
Abstract
CRISPR-based base editor (BE) offer diverse editing options for genetic engineering of microorganisms, but its application is limited by protospacer adjacent motif (PAM) sequences, context preference, editing window, and off-target effects. Here, a series of iteratively improved cytosine base editors (CBEs) are constructed using the FrCas9 nickase (FrCas9n) with the unique PAM palindromic structure (NNTA) to alleviate these challenges. The deaminase domain-inlaid FrCas9n exhibits an editing range covering 38 nucleotides upstream and downstream of the palindromic PAM, without context preference, which is 6.3 times larger than that of traditional CBEs. Additionally, lower off-target editing is achieved when incorporating high-fidelity mutations at R61A and Q964A in FrCas9n, while maintaining high editing efficiency. The final CBE, HF-ID824-evoCDA-FrCas9n demonstrates broad applicability across different microbes such as Escherichia coli MG1655, Shewanella oneidensis MR-1, and Pseudomonas aeruginosa PAO1. Collectively, this tool offers robust gene editing for facilitating mechanistic studies, functional exploration, and protein evolution in microbes.
Similar content being viewed by others


Introduction
Achieving rapid and efficient gene editing in microbes is essential for mechanism study and modification of functional phenotypes1. Among the array of microbial genome editing tools developed to date, base editor (BE) stands out as a highly promising genetic manipulation tool2. Consisting of inactive or partially active Cas proteins fused with deaminases, the BE enables precise and efficient targeted modifications of microbial genomic DNA while avoiding double-strand breaks3. Of these, cytosine base editors (CBEs) and adenine base editors (ABEs) are the two primary categories of BE, facilitating the conversion of cytosine (C) to thymine (T) and adenine (A) to guanine (G), respectively4. These alterations offer powerful and diverse editing capabilities and application potentials for practical microbial genetic engineering5.
CBEs have become the frontier for microbial gene-editing research, holding the potential for versatile applications and providing powerful solutions for editing various microorganisms6,7,8. For instance, CBEs can integrate premature stop codons (5’-TAG-3’, 5’-TAA-3’, or 5’-TGA-3’) into open reading frames (ORFs), resulting in truncated non-functional proteins and achieving functional gene knockout9. Furthermore, CBEs can also generate endogenous gene mutation libraries, facilitating in situ gene evolution of promoter sequences, drug-resistant proteins, and functional proteins8,10,11. With these benefits, CBEs have been widely applied to various microbes7,12,13. However, the increasingly diverse demands of microbial gene editing have raised new challenges for CBEs in terms of PAM sequence, editing efficiency, editing window, and off-target effects.
Most microbial CBEs are based on the SpCas9 protein, which recognizes the 5’-NGG-3’ PAM sequence but faces challenges in regions with high TA content14,15. Although members of the Cas12 family prefer PAMs rich in T, the lack of a nickase in Cas12 hinders their practical application16. Subsequent Cas9 variants such as iSpyMacCas9 recognize a more relaxed 5’-NNA-3’ PAM sequence but lack stability17. The SpRY variant of SpCas9 (which prefers 5’-NRN-3’ over 5’-NYN-3’, where R is A or G and Y is C or T) nearly eliminated PAM limitations. However, it may interfere with sgRNA transcription on its plasmid and poses certain risks of off-target effects18. Additionally, some deaminases exhibit a preference for the sequence context of the target C during practical use, with minimal editing observed on GC sequences7,19. Crucially, the editing window of CBEs determines the range of target Cs on gene termination and also influences the number of mutated Cs when constructing mutation libraries. The editing window of unmodified SpCas9-BE3 in most microbes ranges from C3 to C8, providing a 6-nt editing range only6,20,21. The existing strategies have broadened the editing window in microbes through Cas9 protein variants22. For optimization of sgRNA length23, deaminase screening24, and structural improvements25, further improvement in the editing efficiency and spectrum in microbes is still needed. Finally, it is essential to note that off-target editing generated during microbial genome editing by CBEs cannot be overlooked26. Therefore, researchers often face a trade-off when using CBEs in microbes and lack a robust and functionally compatible genetic manipulation tool.
In this work, we develop a robust and reliable CBE, based on the FrCas9 derived from Faecalibaculum rodentium, featuring a unique palindrome structure of the PAM sequence 5’-NNTA-3'27. Through a series of optimizations including deaminase screening, protein fusion, and high-fidelity mutations, we develop the HF-ID824-evoCDA-FrCas9n. This tool features wide-window, high-efficiency, and unbiased C-to-T editing at positions C1 to C19 in the microbial genome, with lower off-target effects. Meanwhile, by utilizing the unique palindromic structure of FrCas9’s PAM, the editing range spans both 19 nt upstream and downstream, covering approximately 38 nt of the sequence. To validate the broad applicability of the HF-ID824-evoCDA-FrCas9n in microbes, we apply it to three distinct microbial models (Escherichia coli MG1655, Shewanella oneidensis MR-1, and Pseudomonas aeruginosa PAO1) to explore the molecular mechanisms of antibiotic resistance, enhance extracellular electron transfer capacity, and probe pivotal genes for adaptive evolution, respectively. Overall, the HF-ID824-evoCDA-FrCas9n can serve as a powerful and efficient tool for versatile microbial genome manipulations, contributing significantly to the expansion of the genetic editing toolbox in microbial genome engineering.
Results
Construction of base editor CBE based on FrCas9 in E. coli MG1655
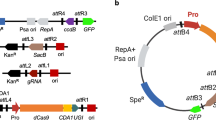
Distinguishing from the commonly used SpCas9 base editor with 5’-NGG-3’ PAM sequences, we chose the FrCas9 derived from Faecalibaculum rodentium, which has a palindromic 5’-NNTA-3’ PAM sequence (a complementary PAM sequence exists on the NTS, 3’-ATNN-5’) with a spacer region length of 22 bp in the sgRNA27. To effectively convert target C-to-T in microbial genomes, we designed a single plasmid-mediated base editing system. This system included constitutively expressed FrCas9n (E795A)-sgRNA, with rat-derived cytidine deaminase rAPOBEC1 positioned at the N-terminus of FrCas9n, and uracil glycosylase inhibitor (UGI) at the C-terminus of FrCas9n. We refer to this as the first-generation FrCas9n-CBE. The FrCas9n-CBE was introduced into the target strain, followed by Sanger sequencing and EditR software analysis to assess the efficiency of cytosine (C) editing to thymine (T) at the target site (Fig. 1a)28,29.

a A schematic illustration of FrCas9n-CBE editing in MG1655. b The C-to-T editing frequencies at each cytosine position for APOBEC-SpCas9n-UGI, APOBEC-FrCas9n-UGI, and APOBEC-SPRY-UGI are quantified and summarized in the light red, blue, and pink lines, respectively. c Schematic of premature stop codon induction by the 5’-NNTA-3’ palindromic PAM sequence over a wider range. d The Venn diagram of the quantity and differences for genes termination in MG1655 by APOBEC-FrCas9n-UGI and SpCas9n-BE3. e Using four different deaminases to construct FrCas9n-CBE, selecting nine 22-nt original spacers containing each motif (TC, CC, AC, GC) at different genomic positions to comprehensively evaluate the impact of sequence context on targeted C editing efficiency. f–i Editing efficiency of four deaminases on targeted Cs in the range of C3-C11 with different motifs (TC, CC, AC, GC). (f) APOBEC; (g) evoAPOBEC; (h) hAID and (i) evoCDA, respectively (n = 4 biological replicates, data represent mean ± SD). j Quantitation and summary of C-to-T editing frequencies at each cytosine position of twelve endogenous loci in E. coli by four FrCas9-CBEs. k Comparing the off-target editing of four FrCas9-CBEs and SpCas9n-BE3 on predicted mismatched target sequences by Cas-offinder (n = 4 biological replicates, data represent mean ± SD). Statistical significance was analyzed using a two-tailed t-test: ns, p > 0.05.
We initially assessed the capability of the rAPOBEC-FrCas9n-UGI to achieve effective C-to-T gene editing in the genome of E. coli MG1655. We evaluated seven target sites within the spacer region by examining each cytosine from position 1 to 13 within the spacer region (with the 5’ end of the spacer designated as position 1) (Supplementary Fig. 1). The rAPOBEC-FrCas9n-UGI facilitated efficient C-to-T conversions at multiple sites ranging from C3 to C12, demonstrating that the FrCas9n-CBE could be used for gene editing in MG1655. To evaluate the microbial genome editing performance of APOBEC-FrCas9n-UGI, we constructed two other base editors: APOBEC-SpCas9n-UGI and a flexible PAM variant of SpCas9n, named SPRY30. The editing efficiencies of these three CBEs at 9 different target C sites in the MG1655 genome were evaluated by selecting specific PAM sequences. We summarized the editing efficiencies of different locations and defined the locations with an average efficiency greater than 15% as editing windows. Sequencing results of the colonies show that the editing window for APOBEC-SpCas9n-UGI in MG1655 ranged from C3 to C8 with editing efficiencies between 54.96% and 79.31%. In contrast, APOBEC-FrCas9n-UGI exhibited a preliminary editing window of C3 to C12 with efficiencies ranging from 17.50% to 79.18%. Additionally, the flexible PAM base editor APOBEC-SPRY-UGI had an editing window of C3 to C9 with efficiencies ranging from 20.04% to 70.75% (Fig. 1c and Supplementary Fig. 2a). Overall, the APOBEC-SPRY-UGI exhibited a lower editing efficiency than the other two base editors. APOBEC-FrCas9n-UGI demonstrated a comparable or slightly lower C-to-T conversion efficiency relative to APOBEC-SpCas9n-UGI (Supplementary Fig. 2b). Importantly, FrCas9n-CBE had a broader editing window than SpCas9-BE3, as C-to-T editing could still occur at target C positions C9 ~ C12.
Functional gene knockout is a primary application of CBEs in microbial genetic engineering, relying on distinct editing windows and available PAMs14. Unlike the widely used SpCas9-BE3 with a 5’-NGG-3’ PAM, the developed FrCas9-CBEs had a 5’-NNTA-3’ PAM sequence with a unique palindromic structure. Therefore, it enabled the design of the sgRNAs on both the sense and antisense strands of a PAM with a broader editing window (Fig. 1c), implying a greater potential for prematurely terminating functional genes within microbial genomes. We utilized the bioinformatics tool “autocbei” to predict the number, distribution, and sequence of sgRNAs causing gene inactivation using rAPOBEC-FrCas9n-UGI in MG165531. Among the 4639 genes in MG1655, we identified the 32228 sgRNAs, which could target 87.9% of the genes (4077 out of 4639) by introducing premature stop codons, representing an improvement over SpCas9-BE3, which targets 84.0% of genes (Supplementary Fig. 3a, b). Moreover, rAPOBEC-FrCas9n-UGI targeted 264 genes that SpCas9-BE3 cannot precociously terminate (Fig. 1d).
We further supplemented 9 common microbial genomes to comprehensively evaluate the gene termination capability of the rAPOBEC-FrCas9n-UGI and SpCas9-BE3. These microbes includeg Acinetobacter baumannii ATCC19606, Staphylococcus epidermidis ATCC14990, Clostridium butyricum DSM 10702, Pseudomonas putida KT2440, Streptococcus pneumoniae NCTC7465, Corynebacterium glutamicum ATCC 13032, Bacillus subtilis 168, Clostridium tetani E88 and Clostridium autoethanogenum DSM 10061. Using CBEI, we predicted the potential of FrCas9n-CBE and SpCas9-BE3 to induce premature stop codons in these microbial genomes. Among the selected 10 microbes, FrCas9n-CBE was able to generate premature stop codons in over 90% of the genes in 8 species, and in over 95% of genes in 7 of these species. In contrast, SpCas9-BE3 achieved premature termination in 63.67 ~ 89.03% only of the genes in 7 microbes (Supplementary Fig. 4). Furthermore, when the translation was prematurely terminated at the first 50% position of functionally targeted genes across different species, FrCas9n-CBE demonstrated a significant advantage. Its flexible palindromic NNTA PAM sequence allowed for a large selection of sgRNAs than SpCas9-BE3. Among the 10 selected microbes, FrCas9n-CBE could achieve premature termination in approximately 80% of the genes within the first 50% position across 8 of these species. In contrast, SpCas9-BE3 showed 43.58 ~ 87.66% less genes targeted for premature termination in most species (Supplementary Fig. 5). Therefore, the first-generation FrCas9n-CBE demonstrated a high potential for functional gene knockout in microbial genomes.
Screening different types of deaminases to eliminate base editing preferences
During microbial genome base editing, the rat-derived cytidine deaminase rAPOBEC exhibited a certain preference, achieving little to no editing for target Cs preceded by a G base in practical application7. Analysis of all inactivation gene sgRNAs in MG1655 shows that the proportion of target Cs preceded by a G base reached 29% (Supplementary Fig. 3c, d). Therefore, it is necessary to eliminate the editing preference of base editors. To systematically investigate the impact of upstream bases on the target C of the base editor, we designed a target matrix array within the editing window. This array evaluated the editing efficiency of all four NCs (where N represents any nucleotide and the second nucleotide C is the target nucleotide) at positions 3-11 away from the PAM end spacer (Fig. 1e). We initially tested the editing of the target matrix array by rAPOBEC-FrCas9n-UGI. Sequencing results show a clear preference of rAPOBEC-FrCas9n-UGI within the editing window of positions 3-11, suggesting a trend in the selection of preceding bases for target Cs as TC > AC/CC > GC (Fig. 1f). Such a base preference was unfavorable for the efficient genome editing, prompting us to explore different deaminases from various sources to mitigate this bias. We selected the evolutionarily engineered rat APOBEC (evoAPOBEC), human AID (hAID) and the evolved version of lamprey CDA (evoCDA), and examined their respective base preferences for target matrix array (Fig. 1e)32,33.
Sequencing results show that evoAPOBEC effectively improved editing efficiency on GC sequences, with an average editing efficiency of 37.8% (Fig. 1g). Unexpectedly, evoAPOBEC exhibited a lower average editing efficiency of 8.2% only on AC sequences. When constructing the base editor with hAID, we observed a notable reduction in its bias, resulting in an increased average editing efficiency of AC sequences to 38.8% (Fig. 1h). We attempted a dual deaminase strategy to enhance editing efficiency of target Cs, but the outcomes were generally lower (Supplementary Fig. 6a,b). The editing efficiency decline might be due to the potential steric hindrance that incurs deaminase competition for substrates. Furthermore, evoCDA-FrCas9n-UGI could effectively edit target Cs at various positions within C3-C11. The average editing efficiencies for TC, CC, GC, and AC were 79.2%, 79.3%, 66.6%, and 54.5%, respectively (Fig. 1i), with C-to-T efficiencies exceeding 50% in all cases. These results demonstrate that the evoCDA-FrCas9n-UGI base editor could largely mitigate the sequence preference for editing target Cs upstream.
To comprehensively characterize the editing windows of the four different types of base editors, we selected 12 target sequences in the MG1655 genome (Supplementary Fig. 7). Sequencing results show that the editing window of rAPOBEC-FrCas9n-UGI spanned from C3 to C13. The evoAPOBEC-FrCas9n-UGI’s editing window was expanded from C3 to C14. The AID-FrCas9n-UGI exhibited an editing window from C2 to C14. The most effective editing was achieved by evoCDA-FrCas9n-UGI with an editing window from C2 to C15 (Fig. 1j). Within these editing windows, efficient C-to-T base changes were realized, demonstrating that evoCDA-FrCas9n-UGI was an unbiased and broad-window base editor (Fig. 1j and Supplementary Fig. 8). Furthermore, we compared the editing efficiencies of evoCDA-FrCas9n-UGI and evoCDA-SpCas9n-UGI at 6 identical target C sites in E. coli MG1655. The results show that the editing windows for evoCDA-FrCas9n-UGI and evoCDA-SpCas9n-UGI were C2–C15 and C1–C11, respectively, with average editing efficiencies of 61.65% and 57.14% (Supplementary Fig. 9). This result demonstrates that evoCDA-FrCas9n-UGI exhibited superior editing capabilities.
To evaluate the specific off-target activity of the base editor, we chose a challenging site for specificity testing of base editor, and the predicted off-target site had a mismatch of 2-3 bases only compared to the on-target site. In this way, the ability of SpCas9-BE3 and FrCas9-CBEs to discriminate between mismatched sites was evaluated. SpCas9-BE3 exhibited noticeable off-target editing at positions C3 in off-target 1 (OT1) and OT3. Conversely, the 4 different deaminase base editors constructed from FrCas9n showed no significant off-target editing at the three off-target sites. These results indicate that FrCas9n had a higher specificity for microbial genome editing (Fig. 1k). Overall, we demonstrated that the second-generation FrCas9n-CBE, evoCDA-FrCas9n-UGI, could offer efficient, unbiased, and broad C-to-T base conversions with low off-target editing rates.
Construction of a wider range of base editors with a deaminase embedded in FrCas9n protein
Previous studies in eukaryotic cells have demonstrated that embedding a deaminase into SpCas9, SaCas9, Nme2Cas9 or Cas12f can change the editing scope or reduce off-target effects34,35,36,37,38. Therefore, to further broaden the editing window, we tried to embed cytidine deaminase into FrCas9n. To select suitable embedding sites, we initially used AlphaFold2 for predicting the protein structure of FrCas9n39. The predicted FrCas9n consisted mainly of 4 distinct structural domains: REC, HNH, RUC, and PI domains (Fig. 2a). Within these domains, we selected 8 sites from unstructured loops for embedding hAID, with 16 amino acids of XTEN flexible linkers connecting the ends of hAID (Fig. 2b and Supplementary Fig. 10). Two endogenous target sites (ptrB_1 and yggR_1) were selected to test the C-to-T efficiency of the embedded deaminase-based base editor. Sequencing results show that, in addition to N-terminal and C-terminal editors, internally embedded ID822-AID-FrCas9n and ID838-AID-FrCas9n exhibited a broader editing range on the two targets, albeit with a lower editing efficiency (Fig. 2c). Removing the linker region linked to ID822-AID-FrCas9n and extending the spacer region of sgRNA did not substantially improve the editable range (Supplementary Figs. 11, 12).

a Diagram displaying the structural domains of FrCas9 and internal fusion sites (orange lines) for generating potential wide-window CBEs. b The FrCas9 structural domains predicted by Alphafold2 correspond to colors for (a) REC, HNH, RuvC, and PI domains. Eight IDs and N-/C-terminal positions (marked in red) were selected as hAID integration sites. c, d Fusing hAID to the N-, C-terminal, or internal sites of FrCas9n to generate various potential CBEs, and assessing the C-T conversion frequency of each cytosine nucleotide in two endogenous gene sites prtB_1(c) and yggR_1(d). With the target cytosines marked in red, the editing outcomes were quantified using EditR. All Data are presented as mean ± standard deviation from four independent experiments.
To further enhance editing efficiency, we replaced the embedded hAID with a more efficient and unbiased evoCDA. Sequencing results show that at sites where hAID exhibited minimal editing, the incorporation of evoCDA also failed to produce effective edits. This result suggests that embedding at these sites might disrupt the functional structure of FrCas9-sgRNA (Supplementary Fig. 13a). Conversely, at sites ID248, ID822, and ID838, where hAID demonstrated some editing efficiency, the inclusion of evoCDA similarly enhanced the editing efficiency and broadened the editing window, confirming that such substitution did not affect the selection of embedding sites (Supplementary Fig. 13a). By progressively adjusting the fusion evoCDA positions around ID822 (ID819-ID826), we identified the optimal base editor, ID824-evoCDA-FrCas9n (Fig. 3a, b and Supplementary Fig. 13b). In addition, we compared the editing efficiencies of the three base editors-N/ID822/ID824-evoCDA-FrCas9n-using ptrB_1 and yggR_1 as targets reinforced that ID824-evoCDA-FrCas9n exhibited the highest editing efficiency at the C14-C18 (Fig. 3c). Furthermore, we conducted a statistical analysis of the editing range of ID824-evoCDA-FrCas9n at six endogenous target sites in MG1655. Notably, ID824-evoCDA-FrCas9n exhibited an editing window of 19 nt, covering almost all target Cs on the sgRNA except for the 3 nt near the PAM site (Fig. 3d, e). Such a wide editing range enables extensive C-to-T base changes.

a Schematic diagram illustrating the stepwise movement of evoCDA insertion sites within FrCas9n. b Achievement of C-to-T editing activity at target sites by different CBEs (n = 4 biological replicates, data represent mean ± SD). c Comparing the editing activity of three CBEs (N-, ID822, and ID824) at two endogenous gene loci (n = 4 biological replicates, data represent mean ± SD). d The C-T conversion frequencies of each cytosine nucleotide were measured for ID824-evoCDA-FrCas9n at six endogenous gene loci, and the editing results were quantified and displayed in a heat map using EditR (n = 4 biological replicates, data represent mean ± SD). e Editing capability of N-evoCDA-FrCas9n and ID824-evoCDA-FrCas9n in 22-bp spacer region (n = 4 biological replicates, data represent mean ± SD). f Schematic illustrating the broad-range C-to-T editing induced by ID824-evoCDA-FrCas9n on both sides of a palindromic PAM. sgRNA1 targets C sites on the upstream sense strand, sgRNA2 targets C sites on the downstream antisense strand. g ID824-evoCDA-FrCas9n achieves ultra-wide-range editing across nearly 38 bases in the genome. C1–C22 represent target C sites on the sense strand upstream of the PAM, while G'1–G'22 represent the corresponding G sites on the complementary sense strand downstream of the PAM (n = 4 biological replicates, data represent mean ± SD).
We also tested the embedded deaminase sites in SpCas9 reported in other studies, which have been shown to enhance editing efficiency and expand the editing window34,37. We integrated evoCDA at positions ID1058 and ID1246 of SpCas9, resulting in two embedded SpCas9-CBEs: ID1058-evoCDA-SpCas9n and ID1246-evoCDA-SpCas9n (Supplementary Fig. 14a). The results show that the editing window of ID1058-evoCDA-SpCas9n was expanded to C1–C14, while the editing window for ID1246-evoCDA-SpCas9n was C1–C13 (Supplementary Fig. 14b, c). These were narrower than the C1–C19 editing window exhibited by our developed ID824-evoCDA-FrCas9n. To evaluate the efficiency of broad-range base editing, we selected mixed bacterial strains with specific base edits for next-generation sequencing to quantify the editing results. The sequencing results show that ID824-evoCDA-FrCas9n effectively induced widespread C-to-T base conversions across a broad range of positions (C1 to C19) within the spacer sequence, with minimal mutations such as C-to-A, C-to-G, and indels (Supplementary Fig. 15a). This broad editing effect was also consistent in individual clones (Supplementary Fig. 15b).
Therefore, we developed the third-generation version of FrCas9n-CBE: ID824-evoCDA-FrCas9n to effectively broaden the editing window to a maximum of 19 nt. Due to FrCas9n’s unique 5’-NNTA-3’ PAM sequence, targeting a PAM on the antisense strand allowed for another downstream editable range of 19 nt (Fig. 3f). This means, a palindromic 5’-NNTA-3’ PAM sequence included an editing window of 19 nt upstream and 19 nt downstream. Thus, a total editing range of approximately 38 nt could be provided (Fig. 3g and Supplementary Fig. 16). Such a wide-window, high-efficiency CBE with flexible PAM sequences not only expanded the selection of suitable target C sites for early gene termination, but also enhanced the accessibility of mutation libraries.
Improving editing specificity of fusion base editors by high-fidelity mutations
The specificity of base editors is a key guarantee for their safe and reliable genome editing application40. Despite the capability for efficient and broad-window base editing, however, several base editors embedded in FrCas9n (i.e., from ID821 to ID826) still exhibited off-target editing at mismatched sites in internal target specificity testing (Supplementary Fig. 17). To reduce the off-target effects of base editors, we analyzed the FrCas9n structure and identified seven potentially high-fidelity mutation sites: R61A, N532A, N694A, K724A, Q728A, N892A, and Q964A (Fig. 4a and Supplementary Fig. 18). Based on structural comparisons between SpCas9 and FrCas9, we proposed that FrCas9 residues N532, N694, Q728, and Q964 might directly form hydrogen bonds with the phosphate backbone of the target DNA41. Additionally, K724 is located near the phosphate backbone domain of the protein-DNA complex. Disruption of these residues might directly affect the thermodynamics of the FrCas9-sgRNA complex, potentially weakening its ability to recognize mismatched off-target sites42. R61 is located in a conserved arginine-rich bridge helix, where it interacts with the phosphate backbone of sgRNA in the specificity-determining region. Mutating this residue to alanine would increase FrCas9’s sensitivity to mismatches in the PAM-proximal region of the target43. N892 resides in a positively charged groove of a distinct FrCas9 domain44. Neutralizing positively charged residues within the non-target groove may weaken binding to the non-target strand, necessitating more stringent Watson-Crick base pairing between the RNA guide and the target DNA.

a Illustration of seven amino acid mutations to alanine in the active pocket of FrCas9n. b Schematic of sgRNA-dependent off-target editing by FrCas9-CBE. c, d Editing outcomes of Single, double, and triple mutations of evoCDA-FrCas9n in the endogenous target sites. Specific editing C-T conversion rate (c) and off-target editing at 2-3 mismatch sites (d), with editing outcomes quantified using EditR and displayed in a heatmap (n = 4 biological replicates, data represent mean ± SD). e Schematic of orthogonal R-loop analysis for testing sgRNA-independent off-target effects of FrCas9-CBE. f C-to-T conversion frequencies of sgRNA-independent off-targets detected at six R-loop sites using dCas12a and corresponding sgRNAs. Bars represent the highest C-to-T activity at each R-loop site (n = 4 biological replicates, data represent mean ± SD). Statistical significance was analyzed using a two-tailed t-test: ns, p > 0.05. g Diagram illustrating genome-wide SNV mutations induced by CBEs in the E. coli Genome. h Bar chart showing the number of unpredictable SNVs and indels generated in the E. coli genome following base editing with different CBEs, as determined by whole-genome sequencing.
The off-target test sites selected here were challenging regions in E. coli, with the predicted 5 off-target sites containing 2 to 3 mismatches only (Supplementary Fig. 19a). We evaluated the on-target and off-target editing efficiencies of evoCDA-FrCas9n UGI and its variants at these sites. The mutated base editors led to decreased editing efficiency for the target site, with K724A and Q728A showing the most significant reductions (Fig. 4c). Additionally, among the 5 off-target sites, only OT2 and OT5 at the C10 position were the hotspots for off-target effects, while the overall average off-target editing was effectively reduced (Fig. 4d and Supplementary Fig. 19b, c). We selected single mutants R61A, N532A, and Q964A, which maintained high on-target editing capabilities while effectively reducing off-target effects. We further constructed double mutants R61A/N532A, R61A/Q964A, N532A/Q964A, and the triple mutant R61A/N532A/Q964A for off-target editing tests. The double mutant R61A/Q964A exhibited the lowest average off-target levels. However, it maintained the lowest off-target/on-target ratio in spite of the decreased overall editing efficiency, demonstrating a strong specificity (Fig. 4d and Supplementary Fig. 19c, d). Next, we introduced double and triple mutations to enhance the base editing specificity of the wide-range CBE, ID824-evoCDA-FrCas9n. The embedded ID824 base editor enhanced the editing efficiency for double mutations, although this also increased the off-target efficiency (Supplementary Fig. 20a, b). After carefully considering both on-target and off-target efficiencies, we selected the combination of ID824-evoCDA-FrCas9n with the R61A/Q964A double mutation. While this combination showed a slight decrease in on-target efficiency compared to ID824-evoCDA-FrCas9n, it substantially reduced off-target editing (Supplementary Fig. 20c, f). We refer to the R61A/Q964A double mutant FrCas9 protein as HF-FrCas9, ultimately resulting in our fourth-generation FrCas9-CBE: HF-ID824-evoCDA-FrCas9n.
Next, we employed a mutated sgRNA approach to examine the tolerance of FrCas9-CBEs to sgRNA mutations. We designed a 22 bp sgRNA targeting the gfp gene and created a set of sgRNAs with dinucleotide mismatch sites distributed across the sgRNA sequence. Under normal circumstances, the wild-type on-target sgRNA directs the base editor to convert the glutamic acid codon CAG into the stop codon TAG, resulting in premature termination of green fluorescent protein translation (Supplementary Fig. 21a, b). Flow cytometry results indicate that the sgRNA of FrCas9 exhibited higher tolerance to mutations at the distal PAM site, but it had lower tolerance at the proximal PAM site. Compared to evoCDA-FrCas9n and ID824-evoCDA-FrCas9n, the high-fidelity versions HF-evoCDA-FrCas9n and HF-ID824-evoCDA-FrCas9n displayed lower overall off-target editing levels, except for the G1112 mutation associated with evoCDA-FrCas9n (Supplementary Fig. 21c, f). Although subsequent sequencing results show elevated off-target editing efficiency, likely due to the extended editing times, there was still a notable reduction in off-target editing at the proximal PAM site, consistent with the flow cytometry findings (Supplementary Fig. 21g, j). This result demonstrates that the high-fidelity mutations R61A/Q964A indeed enhanced the specificity of base editing.
To evaluate the non-gRNA-dependent off-target effects of base editors on the genome, we employed CRISPR-dCas12a to generate orthogonal R-loops at 6 targets within the MG1655 genome. This approach allowed us to assess the off-target editing activity of various CBEs at these R-loops45,46,47 (Fig. 4e). Based on next-generation sequencing results, among the 7 tested CBEs, APOBEC-SPRY exhibited the highest off-target effects independent of gRNA. In contrast, APOBEC-FrCas9 showed a slightly lower off-target editing activity, while the modified HF-evoCDA effectively reduced the average off-target rate compared to evoCDA-FrCas9n. Notably, the embedded ID824-evoCDA and HF-ID824-evoCDA exhibited negligible off-target editing across all 6 sites (Fig. 4f). This might be attributed to the chimeric structure that spatially restricts the enzyme’s access to the Cas9-bound DNA strand, thereby effectively reducing gRNA-independent off-target effects. Furthermore, we assessed the off-target effects of CBEs across the genome of E.coli MG1655 (Fig. 4g). Whole-genome sequencing reveals that the primary off-target mutation observed across the genomes was C > T, along with negligible off-target effects and minimal Indels. SPRY Cas9 exhibited the highest rate of single nucleotide variations (SNVs), while the off-target editing associated with APOBEC-SpCas9, evoCDA-FrCas9, and APOBEC-FrCas9 progressively decreased. Notably, HF-evoCDA effectively reduced off-target editing across the genome. Importantly, the embedded base editors ID824-evoCDA and HF-ID824-evoCDA displayed negligible SNVs in the genome (Fig. 4h and Supplementary Data 4). All these results demonstrate that our embedded base editor maintained high fidelity at the whole-genome level.
The above results demonstrate that the fourth-generation version of FrCas9n-CBE: HF-ID824-evoCDA-FrCas9n, obtained through a series of screening optimizations and protein modifications, is an efficient, unbiased, wide-window, and low off-target base editor (Table 1 and Supplementary Fig. 22). This advancement could facilitate its practical application in microbial genome engineering.
Construction of rpoB mutant library using HF-ID824-evoCDA-FrCas9n
Given the efficient C-to-T base conversion within a 38 nt editing range in the E. coli genome by our base editor HF-ID824-evoCDA-FrCas9n, we applied it to construct an rpoB gene mutant library (Fig. 5a)10. The rpoB gene is highly conserved in microbes and encodes the β subunit of DNA-dependent RNA polymerase48. Several single amino acid mutations in RpoB protein have been extensively documented to confer rifampicin resistance (RifR) in MG165510. Studying the mutations in the rpoB gene that led to RifR provided valuable insights into the clinical antibiotic resistance of E. coli and other pathogenic microorganisms.

a Generating rpoB mutations targeting different mutation positions using base editors and screening for RifR mutants through spot plating. b, c Variants of rifampicin-resistant rpoB alleles generated at different target sites. Multiple alleles with the same genotype are described only once, with the wild-type amino acid sequence shown in the first row as a reference. Modified amino acids are highlighted in red bold and blue shading, while silent mutations at the DNA level that do not change the amino acid are highlighted in gray shading. d Complementation experiment validating identified rifampicin-resistant rpoB mutants. The three discovered variants, H526Y, P564L, and S574F, were individually constructed in the rpoB gene of E. coli, and test their rifampicin resistance (n = 3 biological replicates). e Distribution of mutations in the 520–560 amino acid region of the RpoB protein. Synonymous mutations with nucleotide changes but no amino acid alteration was shown in black font. Reported RpoB amino acid mutations conferring rifampicin resistance were highlighted in red font. Mutations conferring rifampicin resistance (previously unknown) were highlighted in green font.
We selected six sites distributed across three RifR-determining regions in the MG1655 genome as targets. Clones with RifR were further screened, and the genotypes of independent RifR colonies appearing on rifampicin-containing plates were determined using Sanger sequencing. In the tested region, a wide range of C-to-T base mutations occurred (Fig. 5b, c) and the most target sites exhibited rifampicin resistance (Supplementary Fig. 23). We identified several previously reported rpoB gene rifampicin resistance mutation sites and some rifampicin resistance mutation sites that had not been previously addressed, such as H526Y、P564L and S574F10,49. We conducted separated experiments to introduce single mutations into the rpoB gene for examining whether these identified mutations conferred rifampicin (50 μg/mL) resistance to E. coli. The results show that both wild-type and mutant E. coli strains grew normally on plates without rifampicin. Single mutation H526Y or P564L in the rpoB gene was sufficient to confer rifampicin resistance to E. coli, indicating that these two mutations represented previously unreported rpoB gene resistance sites (Fig. 5d). In contrast, neither the wild-type rpoB nor the rpoB-S574F single mutation produced observable colonies (Fig. 5d), suggesting that the numerous synonymous mutations surrounding S574F might have caused uncharacterized changes during initial screening and led to rifampicin resistance. Therefore, the HF-ID824-evoCDA-FrCas9n efficiently identified clusters of single or multiple amino acid replacements responsible for RifR in E. coli (Fig. 5e), demonstrating its potential for saturation mutagenesis of targeted loci and facilitating rapid functional characterization of induced mutations.
Enhancing the extracellular electron transfer efficiency of S. oneidensis MR-1 with HF-ID824-evoCDA-FrCas9n
To expand the broad-spectrum applicability of our base editor, we also analyzed its performance for base editing in an electroactive bacterium- Shewanella oneidensis MR-1(Fig. 6a)13. MR-1 has garnered great interests because of its unique extracellular electron transfer (EET) capability and wide application for energy recovery and pollution remediation50. Therefore, an efficient and versatile gene editing tool for rapid manipulation of the Shewanella genome was on urgent demand to elucidate its distinctive extracellular electron transfer behaviors. Our HF-ID824-evoCDA-FrCas9n-UGI base editor maintained a wide editing window from C1 to C18 for the genome target of MR-1, expanding the previously reported 6-nucleotide range of APOBEC-SpCas9n by threefold (Fig. 6b and Supplementary Fig. 24)13. To further compare the editing capabilities with APOBEC-SpCas9n, we selected the gene SO_3350, which is associated with biofilm formation13. When using APOBEC-SpCas9n, practical application was limited by PAM selection, editing window, and sequence context, resulting in ineffective inactivation of the SO_3350 (0/12). However, with HF-ID824-evoCDA-FrCas9n, the editing efficiency reached 100% (12/12) at the same target sites as APOBEC-SpCas9n. Moreover, multiple sgRNAs could be chosen to prematurely terminate the SO_3350 gene (Fig. 6c).

a Schematic diagram of MFCs inoculated with MR-1 to generate electricity. b HF-ID824-evoCDA-FrCas9n editing scope for MR-1 Genome. c Comparative analysis of the inactivation efficiency of SO_3350 in MR-1 by APOBEC-SpCas9n and HF-ID824-evoCDA-FrCas9n. d Photos of dual-chamber MFC. MR-1/SO3350Q88* produced more biofilm than MR-1. e–g EET performances of MR-1 and MR-1/SO3350Q88*. Current density (e), power density (f), and the CV curve (g) of MR-1 and MR-1/SO3350Q88* in the MFCs. h Comparing the protein content on carbon felts after MFCs run to completion. Data obtained from four replicate tests (n = 6 biological replicates, data represent mean ± SD). Statistical significance was analyzed using a two-tailed t-test: ns, p > 0.05.
Subsequently, the dual-chamber microbial fuel cells (MFCs) were used to assess the EET efficiency of MR-1 and MR-1/SO_3350Q88*. Compared to the MR-1, the engineered microbe exhibited apparent biofilm growth on the anodic carbon felt (Fig. 6d). The MFCs inoculated with Shewanella strains reached the current peak of 245.23 mA/m² at 75–80 h, a value 1.49-fold that to MR-1 (Fig. 6e). Also, we found that the MFC with MR-1/SO_3350Q88* achieved a maximum power density of 172.55 mW/m2, 4.98 times higher than that with MR-1(Fig. 6f). Additionally, the cyclic voltammetry (CV) curve of the MR-1/SO_3350Q88* showed two pairs of anodic and cathodic peaks corresponding to outer membrane c-type cytochromes and flavins (centered at −0.25 V vs. Ag/AgCl and −0.43 V vs. Ag/AgCl, respectively). This results indicates that more cytochrome C and electron shuttles were produced by MR-1/SO_3350Q88* on the anode due to biofilm accumulation (Fig. 6g)50. The protein content of the carbon felt of MR-1/SO_3350Q88*, also determined using the BCA method, was 3.93 times higher than that of MR-1 (Fig. 6h). These results further demonstrate the enhanced EET capacity of the MR-1/SO_3350Q88* and phenotypically confirm the functional knockout of the targeted gene using HF-ID824-evoCDA-FrCas9n.
Exploration of polymyxin B resistance mechanism in PAO1 with the optimal base editor
Furthermore, we applied the HF-ID824-evoCDA-FrCas9n to the widely studied pathogen Pseudomonas aeruginosa PAO1. P. aeruginosa is a conditionally pathogenic bacterium that thrives in diverse environments and exhibits high intrinsic resistance to antibiotics51,52. In clinical settings, polymyxin, including polymyxin B (PMB) and polymyxin E (PME), are currently used as the last line of defense against multidrug-resistant P. aeruginosa infections53,54 (Fig. 7b). However, due to the overuse of antibiotics, sub-lethal concentrations of PMB are present in various environmental settings, leading to adaptive evolution of P. aeruginosa to increased PMB resistance55,56. Therefore, investigating the adaptive evolution of P. aeruginosa to PMB in the presence of sub-lethal concentrations of antibiotics is pivotal.

a Schematic representation of the effects of multi-gene inactivation mediated by CBE on the adaptive evolution of PAO1 under sub-MIC PMB conditions. b HF-ID824-evoCDA-FrCas9n editing range on PAO1 genome. c Sanger sequencing results for the inactivation of six genes using HF-ID824-evoCDA-FrCas9n. d The MIC values of PAO1 and the six mutants were compared before exposure and after three days under 1/2 MIC PMB conditions (n = 8 biological replicates, data represent mean ± SD). Statistical significance was analyzed using a two-tailed t-test: ns, p > 0.05.
We initially investigated the editing scope of HF-ID824-evoCDA-FrCas9n at multiple sites in PAO1 genome. Sequencing results show that the HF-ID824-evoCDA-FrCas9n maintained a broad editing window of C1-C18 in PAO1 (Fig. 7a and Supplementary Fig. 25). We utilized it for efficient and rapid inactivation of genes highly associated with drug resistance in PAO1, such as mexA, mexB, oprF, betT1 (encoding four distinct outer membrane proteins of PAO1), algU (encoding a sigma factor leading to excessive alginate production), and ampC (encoding a protein associated with β-lactam resistance)57,58,59. Sequencing results show that the premature stop codons were successfully introduced at the corresponding genomic loci (Fig. 7c). Except for the PAO1/algUQ8*, the minimal inhibitory concentration (MIC) of all the mutants decreased (Fig. 7d). This result highlights the important roles of these outer membrane proteins on PAO1’s tolerance to PMB. The wild-type and mutant strains were subjected to a three-day laboratory adaptive evolution under PMB concentration at 1/2 MIC. After evolution, the MIC values of strains to PMB were tested again. Compared to the wild-type, the MIC values of all the mutants, except for PAO1/betT1W55*, drastically increased after 3-d evolution. It was likely that the loss of these gene products might pre-activate some stress pathways, leading to the stress-induced evolution of resistance of PAO1 when exposed to sub-MIC concentrations of PMB60. Interestingly, the PAO1/betT1W55* mutant lacking choline transporter protein maintained a relatively low MIC after 3-d evolution. The C-to-T mutations in the gene stop regions of 6 different strains remained unchanged following evolution under sub-MIC concentrations of PMB, with no reversion to the wild-type sequence (Supplementary Fig. 26). This result suggests its potential key role in the adaptive evolution of P. aeruginosa under sub-MIC PMB conditions, providing a potential target for future therapeutic interventions against P. aeruginosa (Fig. 7d).
Discussion
Widespread applications of base editors in microbes are hindered by drawbacks such as PAM selection, base preferences, editing scope, and off-target effects. In this work, we addressed these issues by selecting FrCas9 with a unique 5’-NNTA-3’ PAM and further improvement. With deaminase screening and embedding strategies and high-fidelity mutations, we developed a robust CBE capable of simultaneously overcoming these challenges (Table 1). Furthermore, we demonstrated its potential for expanded applications in three representative microbes, including screening gene loci for rifampicin resistance, enhancing extracellular electron transfer capacity, and exploring factors influencing polymyxin B adaptive evolution.
During the process of CBE improvement, we found some interesting phenomena. Initially, to address the issue of rarely editing of GC sequences by rAPOBEC, we employed evoAPOBEC, known for its lack of base preference, to construct FrCas9n-CBE. Surprisingly, the evoAPOBEC-FrCas9n-UGI exhibited drastic editing efficiency decline for target Cs in AC sequences. This shift in upstream base preference is uncommon, suggesting that the sequence context preference of targeted Cs for CBE was influenced jointly by cytidine deaminase and Cas9. Meanwhile, we observed that embedding the deaminase into the FrCas9 protein led to a higher success rate when positioned on the surface of the structural domain compared to interior. This result suggests that internal embedding of the deaminase might interfere with the normal spatial folding of the FrCas9 protein, rendering it incapable of functioning properly and resulting in the ineffectiveness of the base editor. Conversely, selecting suitable fusion sites on the protein surface allows for closer proximity to the active site pocket of Cas9, while minimizing the impact of the deaminase on the proper functioning of the Cas9 protein.
Although the strategy of embedding the deaminase can improve editing efficiency, expand the editing window, and reduce non-specific RNA off-target effects, it also led to significant increase in specific off-target effects. This might be attributed to the looser structure of the embedded Cas9 protein that decreased the recognition ability for both target and mismatched sequences. To address this issue, we effectively reduced off-target editing by introducing high-fidelity mutations into the FrCas9 protein. Remarkably, even after embedding the deaminase, the high-fidelity FrCas9 mutation protein continued to mitigate off-target editing. This underscores the importance of concerning about the off-target editing when using deaminase-based base editors.
In the future, high-efficiency, flexible PAM sequences and broad-window base editors may facilitate a greater selection of sgRNAs and target Cs in microbial gene editing. Achieving this goal often involves applying variant Cas proteins, optimizing sgRNA lengths, and screening deaminases and fusion proteins from different sources23,61,62. In a recent study the small dBhCas12b protein was successfully utilized to reduce site-blocking effects within Cas proteins and deaminases, thereby achieving high conversion efficiency and an extended editing window63. Therefore, spatial hindrance between Cas9 proteins and deaminases largely impacts the editing window. Further efforts should be devoted to improving base editing within the proximal 3 bp of the PAM, thus enabling the development of broader-range CBEs and expanding the microbial gene editing toolkit.
Methods
Strains and culture conditions
The strains used in this study are listed in Supplementary Table 1. E. coli Turbo, E. coli MG1655, and Pseudomonas aeruginosa PAO1 are typically cultured in LB medium at 37 °C, while Shewanella oneidensis MR-1 is cultured in LB medium at 30 °C. Chemicals were added at the following concentrations if necessary: 34 μg/mL chloramphenicol for Turbo, MG1655, and MR-1, and 20 μg/mL gentamycin for PAO1. Homologous recombination of the lac promoter-driven gfp-lacZ expression cassette was integrated into the genome of MG1655 according to a previous study13.
Plasmid construction
FrCas9n (E796A), evoAPOBEC, hAID, evoCDA, and sgRNA expression cassettes were commercially synthesized (General Bio Co., China). The rAPOBEC, UGI, and plasmids for expression were derived from previous work, pCBEso13. The cytidine deaminase was linked to the N-terminus of FrCas9n via a 16-amino acid XTEN linker, while UGI was connected to the C-terminus of FrCas9n via a 4-amino acid linker. For constructing the fusion protein of cytidine deaminase inserted into FrCas9n, cytidine deaminase was linked to each end of the FrCas9n with a 16-amino acid XTEN linker. To construct a high-fidelity FrCas9, seven amino acid residues (R61A, N532A, N694A, K724A, Q728A, N892A, and Q964A) were mutated to alanine through PCR amplification and Gibson assembly. Subsequently, double mutants (R61A/N532A, R61A/Q964A, N532A/Q964A) and the triple mutant (R61A/N532A/Q964A) were similarly constructed. Additionally, all sgRNA spacer information was listed in Supplementary Data 1. Plasmids constructed in this work are listed in Supplementary Data 2. The amino acid sequences of the proteins and base editors used in the text are listed in Supplementary Data 3.
Base editing by the FrCas9n-CBE system
For the base editing in E. coli MG1655, two single-stranded DNA (ssDNA) (26-nt) were designed as follows: (a) 5’-GTGGNNN…NNN (N22) -3’; (b) 5’-AAACNNN…NNN (reverse complement of N22) -3’. Briefly, the primers were annealed and phosphorylated using T4 Polynucleotide Kinase (NEB, Co., LTD, USA) according to the manufacturer’s instructions. The dsDNA was then ligated to the BsaI (NEB, Co., LTD, USA) linearized FrCas9n-CBE plasmid using T4 DNA ligase (NEB, Co., LTD, USA) using the Golden Gate assembly method64. The resulting FrCas9n-CBE plasmids were transformed into E. coli Turbo. Next, a mixture of 100 μL of sucrose-washed MG1655 electrocompetent cells and 1 μg of target CBE plasmid extracted from the corresponding Turbo strains was transferred into a 0.2 cm sterile electroporation cuvette. Electroporation (2.47 kV/mm, 100 Ω, and 25 μF) was performed, followed by immediate supplementation of 1 mL of LB medium to the mixture and incubation at 37 °C for 2 h for recovery. Cells were collected by centrifugation, then plated on LB agar (1.5%) supplemented with 34 μg/mL chloramphenicol. The same procedure was applied for base editing in MR-1 and PAO1 using FrCas9n-CBE plasmids by electroporation. PAO1 was plated on LB agar (1.5%) supplemented with 20 μg/mL kanamycin.
Analysis of sanger sequencing results and plasmid elimination
Individual clones were picked from the plates or all colonies grown on the plates were collected. Primers were designed for PCR amplification targeting 300 bp upstream and downstream of the target site in the microbial genome. The amplified products from the target region were subjected to Sanger sequencing analysis. The sequencing results were analyzed using the online tool EditR28 (https://moriaritylab.shinyapps.io/editr_v10/) to calculate the proportion of edited T at the target site, which is defined as the editing efficiency. After base editing, single-clones with introduced premature stop codons were selected. Plasmid elimination was performed using the sacB gene, which is sensitive to sucrose and was preloaded onto the CBE plasmids. Initially, 1% of the microbial culture was inoculated into antibiotic-free LB medium and incubated for 24 h. Subsequently, the culture was plated onto LB plates containing 10% (w/v) sucrose and incubated for another 24 h. Successful plasmid elimination was verified by PCR amplification of a targeted 500 bp gene fragment.
Predicting the deactivation ability of CBEs and protein structure
To screen for potential gRNA and codons that may cause deactivation of genes in MG1655 genome, we employed the bioinformatics tool “autocbei“31. Data were obtained following the three major criteria: the PAM sequence was 5’-NNTA; mutating C to T within the editing window from C3 to C12 would generate the premature stop codons-TGA, TAG, and TAA in the coding strand. Similarly, the predicted sgRNAs for gene termination in SpCas9-CBE have a PAM of 5’-NGG with an editing window of C3 to C8. In addition, the structure of the FrCas9 was predicted using AlphaFold2 with Google Colab and default settings39. We compared the top five rankings of the outputs and selected the top-ranked one for data preparation. The structural diagram was generated using Pymol v.2.5.2 (http://www.pymol.org/).
Next generation sequencing (NGS)
To perform next-generation sequencing on endogenous target genes to calculate editing efficiency, the target genomic regions of interest were first amplified from bacterial lysates using Phanta Max Super-Fidelity DNA Polymerase (Vazyme Biotech Co., USA). For targeted deep sequencing, PCR reactions were conducted using primers with unique barcodes. The amplified products were purified with a Gel Extraction Kit (Omega) and sequenced on an Illumina NovaSeq 6000 platform (Genewiz) with 150 bp paired-end reads. Fastq data were processed and analyzed using Crispresso2.35 (version 2.0.20b)65. All analysis was based on paired-end reads, and the Fastq sequence data have been deposited in the NCBI Sequence Read Archive (PRJNA1182743).
Off‑target assay
According to Cas-OFFinder, potential Cas9-dependent off-target sites (POTs) with up to 2-nucleotide or 3-nucleotide mismatches in the MG1655 genome were predicted to analyze site-specific editing (http://www.rgenome.net/cas-offinder/)66. The genomic regions around the target and off-target sites were amplified by PCR and subsequently subjected to Sanger sequencing.
Flow cytometry assay
All microbes were resuspended in 1 mL of PBS buffer and diluted by 100-fold. Then, the diluted microbes were analyzed using flow cytometer (CytExpert; Beckman Coulter Inc., USA). MG1655-GFP and MG1655 strains served as GFP-positive and negative controls, respectively.
Orthogonal R-loop assay
As described previously, orthogonal R-loop assays were conducted to assess gRNA-independent off-target editing. First, 0.8 μg of the PYYDT plasmid encoding each of the seven base editor proteins and their respective gRNA, along with 0.8 μg of the dLbCas12a plasmid containing sgRNAs targeting five R-loop sites, were co-electroporated into E. coli. After 48 h of incubation on dual antibiotic selection plates, the strains were collected from the plates, and the R-loop off-target sites were amplified and subjected to targeted deep sequencing. All targeted sequences are provided in Supplementary Data 1.
Whole genome sequencing for DNA off-target editing
After transfecting the base editor into E. coli MG1655 or Pseudomonas aeruginosa PAO1, the strains were incubated on antibiotic selection plates for 24 h. Editing at the target site was confirmed via Sanger sequencing. Plasmid curing was then performed on 10% sucrose plates. The strains lacking plasmids were collected after overnight culture expansion, and genomic DNA was extracted using the MagPure Bacterial DNA Extraction Kit (Magen Co., China). The integrity of the extracted DNA was verified by 1% agarose gel electrophoresis. The library preparation and next generation sequence were finished by Sangon Biotech Co., China. First, 500 ng quantified DNA was randomly fragmented by Covaris (Woburn Co., USA). Next, Endprep enzyme was added to repair end and 3’ end A tail ligation. Then adaptor was ligated by enhancer and Fast T4 DNA ligase. Index primer was added by PCR and the amplified product about 400 bp was selected by DNA selection beads. Then, the libraries were pooled and loaded on Novaseq 6000 (Illumina,San Diego,USA)/DNBseq-T7 (BGI,Shenzhen,China) sequencer by 2×150 bp paired end sequence kit according to the manufacturer’s instructions.
Generation of rpoB mutant library and RifR assay
Six sgRNA expression cassettes were designed to rpoB gene and the HF-ID824-evoCDA-FrCas9n plasmid was transformed into MG1655. After 24 h, these clones were transferred to a 96-well plate with liquid medium and 10 µL of the microbial droplets were spotted on LB plates containing rifampicin (50 μg/mL) to further screen for RifR mutants. Finally, the genotypes of RifR colonies grown on rifampicin plates were confirmed by Sanger sequencing.
Electrochemical tests
A dual-chamber MFC with a working volume of 90 mL was assembled. Carbon felts with a surface area of 4 cm2 (Beijing Yesan Carbon Co., China) were used as the anode and cathode materials, respectively, while a proton exchange membrane (GEFC-10N, GEFC Co., China) served as the separator. The anode electrolyte was prepared using 50 mL of Shewanella mineral medium and 20 mM lactate as the electron donor, while the cathode electrolyte consisted of 50 mM potassium ferricyanide in a 50 mM phosphate buffer solution. The composition of the Shewanella mineral medium was referenced from previous studies, with the following composition: 0.225 g/L KH2PO4, 0.46 g/L NaCl, 0.225 g/L (NH4)2SO4,0.117 g/L MgSO4·7H2O,5 mL/L vitamin mix, 5 mL/L trace minerals, 0.01% casamino acids, 20 mM lactate, and 20 mM HEPES, pH7.250. A Shewanella MR-1 and MR-1/SO3350Q88* were cultured in LB at 30 °C for 16 h. Subsequently, the cultures were centrifuged at 8000 g for 2 min, washed twice, and resuspended in PBS buffer. The microbial suspensions were then inoculated into the anodic chamber at an initial optical density (OD600) of 0.4. The voltage output of the MFCs were recorded every 10 min using a data acquisition system (USB2801, ATD Co., China). The protein content on the carbon felts was determined using a modified Bradford protein assay kit (Sangon Bio Co., China), and analyzed through linear sweep voltammetry (LSV) and cyclic voltammetry (CV)50. Briefly, A 1 kΩ resistor was connected to the external circuit of the MFC to initiate the bioelectrochemical system, and the output voltage was recorded using a digital multimeter. LSV was performed once the voltage stabilized after MFC feeding, using a scan rate of 0.1 mV/s from the open-circuit potential to −0.2 V. The anode served as the working electrode, while the cathode and reference electrode functioned as the counter electrodes. Power densities (P) were calculated as P = V (output voltage) × I (current density), with both current (I) and power density (P) normalized to the projected area of the anode surface.
MIC determination and Short-term ALE assays
MIC of Pseudomonas aeruginosa PAO1 to PMB was determined using the microdilution technique described by the Clinical and Laboratory Standards Institute67. The results were recorded after incubation at 37 °C for 24 h. The MIC is defined as the lowest concentration of antibiotic that completely inhibits microbial growth.
The wild-type and six mutants (PAO1-mexAQ2*R3*, PAO1-mexBW13*, PAO1-oprFQ258*, PAO1-betT1W55*, PAO1-algUQ8*, PAO1-ampCQ155*) were used as parental strains. The adaptive evolution experiments were conducted in the presence of PMB under conditions of 1/2 MIC56 for a period of 3 d. The cultures were diluted (1/100) daily, and 10 μL of microbes were added to 1 mL of fresh Mueller-Hinton broth (MHB) in test tubes containing 1/2 MIC of polymyxin B in MHB67. After 3-d evolution, the strains were re-evaluated for MIC assay.
Statistics & Reproducibility
No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment. Statistical analysis was performed using Microsoft Excel (2016) and GraphPad Prism 10 (version 10.1.2). Significance testing was conducted using a two-tailed t-test, with a significance level set at P < 0.05. All experimental results were derived from at least three biologically independent experiments, and data are presented as the mean ± standard deviation (mean ± s.d.).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are provided in the Source Data File. The sequences of the primers used in this study are provided in the supplementary data. All Saner sequencing results are included in the source data file. All next-generation sequencing and whole-genome sequencing data were deposited to the National Center for Biotechnology Information Sequence Read Archive under accession number PRJNA1182743. We are open to sharing the experimental materials with interested researchers. Please address Dong-Feng Liu or Han-Qing Yu (corresponding author). Source data are provided with this paper.
References
Barrangou, R. & Horvath, P. A decade of discovery: CRISPR functions and applications. Nat. Microbiol. 2, 1–9 (2017).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Cui, L. & Bikard, D. Consequences of Cas9 cleavage in the chromosome of Escherichia coli. Nucleic Acids Res. 44, 4243–4251 (2016).
Gaudelli, N. M. et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Gallagher, L. A. et al. Genome-wide protein–DNA interaction site mapping in bacteria using a double-stranded DNA-specific cytosine deaminase. Nat. Microbiol. 7, 844–855 (2022).
Banno, S., Nishida, K., Arazoe, T., Mitsunobu, H. & Kondo, A. Deaminase-mediated multiplex genome editing in Escherichia coli. Nat. Microbiol. 3, 423–429 (2018).
Volke, D. C., Martino, R. A., Kozaeva, E., Smania, A. M. & Nikel, P. I. Modular (de)construction of complex bacterial phenotypes by CRISPR/nCas9-assisted, multiplex cytidine base-editing. Nat. Commun. 13, 3026 (2022).
Hao, W. et al. Development of a base editor for protein evolution via in situ mutation in vivo. Nucleic Acids Res. 49, 9594–9605 (2021).
Kuscu, C. et al. CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nat. Methods 14, 710–712 (2017).
Shelake, R. M., Pramanik, D. & Kim, J.-Y. Improved dual base editor systems (iACBEs) for simultaneous conversion of adenine and cytosine in the bacterium Escherichia coli. mBio 14, e02296–22 (2023).
Pan, Y. et al. Random base editing for genome evolution in Saccharomyces cerevisiae. ACS Synth. Biol. 10, 2440–2446 (2021).
Zheng, K. et al. Highly efficient base editing in bacteria using a Cas9-cytidine deaminase fusion. Commun. Biol. 1, 1–6 (2018).
Cheng, L. et al. Developing a base‐editing system to expand the carbon source utilization spectra of Shewanella oneidensis MR‐1 for enhanced pollutant degradation. Biotech. Bioeng. 117, 2389–2400 (2020).
Li, K. et al. Powerful microbial base-editing toolbox: from optimization strategies to versatile applications. ACS Synth. Biol. acssynbio.3c00141. https://doi.org/10.1021/acssynbio.3c00141 (2023).
Rajewska, M., Wegrzyn, K. & Konieczny, I. AT-rich region and repeated sequences – the essential elements of replication origins of bacterial replicons. FEMS Microbiol. Rev. 36, 408–434 (2012).
Li, X. et al. Base editing with a Cpf1–cytidine deaminase fusion. Nat. Biotechnol. 36, 324–327 (2018).
Chatterjee, P. et al. A Cas9 with PAM recognition for adenine dinucleotides. Nat. Commun. 11, 2474 (2020).
Wu, Y. et al. Genome-wide analyses of PAM-relaxed Cas9 genome editors reveal substantial off-target effects by ABE8e in rice. Plant Biotechnol. J. 20, 1670–1682 (2022).
Ru-Li He et al. Biomolecular insights into extracellular pollutant reduction pathways of Geobacter sulfurreducens using a base editor system. Environ. Sci. Technol. https://doi.org/10.1021/acs.est.2c02756 (2022).
Wu, J. et al. Controlling pathogenic risks of water treatment biotechnologies at the source by genetic editing means. Environ. Microbiol. 23, 7578–7590 (2021).
Wang, Y. et al. CRISPR-Cas9 and CRISPR-assisted cytidine deaminase enable precise and efficient genome editing in Klebsiella pneumoniae. Appl. Environ. Microbiol. 84, e01834-18 (2018).
Kim, Y. B. et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 35, 371–376 (2017).
Chen, Y. et al. Development of whole genome-scale base editing toolbox to promote efficiency of extracellular electron transfer in Shewanella oneidensis MR-1. Adv. Biol. 6, 2101296 (2022).
Cheng, T.-L. et al. Expanding C–T base editing toolkit with diversified cytidine deaminases. Nat. Commun. 10, 3612 (2019).
Wang, Y., Zhou, L., Liu, N. & Yao, S. BE-PIGS: a base-editing tool with deaminases inlaid into Cas9 PI domain significantly expanded the editing scope. Sig Transduct. Target Ther. 4, 1–3 (2019).
Park, S. & Beal, P. A. Off-target editing by CRISPR-guided DNA base editors. Biochemistry 58, 3727–3734 (2019).
Cui, Z. et al. FrCas9 is a CRISPR/Cas9 system with high editing efficiency and fidelity. Nat. Commun. 13, 1425 (2022).
Kluesner, M. G. et al. EditR: a method to quantify base editing from sanger sequencing. CRISPR J. 1, 239–250 (2018).
Wang, Y., Liu, Y., Zheng, P., Sun, J. & Wang, M. Microbial base editing: a powerful emerging technology for microbial genome engineering. Trends Biotechnol. 39, 165–180 (2021).
Walton, R. T., Christie, K. A., Whittaker, M. N. & Kleinstiver, B. P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368, 290–296 (2020).
Yu, H., Wu, Z., Chen, X., Ji, Q. & Tao, S. CRISPR-CBEI: a designing and analyzing tool kit for cytosine base editor-mediated gene inactivation. mSystems 5, e00350–20 (2020).
Thuronyi, B. W. et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol. 37, 1070–1079 (2019).
Komor, AlexisC. et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T: A base editors with higher efficiency and product purity. Sci. Adv. 3, eaao4774 (2017).
Nguyen Tran, M. T. et al. Engineering domain-inlaid SaCas9 adenine base editors with reduced RNA off-targets and increased on-target DNA editing. Nat. Commun. 11, 4871 (2020).
Zhao, D. et al. Engineered domain-inlaid Nme2Cas9 adenine base editors with increased on-target DNA editing and targeting scope. BMC Biol. 21, 250 (2023).
Li, S. et al. Docking sites inside Cas9 for adenine base editing diversification and RNA off-target elimination. Nat. Commun. 11, 5827 (2020).
Huang, T. P. et al. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat. Biotechnol. 37, 626–631 (2019).
Zhang, S. et al. TadA reprogramming to generate potent miniature base editors with high precision. Nat. Commun. 14, 413 (2023).
Bryant, P., Pozzati, G. & Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 13, 1265 (2022).
Hu, Z. et al. Improving the precision of base editing by bubble hairpin single guide RNA. mBio 12, e00342-21 (2021).
Kleinstiver, B. P. et al. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016).
Vakulskas, C. A. et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 24, 1216–1224 (2018).
Bratovič, M. et al. Bridge helix arginines play a critical role in Cas9 sensitivity to mismatches. Nat. Chem. Biol. 16, 587–595 (2020).
Slaymaker, I. M. et al. Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2016).
Doman, J. L., Raguram, A., Newby, G. A. & Liu, D. R. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 38, 620–628 (2020).
Yu, Y. et al. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat. Commun. 11, 2052 (2020).
Kim, D. Y. et al. Hypercompact adenine base editors based on a Cas12f variant guided by engineered RNA. Nat. Chem. Biol. 18, 1005–1013 (2022).
Rasouly, A. et al. Analysing the fitness cost of antibiotic resistance to identify targets for combination antimicrobials. Nat. Microbiol 6, 1410–1423 (2021).
Garibyan, L. et al. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair (Amst.) 2, 593–608 (2003).
Min, D. et al. Enhancing extracellular electron transfer of Shewanella oneidensis MR-1 through coupling improved flavin synthesis and metal-reducing conduit for pollutant degradation. Environ. Sci. Technol. 51, 5082–5089 (2017).
Vincent, J.-L. Nosocomial infections in adult intensive-care units. Lancet 361, 2068–2077 (2003).
Nolan, L. M. et al. Identification of Tse8 as a Type VI secretion system toxin from Pseudomonas aeruginosa that targets the bacterial transamidosome to inhibit protein synthesis in prey cells. Nat. Microbiol 6, 1199–1210 (2021).
Tamma, P. D. et al. Infectious diseases society of America guidance on the treatment of extended-spectrum β-lactamase producing Enterobacterales (ESBL-E), carbapenem-resistant enterobacterales (CRE), and Pseudomonas aeruginosa with difficult-to-treat resistance (DTR-P. aeruginosa). Clin. Infect. Dis. 72, e169–e183 (2021).
Nicoloff, H., Hjort, K., Levin, B. R. & Andersson, D. I. The high prevalence of antibiotic heteroresistance in pathogenic bacteria is mainly caused by gene amplification. Nat. Microbiol. 4, 504–514 (2019).
Andersson, D. I. & Hughes, D. Microbiological effects of sublethal levels of antibiotics. Nat. Rev. Microbiol. 12, 465–478 (2014).
Sanz‐García, F., Hernando‐Amado, S. & Martínez, J. L. Evolution under low antibiotic concentrations: a risk for the selection of Pseudomonas aeruginosa multidrug‐resistant mutants in nature. Environ. Microbiol. 24, 1279–1293 (2022).
Dötsch, A. et al. Genomewide identification of genetic determinants of antimicrobial drug resistance in Pseudomonas aeruginosa. Antimicrobial Agents Chemother. 53, 2522–2531 (2009).
Li, Y. et al. Resistance elicited by sub-lethal concentrations of ampicillin is partially mediated by quorum sensing in pseudomonas aeruginosa. Environ. Int. 156, 106619 (2021).
Glen, K. A. & Lamont, I. L. β-lactam resistance in Pseudomonas aeruginosa: current status, future prospects. Pathogens 10, 1638 (2021).
Malek, A. A., Chen, C., Wargo, M. J., Beattie, G. A. & Hogan, D. A. Roles of three transporters, CbcXWV, BetT1, and BetT3, in Pseudomonas aeruginosa choline uptake for catabolism. J. Bacteriol. 193, 3033–3041 (2011).
Hua, K., Tao, X. & Zhu, J.-K. Expanding the base editing scope in rice by using Cas9 variants. Plant Biotechnol. J. 17, 499–504 (2019).
Jiang, W. et al. BE-PLUS: a new base editing tool with broadened editing window and enhanced fidelity. Cell Res. 28, 855–861 (2018).
Hao, W. et al. A new-generation base editor with an expanded editing window for microbial cell evolution In vivo based on CRISPR‒Cas12b engineering. Adv. Sci. n/a, 2309767 (2024).
Engler, C., Kandzia, R. & Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PLOS ONE 3, e3647 (2008).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Bae, S., Park, J. & Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
Miller, R. A. et al. Standardization of a broth microdilution susceptibility testing method to determine minimum inhibitory concentrations of aquatic bacteria. Dis. Aquat. Org. 64, 211–222 (2005).
Kozaeva, E., Nielsen, Z. S., Nieto-Domínguez, M. & Nikel, P. I. The pAblo · pCasso self-curing vector toolset for unconstrained cytidine and adenine base-editing in Gram-negative bacteria. Nucleic Acids Res. gkad1236. https://doi.org/10.1093/nar/gkad1236 (2024).
Wang, Y. et al. Expanding targeting scope, editing window, and base transition capability of base editing in Corynebacterium glutamicum. Biotechnol. Bioeng. 116, 3016–3029 (2019).
Yu, S. et al. CRISPR-dCas9 mediated cytosine deaminase base editing in Bacillus subtilis. ACS Synth. Biol. 9, 1781–1789 (2020).
Acknowledgements
The authors wish to thank the National Key Research and Development Program of China (2019YFA0905504-D-F.L.), the National Natural Science Foundation of China (52192684-H.-Q.Y, 51821006-H.-Q.Y, 52322002-D-F.L. and 52400255-Z-H.C.), the Major Science and Technology Projects of Anhui Province (202203a07020015-D-F.L.), the Science Fund for Distinguished Young Scholars of Anhui Province (2208085J36-D-F.L.), the Natural Science Foundation of Anhui Province (2408085QB055-Z-H.C.) and the Postdoctoral Fellowship Program of CPSF under Grant Number GZC20241642-Z-H.C. for supporting this work.
Author information
Authors and Affiliations
Contributions
Z.H.C., D.F.L. and H.Q.Y. designed this work. L.H., J.H., Z.H.C., D.F.L., and H.Q.Y. performed the experimental investigations. L.H., Z.H.C., H.D.W., C.C.L., and J.H. performed the data analyses. All authors contributed to the interpretation of the findings. L.H., Z.H.C., D.F.L., and H.Q.Y. wrote and edited. All the authors contributed to discussion of the results and the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Daniel Volk and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, L., Han, J., Wang, HD. et al. A universal and wide-range cytosine base editor via domain-inlaid and fidelity-optimized CRISPR-FrCas9. Nat Commun 16, 1260 (2025). https://doi.org/10.1038/s41467-025-56655-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56655-7
