
Abstract
Genome-wide association studies have identified thousands of genetic variants associated with non-small cell lung cancer (NSCLC), however, it is still challenging to determine the causal variants and to improve disease risk prediction. Here, we applied massively parallel reporter assays to perform NSCLC variant-to-function mapping at scale. A total of 1249 candidate variants were evaluated, and 30 potential causal variants within 12 loci were identified. Accordingly, we proposed three genetic architectures underlying NSCLC susceptibility: multiple causal variants in a single haplotype block (e.g. 4q22.1), multiple causal variants in multiple haplotype blocks (e.g. 5p15.33), and a single causal variant (e.g. 20q11.23). We developed a modified polygenic risk score using the potential causal variants from Chinese populations, improving the performance of risk prediction in 450,821 Europeans from the UK Biobank. Our findings not only augment the understanding of the genetic architecture underlying NSCLC susceptibility but also provide strategy to advance NSCLC risk stratification.
Similar content being viewed by others


Introduction
Lung cancer has a high incidence and mortality rate in both China and worldwide. Non-small cell lung cancer (NSCLC) accounts for 85% of total lung cancer cases and poses a significant threat to public health1,2. NSCLC is a multifactorial disease driven by environmental exposures, especially cigarette smoking and inherited germline genetic variants3. Genome-wide association studies (GWASs) are powerful approaches for identifying genetic factors for NSCLC. Large-scale GWASs have thus far identified thousands of genetic variants for NSCLC at genome-wide significance across various populations4,5,6,7,8,9,10,11,12,13,14,15,16,17. However, determining causal variants that manifest phenotypes in a GWAS remains difficult. More than 90% of the identified genetic variants reside in noncoding regions of the genome with unknown function. In addition, due to linkage disequilibrium (LD), each of these loci can encompass hundreds of disease-associated single-nucleotide polymorphisms (SNPs), which render it extremely difficult to pinpoint those genetic variants that functionally contribute to the phenotype18.
A variety of fine-mapping strategies have been developed to determine these disease-causing variants18,19,20,21,22,23. One prevalent strategy is to propose hypotheses of genetic mechanisms by integrating statistical association containing LD information with epigenetic annotations. Then, a small number of candidate variants are nominated for validation via low-throughput experiments. By employing the strategy, recent studies have successfully identified a series of causal variants for multiple diseases24,25,26,27, but most of them investigate one locus at a time, and can hardly elucidate all causal variants or clarify the genetic architecture at each locus due to the limited number of functionally evaluated variants. Massively parallel reporter assays (MPRAs) enable the high-throughput experimental evaluation of the transcriptional regulatory potential of noncoding DNA sequences28. In an MPRA, multiple regulatory elements are cloned into an expression vector containing a reporter gene and a unique DNA barcode, creating an expression library. This library is then analyzed using high-throughput sequencing to assess the regulatory activity of the cloned elements29,30. To date, MPRAs have successfully identified causal variants for multiple traits and disorders, including red blood cell traits, autoimmune disease, and neurodegenerative disease29,30,31,32, yet they have not been systematically adopted for lung cancer studies.
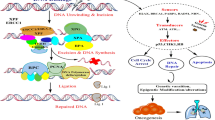
In this work, we designed and applied an MPRA to systematically characterize causal variants underneath NSCLC GWAS loci from our previous study, which included 14,240 cases and 14,813 control individuals in the Chinese populations4. We totally evaluated 1249 genetic variants and identified 82 functional regulatory variants (frVars) in three lung-related cell types. Through the integration of MPRA and lung-specific transcriptional regulatory annotations, we identified 30 potential causal variants within 12 loci, revealing the distinct genetic architectures underlying NSCLC susceptibility. We also determined the target genes of these variants using a lung tissue-specific expression quantitative trait loci (eQTL) database and elucidated the genetic mechanisms at three representative loci (4q22.1, 5p15.33, and 20q11.23). Finally, we incorporated the potential causal variants into the construction of polygenic risk score (PRS) and evaluated it in a cohort of 450,821 Europeans from UK Biobank (UKB) datasets, with the aim to improve the cross-ancestry performance of polygenic risk prediction (Fig. 1).

a Variant Selection. A total of 1,288 variants with an overall NSCLC P-value smaller than 1 × 10−5 were selected from previous GWAS study. The P-values were calculated using Firth’s logistic regression in 14,240 cases and 14,813 control individuals. These P-values were two-sided and unadjusted. b Massively Parallel Reporter Assay (MPRA). All allelic pairs were barcoded and cloned into an expression library that was transfected into A549、H1299 and BEAS-2B cells. Allelic expression was quantified by next-generation sequencing of associated barcodes and variants with significant allele-specific transcriptional activity were identified. c Variant Function Annotation. Variants with MPRA results were further prioritized using lung-specific genomic annotations. d Causal Variant Identification. By integrating MPRA results with lung-related functional genomic features, we pinpointed potential causal variants at 12 different GWAS loci. The GWAS P-values were same with (a). e PRS Improvement in Cross-ancestry Population. Incorporating the potential causal variants from Chinese into construction of polygenic risk score (PRS) from a total of 450,821 Europeans in UK Biobank improved the cross-ancestry performance of risk prediction. Error bars represented 95% confidence interval (CI). The Cox proportional hazard model was used to calculate the P-value. The P-values were two-sided and unadjusted.
Results
Study design and MPRA quality control
We selected common and low-frequency variants (minor allele frequency [MAF] > 0.5%) with a P-value smaller than 1 × 10−5 in our recent GWAS4, which utilized whole-genome sequencing (WGS) and sequencing-based imputation, and then excluded the variants within the major histocompatibility complex (MHC) region (Fig. 1a). As a result, a total of 1288 variants were included. For each variant, we generated a pair of 120 base pair (bp) DNA oligonucleotides (subsequently referred to as “oligos”) for each allele in both forward and reverse directions, with the variant located in the center and identical flanking genomic sequence across the alleles. A scrambled sequence for its core 20 bases was also generated and this finally resulted in an assay of 7728 total allelic pairs (Fig. 1a and Supplementary Data 1). For barcoding, random 20-bp sequences were coupled to each oligo. The number of unique barcodes had an approximately normal distribution with a median of 6803 barcodes per oligo (Supplementary Fig. 1a). After reporter gene insertion, the plasmid library was separately transfected into lung or lung cancer epithelial-like cells (A549, H1299 and BEAS-2B cell lines) in six independent technical replicates, obtaining activity measurements from at least five unique barcodes for both alleles of 1249 of 1288 (96.8%) different variants (Fig. 1b). Those variants consisted of median library complexities of 103, 80, and 89 barcodes per allele in different cell types respectively (Supplementary Fig. 1b). Quality assessment also showed that barcode prevalence in DNA and complementary DNA (cDNA) replicates were tightly correlated (Supplementary Fig. 1c) and that some barcodes were more present in cDNA than in plasmid libraries, suggesting higher expression of some active regulatory elements (Supplementary Fig. 1d).
Identification of frVars and the evaluation in lung-related functional annotations
Then, we used negative binomial regression to measure the regulatory effects of each variant (Methods). Overall, we first observed that 387 library elements (23.0%) were transcriptionally active (Fig. 2a). Relative to non-active elements, transcriptionally active elements were enriched within lung-related epigenomic annotations, including DNase and ATAC (open and active chromatin), H3K27Ac and H3K4me1 (active enhancers), and H3K4me3 and H3K9ac (active promoters) (Figs. 1c and 2b), but not in repressive signals such as H3K36me3 and H3K9me3. We also identified significant enrichment of transcription factor binding sites (TFBSs) within active elements, including SP/KLF and E2F family members (Supplementary Fig. 2a). We then defined frVars with a stringent threshold, requiring both significant regulatory effect on the elements and significantly different transcriptional efficacy between alleles. In total, we identified 82 frVars distributed across 15 loci including four novel loci, with a median of four frVars per locus (Fig. 2c, Table 1 and Supplementary Data 2). As expected, effect sizes of these frVars were generally modest (mean absolute Log2FoldChange = 0.36; Fig. 2d). We then characterized frVars using functional annotations from lung-specific expression quantitative trait loci (eQTLs), predicted disruption of TFBSs, lung-specific open chromatin data and ChromHMM active states. Of 82 frVars, all except three (79/82, 96.3%) had at least one functional regulatory annotation, and the majority overlapped two or more functional annotations; 55 (67.1%) overlapped transcriptionally active regions in human lung; 74 (90.2%) regulated gene expression of lung normal tissue; and 56 (72.0%) were predicted to significantly alter transcription factor binding (Fig. 2e, f). The magnitude and directionality of predicted TFBSs disruption correlated with MPRA effect sizes for frVars in our dataset (Pearson’s rho = 0.37, P = 1.2 × 10−5, Supplementary Fig. 2b). Furthermore, we developed LungENN (Lung Effect Neural Network), a neural network-based epigenomic effects model. It was an extension of DeepSEA33 that utilized the convolutional neural network architecture to predict regulatory effects of genome-wide variants based on the lung-specific chromatin profiles available in public databases (Supplementary Data 3). LungENN predicted functional features with high accuracy, with a median area under the curve (AUC) of 0.948 (Supplementary Fig. 3). Of note, we found frVars identified in this study had good concordance with functional predictions from LungENN (Fig. 2g, h), but not with other popular computational algorithms (i.e., CADD and LINSIGHT) (Supplementary Fig. 4). In addition, we observed a poor correlation between the LungENN score and the allelic effects of non-frVars (Fig. 2i). Overall, the collective evidence presented above strongly supported the validity and reliability of our MPRA results and most of the regulatory variants identified by our assay likely had an active regulatory role in human lung tissues.

a Quantification of MPRA elements transcriptional activity (n = 6 replicates). Significantly (FDR-adjusted q < 0.01) increased (active) and decreased (repressed) library elements highlighted on the volcano plot in orange and blue respectively. The Wald test was used to calculate the P-value in a nested fixed model. The P-values were two-sided and adjusted for multiple comparisons using the False Discovery Rate (FDR) method. b Active elements were enriched for relevant genomic features. A total of 1249 variants were included in the analysis. Enrichment log2 odds ratios and P-values (Fisher’s exact test) of active and repressed elements within lung-related chromatin accessibility and histones were presented. The P-values were two-sided and unadjusted. Error bars indicated 95% confidence interval (CI). c Histogram of the number of frVars identified per GWAS locus (median = 4). d Volcano plot showed log2 allelic effect sizes and −log10 P values for 1249 different variants evaluated by MPRA. The P-values were generated using the same statistical methods as (a). e Upset plot showed the number of frVars (bars) overlapping combinations (dots and lines below bars) of different functional genomic annotations. Marginal values of each specific annotation were shown in the graphic (lower left). f Pie chart depicted frVars binned by total number of overlapping genomic annotations (percentages and counts). g All variants evaluated by MPRA were scored using LungENN. Violin plots showed algorithm prediction scores for the frVars and non-frVars. The P-values were obtained from Wilcoxon test and were two-sided and unadjusted. h Allelic effect of frVars was highly correlated with LungENN score (Pearson’s correlation). The error bands were estimated based on the standard error calculated from the linear regression model, representing a 95% confidence interval around the regression line. The P-values were two-sided and unadjusted. i Allelic effect of non-frVars was poorly correlated with LungENN score (Pearson’s correlation). The error bands were estimated based on the standard error calculated from the linear regression model, representing a 95% confidence interval around the regression line. The P-values were two-sided and unadjusted.
Functional fine-mapping of NSCLC GWAS loci
By integrating MPRA results with lung-related functional genomic features, including chromatin accessibility of the element and the potential for variants to regulate gene expression, our analysis was enhanced and this enabled us to pinpoint causal variants in different GWAS loci (Fig. 1d). We identified potential causal variants in a total of 12 GWAS loci, comprising nine loci that had been previously reported (accounting for 64.3% of all assessed reported GWAS loci) and three novel loci (Table 2). Of these 12 GWAS loci, seven of them had two or more potential causal variants. Utilizing the identified potential causal variants and their LD relationships, we further classified these 12 loci and discerned three distinct genetic architectures: multiple causal variants in a single haplotype block (4q22.1, 3q28, 14q13.1, 10q25.2, and 17q24.2), multiple causal variants in multiple haplotype blocks (5p15.33 and 11q23.3), and a single causal variant (20q11.23, 6p21.2, 8p12, 15q23, and 4p15.31) (Table 2).
Characterization of loci that included multiple causal variants in a single haplotype block
We first characterized the chromosome 4q22.1 locus, as one of the frVars within it displayed the strongest allelic biases in reporter activity in the MPRA (rs2904259: log2FoldChange = −2.64, q = 7.79 × 10−292, Supplementary Data 2) and it was a newly identified suggestive loci for NSCLC (Pgwas = 7.26 × 10−8). The 4q22.1 locus only involved a single haplotype block associated with NSCLC and two potential causal variants were identified in the block (Fig. 3a). The transcription-directing activity of the two variants was immediately validated using a luciferase assay in H1299 cells (Fig. 3b). Among them, the C allele of rs2904259 was predicted to disrupt binding motifs of multiple transcription factors, especially the FOS/JUN family (Fig. 3c), which usually form dimeric complexes in the nucleus that bind to AP1 elements and regulate gene transcription34. The public lung-specific ChIP experiments also confirmed the binding of the FOS and FOSL2 transcription factors at this position (Fig. 3c). Furthermore, we determined the candidate causal genes of this locus with our previous lung tissue-specific eQTL support4. Both two causal variants were within the intron of FAM13A and can regulate the expression of FAM13A in lung tissues (Fig. 3a, d). A locus-level GWAS-eQTL colocalization was also observed (Fig. 3e). We separately confirmed the regulations of two different elements where the causal variants were located using CRISPR-mediated excision (Fig. 3f). We also conducted a CRISPR knock-in experiment on rs2904259 using the adenine base editor (ABE), NG-ABE8e35. As a result, we observed a significant decrease in FAM13A gene expression after base editing (Fig. 3g). Since 4q22.1 has been reported to be a risk locus for chronic obstructive pulmonary disease (COPD)36, we further observed a strong correlation between the GWAS P values of lung cancer and COPD at the locus based on the BioBank Japan (BBJ) cohort data37 (Fig. 3h), suggesting it plays a crucial role in mediating the related pathogenic mechanisms underlying the development of both lung cancer and COPD.

a Top: All evaluated variants at 4q22.1 plotted by position and MPRA significance (−log10 P values; the shaded rs2904259 and rs2464522 indicated potential causal variants). Middle: GWAS regional plot of 4q22.1 was shown and the same colors indicated the same haplotype blocks (r2 > 0.2 with rs2904259). Bottom: Lung-related functional annotations of variants were shown. The vertical line indicated that the variants coincide with the annotation, and the red line marked the potential causal variants. The GWAS P-values were calculated using Firth’s logistic regression in 14,240 cases and 14,813 control individuals. These P-values were two-sided and unadjusted. The MPRA P-values were calculated using the Wald test in a nested fixed model. The P-values were two-sided and adjusted for multiple comparisons using the False Discovery Rate (FDR) method. b Luciferase assays for the activity of rs2904259:T > C and rs2464522:G > A in the H1299 cell lines. The P-values were obtained using the Student’s T-test and were two-sided and unadjusted. The error bars represented mean ± SD. A total of 6 biological replicates were used in each group. c The alternate allele of rs2904259 was predicted to disrupt FOS/JUN family TFBSs. d eQTL signals were presented for FAM13A with rs2904259 and rs2464522 respectively. The P-values were obtained using the linear regression model from 116 Chinese individuals. The P-values were two-sided and unadjusted. The bottom, middle, and top of each box plot represented the 25th, 50th, and 75th percentiles. The solid line ends represented the minimum and maximum values, respectively. e The Pearson correlation between log-transformed P values of eQTL and GWAS was shown. Green dots indicated evaluated MPRA variants and red dots indicated two potential causal variants. The reported P-values were two-sided and unadjusted. f CRISPR-mediated excisions of two different genomic elements containing rs2904259 or rs2464522 both changed FAM13A expression. The P-values were generated using the same statistical methods as (b). The error bars represented mean ± SD. A total of 6 biological replicates were used in the rs2904259, and 9 biological replicates were used in the rs2464522. g Transcriptional analysis of FAM13A gene following base editing of rs2904259 in H1299 cells. The P-values were generated using the same statistical methods as (b). The error bars represented mean ± SD. A total of 9 biological replicates were used in each group. h The Pearson correlation between log-transformed P-values of COPD GWAS in BBJ dataset and NSCLC GWAS in our study. The P-values were two-sided and unadjusted. COPD: chronic obstructive pulmonary disease; BBJ: BioBank Japan.
Similar to 4q22.1, some reported loci such as 3q28, 10q25.2, 17q24.2, and 14q13.1, also showed the same genetic architecture, that is, multiple causal variants in a single haplotype block. In this study, we tagged the causal effect of rs34951828, as well as another variant of high LD, rs7619517, identifying TP63 as a strong candidate risk gene, at the 3q28 locus (Supplementary Fig. 5). At 10q25.2, two variants were identified as causal variants, which we show regulate ZDHHC6 and ACSL5 expression, respectively (Supplementary Fig. 6). Besides, potential causal variants at 14q13.1 and 17q24.2 exhibited pleiotropic regulatory effects, thereby identifying multiple potential risk genes, such as WIPI1, CACNG1, BPTF and KPNA2 at 17q24.2 (Supplementary Fig. 7 and 8), and KIAA0391, EAPP, and FAM177A1 at 14q13.1 (Supplementary Fig. 9). In addition, potential causal variants located at 3q28, 14q13.1 and 10q25.2 displayed considerable heterogeneity across different histological subtypes, demonstrating stronger genetic effects in lung adenocarcinoma (LUAD) (Supplementary Data 4).
Characterization of loci that included multiple causal variants in multiple haplotype blocks
The 5p15.33 locus harbors the strongest common genetic association with lung cancer and it contains complex haplotype blocks and extensive LD, hampering interrogation with traditional statistical genetics approaches. We leveraged the ability to functionally dissect this locus with MPRA, ultimately identifying four distinct haplotype blocks (Fig. 4a, Table 2, and Supplementary Data 5). The variants in block 1 were within the intron of TERT gene and were in strong LD with the lead SNP rs7705526 (Fig. 4a). We did not identify any frVar in block 1, indicating that they may not operate through a lung cell-specific mechanism. Previous studies, mainly in European and African ancestry, have reported that 5p15.33 locus was associated with leukocyte telomere length (TL)38. We validated the associations between variants of block 1 and TL by using our WGS dataset4, and found that the other three blocks were not associated with TL, which suggested multiple distinct mechanisms at this locus (Fig. 4b and Supplementary Data 6). We also found variable-sized associations between block 1 and the risk of various tumors by searching the GWAS catalog (Supplementary Data 7). In addition, we observed risk of variants in block 1 showed high degree heterogeneities in different subgroups of smoking and histology status (Fig. 4c). These data indicated that effect allele of variants in block 1 could lengthen TLs and thereby increase the risk of various tumors, including lung cancer.

a Upper: All evaluated variants at 5p15.33 plotted by position and MPRA significance (−log10 P values; the shaded variants indicated potential causal variants). Middle: GWAS regional plot of 5p15.33 was shown and different colors indicated the different haplotype blocks. Blocks 1 to 4 indicated variants had r2 greater than 0.2 with rs7705526, rs528894327, rs2735846 and rs459961 respectively. Bottom: Lung-related functional annotations of variants were shown. The vertical line indicated that the variants coincided with the annotations, and the red line marked the potential causal variants. The GWAS P-values were calculated using Firth’s logistic regression in 14,240 cases and 14,813 control individuals. These P-values were two-sided and unadjusted. The MPRA P-values were calculated using the Wald test in a nested fixed model. The P-values were two-sided and adjusted for multiple comparisons using the False Discovery Rate (FDR) method. b All evaluated variants at 5p15.33 plotted by position and telomere length significance and colors of the dots were consistent with figure a, indicating the different blocks. The P-values were calculated using linear regression in 2984 cases and 3020 control individuals. These P-values were two-sided and unadjusted. c Risk of rs7705526 showed high degree heterogeneities in different subgroups of smoking and histology status. A total of 9566 LUAD and 3969 LUSC cases were included for analysis. A total of 13,089 smokers and 15,964 non-smokers were included for analysis. The odds ratios were calculated using Firth’s logistic regression and the error bars indicated 95% confidence interval (CI). LUAD: lung adenocarcinoma; LUSC: lung squamous cell carcinoma. d Luciferase assays for the activity of rs2735846:C > G, rs528894327:G > A, rs2735948:G > A and rs459961:T > A in the H1299 cell lines. P-values were obtained using the Student’s T-test. The P-values were two-sided and unadjusted. The error bars represented mean ± SD. In each group, 4 biological replicates were used for the rs2735846, 9 for the rs528894327, 6 for the rs2735948, and 3 for the rs459961, with both experimental and control groups having the corresponding biological replicates. e–h eQTL signals and CRISPR results were presented for LPCAT1 with rs2735846, SLC12A7 with rs528894327, SLC6A3 with rs2735846 and NKD2 with rs459961, respectively. The P-values for eQTL signal were obtained using the linear regression model from 116 Chinese individuals. The bottom, middle, and top of each box plot represented the 25th, 50th, and 75th percentiles. The solid line ends represented the minimum and maximum values, respectively. The P-values for the bar charts were obtained using the Student’s T-test. These P-values were all two-sided and unadjusted. In each group, 9 biological replicates were used for rs2735846 and rs528894327, and 6 biological replicates were used for both rs2735948 and rs459961, with corresponding replicates in both the experimental and control groups. The error bars represented mean ± SD.
We next characterized the remaining three blocks of 5p15.33, each of which unveiled lung cancer causal variants that were implicated in lung cells, including rs2735846 in block 2, rs528894327 in block 3, and rs2735948 and rs459961 in block 4. Their transcriptional activities were all validated with luciferase assays in H1299 cells (Fig. 4d). Rs2735846 and rs528894327 both demonstrated specific associations with the risk of LUAD rather than with lung squamous cell carcinoma (LUSC) (rs2735846: ORLUAD = 1.15, PLUAD = 5.81 × 10−12, ORLUSC = 1.03, PLUSC = 0.346, Pheterogeneity = 0.002; rs528894327: ORLUAD = 2.62, PLUAD = 3.20×10−10, ORLUSC = 1.48, PLUSC = 0.137, Pheterogeneity = 0.05). Notably, rs528894327 was a low-frequency variant and was only observed in the Asians (MAFAsian=0.1%, MAFEuropean = 0), with an MAF of 0.5% in our study. We found that variants in these two blocks may regulate the gene expression of LPCAT1 and SLC12A7 respectively, and successfully verified the corresponding regulatory activities of the two elements through CRISPR experiments (Fig. 4e, f). We also identified a separate haplotype block harboring five frVars, and two of which were further prioritized as potential causal variants by regulatory annotations: rs2735948, which falls between CLPTM1L and TERT (disrupts ETV family, Supplementary Fig. 10); and rs459961, which falls within the CLPTM1L intron (predicted to disrupt STAT1 binding, Supplementary Fig. 10). Notably, neither variant regulated the gene in which it was located nor the nearest. By integrating eQTL data and CRISPR experiments validation, we have identified the underlying target genes, SLC6A3 and NKD2, respectively (Fig. 4g, h). A borderline multiplicative interaction was observed between rs459961 and smoking status on the risk of lung cancer (Supplementary Data 8), implicating a potential smoking-related mechanism for this block. Our findings indicated that 5p15.33 demonstrated the genetic architecture with multiple causal variants in different haplotype blocks. A similar architecture was also observed at 11q23.3. In two different blocks, five and one potential causal variants were identified respectively, which were associated with different potential risk genes (Supplementary Fig. 11 and 12). In addition, previous public lung single-cell eQTL data39 showed that rs10892229 at the 11q23.3 locus regulates the expression of the gene MPZL3 specifically in epithelial cell types, such as alveolar type 2 cells, ciliated cells, and SCGB1A1+/MUC5B+ secretory cells, but not in other cell types (Supplementary Data 9).
Characterization of loci that included a single causal variant
We next highlighted the classic genetic architecture, characterized by the presence of a single causal variant at the locus. For instance, the 20q11.23 locus was first reported in our recent study4. We observed rs6130139, rather than the lead SNP rs11466931, showed significant MPRA-allele-specific activity and was identified as a potential causal variant (Fig. 5a). Thus, we chose to further dissect the regulatory capacity of rs6130139 in lung-related cells. Its transcriptional regulatory activity was further confirmed via a luciferase assay in the H1299 cell line (Fig. 5b). We found and validated rs6130139 as a regulating variant of distal SRC gene rather than the adjacent genes (Fig. 5c, d), with the disease-protected allele (C) predicted to disrupt binding of SOX9/SOX5/SRY and decreased the expression of SRC (Fig. 5e).

a Top: All evaluated variants at 20q11.23 plotted by position and MPRA significance (−log10 P values; the shaded rs6130139 indicated the potential causal variant). Middle: GWAS regional plot of 20q11.23 was shown and the same color indicated the same haplotype blocks (r2 > 0.2 with rs6130139). Bottom: Lung-related functional annotations of variants were shown. The vertical line indicated that the variant coincides with the annotation, and the red line marked the potential causal variants. GWAS P-values were from a previous GWAS study. MPRA P-values were calculated using the Wald test in a nested fixed model. The MPRA P-values were two-sided and adjusted for multiple comparisons using the False Discovery Rate (FDR) method. b Luciferase assay for the activity of rs6130139:T > C in the H1299 cell lines. The P-values were obtained using the Student’s T-test. The reported P-values were two-sided and unadjusted. A total of 4 biological replicates were used in each group. The error bars represented mean ± SD. c eQTL signal was presented for SRC with rs6130139. Lung specific eQTL database is derived from 116 Chinese individuals4 P-values were obtained using the linear regression model. The reported P-values were two-sided and unadjusted. The bottom, middle, and top of each box plot represented the 25th, 50th, and 75th percentiles. The solid line ends represented the minimum and maximum values, respectively. d CRISPR-mediated excision of genomic element containing rs6130139 decreased SRC expression. The P-values were generated using the same statistical methods as (b). A total of 9 biological replicates were used in each group. The error bars represented mean ± SD. e The alternate allele of rs6130139 was predicted to disrupt SOX9/SOX5/SRY TFBSs.
Similarly, at both 8p12 and 15q23, we identified single causal variants, rs2466066 and rs28750234 respectively, neither of which were lead SNPs at the locus (Supplementary Fig. 13 and 14). The NRG1, a classic tumor susceptibility gene, was identified as the target regulatory gene for the 8p12 (Supplementary Fig. 13). The 15q23 was a newly identified locus whose potential causal variant, rs28750234, was located in the promoter region of PAQR5, and population-based eQTL data supported its transcriptional regulation of PAQR5 and nearby gene KIF23 (Supplementary Fig. 14). Besides, we also identified that the single causal variants at other two loci, rs2610989 at 4p15.31 and rs2436733 at 6p21.2. Both the two variants were lead SNPs. Among them, 4p15.31 was a novel locus that has never been reported and the putative risk gene was DCAF16 (Supplementary Fig. 15). At 6p21.2, our eQTL data supported that rs2436733 could regulate the expression of TREML2 and UNC5CL (Supplementary Fig. 16).
A polygenic risk score based on functional variants in Chinese improves the prediction of lung cancer in the European populations
To demonstrate the potential of causal variants in improving cross-ancestry PRS performance, we further incorporated the lung cancer causal variants identified in this study from the Chinese populations into the construction of lung cancer PRS from the 450,821 Europeans in UKB datasets (Fig. 1e). We first built the PRSEUR using 17 lead SNPs at previous lung cancer susceptibility loci in European populations8, and then substituted the variants in four loci shared by Chinese and European populations with the potential causal variants identified in this study to create a new PRSEUR-substituted, which involved 25 variants (Supplementary Data 10). The result showed that our new PRSEUR-substituted improved lung cancer prediction compared with the previous PRSEUR (Fig. 6). Subsequently, we integrated all identified NSCLC potential causal variants in the Chinese populations with PRSEUR to create an expanded PRS named PRSEUR-expanded, which involved 46 variants (Supplementary Data 10). Members of the top 5% PRSEUR-expanded had the highest lung cancer risk compared to the other two PRSs (Fig. 6, hazard ratio [HR] = 2.48, 95% confidence interval [CI], 1.84-3.34; P = 2.04 × 10−9).

PRSEUR was built using the previous lead SNPs of lung cancer susceptibility loci in European GWASs; PRSEUR-substituted was built by substituting the variants in the loci shared by Chinese and European populations with the potential causal variants identified in this study; PRSEUR-expanded was built by integrating all identified NSCLC causal variants in the Chinese populations with PRSEUR. A total of 450,821 Europeans from the UK Biobank were included for analysis. Error bars represented 95% confidence interval. The Cox proportional hazard model was used to calculate the P-value. The P-values were two-sided and unadjusted. HR: hazard ratio; CI: confidence interval.
Discussion
In this study, we perform a systematic functional evaluation of common and low-frequency susceptibility variants in NSCLC, determining 30 potential causal variants at 9 known and 3 new loci, with 90% of these variants not being lead SNPs. Moreover, we observe that the majority of loci contained more than one potential causal variant. We further conclude three different genetic patterns and elucidate the underlying genetic mechanisms at the representative loci. We also include the causal information in the PRS construction and improve risk prediction performance across ancestry.
Previous fine-mapping studies usually hypothesized that a GWAS hit was driven by a single causal variant18; however, increasing evidence suggests that a disease-associated locus can harbor multiple causal variants, which collectively contribute to the susceptibility of complex traits30,40,41. In our study, 7 out of the 12 loci harbored more than one potential causal variant, including two distinct genetic architectures: (I) multiple causal variants in a single haplotype block and (II) multiple causal variants in multiple haplotype blocks. Architecture (I) was the predominant genetic architecture of susceptibility loci of NSCLC, involving 5 loci (i.e., 4q22.1, 3q28, 14q13.1, 10q25.2, and 17q24.2). These causal variants could not be fully identified in prior fine-mapping studies, primarily because experiment validation was restricted to variants with the highest probability based on bioinformatics annotation, thereby leaving a large number of potential candidates unevaluated. Consequently, the effect of the block was frequently misinterpreted as the sole effect of one or a few variants with the highest annotation probability, leading to an incomplete elucidation of the genetic mechanism42.
In this genetic architecture, multiple causal variants located in a haplotype block typically collaborated to contribute to the genetic effect of the block estimated by the GWASs42,43. In our study, for the two potential causal variants at the 4q22.1 locus, we observed that the effect size increased with an increase in the number of risk alleles (P for trend = 8.43 × 10−6, Supplementary Fig. 17), implying that the variants may also collectively increased the risk of lung cancer in an additive manner. In addition, some functional variants may influence phenotype by regulating different genes and affecting distinct biological processes, despite being genetically linked within the same haplotype block. For instance, at the 10q25.2 locus, the two causal variants within a haplotype block, rs1885281 and rs138322120, regulate ACSL5 and ZDHHC6 respectively, as confirmed by CRISPR-mediated excisions (Supplementary Fig. 6 and 18). Furthermore, given that neither ZDHHC6 nor ACSL5 are recognized as known causal genes for lung cancer, further well-designed mechanistic studies will be necessary to elucidate the genetic causal effect driving this association in the future.
In our study, two previously reported loci (i.e., 5p15.33 and 11q23.3) exhibited genetic architecture (II), including 4 and 2 causal blocks, respectively. The 5p15.33 locus was one of the earliest susceptibility loci for lung cancer9. Two fine-mapping studies based on statistical association have reported two independent blocks44,45, namely blocks 1 and 4 in our study. A prior study has characterized rs36115365 as a causal variant in block 1 in 4 different cell lines, including lung cancer cells46. Despite the absence of significant allelic effects after multiple testing corrections, rs36115365 exhibited nominal significance in the combined data of three lung-related cell lines (MPRA: log2FoldChange = 0.12, Pnominal = 0.03). The block was recognized for its association with TL46,47,48 and its influence on the onset of various tumors9,49,50. The effect of block 4, however, was found to be independent of TL, with the underlying genetic mechanisms remaining elusive. In this study, we identified the potential causal variants and the target gene SLC6A3, which was involved in nicotine dependence51,52. It was consistent with our finding that the effect of the variant in this block was stronger in smokers (Supplementary Data 8). In contrast to the aforementioned blocks, two novel blocks were specifically identified in the Chinese populations. The causal variant in block 2 can influence the expression of LPCAT1, which has been reported to influence the transcription of MYC through the PI3K/AKT signaling pathway, thereby impacting the progression of lung adenocarcinoma53. In block 3, the causal variant was low-frequency among Asians but was absent in European populations (MAFAsian=0.1%, MAFEuropean = 0). These results suggested that nearby variants in the genome can form distinct haplotype blocks and potentially operate in different manners. Therefore, it is necessary to perform refined interpretations of the functional genomic structure in the vicinity of the previously reported SNPs.
This study also unveiled three loci (i.e. 4q22.1, 4p15.31, and 15q23) that failed to reach genome-wide significance, and determined the potential causal variants at these loci. The 4q22.1 locus has been previously reported in GWAS for COPD and lung-function36,54,55, yet its association with lung cancer risk has not been documented. The block identified in our study was also associated with lung-function54, suggesting shared genetic mechanisms of lung-function and lung cancer of this block56. The target gene, FAM13A, was supported by robust evidence from both populations’ eQTL data and the CRISPR experiments. Elevated expression of FAM13A has been demonstrated to affect the repair and regeneration of alveolar epithelial cells, potentially via activation of the Wnt pathway57, thereby altering the proliferation and differentiation of alveolar epithelial progenitor cells. It is worth noting that there was a signal, rs7690881, associated with lung function independent of our potential causal variants54. The signal was not associated with the expression of FAM13A (Supplementary Fig. 19), indicating that the effect of this locus was specific to lung-function. Therefore, there may be additional causal variants located at 4q22.1, which influence lung function through different mechanisms and warrant further investigation. The remaining two loci, 4p15.31 and 15q23, each harbored a single causal variant, respectively regulating the target genes DCAF16 and PAQR5. DCAF16 has been identified as a lung cancer susceptibility gene58 through a transcriptome-wide association study, with changes in its expression levels influencing the proliferation and migration capabilities of cancer cells. PAQR5 has been reported to suppress the growth and metastasis of clear cell renal cell carcinoma by inhibiting the JAK/STAT3 signaling pathway59. Thus, MPRA may facilitate the identification of novel loci, even if they do not achieve genome-wide significance in a GWAS.
PRSs have emerged as a valuable tool for quantifying the aggregate genetic predisposition to a trait or disease, holding significant potential in risk assessment and precision medicine60,61,62. However, several challenges remained to be addressed, such as enhancing the prediction accuracy and improving the transferability of PRS across different populations63,64. Firstly, the prediction accuracy of PRS hinges on the choice of SNPs used for its construction. Previous studies have indicated that incorporation of functional annotation data can enhance the prediction capability of PRS65,66,67,68. In this study, the performance of PRS was immediately improved when we replaced the original variants with potential causal variants at four loci shared by Chinese and European GWASs. This suggests that causal variants identified through MPRA may contribute to refining the predictive accuracy of PRS. Secondly, disparities in LD patterns across populations hinder the efficient transferability of PRS across ancestries68,69,70,71, as conventional PRSs were primarily constructed by lead SNPs. Emerging studies proposed that the allelic effects of causal variants should be universally applicable across populations72,73. Our study provided evidence that the potential causal variants determined by MPRA can improve the performance of PRS in a disparate population. A study also found that incorporating functional annotations to PRS could improve its trans-ancestry application71. As current efforts primarily focused on enhancing the transition from European population-based PRS to non-European populations71,74,75,76, our study suggested that causal variants identified in non-European populations might also refine the predictive accuracy for lung cancer PRS in the European populations.
Our study did not identify any causal variants at the remaining five reported susceptibility loci included in this study, namely 3q26.2, 9p21.3, 9q33.2, 2p14, and 15q21.1. A potential explanation is that MPRA was only performed in the lung epithelial cells. Consequently, causal variants that influence lung cancer susceptibility by affecting cell types other than lung epithelial cells, such as immune cells, were difficult to identify. This is also why we did not include the MHC region in our study. In addition, the causal variants that exert their influence through mechanisms other than direct transcriptional regulation (e.g., alternative splicing or alternative polyadenylation77,78) were not discernible within the scope of this study, and would necessitate the employment of other high-throughput methodologies in the future study79.
In conclusion, we augment the understanding of the functional genomic architecture required for NSCLC susceptibility by performing a high-throughput evaluation of variant regulatory activity, determining distinct genetic architecture for different loci and advancing lung cancer risk stratification across different populations. These findings greatly broaden our understanding of the genetic etiology of lung cancer and will ultimately improve the precise prevention of lung cancer.
Methods
Variant selection and DNA sequence generation
We selected common and low frequency (MAF > 0.5%) variants with an overall NSCLC P-value smaller than 1 × 10−5 from our previous study and excluded the variants within MHC region. For SNVs, we pulled 120 base pair (bp) of GRCh37-flanking DNA sequences for every allele, with the variant located in the center (59 bp upstream and 60 bp downstream of the variant). For the other types of variants (indels), we designed the flanking sequences to ensure that the longest allele has 120 bp. Adapters (15 bp) were added to each sequence at either end (5’- ACTGGCCGCTTGACG - [120 bp oligo] - CACTGCGGCTCCTGC -3’) to make a 150 bp DNA sequence (Supplementary Data 1). A scrambled sequence (core 20 bases encompassing the SNP with the reference allele were shuffled) was also generated. For all resulting sequences, we created a forward and reverse complement sequence to compensate for possible DNA synthesis errors. Finally, a total of 7728 oligos (1288 variants, 2576 alleles) were obtained.
Reporter plasmid library construction
Unique 20 bp barcodes were attached to each oligo fragment by PCR in 28 parallel reactions using Barcode_F and Barcode_R primers. Then all PCR reactions were pooled together and cleaned up in column. To create our mpra∆orf library, the purified PCR products were cloned into pMPRA1 (MiaoLingBio, P31645) plasmid which was digested with SfiI-HF (NEB, R0123) in advance. Then the ligated library was electroporated into DH10B-Plus Electroporation-Competent Cell (DE1072) in six parallel transformations with BioRad Gene Pulser Xcell Eukaryotic System, followed by independent outgrowth in 20 mL LB media with 100 ug/mL carbenicillin for 14–16 h. After growth, cultures were pooled and the plasmids were extracted by maxiprep according to standard protocols (Qiagen, 12362). All primers in MPRA experiments were shown in Supplementary Data 11.
After constructing the mpra∆orf library, we performed sequencing to create a lookup table mapping barcodes to oligos. Illumina libraries were prepared by performing 6 parallel PCR reactions with the following composition: [200 ng of plasmid, 200 µL High-Fidelity 2× PCR Master Mix (NEB, M0541), 10 µL pMPRA1_∆orf_F2, 10 µL pMPRA1_∆orf_R primers] and cycled for [95 °C for 2 min, (95 °C for 20 s, 62 °C for 15 s, 72 °C for 30 s) × 6, 72 °C for 2 min]. Amplified material was purified with DNA clean beads using a 0.8× bead/sample ratio and eluted with 30 µL of water. For indexing and sequencing of all libraries, multiplex adapters were added using the following PCR reaction: [50 µl High-Fidelity 2× PCR Master Mix, 10 μl illumia_P70X_F, 10 μl pMPRA1_∆orf_R, 10 µl water, 30 µl sample] and cycled for [95 °C 2 min, (95 °C 20 s, 60 °C 30 s, 72 °C 30 s) ×6, 72 °C 2 min]. After amplification, samples were purified from agarose gels. The library was sequenced using 2 × 150 bp chemistry on Illumina Novaseq 6000.
To prepare our final library, the minP:luciferase fragment was amplified from the pGL4.23 vector (MiaoLingBio, P0696) using minP_luciferase_F and minP_luciferase_R primers. The purified products were then cloned into mpra:∆orf which linearized with AsiSI-HF (NEB, R0630). The final plasmid pool was electroporated as before except in ten parallel transformations followed by every five cultures recovery in 200 mL LB with carbenicillin. After outgrowth, each 200 mL cultures were purified by maxiprep, resuspended in nuclease-free water and quantified by NanoDrop.
Cell culture and transfection
The human cell lines A549, H1299 and BEAS-2B were purchased from the American Type Culture Collection (ATCC). A549 and H1299 cells were cultured in completed RPMI 1640 medium (Gibco, Carlsbad, CA), while BEAS-2B cell line was cultured in completed DMEM high glucose medium (Gibco, Carlsbad, CA). all supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/ streptomycin at 37 °C with 5% CO2.
Cells were cultured in 10 cm dishes maintaining a density of 0.7–1.2 × 106 cells/mL and grown to 70–90% confluence. The next day, media was replaced with Opti-MEM followed by transfection with 15 µg of plasmid library using X-treme GENE HP DNA Transfection System Removed the transfection mixture after 6 h and cultured the cells for another 24 h. Six biological replicates were performed on separate cultures.
Reporter mRNA isolation and normalization
After recovery, total RNA was extracted using Qiagen Mini RNeasy (Qiagen) following the manufacturer’s protocol including the on-column DNase digestion. Total RNA was subjected to mRNA selection (Vazyme, N403) and first-strand cDNA was synthesized from purified RNA with HiScript II Enzyme Mix (Vazyme) and a gene-specific primer (RT_primer).
To minimize amplification bias during the creation of cDNA tag sequencing libraries, samples were normalized by qPCR using [1 µL of sample, 5.2 µL ChamQ SYBR Master Mix (Vazyme), 0.2 ul mpra:miniP:luciferase_F2 and 0.2ul pMPRA1_ΔORF_R primers]. Samples were amplified with the following conditions: [95 °C for 20 s, 40 cycles (95 °C for 20 s, 65 °C for 20 s, 72 °C for 30 s), 72 °C for 2 min].
For sequencing of barcodes from plasmid or cDNA, the following PCR reaction was performed with each sample: [25 µl NEBNext High-Fidelity 2× PCR Master Mix, 2.5 µl mpra:miniP:luciferase_F2 (including UMIs 10 bp in length), 2.5 µl pMPRA1_ΔORF_R, 10 µl water, 10 µl sample] and cycled for [95 °C 2 min, [95 °C 20 sec, 60 °C 15 s, 72 °C 30 s] × 12, 72 °C 2 min]. Amplified material was purified with DNA clean beads using a 0.8x bead/sample ratio and eluted with 30 µL of water. Indexes were added to each sample by amplifying the entire 30 µL elution in a 100 µL NEBNext reaction: [50 µl High-Fidelity 2× PCR Master Mix, 10 μl illumia_P70X_F, 10 μl pMPRA1_ΔORF_R, 10 µl water, 30 µl sample] and cycled for [95 °C 2 min, [95 °C 20 s, 60 °C 30 s, 72 °C 30 s] × 6, 72 °C 2 min]. Indexed libraries were DNA clean beads purified. Samples were sequenced using 2 × 150 bp Illumina Novaseq 6000.
MPRA data analysis
Analysis of sequencing data from MPRA experiments mainly drew on previous study43. Basically, we used FLASH80 to merge paired-end reads from NovaSeq data and then used STAR v2.7.1a81 to align the merged reads against a reference index created from the designed library sequences. Following the filtration of reads that did not uniquely map to a designed sequence or had low-quality alignment scores, we extracted the resulting barcode-oligo pairs and removed any sequences detected on multiple oligos. To quantify oligo-level counts from barcodes, we further used Bartender v1.182 to obtain barcode clusters on each sample individually and correct for sequencing errors. After clustering, we computed oligo counts by mapping each barcode to its corresponding parent oligo with an exact match, and then aggregated all barcode counts within each oligo. Oligos were excluded from the analysis if activity measurements could not be obtained from at least five unique barcodes, or if the mean raw count was less than 150 across all samples. The final oligo count matrix included measurements for 2383 allelic pairs of 1249 variants, encompassing a combination of three cell types (A549, H1299 and BEAS-2B) and different stands (forward or reverse direction).
To account for variation due both sequencing depth and allelic ratios, we applied a nested fixed model using DESeq283 described for high-depth allele-specific expression analysis that accounts for the intrinsically paired allelic design, after adjusting the effect of cell types as a categorical covariate. After fitting, we tested for expression effects using a Wald test and tested for allele effects using a linear contrast between DNA and RNA levels to test the null hypothesis that the nested allelic coefficients were equal. For both sets of summary statistics, P-values were adjusted for multiple testing using the Benjamini-Hochberg procedure. We required our final MPRA regulatory variant set to have significant expression and allelic adjusted P-values (FDR < 0.01) in either strand, and then further selected those with either allele displaying a significant departure (FDR < 0.01) from the scrambled core sequence. According to these criteria, a total of 82 variants were ultimately identified as frVars.
Functional genomic data and annotation
Functional annotations and enrichment of regulatory elements: We obtained narrowPeak files for lung-related DNase-seq, ATAC-seq, active histone ChIP-Seq (H3K4me3, H3K9ac, H3K27ac, and H3K4me1), and TF-ChIP-seq marks from the ENCODE project server (https://www.encodeproject.org/), specifically for the bronchial epithelial cell, lung fibroblast, lung tissues, and lung-related cell lines such as A549, AG04450, IMR90, PC9, and WI38. We also annotated the frVars with the core 15-state model learned using ChromHMM from the ROADMAP epigenetics consortium. We considered the following as “active” transcriptional states from the 15-state model: 1_TssA, 2_TssAFlnk, 3_TxFlnk, 4_Tx, 5_TxWk, 6_EnhG, 7_Enh, 8_ZNF/Rpts, 10_TssBiv, 11_BivFlnk, 12_EnhBiv. Subsequently, we assessed the overlap between these marks and MPRA “active” and “repressive” elements, requiring a minimum of 1 bp overlap between the 120 bp oligonucleotide and the chromatin mark using bedtools84. Enrichment for active or repressive elements was then calculated against a background set of all other evaluated oligos using a Fisher’s exact test. Predicted TFBSs enrichment for active elements was calculated using the HOMER (4.11)85 against a background set of all other oligos, after prefiltering for oligos containing reference alleles.
Functional annotations of frVars: The frVars from this study were annotated for TFBSs disruption and overlapped with functional lung-related annotations. We calculated TFBSs disruptions using the motifbreakR package86 based on the HOCOMOCO v1187 and JASPAR 201888 TF binding database (filtered for a binding threshold of P < 1 × 10−4 and “strong” predicted effects). TFBSs disruptions were also scored using the SNP2TFBS webtool89 and an enrichment odds ratio for TFBSs-disrupting variants amongst frVars was calculated using Fisher’s exact test against a background set of all evaluated variants. For frVars predicted to disrupt TFBSs, we tested the correlation between allelic effect sizes from our MPRA and the predicted TFBSs disruption scores.
Identification of potential causal variants: We identified potential causal variants in each locus by assigning an integrative score to these frVars defined by MPRA results, using lung-specific epigenomic and gene expression regulation data. Each variant was initially scored within specific categories (score 0 for no hit, score 1 for a hit), and the scores from all categories were then added up to form a total score. Variants with a total score of 3 were identified as potential causal variants. The detailed categories were listed as below: (1) MPRA score: Functional regulatory variants defined by MPRA results were considered as a hit; (2) Chromatin annotation score: Overlap with an accessible chromatin region reported in at least one dataset (datasets from ENCODE, including genomic peaks from DNase-seq, ATAC-seq, H3K4me3, H3K9ac, H3K27ac, and H3K4me1 of the lung tissues or lung-related cell lines [normal lung tissue, the fibroblast of lung, the bronchial epithelial cell, A549, AG04450, IMR90, PC9 and WI38]) was considered as a hit; (3) Gene expression regulation score: Variants displaying significant eQTL associations (P < 0.05) in lung tissue or significant matches with TF-binding disruptions for either allele predicted by motifbreakR86 analysis (filtered for a binding threshold of P < 1 × 10−4 and “strong” predicted effects), were considered as a hit. We identified target genes regulated by frVars using our previous lung specific eQTL database with 116 Chinese individuals4. In addition, for the causal variants that did not identify any regulatory genes that met the nominal threshold (P < 0.05), including the rs2735846, rs528894327, and rs459961 variants at 5p15.33, we selected the genes with suggestive threshold (P < 0.1).
Comparison with computational predication algorithms: We scored all evaluated variants using the LINSIGHT90, CADD91 and LungENN algorithms. LungENN was a neural network-based epigenomic effects model, using the convolutional neural network architecture to predict regulatory effects of genome-wide variants based on the lung-specific chromatin profiles available in public database. The architecture of the LungENN model was implemented using PyTorch and the Selene library92. Specifically, during the training process, each training sample consists of a 1000 bp sequence from the human GRCh37 reference genome centered on each 200 bp bin, paired with a label vector for 291 lung-specific chromatin features (Supplementary Data 3). These features include histone marks, transcription factors, and DNase or ATAC accessibility data, which were processed from Cistrome93, ENCODE94 and Roadmap Epigenomics95 projects. The training and testing sets were split by chromosomes and were strictly non-overlapping. Chromosomes 8 and 9 were excluded from training in order to evaluate chromatin feature prediction performance, while the remaining autosomes were utilized for training and validation. For performance evaluation on the test set, we used the area under the receiver operating characteristic curve (AUC). The predicted probability for each sequence was calculated as the average of the probability predictions for the forward and complementary sequence pairs. Finally, the prediction of variants requires only the genomic position in GRCh37 and the allele changes. The maximum functional difference score observed across any feature was assigned to each variant in this study. We used the Wilcox.test to compare the differences in predicted scores between the two groups of frVars and non-frVars. Pearson’s correlation of frVars was calculated between allelic effect-sizes and computational predicted scores.
Haplotype block
To calculate LD between variants within the susceptibility loci, we utilized whole-genome sequencing data from 3020 unrelated cancer-free controls of Chinese descent, which were included in the OMICS database we previously established4. At each locus, we used PLINK to calculate the LD relationship between all evaluated variants and causal variants. If the r2 between variants was greater than 0.2, they were divided into the same haplotype block.
Leukocyte telomere length
Leukocyte telomere length (TL) was estimated from our WGS data4 using Telseq96. In short, the reads were classified as telomere, if they contain more than k occurrences of the telomeric nucleotide motif TTAGGG. Given that the WGS data were sequenced using reads length of 150 bp, a k of 12 was recommended by previous studies38,97. Then, telomeric read counts were normalized to the number of reads in the WGS data having GC content between 48% and 52%, and this fraction was multiplied by a constant equal to the number of base pairs of a reference genome (GRCh37) with GC content between 48% and 52% and divided by the number of chromosome ends, resulting in the estimated average telomere length in kb. We used age-adjusted TL, calculated as the residuals from linear regressions of TL on age, for further analysis. Associations between variants at 5p15.33 and TL were performed in 2984 NSCLC cases and 3020 controls, respectively. After adjusting for sex, smoking status, and the first two principal components, per-allele effects (Beta) and standard errors (SE) were calculated using linear regression analysis. Finally, a fixed-effect meta-analysis was performed to combine association estimates from different groups.
Plasmids and reporter assays
Using normal germline DNA, a total of seven selected variants was cloned into the multiple cloning site of pGL3-Basic upstream of the firefly luciferase gene with primers flanked with KpnI or Xho1 sites to create wild-type construct. Site-directed mutagenesis was performed on the wild-type construct to produce the variant. Constructs were verified by Sanger sequencing. H1299 cells were cultured in RPMI 1640 medium, containing 10% fetal bovine serum. Cells were seeded in 24-well plates and transfected using X-tremeGENE HP DNA Transfection Reagent (Roche) with 2 mg of the variants-luc construct and 0.02 mg of pRL-TK (Promega), a control Renilla luciferase vector. 48 h later cells were lysed and luciferase activity was assayed with the Dual Luciferase Reporter (Promega) assay in a 96-well format according to manufacturer instructions. Experiments were performed in triplicate wells. Relative luciferase activity was calculated as the ratio of firefly to Renilla luciferase.
CRISPR excision experiments
We excised gene containing rs2904259, rs2464522, rs2735846, rs528894327, rs2735948, rs459961, rs6130139, rs1885281 and rs138322120: Three pairs of guide RNAs targeting upstream (5’) and downstream (3’) flanking sequences were designed to span a -300 to +300 base region around each variant locus and cloned into pSpCas9 BB-2A-Puro (PX459) V2.0 (Plasmid #62988). The guide RNAs were finally synthesized by the GenScript Biotech Corporation (Supplementary Data 12). Three pairs of guide RNAs were used in conjunction with X-tremeGENE™ HP DNA Transfection Reagent (Roche, 6365779) for transfection. These gRNA were employed to infect 80% confluent six-well plates of H1299 cells. Culture media was replaced 24 h later and cells were incubated for two days post-infection. For each replicate we collected total RNA using the TRIZOL method. The cDNA was reversed transcribed using Hiscript lll Reverse Transcriptase (Vazyme, R323-01) and 1 ug of total RNA. We performed qPCR using the 2× ChamQ SYBR gPCR Master Mix (Vazyme, 0711-02) and 200 nM qPCR primers (Supplementary Data 13). Relative transcript abundance was quantified using the 2-ΔΔCT method normalized to the geometric mean of the GAPDH reference genes.
CRISPR base editing
To introduce the rs2904259:T > C into lung cancer cells, we employed a system utilizing the NG-ABE8e35 adenine base editor (RRID: Addgene_138491) together with the guide RNA (gRNA) plasmid. We purchased the gRNA expression plasmid (gRNA sequence: GGTGATTCATTATGACTAGA) from Tsingke Biotechnology. H1299 cells were seeded in six-well plates at a density of 3.5 × 105 cells per well, incubated overnight at 37 °C, and transfected with 3 µg ng of NG-ABE8e plasmid, along with 1 µg of sgRNA expression plasmid, following the manufacturer’s protocol. Following transfection, the cells underwent selection with 2.5 µg/mL puromycin for two days. Total RNA was then extracted from each replicate using the TRIZOL method. The cDNA was reversed transcribed using Hiscript III Reverse Transcriptase (Vazyme) with 1 µg of total RNA. We performed qPCR using the 2×ChamQ SYBR qPCR Master Mix (Vazyme) and 200 nM qPCR primers. Relative transcript abundance was quantified using the 2-ΔΔCT method normalized to the geometric mean of the GAPDH reference gene. Three independent biological replicates were conducted.
PRS construction
In the present study, PRS was generated by multiplying the genotype dosage of each risk allele for each variant by its respective weight (ie, the Ln of the odds ratio [OR]), summing all included variants together into a PRS. We built the PRSEUR for the European populations in the UKB by using the lead SNPs in previous reported lung cancer susceptibility locus of the largest available genome-wide association studies dataset of European descent8. By using the same exclusion criteria as before98, we included a total of 450,821 individuals for the final analysis. The UKB had obtained ethics approval from the North West Multi-centre Research Ethics Committee, which covers the UK (approval number 11/NW/0382) and had obtained written informed consent from all participants. PRSEUR-substituted was built by substituting the variants in the loci shared by Chinese and European lung cancer GWASs with the potential causal variants identified in this study and PRSEUR-expanded was built by integrating all identified NSCLC causal variants in the Chinese populations with PRSEUR. To ensure the comparability of the different models, effect sizes for all included variants were derived from the association of patients with NSCLC of European descent in the previous OncoArray Project15, which were all flipped to risk alleles, where appropriate, for consistency. After excluding redundant SNPs with a minor allele frequency of less than 0.5%, the final variants used for building various PRS models were shown in Supplementary Data 10. The PRSs were categorized as low (<5%), intermediate (5%–95%), or high (>5%) genetic risk, where percentages were calculated on the basis of the distribution of the PRS among participants without lung cancer.
Statistical analysis
MPRA data analysis has been described in detail above. All enrichment was determined using a Fisher’s exact test, except when explicitly stated. A Wilcoxon rank-sum test was used to assess the difference in algorithm prediction scores between the frVars and non-frVars. For reporter assays experiments, a two-sided Student’s t-test was performed by comparing the average value of ref-allele against the average value of alt-allele conditions. We applied a Cox proportional hazard model to evaluate the association between the different PRSs and the risk of lung cancer, and estimated the HRs and 95% CIs. We adjusted for age (continuous; age at assessment in UKB), age squared (continuous), sex (categorical), smoking pack-years (continuous), DNA source (categorical), and top ten principal components (continuous). All reported P values were two-sided. All the analyses were performed using the R software (version 3.6.3).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequencing data of MPRA have been deposited in the Genome Sequence Archive (GSA) in National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences, under accession number CRA020239 (https://ngdc.cncb.ac.cn). All other data generated in this study are available within the paper and/or the Supplementary Information. Source data are provided with this paper.
References
Sampson, J. N. et al. Analysis of Heritability and Shared Heritability Based on Genome-Wide Association Studies for Thirteen Cancer Types. J. Natl Cancer Inst. 107, djv279 (2015).
Chen, W. et al. Cancer statistics in China, 2015. CA: a cancer J. Clin. 66, 115–132 (2016).
Malhotra, J., Malvezzi, M., Negri, E., La Vecchia, C. & Boffetta, P. Risk factors for lung cancer worldwide. Eur. Res. J. 48, 889–902 (2016).
Wang, C. et al. Analyses of rare predisposing variants of lung cancer in 6,004 whole genomes in Chinese. Cancer Cell 40, 1223–1239.e1226 (2022).
Shiraishi, K. et al. A genome-wide association study identifies two new susceptibility loci for lung adenocarcinoma in the Japanese population. Nat. Genet. 44, 900–903 (2012).
Shi, J. et al. Genome-wide association study of lung adenocarcinoma in East Asia and comparison with a European population. Nat. Commun. 14, 3043 (2023).
Miki, D. et al. Variation in TP63 is associated with lung adenocarcinoma susceptibility in Japanese and Korean populations. Nat. Genet. 42, 893–896 (2010).
McKay, J. D. et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 49, 1126–1132 (2017).
McKay, J. D. et al. Lung cancer susceptibility locus at 5p15.33. Nat. Genet. 40, 1404–1406 (2008).
Li, Y. et al. Genetic variants and risk of lung cancer in never smokers: a genome-wide association study. Lancet Oncol. 11, 321–330 (2010).
Lan, Q. et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat. Genet. 44, 1330–1335 (2012).
Hung, R. J. et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 452, 633–637 (2008).
Hu, Z. et al. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nat. Genet. 43, 792–796 (2011).
Dong, J. et al. Association analyses identify multiple new lung cancer susceptibility loci and their interactions with smoking in the Chinese population. Nat. Genet. 44, 895–899 (2012).
Dai, J. et al. Identification of risk loci and a polygenic risk score for lung cancer: a large-scale prospective cohort study in Chinese populations. Lancet Res. Med. 7, 881–891 (2019).
Byun, J. et al. Cross-ancestry genome-wide meta-analysis of 61,047 cases and 947,237 controls identifies new susceptibility loci contributing to lung cancer. Nat. Genet. 54, 1167–1177 (2022).
Amos, C. I. et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat. Genet. 40, 616–622 (2008).
Schaid, D. J., Chen, W. & Larson, N. B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 19, 491–504 (2018).
Westra, H. J. et al. Fine-mapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat. Genet. 50, 1366–1374 (2018).
Tin, A. et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 51, 1459–1474 (2019).
Qin, N. et al. Comprehensive functional annotation of susceptibility variants identifies genetic heterogeneity between lung adenocarcinoma and squamous cell carcinoma. Front. Med. 15, 275–291 (2021).
Benner, C. et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501 (2016).
Kichaev G. et al. Integrating Functional Data to Prioritize Causal Variants in Statistical Fine-Mapping Studies. Plos Genet. 10, e1004722 (2014).
Kycia, I. et al. A Common Type 2 Diabetes Risk Variant Potentiates Activity of an Evolutionarily Conserved Islet Stretch Enhancer and Increases C2CD4A and C2CD4B Expression. Am. J. Hum. Genet. 102, 620–635 (2018).
Roman, T. S. et al. A Type 2 Diabetes-Associated Functional Regulatory Variant in a Pancreatic Islet Enhancer at the ADCY5 Locus. Diabetes 66, 2521–2530 (2017).
Shao, L. et al. The inherited variations of a p53-responsive enhancer in 13q12.12 confer lung cancer risk by attenuating TNFRSF19 expression. Genome Biol. 20, 103 (2019).
Wang, H. et al. Identification of specific susceptibility loci for the early-onset colorectal cancer. Genome Med. 15, 13 (2023).
Melnikov, A. et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 30, 271–277 (2012).
Tewhey, R. et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 165, 1519–1529 (2016).
Cooper, Y. A. et al. Functional regulatory variants implicate distinct transcriptional networks in dementia. Science 377, eabi8654 (2022).
Ulirsch, J. C. et al. Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell 165, 1530–1545 (2016).
Klein, J. C. et al. Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 10, 2434 (2019).
Zhou, J. & Troyanskaya, O. G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 12, 931–934 (2015).
Rauscher, F. J. 3rd, Voulalas, P. J., Franza, B. R. Jr. & Curran, T. Fos and Jun bind cooperatively to the AP-1 site: reconstitution in vitro. Genes Dev. 2, 1687–1699 (1988).
Richter, M. F. et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 38, 883–891 (2020).
Cho, M. H. et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat. Genet. 42, 200–202 (2010).
Ishigaki, K. et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 52, 669 (2020).
Taub M. A. et al. Genetic determinants of telomere length from 109,122 ancestrally diverse whole-genome sequences in TOPMed. Cell Genom. 2, 100084 (2022).
Natri, H. M. et al. Cell-type-specific and disease-associated expression quantitative trait loci in the human lung. Nat. Genet. 56, 595–604 (2024).
Hua, J. T. et al. Risk SNP-Mediated Promoter-Enhancer Switching Drives Prostate Cancer through lncRNA PCAT19. Cell 174, 564–575 e518 (2018).
Tian, J. et al. Systematic Functional Interrogation of Genes in GWAS Loci Identified ATF1 as a Key Driver in Colorectal Cancer Modulated by a Promoter-Enhancer Interaction. Am. J. Hum. Genet. 105, 29–47 (2019).
Burgess, D. J. Fine-mapping causal variants - why finding ‘the one’ can be futile. Nat. Rev. Genet. 23, 261 (2022).
Abell, N. S. et al. Multiple causal variants underlie genetic associations in humans. Science 375, 1247–1254 (2022).
Dong, J. et al. Fine mapping of chromosome 5p15.33 identifies novel lung cancer susceptibility loci in Han Chinese. Int. J. Cancer 141, 447–456 (2017).
Li, Z. et al. Fine mapping in TERT-CLPTM1L region identified three independent lung cancer susceptibility signals: A large-scale multi-ethnic population study. Mol. Carcinogenesis 57, 1289–1299 (2018).
Chen, H. et al. Large-scale cross-cancer fine-mapping of the 5p15.33 region reveals multiple independent signals. HGG Adv. 2, 100041 (2021).
Dorajoo, R. et al. Loci for human leukocyte telomere length in the Singaporean Chinese population and trans-ethnic genetic studies. Nat. Commun. 10, 2491 (2019).
Codd, V. et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat. Genet. 45, 422–427 (2013). 427e421-422.
Telomeres Mendelian Randomization C. et al. Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol. 3, 636–651 (2017).
Fang, J. et al. Functional characterization of a multi-cancer risk locus on chr5p15.33 reveals regulation of TERT by ZNF148. Nat. Commun. 8, 15034 (2017).
Ling, D., Niu, T., Feng, Y., Xing, H. & Xu, X. Association between polymorphism of the dopamine transporter gene and early smoking onset: an interaction risk on nicotine dependence. J. Hum. Genet. 49, 35–39 (2004).
Lerman, C. et al. Evidence suggesting the role of specific genetic factors in cigarette smoking. Health Psychol.: Off. J. Div. Health Psychol., Am. Psychological Assoc. 18, 14–20 (1999).
Wei, C. et al. LPCAT1 promotes brain metastasis of lung adenocarcinoma by up-regulating PI3K/AKT/MYC pathway. J. Exp. Clin. Cancer Res.: CR 38, 95 (2019).
Shrine, N. et al. Multi-ancestry genome-wide association analyses improve resolution of genes and pathways influencing lung function and chronic obstructive pulmonary disease risk. Nat. Genet. 55, 410–422 (2023).
Zhu Z. et al. A large-scale genome-wide association analysis of lung function in the Chinese population identifies novel loci and highlights shared genetic aetiology with obesity. Eur.Res. J. 58, 2100199 (2021).
Qin, N., Wang, C. & Hu, Z. Genetic insights into lung function inform better management of respiratory diseases. Cell Rep. Med. 4, 101041 (2023).
Lin, X. et al. Tempo-spatial regulation of the Wnt pathway by FAM13A modulates the stemness of alveolar epithelial progenitors. EBioMedicine 69, 103463 (2021).
Zhu, M. et al. A cross-tissue transcriptome-wide association study identifies novel susceptibility genes for lung cancer in Chinese populations. Hum. Mol. Genet. 30, 1666–1676 (2021).
Wang, L. et al. PAQR5 inhibits the growth and metastasis of clear cell renal cell carcinoma by suppressing the JAK/STAT3 signaling pathway. Cell. Oncol. 46, 1317–1332 (2023).
Choi, S. W., Mak, T. S. & O’Reilly, P. F. Tutorial: a guide to performing polygenic risk score analyses. Nat. Protoc. 15, 2759–2772 (2020).
Cho, S. M. J. et al. Measured Blood Pressure, Genetically Predicted Blood Pressure, and Cardiovascular Disease Risk in the UK Biobank. JAMA Cardiol. 7, 1129–1137 (2022).
Chatterjee, N., Shi, J. & Garcia-Closas, M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat. Rev. Genet. 17, 392–406 (2016).
Kachuri, L. et al. Principles and methods for transferring polygenic risk scores across global populations. Nat. Rev. Genet. 25, 8–25 (2024).
Lennon, N. J. et al. Selection, optimization and validation of ten chronic disease polygenic risk scores for clinical implementation in diverse US populations. Nat. Med. 30, 480–487 (2024).
Torkamani, A., Wineinger, N. E. & Topol, E. J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 19, 581–590 (2018).
Speed, D. & Balding, D. J. MultiBLUP: improved SNP-based prediction for complex traits. Genome Res. 24, 1550–1557 (2014).
Marquez-Luna, C. et al. Incorporating functional priors improves polygenic prediction accuracy in UK Biobank and 23andMe data sets. Nat. Commun. 12, 6052 (2021).
Lewis, C. M. & Vassos, E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 12, 44 (2020).
Martin, A. R. et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 51, 584–591 (2019).
Ding, Y. et al. Polygenic scoring accuracy varies across the genetic ancestry continuum. Nature 618, 774–781 (2023).
Amariuta, T. et al. Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-type-specific regulatory elements. Nat. Genet. 52, 1346–1354 (2020).
Marigorta, U. M. & Navarro, A. High trans-ethnic replicability of GWAS results implies common causal variants. Plos Genet 9, e1003566 (2013).
Hou, K. et al. Causal effects on complex traits are similar for common variants across segments of different continental ancestries within admixed individuals. Nat. Genet. 55, 549–558 (2023).
Ruan, Y. et al. Improving polygenic prediction in ancestrally diverse populations. Nat. Genet. 54, 573–580 (2022).
Miao, J. et al. Quantifying portable genetic effects and improving cross-ancestry genetic prediction with GWAS summary statistics. Nat. Commun. 14, 832 (2023).
Duncan, L. et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun. 10, 3328 (2019).
Chen, H. et al. A distinct class of pan-cancer susceptibility genes revealed by an alternative polyadenylation transcriptome-wide association study. Nat. Commun. 15, 1729 (2024).
Wang, Y. et al. Integrative splicing-quantitative-trait-locus analysis reveals risk loci for non-small-cell lung cancer. Am. J. Hum. Genet. 110, 1574–1589 (2023).
Soemedi, R. et al. Pathogenic variants that alter protein code often disrupt splicing. Nat. Genet. 49, 848–855 (2017).
Magoc, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Zhao, L., Liu, Z., Levy, S. F. & Wu, S. Bartender: a fast and accurate clustering algorithm to count barcode reads. Bioinformatics 34, 739–747 (2018).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Coetzee, S. G., Coetzee, G. A. & Hazelett, D. J. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 31, 3847–3849 (2015).
Kulakovskiy, I. V. et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 46, D252–D259 (2018).
Khan, A. et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D260–D266 (2018).
Kumar, S., Ambrosini, G. & Bucher, P. SNP2TFBS - a database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Res. 45, D139–D144 (2017).
Huang, Y. F., Gulko, B. & Siepel, A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet. 49, 618–624 (2017).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Chen, K. M., Cofer, E. M., Zhou, J. & Troyanskaya, O. G. Selene: a PyTorch-based deep learning library for sequence data. Nat. Methods 16, 315–318 (2019).
Zheng, R. et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 47, D729–D735 (2019).
Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Roadmap Epigenomics, C. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Ding, Z., Mangino, M., Aviv, A., Spector, T. & Durbin, R. Consortium UK. Estimating telomere length from whole genome sequence data. Nucleic Acids Res. 42, e75 (2014).
Lee, M. et al. Comparative analysis of whole genome sequencing-based telomere length measurement techniques. Methods 114, 4–15 (2017).
Qin, N. et al. Association of the interaction between mosaic chromosomal alterations and polygenic risk score with the risk of lung cancer: an array-based case-control association and prospective cohort study. Lancet Oncol. 23, 1465–1474 (2022).
Acknowledgements
This work was supported by the National Natural Science of China (82388102 and 82322032), the Outstanding Youth Foundation of Jiangsu Province (BK20220050), and Research Unit of Prospective Cohort of Cardiovascular Diseases and Cancer, Chinese Academy of Medical Sciences (2019RU038). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
H.S. and C.W. initiated, conceived the study, and supervised the study with H.M. C.C. and C.W. performed bioinformatics/statistical analyses and prepared the manuscript, along with Y.L., Y.G. and J.Xiang. Y.L., Q.Z. and C.L. conducted the MPRA experiments. M.A., Q.Z., S.G., and Y.S. conducted the plasmids and reporter assays and CRISPR excision experiments. Y.X., N.Q., L.Y., J.Z., X.X., Z.X. and K.W. contributed to the visualization of the original draft. Z.H., G.J., J.D., J.Xu, R.Y., L.C., L.X, Y.H., Y.J. and M.Z. revised and proofread the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, C., Li, Y., Gu, Y. et al. Massively parallel variant-to-function mapping determines functional regulatory variants of non-small cell lung cancer. Nat Commun 16, 1391 (2025). https://doi.org/10.1038/s41467-025-56725-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56725-w
