ 0.01, *** for p ≤ 0.001, and **** for p ≤ 0.0001; all plots show mean ± SD for n = 3 technical replicates). d The diagram illustrates the clinical sampling of NB cancer and the subsequent RNA extraction process. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/j94m460]. Detection results (e) and summarization (f) of NAPTUNE from clinical samples of neuroblastoma cancer patients. The results were categorized into two groups based on qRT-PCR findings: one negative and the other positive. The outcomes from both detection methods were consistent across all samples, except for sample 24, as indicated by the black arrow. Cut-off value (1.33) is marked as dashed black line. All the clinical samples of (e) were conducted in three technical replicates and error bars represent mean value ± SD (n = 3). g ROC curves generated from the diagnostic results of a total of 40 clinical samples. h Evaluation of NAPTUNE sensitivity and specificity compared to standard qRT-PCR using a confusion matrix. The sensitivity was 95.0%, its specificity was 100.0%, and the accuracy was 97.5%. i Signal readings for each group from qRT-PCR and NAPTUNE reveal marked discrepancies and a wider range of results between the negative and positive groups of NAPTUNE. For qRT-PCR, negative groups show a cycle threshold (Ct) of ~37, while positive samples display a Ct around 25, resulting in a relative difference of ~0.48 ((Ct positive − Ct negative)/Ct negative). In contrast, NAPTUNE positive results average around 8000 arbitrary units (a.u.) and negative around 400 a.u., with a difference value of 19 times ((Fluorescence Positive − Fluorescence Negative)/ Fluorescence Negative). Each dot represents a reported test result (N = 20 samples). Statistical significance was determined as follows: **** for p ≤ 0.0001, two-tailed t-test.
We further explored the potential utility of the NAPTUNE assay for miRNAs in clinical specimens. Notably, previous studies have highlighted the overexpression of miR-21 in NB cancer patients, attributing to the invasion, migration, and metastasis of NB cancer35,43,44,45. In our analysis, we measured the expression levels of miR-21 in tissue samples collected from children diagnosed with NB using the NAPTUNE (Fig. 5d). As detailed in Fig. 5f, a total of 40 children’s samples (detailed information shown in Supplementary Data 1) were meticulously examined using the qRT-PCR method (Ct threshold: 35), confirming 20 positive samples and 20 negative samples (Ct threshold: 35). Subsequent testing with our NAPTUNE assay ((cut-off = mean fluorescence value of 10 negative samples + 5 × SD), Supplementary Fig. 6b,c) exhibited a remarkable agreement, with 95.0% sensitivity, 100.0% specificity, and 97.5% accuracy negative when compared to the qRT-PCR method (Fig. 5e–g). Additionally, the receiver operating characteristic analysis of NAPTUNE displayed an area-under-the-curve value of 0.985 (Fig. 5h), further underscoring its potential for the detection of miR-21 in clinical samples. This analysis reveals that NAPTUNE exhibits performance comparable to qRT-PCR when analyzing clinical samples. Crucially, NAPTUNE demonstrates a greater distinction between negative and positive samples. While qRT-PCR differentiates positive from negative results based on cycle threshold (Ct) values—with negative samples typically showing a Ct of around 37 and positive samples around 25, resulting in a difference value change of ~0.48 times ((Ct positive − Ct negative)/Ct negative) (Fig. 5i). NAPTUNE utilizes a fluorescence reader that shows more pronounced differences. Specifically, fluorescence readings for negative results average around 400 arbitrary units (a.u.), while positive results average ~8000 a.u., yielding a difference value change of 19 times (Fluo. Positive-Fluo. Negative)/Fluo. negative). This significant discrepancy markedly improves the ease of distinguishing between negative and positive results compared to qRT-PCR (Fig. 5i). Moreover, the broad range of fluorescence values observed in the positive samples suggests that NAPTUNE can effectively quantify the concentration of target molecules, further supporting its utility in clinical settings (Fig. 5i).
In summary, these comprehensive findings underscore the clinical viability of the NAPTUNE platform for the diagnosis of ncRNAs, such as miR-21. Notably, this platform offers several advantages, including ease of operation, no need for expensive equipment, and a rapid turnaround time (45 min vs 160 min of qRT-PCR, Fig. 5b), positioning it as a promising tool for streamlined and efficient clinical diagnostics.
Harnessing NAPTUNE for protein biomarkers testing
Cancer diagnosis and prognosis are often associated with the abnormal expression of certain proteins that are produced either directly by the cancer cells or in response to their presence46,47. Such protein biomarkers are generally present in blood and occasionally in urine. In this study, the NAPTUNE assay demonstrated robust capability for nucleic acids detection. Additionally, the core enzyme APE1 has been identified as a protein biomarker in numerous cancers. Consequently, we selected APE1 and its functional analog, flap endonuclease 1 (FEN1), as potential biomarkers to explore whether the NAPTUNE assay could effectively identify the presence of these enzymes.
APE1 and FEN1 were found in breast cancer, gastric cancer, and lung cancer, suggesting the essential role of them in tumorigenesis48,49,50,51. Protein expression analyses revealed that APE1 and FEN1 levels are significantly elevated in a broad spectrum of cancerous and tumorigenic cell types, with the highest expression observed in chronic lymphocytic leukemia (Fig. 6a). This indicates that APE1 and FEN1 may serve as effective biomarkers for lymphoid malignancies. To further substantiate this, we compared mRNA transcription levels of APE1 and FEN1 in cells/tissues from normal lymph nodes and those in lymphoma cell lines. Notably, the transcription levels of APE1 and FEN1 mRNA were elevated approximately threefold in the lymphoma cell lines, underscoring their potential as biomarkers in cancer diagnostics (Fig. 6b). Given the challenges associated with direct sampling of cancerous tissues, we chose to assess these biomarkers in blood samples. We checked the mass spectrometry database to quantify the protein levels of FEN1 and APE1 in human plasma, revealing concentrations of 0.075 ng/ml and 0.23 ng/ml, respectively (Supplementary Fig. 7a, b). These findings enabled the use of blood samples for further testing using the NAPTUNE platform. By extracting plasma from patient samples, we can apply the NAPTUNE assay to determine whether the levels of APE1 and FEN1 are sufficiently high to trigger the assay’s nuclease cascade, providing an approach for the molecular diagnosis of lymphoid cancers (Fig. 6c).

a Diagram illustrating the normalized protein expression levels across various cancers for APE1 and FEN1. These data were obtained from the open-access database, the Human Protein Atlas. The results highlight a significant expression increase of both protein biomarkers in chronic lymphocytic leukemia. Each dot represents a patient. The center line represents the median expression level, the bounds of the box indicate the interquartile range, and the whiskers indicate the maximum and minimum values. b The mRNA normalized transcripts per million (nTPM) distribution of APE1 and FEN1 in normal lymph nodes and lymphama cell lines, where each dot represents a reported test result (N = 107, 113, 130, and 104 samples, respectively). The black line represents the median expression level. Statistical significance was determined as follows: * for p ≤ 0.05, ** for p ≤ 0.01, *** for p ≤ 0.001, and **** for p ≤ 0.0001, two-tailed t-test. c The diagram outlines the testing of protein biomarkers using NAPTUNE. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/j94m460]. Native-PAGE showing the feasibility of NAPTUNE in the presence of FEN1 (d) and APE1 (e), respectively. S2, S3, and S4 are labeled with Cy3 (Blue), Cy5 (Red), and FAM (Green). P1 of the dsDNA, P2, and P3 are labeled with FAM (Red), Cy3 (Blue), and Cy5 (Green) Red + Blue = Pink; Blue + Green = Cyan. All Denaturing PAGE experiments were repeated three times. Calibration curves for quantitative analysis of FEN1 protein biomarker using NAPTUNE (f) and ELISA method (g), respectively. h Specificity evaluation of NAPTUNE for recognition of FEN1, from BSA, ExoII, ExoI, APE1. i Signal intensities of NAPTUNE profiling of FEN1 for negative and spiked groups. The black line represents the median expression level, where each dot represents a reported test result (N = 22 samples). Statistical significance was determined as follows: * for p ≤ 0.05, ** for p ≤ 0.01, *** for p ≤ 0.001, and **** for p ≤ 0.0001, two-tailed t-test.
As shown in Fig. 6c, an overlap flap structure was rationally designed (Supplementary Fig. 7c) that allows FEN-1 to cleave the 5′-flap containing the fluorophore, exposing a 5′phosphate group and yielding a detectable fluorescence signal. The FEN-1-cleaved products from the S1 probe can be regarded as gDNA, activating the PfAgo-mediated cleavage of the S2 probe. The renewed gDNA then cuts the additional S3 probe. Following this diagram, more abundant FEN1 leads to higher fluorescence, manifesting an activatable biosensor for FEN1 detection. We utilized non-denaturing PAGE (Fig. 6d) and denaturing PAGE (Supplementary Fig. 7d) to validate this proposed sensing scheme. As shown in Fig. 6d, lane 1 exhibited the DNA duplex with overlap flap structure with S2 containing the Cy3 fluorophore and S3 with the Cy5 fluorophore. Upon the introduction of FEN1, lane 2 displayed a diminished blue band, indicating FEN1-mediated cleavage of S1. As anticipated, the FEN1-induced product triggered the pfAgo cleavage of the S2 probe, resulting in a short red band in lane 4, whereas it remained intact in lane 3. Furthermore, lane 6 demonstrated the complete digestion of the S3 probe. These findings strongly suggest the feasibility of an in-situ cascade behavior between protein biomarkers, such as FEN1 and pfAgo. Additionally, we designed a double-stranded DNA probe (22 bp) with an AP site for APE1 activity testing, as detailed in Supplementary Fig. 7e. Figure 6e clearly underscored the potential for detecting APE1 activity.
We further delved into evaluating the performance of NAPTUNE for sensing FEN1. In contrast to the conventional enzyme-linked immunosorbent assay (ELISA) method, which entails intricate steps and is time-consuming (Supplementary Fig. 7f), our observations revealed that NAPTUNE exhibited strong analytical prowess, boasting a LOD as low as 1.25 ng/ml (equal to ~0.5 mU/mL) (Fig. 6f), showing similar performance with ELISA method (Fig. 6g). Moreover, we examined the selectivity of the biosensor by testing it against several other crucial protein enzymes or proteins, namely bovine serum protein (BSA), Exonuclease I (EXO I), Exonuclease II (EXO II), and APE1. As shown in Fig. 6h, these four interference agents exhibited minimal fluorescence intensity, underscoring the remarkable specificity of our biosensor for detecting FEN1. We next assessed its applicability and specificity for detecting the FEN1 target in biological samples. Due to the lack of clinical samples for this test, we evaluated the potential matrix effects of the method using serum-mimicking samples spiked with FEN1. Significant differences were found between the spiked group and negative control (p < 0.001, n = 22) (Fig. 6i). In summary, NAPTUNE offers a simple, reliable, rapid (45 min), and cost-effective approach for detecting protein biomarkers, enabling effective diagnosis in the early stages of related cancers.
Portable device enables on-site NAPTUNE testing
Smartphone-based POCT is quickly emerging as a promising alternative to traditional laboratory-based diagnostic testing due to economic factors and the accessibility of medical equipment, particularly in resource-limited settings52,53. Based on the robust performance for testing of nucleic acids and protein biomarkers, our next objective was to transform our proposed NAPTUNE into a point-of-care (POC) detection assay by designing a portable, affordable, and easy-to-use alternative to the need for specialist lab equipment (Fig. 7a). To this end, we skillfully designed a small and lightweight device (40 × 30 × 55 mm, 50 g) by employing a mobile phone to display test results (Supplementary Fig. 8a, b). We then assessed the device in comparison to the qRT-PCR thermocycler. This simple heating method was found to preserve ~91% of the amplification efficiency obtained with the sophisticated PCR thermocycler (Supplementary Fig. 8c), indicating that the portable device is an ideal choice and suit for POC platform. We further validated the device by recognizing nucleic acids and protein biomarkers in clinical samples. Figure 7b illustrates the ability of the smartphone-based POC device to detect varying levels of miR-21 in clinical samples, aligning consistently with the qRT-PCR method. In a parallel endeavor, Fig. 7c demonstrates the POC device’s proficiency in detecting differential levels of FEN1 in sparked samples, showcasing a robust linear correlation with the qRT-PCR method. Collectively, the smartphone-based POC testing makes NAPTUNE a versatile tool for the development of new, cost-effective POC diagnostic tests, highlighting the commercial potential of the NAPTUNE for nucleic acids and protein biomarkers with the advantages of simple operation, no need for expensive equipment, and timesaving. More practically, Fig. 7d outlines the main equipment required for NAPTUNE, which includes a white tube for nucleic acids detection, a pink tube for protein biomarkers detection, a portable device, and a handheld centrifuge.

a In-field diagnosis of nucleic acids and protein biomarkers using NAPTUNE. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/j94m460]. b Signal intensities of NAPTUNE profiling of miR-21 for negative control and neuroblastoma (NB) cancer group based on portable device and smartphone. Statistical analysis was conducted using a two-tailed t-test, *** for p ≤ 0.001. all plots show mean ± SD for n = 4 replicates). c Correlation between the parallel measurements by portable device and qRT-PCR for FEN1 detection. Each data point represents the meaning of three replicates obtained through each method. The linear Deming fitting was applied to the data points to produce the linear correlation curves at the 95% confidence level. The pink area represents the 95% confidence band. d Main laboratory equipment required for NAPTUNE (the white arrow indicates portable device. mini centrifuge, tips, and pipette). e Potential protein biomarkers detection using NAPTUNE. [Created in BioRender. Dbs, D. (2025) https://BioRender.com/j94m460]. f NAPTUNE showing the potential in future applications.
Discussion
Biomarker detection methods, including qRT-PCR and immunological methods, still encounter the problem of achieving rapid, specific, and sensitive detection. We provide a comprehensive solution through the development of NAPTUNE. This innovation marks a significant advancement in the field of molecular diagnostics, offering a rapid, sensitive, and versatile platform for the detection of nucleic acids and protein biomarkers. Our approach harnesses the collaborative enzymatic activities of the APE1 and PfAgo endonucleases, enabling amplification-free detection within a remarkably short timeframe. By strategically designing probe structures and optimizing reaction conditions, we achieve ultra-sensitive detection of nucleic acids at the aM level in a one-tube reaction within 45 min, showing some significant advantages over other methods in terms of detection time, convenience, and on-site detection. In addition, NAPTUNE was easily extended to detect protein biomarkers, showing advantageous portability, easy-to-implement operation, and short assay time, avoiding the possibility of open-lid contamination. We employed NAPTUNE platform for highly sensitive and selective detection of FEN1 at levels as low as 1.25 ng/ml.
In this research, we have introduced an enzyme cascade for the detection of nucleic acids and protein biomarkers, where the product of one enzyme serves as the substrate for the next enzyme, thereby initiating a continuous reaction chain. By integrating the cascade signal amplification method, our approach offers several notable advantages: (1) Enhanced sensitivity: NAPTUNE platform can amplify the initial signal multiple times without the need for complex laboratory equipment or specialized operating skills, greatly improving the sensitivity of detection; (2) Reduced background noise: by exclusively utilizing specific substrates for each subsequent reaction, our method effectively minimizes background noise, thereby leading to improved signal-to-noise ratios; (3) Efficient utilization of in situ gDNA products: an in situ generation of gDNA products serves as an efficient activator for the Ago system, further augmenting the detection sensitivity. (4) Another advantageous feature of our assay lies in the distinct activity temperatures of APE1 and FEN1 with PfAgo utilized in a one-tube reaction. Before PfAgo is activated, APE1 or FEN1 has been inactivated by high temperature, effectively avoiding mutual interference between the two enzymes, ensuring their independent and precise functionality. Such compatibility enhances the robustness of the assay and expands its potential applications under various environmental conditions. Importantly, the platform’s compatibility with clinical samples underscores its potential for real-world applications in cancer diagnosis and monitoring.
An early and accurate diagnosis of a diverse array of nucleic acids and protein biomarkers is essential in modern diagnostics because of its high sensitivity, specificity, and ability to provide rapid results. For nucleic acid detection, qRT-PCR is considered the gold standard for viral nucleic acid detection. It offers high sensitivity and specificity, but requires complex equipment and heat cycling, which makes it time-consuming, prone to contamination, and requires skilled operation. Other methods like microarrays and sequencing have powerful capabilities for highly multiplexed RNA profiling, providing both high throughput and flexibility. However, these methods are limited by turnaround time, labor intensity, and the requirement for sophisticated devices. Isothermal amplification shows great potential in resource-limited settings due to its applicability and speed features, as well as the fact that it does not require heat cycling and simplifies equipment requirements. CRISPR-Cas technology, known for its high specificity and adaptability, makes it a promising candidate for nucleic acid detection. However, it faces challenges, including cost and instability associated with guide RNA, constraints imposed by the PAM on detection sequences, and complications in multiplex target detection. In this work, NAPTUNE is versatile and can detect a variety of nucleic acid molecules by simply designing a DNA sequence within AP site (Fig. 4a, b). Additionally, we have provided a more detailed comparison with existing diagnostic technologies, evaluating key aspects such as sensitivity, specificity, time-to-result, and other critical performance metrics (Supplementary Table 1). This comparison highlights how our approach aligns with or surpasses current standards, offering a comprehensive perspective on its advantages and potential limitations.
The identification of protein biomarkers is of paramount importance in biomedical research, clinical diagnostics, and personalized medicine. Currently, high-sensitivity methods like ELISA and mass spectrometry provide accurate quantification and identification but may require expensive equipment and complex protocols. Techniques like western blot offer specificity and spatial localization but are time-consuming and semi-quantitative. Flow cytometry excels in multiparametric, high-throughput analysis but is costly. Immunofluorescence enables real-time detection but can be technically demanding. Like nucleic acid detection, NAPTUNE offers several significant advantages over other diagnostic methods. These advantages include shorter testing time, ease of use, low cost, and other merits, making it a highly competitive option in the field of diagnostics (Supplementary Table 2). BER has undergone evolutionary refinement to maintain the integrity of DNA in response to cellular oxidative stress and external insults. This pathway constitutes a meticulously orchestrated, sequential process involving at least 30 proteins, with single-strand breaks serving as intermediates during the repair cascade. Dysregulation of BER levels in human cells is implicated in a spectrum of diseases, including cancers, cardiovascular disorders, neurological ailments, and certain inflammation-related conditions34,54,55,56,57. Through meticulous scrutiny of over 30 functional attributes associated with BER-related enzymes, we postulate that the NAPTUNE platform has the potential to enable simultaneous detection of multiple enzymes. As illustrated in Fig. 7e, we consolidate these attributes into three distinct enzyme categories: (1) Enzymes directly or indirectly involved in the generation of AP sites; (2) Enzymes with specialized structure-cleavage properties; and (3) Enzymes involved in mismatch recognition. Notably, our investigation reveals that products cleaved by these enzymes yield 5’ phosphate groups, which can be readily identified utilizing the NAPTUNE platform concept we have pioneered.
The NAPTUNE platform we developed can not only detect proteins and nucleic acids but also has the potential to evaluate gene editing efficiency (Fig. 7f). Evaluating gene editing efficiency is crucial for assessing the success and accuracy of genetic modifications introduced by gene editing technologies. Mismatch-sensitive endonucleases can recognize and cleave double-stranded DNA fragments containing a mismatch, such as T7 Endo I and authenticase. Approaches employing these endonucleases are widely used because they are simple, fast, low-cost, and relatively sensitive. If the editing is successful, the target sequence will be cleaved by the enzyme and generate a 5′ phosphate end, from which NAPTUNE can be used to evaluate the editing efficiency.
Further efforts are needed to enhance NAPTUNE and make it a truly “plug-and-play” POCT method. It is essential to acknowledge certain limitations compared to the established qRT-PCR method, particularly in sensitivity improvement. Addressing this limitation could be achieved by enhancing the performance of portable instruments and incorporating microfluidic technology, potentially elevating the sensitivity of our assay and making it more adaptable to various diagnostic needs. Looking towards the future, there are promising avenues for further development. Scaling the platform for broader use, particularly in high-throughput environments or large-scale clinical trials, may pose challenges. This will necessitate optimizing assay protocols, integrating automated systems, and adjusting the platform to handle larger sample volumes. Addressing these scalability issues is essential to adapting the platform for diverse and large-scale applications. In addition, the reproducibility of the NAPTUNE assay across different clinical settings and with various types of samples remains to be fully established. Variations in sample types, handling procedures, and environmental conditions could potentially impact assay performance. We are actively working on standardizing the assay conditions and developing robust protocols that can be consistently applied across different laboratories and clinical environments.
Detecting low-abundance biomarkers in certain clinical samples was challenging, especially when the target concentration approached the detection limit. However, by employing magnetic bead separation, researchers can effectively enrich these low-abundance RNA targets, significantly improving the sensitivity and accuracy of subsequent molecular analyses. Certain components in clinical samples, such as proteins or other biomolecules, can also interfere with the detection process, potentially leading to false negatives or reduced assay performance. To mitigate these matrix effects, we have incorporated additional purification steps and optimized buffer formulations to minimize the impact of interfering substances. The system’s capability for multiplex detection, including both nucleic acids and protein detection, could significantly enhance its versatility and utility. Additionally, exploring the potential of mesophilic Ago protein as a signaling tool holds promise for expanding its applications in the diagnostic realm, paving the way for innovative approaches in ncRNA detection.
In conclusion, NAPTUNE demonstrates remarkable versatility through its ultra-sensitive and specific detection of various ncRNAs and protein biomarkers. Furthermore, we have engineered a smartphone-based portable device for signal quantification, rendering NAPTUNE an attractive option for biosensing and POC diagnostics, with far-reaching implications for the field of molecular diagnostics.
Methods
Ethical statement
This study involved human participants and was approved by Institutional Review Board and Ethics Committee of the Children’s Hospital, Zhejiang University School of Medicine (2024-IRB-0020-P-01). All procedures performed were in accordance with the ethical standards of institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Written informed consent required from their legal guardians for the reporting and sharing of individual-level data was obtained and all eligible participants completed a one-time medical history questionnaire.
Materials
RNA and DNA oligos were purchased from General Biol (Anhui) Co., Ltd (Chuzhou, China) and sequences are listed in Supplementary Data 2. APE1 and FEN1 were supplied by New England Biolabs (MA, U.S.A.). FEN1 ELISA kit (YQ-737140E) was provided by Shanghai Yanxi Biotechnology Co., Ltd (Shanghai, China). MiRNA 1st Strand cDNA Synthesis Kit (by stem-loop), HiScript III 1st Strand cDNA Synthesis Kit (+gDNA wiper). MiRNA Universal SYBR qPCR Master Mix and ChamQ Universal SYBR qPCR Master Mix were obtained from Vazyme (Nanjing, China). Ultrapure water was obtained from Millipore Synergy system (MA, U.S.A.) All other chemicals and reagents were purchased from Sigma-Aldrich (MO, USA), Macklin Biochemical Co., Ltd (Shanghai, China), and Beyotime Biotechnology (Shanghai, China).
Protein expression and purification
PfAgo and CbAgo were expressed in Escherichia coli and purified with the following procedures as reported previously.24,48 Briefly, Escherichia coli BL21(DE3) cells were transformed with the desired plasmids by heat shock. Then the transformant was inoculated in 10 mL Lysogeny Broth with appropriate antibiotics (100 μg/mL ampicillin for PfAgo and 50 μg/mL kanamycin for CbAgo) and incubated at 37 °C, 250 rpm for 16 h. After that, the small-scale culture was added to 1 L Lysogeny Broth with antibiotics and incubated at 37 °C until the OD600 reached 0.6–0.8. Protein expression was then induced with 1 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) at 16 °C for 16–18 h. The cells were collected by centrifugation (3500 × g, 15 min at 4 °C) and resuspended at 4 °C in lysis buffer (20 mM Tris/HCl pH 8.0, 500 mM NaCl for pfAgo and CbAgo), followed by disruption with Ultrasonic Homogenizer. The crude cell lysate was centrifuged (12,000 × g, 25 min at 4 °C) to remove the cell debris. The supernatant of pfAgo was collected, heated at 80 °C for 30 min, and was centrifuged (12,000 × g, 25 min at 4 °C) to remove the denatured protein. The supernatant was then applied to Ni-NTA affinity purification with 30 mL of wash buffer (20 mM Tris/HCl pH 8.0, 300 mM NaCl, 2 mM MnCl2, 50 mM imidazole for pfAgo and 20 mM Tris/HCl pH 7.5, 250 mM NaCl, 30 mM imidazole for CbAgo) and 20 mL of elution buffer (20 mM Tris/HCl pH 8.0, 1 M NaCl, 200 mM imidazole for pfAgo and 20 mM Tris/HCl pH 7.5, 250 mM NaCl, 250 mM imidazole for CbAgo). The eluted sample was concentrated to 100 μL using Amicon Ultra filter unit (Millipore, 30 kDa). The sample was then loaded onto a Superdex 200 16/60 size-exclusion column (Cytiva) equilibrated with buffer (20 mM HEPES pH 7.5, 150 mM NaCl), and were subsequently snap-frozen and stored at −80 °C.
Non-denaturing polyacrylamide gel electrophoresis
It was performed to explore the feasibility of NAPTUNE to detect nucleic acids and protein biomarkers, as well as the evaluation of cleavage efficiencies of NAPTUNE platform. Samples were mixed with the same volume of formamide loading dye, heta-denatured at 95 °C for 5 min, and immediately loaded into 20% ureal-polyacrylamide gel. The gel electrophoresis was performed at 140 V for 60 min in 0.5 X TBE buffer, which is used as the electrophoresis solution. The gels were visualized by ChemiDoc MP imaging system (Bio-Rad, Richmond, CA, USA).
Native polyacrylamide gel electrophoresis
It was conducted to explore the feasibility of NAPTUNE to detect APE1 and FEN1. 10 µL of samples with loading buffer (6×) was loaded onto 20% native PAGE gel. Electrophoresis at a voltage of 140 V for 40–60 min using 0.5 X TBE buffer as the electrophoresis buffer. The gels were then imaged with ChemiDoc MP imaging system (Bio-Rad, Richmond, CA, USA).
Procedures of two-step and one-pot method
In the two-step method, the 8 μL of mixture (50 mM Tris/HCl pH 7.4, 100 mM KCl, 5 mM MgCl2, 2 mM ATP, and 0.05 mg/mL BSA) containing probe 1 (100 nM), APE1 (0.1 U/ml), and 2 μL of target RNA (0.1 pM) were incubated in a tube at 42 °C for 20 min. After the APE1 reaction, the mixture was transferred to another tube containing 10 μL of PfAgo reaction mixture (250 nM PfAgo, 200 nM probe 2, 200 nM probe 3, 20 mM HEPES pH 7.5, 250 mM NaCl, 2.5 mM MnCl2) and incubated together at 90 °C for 25 min. In the one-pot method, all the reagents were mixed and incubated at 42 °C for 20 min, and then heated to 90 °C for another 25 min. In the one-tube method, 10 μL of the above APE1 reaction mixture was prepared in a routine 0.2 mL reaction and incubated at 42 °C for 20 min. Dropped the PfAgo reaction solution (10 μL) by a brief spin, Then, the tube was heated to 90 °C for another 25 min.
Probe preparation for FEN1 and APE1 detection
For the detection of APE1, 5 μL of probe 1 (2 μM), 5 μL complementary ssDNA (2 μM), and 40 μL buffer (20 mM HEPES pH 7.5, 150 mM NaCl) were denatured at 95 °C for 2 min and then cooled down to room temperature to obtain the prepared dsDNA probe for further usage. For the detection of FEN1, 4 μL probe S1 (2 μM), 4 μL probe S2 (2 μM), 4 μL probe S3 (2 μM), and TE buffer (38 μL) were first denatured at 90 °C for 1 min and then cooled down to room temperature to obtain the double-flap structure for further usage.
Procedure of NAPTUNE assay for miR-21 in clinical samples
Two microliters of extracted sample were first added into a pink tube containing 8 μL of APE1 reaction solution (50 mM Tris/HCl pH 7.4, 100 mM KCl, 5 mM MgCl2, 2 mM ATP, and 0.05 mg/mL BSA, 100 mU/mL of APE1, 100 nM of probe 1). 10 μL of PfAgo reaction mixture (250 nM PfAgo, 200 nM probe 2, 200 nM probe 3, 20 mM HEPES pH 7.5, 250 mM NaCl, 2.5 mM MnCl2) was loaded to the mini tube. The tube was incubated at 42 °C for 20 min for APE1 reaction and heated to 90 °C. After that, the PfAgo reaction solution was dropped to the lower chamber by a brief spin. The mixed solution was incubated together at 90 °C for 25 min. The fluorescence intensity was recorded by a microplate reader (Tecan Spark GmbH, Austria). The real-time fluorescence signal was detected by the qPCR device (Bio-Rad, CFX connect real-time system).
Procedure of NAPTUNE assay for FEN1 in mimic clinical samples
Two microliters of the obtained mimic samples solution was introduced into the white tube containing 1 μL of double-flap structure probe, 7 μL of H2O, and 1 μL of 10 x ThermoPol reaction buffer at 65 °C for 20 min and the pfAgo reaction solution (250 nM S4 probe, 40 mM HEPES pH 7.5, 250 mM NaCl, 2.5 mM MnCl2) was introduced into the tube with rapid and brief centrifuge. The above-mixed solutions were incubated together at 90 °C for 25 min. The fluorescence intensity was recorded by a microplate reader.
Fabrication of smartphone device
We collaborated with Hangzhou EzDx Technology Co., Ltd to design and develop this portable device and its software. The original copyright of the software belongs to the company, while our usage rights are authorized by the company. We and collaborator utilized advanced three-dimensional (3D) printing technology with durable nylon material to fabricate all the brackets and enclosures for the device, ensuring both robustness and lightweight portability. The core components of this mini-portable device are several components: an optical module, a heating module, and a PCB control module. The optical module is equipped with a 480 nm light-emitting diode (LED) and a dichroic mirror, enabling precise excitation and accurate detection of fluorescence signals. The heating module is controlled by a microcontroller via General-purpose input/output (GPIO), ensuring stable and optimal reaction conditions throughout the assay. The PCB control module, equipped with the PB-02 Bluetooth module, integrates and manages all device operations. This setup allows for seamless wireless control, data processing, and communication with external smartphones, enabling real-time monitoring and analysis.
Operation process for smartphone-based detection: (a) The program was set by smartphone for the APE1-pfAgo two-step reaction-typically set at 42 °C for 20 min and 90 °C for 25 min. (b) The tubes containing APE1 reaction mixture in the lower chamber and pfAgo reaction solution in the upper chamber were placed in the detector. (c) Then, the tubes were heated to 42 °C for 20 min by running the program, the tubes were then taken out for brief spin and quickly replaced in the detector with the temperature heated to 90 °C. (d) All the fluorescence intensity was continuously monitored, with the data collected by the detector seamlessly transmitted to the smartphone via Bluetooth, and the results were displayed in real time on the smartphone screen.
Cell culture and total RNA extraction
The human normal hepatocytes LO2, the human breast cancer cell line MCF-7, and the human cervical cancer cell line HeLa were all cultured in DMEM medium with 10% FBS and 1% double-antibody at 37 °C with 5% CO2. One milliliter Trizol was added to 7 × 106 cells and total RNA was extracted and stored at −80 °C according to the manual.
Enzyme-linked immunosorbent assays
Different concentrations of FEN1 were added into a 96-well strip plate pre-coated with FEN1 antibodies for ELISA analysis. Measurements were carried out using a spectrophotometer (Thermo Scientific, USA) at the wavelength of 450 nm (Tecan Spark GmbH, Austria).
QRT-PCR
MiRNA cDNA synthesis was performed using miRNA 1st Strand cDNA Synthesis Kit (by stem-loop), circRNA and lncRNA’s cDNA synthesis was performed using HiScript III 1st Strand cDNA Synthesis Kit (+gDNA wiper). MiRNA Universal SYBR qPCR Master Mix was used for miRNA qPCR. ChamQ Universal SYBR qPCR Master Mix was used to detect circRNA and lncRNA levels for qPCR quantification according to the manufacturer’s instructions. Reverse transcription was performed with 20 μL of input, synthetic RNA standards (from 10,000 to 0.1 fM and non-targeting control, prepared in 1 ng/μL of PolyA carrier), or miRNAs extracted from clinical samples in 200 μL PCR tubes. QRT-PCR was performed on the CFX Opus 96 Real-Time PCR System with the amplification program including 40 cycles of 95 °C for 10 s and 60 °C for 30 s after initiation at 95 °C for 30 s.
Tissue and plasma collection
Tissue samples: a total of 40 samples, including 20 samples of normal tissue and 20 NB samples (detailed information see Supplementary Data 1) were obtained from patients treated at the Children’s Hospital, Zhejiang University School of Medicine (Hangzhou, China). Tumor samples were collected during surgical resection and samples were placed in RNAlater, and transported to the laboratory within 1 h. Total RNA was isolated from the tissues using BeyoMag™ Animal RNA Isolation Kit with Magnetic Beads according to the manual (Shanghai, China). A Nanodrop spectrophotometry instrument was used for assessing the quality of RNA and RNAs with an RNA Integrity Number greater than 7 and A260/A280 ratio between 1.8 and 2.0 were selected for detection. The RNA samples were stored at −80 °C in RNase-free tubes to prevent degradation before further use.
Ethical approval was provided by 2024-IRB-0020-P-01, and informed consent was obtained from all participants. (1) Blood samples were collected in plasma collection tubes which contained EDTA. The isolation process was conducted within 1.5 h on ice. The EDTA tube was first centrifuged at 1900 × g for 15 min at room temperature, and then the supernatant was stored at −80 °C until further processing. (2) We chose the ach sample which was thawed only once prior to analysis to preserve the stability of the protein markers. (3) The detection of tumor protein markers was performed using mass spectrometry, which is well-established, sensitive, and specific for quantifying low-abundance proteins in serum.
Protein and mRNA expression level analysis
In the present study, we conducted a comprehensive analysis of protein and mRNA expression levels for apurinic/apyrimidinic endonuclease 1 (APEX1) and FEN1. Our primary data source was The Human Protein Atlas, an accessible online database that provides extensive information on the expression profiles of proteins and genes in various human tissues and cells. For each target protein, specific database entries were referenced. The detailed information for APEX1 was accessed via https://www.proteinatlas.org/ENSG00000100823-APEX1, and for FEN1 via https://www.proteinatlas.org/ENSG00000168496-FEN1. Within the database, we retrieved quantitative and qualitative data detailing the expression levels of both APEX1 and FEN1 mRNA across a variety of cell lines. This included a comparison of expression profiles in both normal and cancerous cells, providing a broad overview of expression dynamics. In addition to mRNA data, we extracted information on protein expression levels as detected by mass spectrometry in plasma samples. This data was critical for assessing the prevalence and distribution of APEX1 and FEN1 in the circulatory system, which can provide insights into their potential as biomarkers.
Statistics and reproducibility
We used GraphPad Prism version 9.5 (GraphPad Software Inc.) for statistical analysis. For two-group comparisons, we used an unpaired, two-tailed t-test. For all statistical tests, p-values less than 0.05 were considered significant. Details on data presentation and the sample size are included in figure legends. In the study of clinical samples, no statistical method was used to predetermine the sample size, and the experiments were not randomized. No data were excluded from the analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are included in this article and its Supplementary Information file. Source data are provided with this paper.
References
Nemeth, K., Bayraktar, R., Ferracin, M. & Calin, G. A. Non-coding RNAs in disease: from mechanisms to therapeutics. Nat. Rev. Genet. 25, 211–232 (2024).
Crosby, D. et al. Early detection of cancer. Science 375, eaay9040 (2022).
Broughton, J. P. et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38, 870–874 (2020).
Vogels, C. B. F. et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat. Microbiol. 5, 1299–1305 (2020).
Rissin, D. M. et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 28, 595–599 (2010).
Roescher, N., Kingman, A., Shirota, Y., Chiorini, J. A. & Illei, G. G. Peptide-based ELISAs are not sensitive and specific enough to detect muscarinic receptor type 3 autoantibodies in serum from patients with Sjogren’s syndrome. Ann. Rheum. Dis. 70, 235–236 (2011).
Jang, H. et al. ANCA: artificial nucleic acids circuit with argonaute protein for one-step isothermal detection of antibiotic-resistant bacteria. Nat. Commun. 14, 8033 (2023).
Zhao, Y. et al. A universal CRISPR/Cas12a-powered intelligent point-of-care testing platform for multiple small molecules in the healthcare, environment, and food. Biosens. Bioelectron. 225, 115102 (2023).
Yang, H., Ledesma-Amaro, R., Gao, H., Ren, Y. & Deng, R. CRISPR-based biosensors for pathogenic biosafety. Biosens. Bioelectron. 228, 115189 (2023).
Wu, Y. et al. CRISPR-Cas12-based rapid authentication of halal food. J. Agric. Food Chem. 69, 10321–10328 (2021).
Karlikow, M. et al. CRISPR-induced DNA reorganization for multiplexed nucleic acids detection. Nat. Commun. 14, 1505 (2023).
Weng, Z. et al. CRISPR-Cas biochemistry and CRISPR-based molecular diagnostics. Angew. Chem. Int. Ed. 62, e202214987 (2023).
Kulkarni, A., Tanga, S., Karmakar, A., Hota, A. & Maji, B. CRISPR-based precision molecular diagnostics for disease detection and surveillance. ACS Appl. Bio Mater. 6, 3927–3945 (2023).
Yang, J. et al. Engineered LwaCas13a with enhanced collateral activity for nucleic acids detection. Nat. Chem. Biol. 19, 45–54 (2023).
Yang, Y. et al. Improving trans-cleavage activity of CRISPR-Cas13a using engineered crRNA with a uridinylate-rich 5’-overhang. Biosens. Bioelectron. 255, 116239 (2024).
Li, H. et al. Amplification-free CRISPR/Cas detection technology: challenges, strategies, and perspectives. Chem. Soc. Rev. 52, 361–382 (2023).
Hu, C. et al. Allosteric control of type I-A CRISPR-Cas3 complexes and establishment as effective nucleic acids detection and human genome editing tools. Mol. Cell 82, 2754–2768 e2755 (2022).
Kellner, M. J., Koob, J. G., Gootenberg, J. S., Abudayyeh, O. O. & Zhang, F. SHERLOCK: nucleic acids detection with CRISPR nucleases. Nat. Protoc. 14, 2986–3012 (2019).
Gootenberg, J. S. et al. Multiplexed and portable nucleic acids detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444 (2018).
Chen, J. S. et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439 (2018).
Harrington, L. B. et al. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 362, 839–842 (2018).
Swarts, D. C. et al. The evolutionary journey of Argonaute proteins. Nat. Struct. Mol. Biol. 21, 743–753 (2014).
Koopal, B., Mutte, S. K. & Swarts, D. C. A long look at short prokaryotic Argonautes. Trends Cell Biol. 33, 605–618 (2023).
Wang, L. et al. Molecular mechanism for target recognition, dimerization, and activation of Pyrococcus furiosus Argonaute. Mol. Cell 84, 675–686 e674 (2024).
Peters, L. & Meister, G. Argonaute proteins: mediators of RNA silencing. Mol. cell 26, 611–623 (2007).
Tolia, N. H. & Joshua-Tor, L. Slicer and the argonautes. Nat. Chem. Biol. 3, 36–43 (2007).
Qin, Y., Li, Y. & Hu, Y. Emerging Argonaute-based nucleic acids biosensors. Trends Biotechnol. 40, 910–914 (2022).
Xun, G., Lane, S. T., Petrov, V. A., Pepa, B. E. & Zhao, H. A rapid, accurate, scalable, and portable testing system for COVID-19 diagnosis. Nat. Commun. 12, 2905 (2021).
Swarts, D. C. et al. Argonaute of the archaeon Pyrococcus furiosus is a DNA-guided nuclease that targets cognate DNA. Nucleic Acids Res. 43, 5120–5129 (2015).
Lisitskaya, L., Aravin, A. A. & Kulbachinskiy, A. DNA interference and beyond: structure and functions of prokaryotic Argonaute proteins. Nat. Commun. 9, 5165 (2018).
Marsic, T. et al. Programmable site-specific DNA double-strand breaks via PNA-assisted prokaryotic Argonautes. Nucleic Acids Res. 51, 9491–9506 (2023).
He, R. et al. Pyrococcus furiosus Argonaute-mediated nucleic acids detection. Chem. Commun. 55, 13219–13222 (2019).
Song, J. et al. Highly specific enrichment of rare nucleic acids fractions using Thermus thermophilus argonaute with applications in cancer diagnostics. Nucleic Acids Res. 48, e19 (2020).
Jenike, A. E. & Halushka, M. K. miR-21: a non-specific biomarker of all maladies. Biomark. Res. 9, 18 (2021).
Jiang, Z. et al. miR-21 targets long noncoding RNA PCAT29 to promote cell proliferation in neuroblastoma. Crit. Rev. Eukaryot. Gene Expr. 32, 1–8 (2022).
Kumarswamy, R., Volkmann, I. & Thum, T. Regulation and function of miRNA-21 in health and disease. RNA Biol. 8, 706–713 (2011).
Goodall, G. J. & Wickramasinghe, V. O. RNA in cancer. Nat. Rev. Cancer 21, 22–36 (2021).
Kristensen, L. S., Jakobsen, T., Hager, H. & Kjems, J. The emerging roles of circRNAs in cancer and oncology. Nat. Rev. Clin. Oncol. 19, 188–206 (2022).
Stelekati, E. et al. MicroRNA-29a attenuates CD8 T cell exhaustion and induces memory-like CD8 T cells during chronic infection. Proc. Natl. Acad. Sci. USA 119, e2106083119 (2022).
Anastasiadou, E., Jacob, L. S. & Slack, F. J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 18, 5–18 (2018).
Slack, F. J. & Chinnaiyan, A. M. The role of non-coding RNAs in oncology. Cell 179, 1033–1055 (2019).
Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 12, 861–874 (2011).
Li, H. et al. MicroRNA-21 lowers blood pressure in spontaneous hypertensive rats by upregulating mitochondrial translation. Circulation 134, 734–751 (2016).
Li, Y., Shang, Y. M. & Wang, Q. W. MicroRNA-21 promotes the proliferation and invasion of neuroblastoma cells through targeting CHL1. Minerva Med. 107, 287–293 (2016).
Hammad, R. et al. Contribution of plasma MicroRNA-21, MicroRNA-155 and circulating monocytes plasticity to childhood neuroblastoma development and induction treatment outcome. Pathol. Res. Pract. 254, 155060 (2024).
Wu, L. & Qu, X. Cancer biomarker detection: recent achievements and challenges. Chem. Soc. Rev. 44, 2963–2997 (2015).
Jardim, D. L., Goodman, A., de Melo Gagliato, D. & Kurzrock, R. The challenges of tumor mutational burden as an immunotherapy biomarker. Cancer Cell 39, 154–173 (2021).
Tao, J., Zhang, H., Weinfeld, M. & Le, X. C. Development of a DNAzyme walker for the detection of APE1 in living cancer cells. Anal. Chem. 95, 14990–14997 (2023).
Li, B. et al. Signal-amplified detection of the tumor biomarker FEN1 based on cleavage-induced ligation of a dumbbell DNA probe and rolling circle amplification. Anal. Chem. 93, 3287–3294 (2021).
Chai, Q. et al. Closed cyclic DNA machine for sensitive logic operation and APE1 detection. Small 19, e2207736 (2023).
Cui, C. et al. Multimodal detection of flap endonuclease 1 activity through CRISPR/Cas12a trans-cleavage of single-strand DNA oligonucleotides. Biosens. Bioelectron. 220, 114859 (2023).
Nguyen, Q. H. & Kim, M. I. Nanomaterial-mediated paper-based biosensors for colorimetric pathogen detection. Trends Anal. Chem. 132, 116038 (2020).
Macchia, E. et al. Point-of-care ultra-portable single-molecule bioassays for one-health. Adv. Mater. 36, e2309705 (2024).
Vitale, I., Manic, G., De Maria, R., Kroemer, G. & Galluzzi, L. DNA damage in stem cells. Mol. Cell 66, 306–319 (2017).
Wallace, S. S., Murphy, D. L. & Sweasy, J. B. Base excision repair and cancer. Cancer Lett. 327, 73–89 (2012).
Grundy, G. J. & Parsons, J. L. Base excision repair and its implications to cancer therapy. Essays Biochem. 64, 831–843 (2020).
Zheng, L. et al. Loosely-packed dynamical structures with partially-melted surface being the key for thermophilic argonaute proteins achieving high DNA-cleavage activity. Nucleic Acids Res. 50, 7529–7544 (2022).
Acknowledgements
We would like to express our appreciation to the Natural Science Foundation of Zhejiang Province of China (Grant Nos. LQ23H200005 and LTGC24H200001) and the National Natural Science Foundation of China (Grant No. 22304157) for their generous support, which contributed significantly to the research presented herein. Additionally, we acknowledge the Presidential Young Professorship (PYP) start-up funding (Grant No. 23-0178-0002), Singapore MOE Tier 1 (23-0443-A0001) and National Centre for Infectious Diseases (NCID) PREPARE funding (24-0994-P0001) provided by the National University of Singapore, which has played a pivotal role in facilitating our scientific endeavors and advancing our work in this field. We thank Hangzhou EzDx Technology Co., Ltd for supporting the portable device.
Author information
Authors and Affiliations
Contributions
C.Y.H., T.H., C.X.C., and C.L.X. conceived of the project. T.H., X.X.K., and C.Y.H. designed the experiments. T.H., X.X.K., Y.Y.Y, H.M.F., Y.N.C., B.Y. Z., and X.M.F. performed the experiments, while the critical task of handling and processing clinical samples from the hospital was deftly managed by Y.B.T., L.L., Y.L., and M.H. C.Y.H., T.H., C.X.C., S.F.Z., H.M.F., Y.N.C., B.Y. Z., and Q.Q.J. analyzed the results. C.Y.H., T.H., C.X.C., X.X.K., and C.L.X. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hailin Tang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, T., Ke, X., Yu, Y. et al. NAPTUNE: nucleic acids and protein biomarkers testing via ultra-sensitive nucleases escalation. Nat Commun 16, 1331 (2025). https://doi.org/10.1038/s41467-025-56653-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56653-9
