
Abstract
Aspirin is a potent lysine acetylation inducer, but its impact on lysine ubiquitination and ubiquitination-directed protein degradation is unclear. Herein, we develop the reversed-pulsed-SILAC strategy to systematically profile protein degradome in response to aspirin. By integrating degradome, acetylome, and ubiquitinome analyses, we show that aspirin impairs proteasome activity to inhibit proteasomal degradation, rather than directly suppressing lysine ubiquitination. Interestingly, aspirin increases lysosomal degradation-implicated K63-linked ubiquitination. Accordingly, using the major pathological protein of Parkinson’s disease (PD), α-synuclein (α-syn), as an example of protein aggregates, we find that aspirin is able to reduce α-syn in cultured cells, neurons, and PD model mice with rescued locomotor ability. We further reveal that the α-syn aggregate clearance induced by aspirin is K63-ubiquitination dependent in both cells and PD mice. These findings suggest two complementary mechanisms by which aspirin regulates the degradation of soluble and insoluble proteins, providing insights into its diverse pharmacological effects that can aid in future drug development efforts.
Similar content being viewed by others


Introduction
Aspirin, also known as acetylsalicylic acid, is a widely used nonsteroidal anti-inflammatory drug (NSAID) for reducing pain, fever, and inflammation. Numerous other health benefits have been reported for aspirin, including for cancer and age-related diseases, leading to its reputation as a “magic” medicine molecule1,2,3. As its chemical structure indicates, aspirin is a potent acetylating agent that induces acetylation on substrates such as cyclooxygenases, which are believed to be involved in the drug’s primary anti-inflammatory mechanism of action4.
Based on large-scale acetylomic data, aspirin is able to acetylate thousands of substrates5,6. The DAcMS method which combines deuterium-labeled aspirin, Kac-specific antibody enrichment, and mass spectrometry-based proteomics, has identified thousands of aspirin-induced cellular acetylated lysine residues, providing important information for an improved understanding of the drug’s pharmacological mechanisms in different diseases5.
Lysine contains a chemically active ε-amino group, making it a hub residue for a variety of protein post-translational modifications (PTMs), such as methylation, acylation, ubiquitination, and lipidation7. Of all lysine-centric PTMs, acetylation and ubiquitination are two dominant ones, both playing essential roles in regulating protein functions8. Acetylation is involved in numerous biological processes. Notably, histone acetylation acts as a key epigenetic regulator in gene expression and has been implicated in cancer and other human diseases9,10. Ubiquitination is another multifunctional PTM that is recognized as a central signal for protein degradation, crucial for maintaining cellular proteostasis11. Unlike other covalent modifications, ubiquitination comes in different higher-ordered linkages. Different types of ubiquitin chains produce distinct biological signals and functions. Notably, K48 and K63 are two abundant and well-studied types of ubiquitin linkages. K48-linked ubiquitination typically targets proteins for degradation by the proteasome, while K63-linked ubiquitination is involved in lysosome-dependent degradation and some other cellular processes, including protein trafficking, DNA repair, and activation of signaling pathways12,13,14,15,16.
Aspirin is able to induce widespread lysine acetylation on many substrates. However, whether lysine acetylation will interfere with ubiquitination that co-occurs on the same lysine residue and thereby affects protein stability remains an unanswered question. It is essential to thoroughly investigate the crosstalk between these two lysine-centric PTMs and its impact on protein degradation upon aspirin treatment, which will provide a deeper understanding of the drug’s pharmacology.
In this work, we aim to investigate whether aspirin-induced lysine acetylation interferes with the ubiquitination-mediated protein degradation pathway. We develop a stable isotope pulsed labeling-based strategy, termed rp-SILAC, to monitor cellular degradome changes in response to aspirin treatment. Furthermore, we combine acetylome and ubiquitinome experiments to identify potential crosstalk between lysine acetylation and ubiquitination. Our data show that aspirin reduces protein degradation by inhibiting proteasome activities rather than directly suppressing lysine ubiquitination. Interestingly, we also show in cells and with a PD mouse model that aspirin is able to increase K63-linked ubiquitination-mediated lysosomal degradation, a mechanism by which aspirin could promote the clearance of protein aggregates, such as α-syn. Collectively, our data provide critical evidence on how aspirin regulates protein stability, as well as shed light on the drug’s diverse pharmacological mechanisms with implications for future drug development.
Results
Establishing rp-SILAC to analyze protein degradome
Aspirin can induce lysine acetylation on many cellular proteins, which may impact lysine ubiquitination and affect protein degradation. To investigate the aspirin-directed protein degradation change on the proteome scale, we established the rp-SILAC strategy (Fig. 1a). The rp-SILAC approach is a swapped version of p-SILAC, which is used to study protein synthesis17,18. In the rp-SILAC experiments, HeLa cells were first fully labeled in culture media containing “medium-heavy (MH)” or “heavy (H)” arginine and lysine. When aspirin or DMSO was added, the media were switched to normal “light (L)” and cells continued to grow in the L-media for 12 h. The proteins synthesized during the pulsed 12 h were labeled with L arginine and lysine while pre-existing proteins were labeled with MH or H arginine and lysine. After combining aspirin-treated and control cell lysates, the proteomic quantification ratios between MH and H arginine and lysine peptides accurately represented the difference in protein degradation. First, we assessed the impact of aspirin treatment on cell toxicity and found no adverse effects on cell proliferation under this concentration (Supplementary Fig. 1a, b). At the same time, a standard SILAC proteomic experiment was performed to measure the protein abundance changes upon aspirin treatment.

a Experimental scheme of the rp-SILAC strategy. This strategy utilizes quantification between the MS signals representing the pre-existing proteins to analyze degradation. The treatment with aspirin (b) or ATN combo (c) leads to the suppression of protein degradation. Aspirin or ATN combo treatment decreases global degradome slightly, but does not affect proteins’ abundance significantly. The distance (“D”) and p-value were calculated using the two-tailed Kolmogorov-Smirnov test (Proteome: n = 3 independent experiments; Degradome: n = 4 independent experiments). Volcano plots display the change in degradome caused by aspirin (d) or ATN combo (e) treatment. Proteins regulated by degradation (p < 0.05) are highlighted in red (FC > 1) or blue (FC < 1) dots, and the number of these proteins is displayed on the plots. Statistical tests were performed using one-sample two-tailed t-test with Benjamini-Hochberg multiple test correction. Melting temperature of degradation-changed proteins (p < 0.05) and those unchanged (p ≥ 0.05) with aspirin (f) or ATN combo (g) treatment. Boxes represent the interquartile range (25th to 75th percentile) for melting temperature, and whiskers extend from the 2.5th to the 97.5th percentile of the data. Two-tailed Mann-Whitney U test was used to analyze the significance of differences between groups (Aspirin: n = 2214 and 1364; ATN: n = 2281 and 1059). h Gene Ontology analysis of degradation-changed proteins. Fisher’s Exact test, with Benjamini-Hochberg adjusted p-values, is used to measure gene enrichment in annotation terms. Source data are provided as a Source Data file.
Aspirin inhibits proteasomal degradation
First, we found that aspirin treatment does not have a global impact on protein abundances. However, the degradome profile clearly shows a slight reduction in protein degradation with aspirin treatment (Fig. 1b; Supplementary Data 1, 2). To further increase the lysine acetylation levels, a combination of aspirin, trichostatin A (TSA), and nicotinamide (NAM) (ATN combo) was used in the rp-SILAC experiment. The latter two compounds are inhibitors of deacetylases, and can increase lysine acetylation levels substantially (Supplementary Fig. 1c)19. Similarly, the ATN combo can inhibit protein degradation to a greater extent compared to aspirin alone (Fig. 1c; Supplementary Data 3, 4). In total, the quantitative data resulted in 3009 and 3089 proteins whose degradation was changed upon the aspirin or ATN combo treatments, respectively (criteria: p-value < 0.05) (Fig. 1d, e). These degradation-changed proteins tend to have medium to high cellular abundances (Supplementary Fig. 1d, e).
Next, our degradomic data were correlated with the proteins’ thermal stabilities and half-lives20,21, which are two important measurements for protein degradation. Although the difference is marginal, our data show that the inhibitory effects caused by aspirin and ATN combo are more discernible for proteins with higher stabilities and longer half-lives (Fig. 1f, g and Supplementary Fig. 1f, g). Moreover, functional analyses reveal that aspirin or ATN combo affects proteins with functions in protein translation, protein transport, RNA binding and processing, and mitochondrial energy metabolism (Fig. 1h). Collectively, we developed the rp-SILAC strategy that allowed us to show that aspirin and ATN combo could suppress protein degradation on a proteome-wide scale.
Aspirin-induced lysine acetylation does not compete directly with lysine ubiquitination to inhibit protein degradation
Given the fact that aspirin is able to acetylate proteins and inhibit protein degradation, we hypothesized that aspirin-induced acetylation inhibited protein degradation by suppressing lysine ubiquitination. To test this hypothesis, we designed a combined quantitative experiment using triple SILAC to study the crosstalk between lysine ubiquitinome and acetylome (Fig. 2a). ATN combo and MG132, a proteasome inhibitor, were used to stimulate lysine acetylation and ubiquitination, respectively. As expected, the treatment of ATN combo or MG132 significantly increased the global acetylation or ubiquitination levels (Fig. 2b, c; Supplementary Data 5). Meanwhile, analyses on individual acetylation and ubiquitination site also confirmed that ATN or MG132 selectively induced acetylation or ubiquitination, respectively, without influencing the other modification (Fig. 2d). The amino acid motifs containing these two lysine modifications are summarized and are in line with previous omics data22,23 (Supplementary Fig. 2a). Specifically, 74.1% of all acetylation sites and 61.4% of all ubiquitination sites had increased levels upon stimuli treatment (FC > 1.5). However, an upregulated modification did not have a global suppressive effect on the other modification (Fig. 2b, c). We further compared the co-occurring lysine sites that were identified with both acetylation and ubiquitination. The relationship between these two modifications was not negatively correlated, in other words, upon ATN combo treatment, ubiquitination does not decrease as acetylation increases, and vice versa upon MG132 treatment, acetylation does not decrease as ubiquitination increases (Fig. 2e). Although single-shot analysis of triple-SILAC samples offers high quantification accuracy, its analysis depth is often limited. To further strengthen our conclusion, we repeated the experiment using label-free quantification on a sensitive TIMS TOF Pro2 platform, quantifying 15,452 lysine acetylation and 39,227 ubiquitination sites. This large-scale, in-depth data further confirms that these two modifications do not compete at the lysine residues (Supplementary Fig. 2b–d; Supplementary Data 6). In addition, these lysine sites can be divided into two groups. One group contains competitive sites (Quadrant II and IV), and the other group of sites are synergistic (Quadrant I and III) (Fig. 2e). Motif and structural analyses revealed no significant differences between the competitive and synergistic lysine sites (Supplementary Fis. 2e–h). Overall, the combined acetylome and ubiquitinome data indicate that these two modifications both targeting lysine residue do not compete directly for occupancy at the same site.

a Quantitative experimental scheme for elucidating crosstalk between lysine acetylome and ubiquitinome. Triple SILAC-based quantitative proteomics was integrated with antibody-based acetylation and ubiquitination enrichment. Lysine acetylation and ubiquitination do not have a significant effect on each other. Cumulative distributions show that ATN combo treatment significantly increases acetylation level but does not affect ubiquitination (b). Similarly, MG132 treatment only changes ubiquitination and not acetylation (c). The distance (“D”) and p-value were calculated using the two-tailed Kolmogorov-Smirnov test (n = 3 independent experiments). d Comparisons of individual ubiquitination (left) and acetylation (right) sites between ATN and MG132 treatments. e Relationship between co-occurring lysine acetylation and ubiquitination. The Pearson correlation coefficients show that no clear positive or negative correlations were observed between these two modifications. f ATN combo or MG132 treatment regulates ubiquitin linkages. Peptides MQIFVK(GG)TLTGK, TLTGK(GG)TITLEVEPSDTIENVK, TITLEVEPSDTIENVK(GG)AK, AK(GG)IQDK, IQDK(GG)EGIPPDQQR, LIFAGK(GG)QLEDGR, and TLSDYNIQK(GG)ESTLHLVLR are used to represent K6, K11, K27, K29, K33, K48, and K63 ubiquitination linkages, respectively. The quantification MS data retrieved from the triple-SILAC ubiquitinome experiment are shown (n = 3 independent experiments. data presented as mean ± SEM). g The effect of aspirin, TSA, and NAM, and their combination, on ubiquitin linkages. The quantification MS data were obtained from a separate label-free experiment using HeLa cells. The signature peptides representing seven types of ubiquitin linkages were used for quantification (n = 4 independent experiments, One-way ANOVA with Dunnett’s multiple comparisons test, data presented as mean ± SEM). h The impact of aspirin on K63 ubiquitination (left) and K48 ubiquitination (right) in HeLa cells. Recombinant wild-type ubiquitin was introduced to enhance quantification performance. Three independent experiments were conducted. i Aspirin induces K63 ubiquitination in HeLa cells. Recombinant K63-only ubiquitin was introduced to improve quantification performance on K63 specific ubiquitination linkage. Three independent experiments were conducted. j Gene Ontology analysis of proteins containing up-regulated (FC > 1.5) acetylated and ubiquitinated sites with ATN combo treatment. Fisher’s Exact test, with Benjamini-Hochberg adjusted p-values, is used to measure gene enrichment in annotation terms. Source data are provided as a Source Data file.
Aspirin increases K63-linked ubiquitination
The analysis was further focused on the ubiquitin chain linkages using linkage-specific peptides that were retrieved from ubiquitinome data. Not surprisingly, the treatment of MG132 increased almost all types of ubiquitin linkages except K6 and K63. Interestingly, ATN combo treatment increased K11- and K63-linked ubiquitination levels dramatically (Fig. 2f). We questioned whether aspirin alone could have the same effect on ubiquitin linkages and therefore performed a label-free ubiquitinome analysis using HeLa cells. The data confirmed that aspirin, deacetylase inhibitors, or a combination of both, increased K11 and K63 ubiquitin linkages by 60-133% compared to the control group (Fig. 2g). Moreover, using K63-specific and K48-specific antibodies, we examined the levels of K63 and K48 ubiquitination in HeLa cells, where recombinant ubiquitin was overexpressed to enhance quantification performance. The Western blot results indicate that the addition of aspirin treatment indeed induced the K63 ubiquitination levels in a concentration-dependent manner, while the K48 ubiquitination levels remained largely unchanged (Fig. 2h), consistent with the mass spectrometry results. When specifically introducing K63-only ubiquitin, the promoting effect of aspirin treatment on K63 ubiquitination became even more pronounced (Fig. 2i).
The ATN combo-induced acetylome (FC > 1.5) was mainly involved in protein translation and RNA processing, while the ATN combo-altered ubiquitinome (FC > 1.5) played important roles in protein ubiquitination and related catabolic processes, particularly K48 and K63 ubiquitination (Fig. 2j). This finding provides an explanation for how ATN combo and aspirin affect ubiquitin linkages. Altogether, although aspirin-induced acetylation does not directly suppress substrate ubiquitination, it is associated with an increase in K11 and K63 ubiquitin linkages, which could potentially impact protein degradation pathways.
Aspirin impairs proteasome activity
Ubiquitin–proteasome system (UPS) is the dominant machinery to degrade cellular proteins24,25. We questioned whether aspirin or ATN combo could impact the efficiency of UPS by affecting the proteasome itself. To this end, we examined changes in the abundance and acetylation levels of proteasome proteins following treatment with aspirin and ATN combo. Our analysis showed that while protein levels of the detected 29 proteasome subunits remained unchanged, 40 out of all 50 acetylation sites of these subunits were upregulated by at least two-fold (Fig. 3a). Notably, 25 of these acetylation sites had not been previously reported. Furthermore, the three core subunits (PSMB5, PSMB6, and PSMB7) with hydrolytic activity were all found with elevated acetylation levels, prompting the question if the increased acetylation could affect their hydrolytic activity.

a Aspirin or ATN combo treatment induces acetylation on many proteasome subunits. The protein abundances of these proteasome components remain unchanged upon aspirin or ATN combo treatment. 25 acetylation sites were previously unknown and are labeled with asterisks. Aspirin decreases proteolytic activities of three major proteasomal hydrolases from HeLa cell extracts (b) or purified proteasome (c). Epoxomicin is a known potent and selective inhibitor of the proteasome. HeLa cells were treated with 3 mM aspirin, 10 μM TSA, and 10 mM NAM. After incubating proteasome with fluorogenic peptide substrates at 37 °C for 60 min, the fluorescence intensity was recorded (n = 3 independent experiments, One-way ANOVA with Dunnett’s multiple comparisons test, data presented as mean ± SEM). d The number of acetylation sites on the proteasome induced by aspirin in vitro. e The effect of aspirin on the integrity of 26S proteasome. 26S proteasome remains intact under low concentration (3 or 5 mM) of aspirin, but undergoes partial disassembly under high concentration (10 mM) in vitro. 6% native page gels were used to separate the capped proteasome (26S) from 20S proteasome. Source data are provided as a Source Data file.
Using fluorogenic peptide substrates, we measured the activities of three major proteolytic enzymes, including trypsin-like, caspase-like, and chymotrypsin-like proteases, in HeLa cells. These activities in HeLa cells were decreased by 18-30% or 33-44% with aspirin or ATN combo, respectively (Fig. 3b). To exclude indirect effects of aspirin on proteasome activity, we performed the same assay on a 26S proteasome system reconstructed in vitro. In line with the findings from cell extracts, aspirin treatment led to a concentration-dependent decrease in all three proteolytic activities in vitro (Fig. 3c). Furthermore, mass spectrometry analysis revealed that aspirin induced many acetylation events on the proteasome, and the number of acetylation sites was inversely correlated with the proteolysis activities (Fig. 3d).
In addition, we investigated whether aspirin would impact the integrity of the 26S proteasome through native gel analysis. The results indicate that at the concentrations of aspirin treatment utilized in our cellular experiments, there is no apparent disassembly of the 26S proteasome (Fig. 3e). However, at a high concentration of 10 mM aspirin, a partial disassembly of the 26S proteasome occurs (Fig. 3e). Altogether, these findings suggest that aspirin primarily inhibits proteasome activities by directly acetylating proteasome components. However, an extremely high concentration of aspirin may also result in the disassembly of the proteasome, further diminishing its enzymatic activity.
Aspirin reduces α-syn aggregates in cultured primary neurons and cells
Although aspirin can inhibit the proteasomal degradation, its impact on lysosomal degradation remains unclear. Lysosomal degradation plays an important role in clearing insoluble proteins, such as protein aggregates, and this process is closely linked to K63 type ubiquitination26,27. Given that aspirin increases K63 ubiquitination, it is reasonable to hypothesize that this may impact the clearance of insoluble proteins via lysosomal degradation. Thereafter, we chose to use the α-syn aggregate as an example to explore this question.
The aggregated form of α-syn is the major component of Lewy bodies (LB) and Lewy neurites (LN), which are highly prevalent in the brains of PD patients. Previous large-scale population-based studies have suggested potential benefits of aspirin for PD patients28,29,30. Therefore, we hypothesized whether aspirin confers the clinical benefit by modulating the degradation of α-syn aggregates. To this aim, we introduced α-syn preformed fibrils (PFFs) into cultured neurons and other cells. These fibrils can be uptaken and primarily present as an aggregate form in cells and could further template the aggregation of endogenous α-syn during long-lasting culturing31. First, primary mouse neurons were isolated and cultured with α-syn PFFs for 7 days, starting from 7 days in vitro (DIV). Aspirin was then added for 18 h. We found that aspirin significantly reduced the abundance of α-syn (Fig. 4a). Moreover, western blot analysis with H4, HeLa, and BV2 cells demonstrated that aspirin could also decrease the abundance of α-syn protein in a dose-dependent manner, with higher concentrations of aspirin showing more profound effects (Fig. 4b and Supplementary Fig. 3a, b).

Aspirin treatment decreases α-syn aggregates in mouse primary neurons (a) and neuroglioma H4 cells (b). Shown are representative western blot (top) and quantitative results (bottom) (n = 3 independent experiments, One way ANOVA with Tukey’s multiple comparisons test, data presented as mean ± SEM). c Proteolytic activities of three major proteasomal hydrolases from H4 cell extracts decrease upon aspirin treatment (n = 3 independent experiments, One way ANOVA with Dunnet’s multiple comparisons test, data presented as mean ± SEM). d Aspirin treatment alters ubiquitin linkages in H4 cells. Shown are the quantification MS data retrieved from a label-free experiment using H4 cells treated with 3 mM aspirin for 18 h. The quantification was based on the signature peptides representing seven types of ubiquitin linkages. (n = 3 independent experiments, unpaired two-tailed t-test, data presented as mean ± SEM). e Aspirin induces K63 ubiquitination in H4 cells. Recombinant wild-type ubiquitin was introduced to enhance quantification performance. Three independent experiments were conducted. f Quantification of GFP-LC3 puncta number per cell after treatment with varied concentrations of aspirin in H4-GFP-LC3 cells. At least 1000 cells per condition in an independent experiment were quantified (n = 5 independent experiments, One way ANOVA with Tukey’s multiple comparisons test, data presented as mean ± SEM). g Aspirin’s reduction of α-syn is lysosome dependent. Inhibiting lysosome activity with NH4Cl significantly diminishes aspirin’s effectiveness in α-syn clearance. Three independent experiments were conducted. Source data are provided as a Source Data file.
Aspirin-induced clearance of α-syn aggregates is lysosome-dependent
Since autophagy is an intracellular process that facilitates degradation of cytoplasmic materials such as protein aggregates32, we next tested if aspirin has an effect on autophagy-mediated protein degradation. In human neuroglioma H4 cells, we confirmed that the three proteolytic activities of the H4 cell proteasome were decreased by 48-63%, consistent with previous findings in HeLa cells (Fig. 4c). Furthermore, treatment with aspirin resulted in a significant increase in K11- and K63-linked ubiquitination forms (Fig. 4d). Notably, unlike the results observed in HeLa cells, other ubiquitin linkage types, including the K48, were also markedly increased in H4 cells treated with aspirin. The increase of K63-ubiquitination was further confirmed using a K63-specific antibody in H4 cells (Fig. 4e).
Furthermore, H4 cells stably expressing GFP-LC3 were treated with different concentrations of aspirin, and the LC3 positive puncta were examined by using high content imaging (HCI). The results showed that aspirin decreased the puncta formed by GFP-LC3 in a dose-dependent manner (Fig. 4f). We then introduced the autophagy inhibitors, NH4Cl, which inhibits lysosome acidification and causes LC3-II accumulation, and 3-methyladenine (3-MA), serving as the class III PI3K inhibitor to inhibit the conversion of LC3-I to LC3-II. Our findings demonstrate that inhibiting the early stage of autophagy with 3-MA and proteasome activity with MG132 had minimal effects on the degradation of α-syn aggregates. In contrast, inhibiting lysosome activity with NH4Cl, which represents the last step of autophagy, significantly impeded their degradation (Fig. 4g). This data supports the notion that aspirin can degrade α-syn aggregates through a lysosomal pathway rather than through classical autophagy.
Aspirin promotes α-syn aggregates clearance through K63 ubiquitination
We then aimed to explore the mechanistic links between the augmented K63 ubiquitination and the reduced α-syn aggregates following aspirin treatment. As K63 ubiquitin linkage is a primary ubiquitination type induced by aspirin, we asked if K63 ubiquitination mediates α-syn aggregates clearance. To this end, wildtype or mutant (lysine to arginine) ubiquitin was overexpressed in both primary mouse neurons and H4 cells. The mutation at the K63 site significantly weakened the clearance effect of α-syn aggregates caused by aspirin in both cell types (Fig. 5a, b). To further elucidate that aspirin-induced K63 ubiquitination indeed occurs on α-syn, we overexpressed wild-type and mutant ubiquitin with a myc tag in H4 cells. After immunoprecipitation of myc-tagged ubiquitin, we quantified the levels of α-syn using mass spectrometry. Our data demonstrated that the addition of aspirin in the WT and K48R-ubiquitin groups resulted in increased levels of ubiquitinated α-syn, suggesting that aspirin promotes α-syn ubiquitination (Fig. 5c). In contrast, no differences were observed in the K63R-ubiquitin group, indicating that the ubiquitination induced by aspirin on α-syn is primarily in the K63 form. These data further support the conclusion that aspirin can enhance K63 ubiquitination of α-syn, thereby promoting its clearance.

K63 ubiquitination is the key signaling mediator for the degradation of α-syn aggregates in primary neurons (a) and H4 cells (b). Shown are representative western blot (top) and corresponding quantitative results (bottom). The overexpression of K63R ubiquitin variant in both primary neurons and H4 cells diminishes the aspirin-induced clearance of α-syn aggregates (n = 3 independent experiments, One-way ANOVA with Tukey’s multiple comparisons test, data presented as mean ± SEM). c Aspirin induces K63-linked ubiquitination on α-syn. Ubiquitinated proteins were immunoprecipitated using an anti-myc pull-down, and α-syn levels were measured by mass spectrometry (n = 3 independent experiments, unpaired two-tailed t-test, data presented as mean ± SEM). Source data are provided as a Source Data file.
Immunohistochemical staining of H4 cells demonstrated that with aspirin treatment α-syn highly colocalized with Rab5, an early endosome marker. Furthermore, a higher concentration of aspirin promoted more α-syn aggregates to enter early endosome (Supplementary Fig. 4a, b). However, the colocalization of α-syn with Rab7, a late endosome marker (Supplementary Fig. 4c, d), or LAMP2, a late endosome and lysosome marker (Supplementary Fig. 4e, f), did not increase with aspirin treatment. These results suggest that aspirin modulates α-syn aggregates degradation by promoting their entry into the early endosome, upon which the α-syn aggregates might be rapidly degraded through a K63 ubiquitination-dependent endosomal-lysosomal pathway. Altogether, our data suggest that α-syn aggregates are degraded by a K63 ubiquitination-dependent endosomal-lysosomal pathway.
Aspirin reduces α-syn aggregates and ameliorates behavioral deficits in a PD-like mouse model
We further investigated the effects of aspirin treatment on a PD mouse model generated by bilaterally inoculating synthetic mouse α-syn PFFs directly into the striatum (PFF mice, Fig. 6a, See Methods). Aspirin was given orally daily 4 weeks after the surgery when mice have already developed some hyperphosphorylated α-syn aggregates and started to show mild PD-like behavioral deficits33. Aspirin was administered for 8 weeks. Behavioral tests were conducted at two time points, both after 4 and 8 weeks of treatment. We found that the general locomotor activity, as measured by open field test, was not significantly different among groups 8 weeks post-surgery with or without 4 weeks of aspirin treatment. However, after 8 weeks of treatment, mice injected with α-syn PFFs exhibited a notable decline in their locomotor ability, whereas the aspirin treatment resulted in a significant improvement (Fig. 6b). Moreover, aspirin largely rescued the coordination-like behavioral deficit of the PFF mice in a rotarod test. This improvement persisted and was more pronounced after 8 weeks of aspirin treatment (Fig. 6c), providing strong support for the drug’s beneficial effects on PD-like symptoms.

a Experimental scheme for establishing a PD mouse model and aspirin treatment. 10, 11, and 12 mice were used for the PBS, the PFF-control, and the PFF-aspirin treatments, respectively. The figure was created using BioRender.com. Open field test (b) and rotarod test (c) for PD mice. These behavioral tests were repeated twice during the aspirin administration. The results of the rotarod test were normalized to the batch-specific PBS mice in each independent experimental batch. Each dot represents a mouse (One way ANOVA with FDR correction for multiple comparisons, data presented as mean ± SEM). d Aspirin treatment decreases α-syn aggregates in the striatum. Representative immunofluorescence images (scale bar, 100 μm) and zoom-in merged images (scale bar, 10 μm) are shown. Quantitative results are shown in e–g, and the dot represents the mouse brain. Aspirin treatment reduces the intensity (e) and density (f) of α-syn aggregates in the striatum, which is represented by p-α-syn (n = 5 or 6, One way ANOVA with FDR correction for multiple comparisons, data presented as mean ± SEM). g DA neurons in the striatum with aspirin treatment (n = 5 or 6, One way ANOVA with Tukey’s multiple comparisons test). h Aspirin treatment increases the number of DA neurons in the substantia nigra. Representative immunofluorescence images (scale bar, 200 μm) and zoom-in merged images (scale bar, 20 μm) are shown. Quantitative results are shown in i–l, and the dot represents the mouse brain. Intensity (i) and density (j) of α-syn aggregates in the substantia nigra with aspirin treatment (n = 6, One way ANOVA with FDR correction for multiple comparisons, data presented as mean ± SEM). DA neurons in the substantia nigra with aspirin treatment (n = 6, One way ANOVA with FDR correction for multiple comparisons, data presented as mean ± SEM). m Aspirin treatment regulates ubiquitin linkages in mouse brains. The signature peptides representing seven types of ubiquitin linkages were used for quantification (n = 3, unpaired two-tailed t-test, data presented as mean ± SEM). Source data are provided as a Source Data file.
Synucleinopathy and dopaminergic (DA) neuronal loss are two common pathological hallmarks of PD34. To unveil the cellular mechanism of aspirin, we compared these two features between PFF mice treated with or without aspirin for 8 weeks. Strikingly, phosphorylated α-syn at ser129 (p-α-syn), a pathological marker for α-syn aggregates, was significantly reduced in both intensity and aggregates density in the striatum with aspirin treatment (Fig. 6d–g). Additionally, aspirin largely prevented DA neuronal loss in the substantia nigra compacta region induced by PFF despite the similar level of synucleiopathy (Fig. 6h–l). These results suggest that aspirin’s benefit for PD is mediated at least partially by modulating the progression of synucleinopathy and protecting DA neurons in the substantia nigra.
Moreover, we analyzed the ubiquitin linkage forms of PD mouse brain using mass spectrometry. Remarkably, aspirin treatment led to increases in all detected types of ubiquitin linkages (Fig. 6m). Both protein degradation-associated forms, K48 and K63, were significantly upregulated in response to aspirin treatment. Notably, K63-linked ubiquitination was the most strongly induced type of linkage in the mouse brains. This finding is consistent with the results obtained from the cultured cells. Taken together, the data obtained from the PD mouse model strongly support the conclusion that aspirin treatment not only enhances α-syn aggregates degradation through increased K63 ubiquitination, but also exhibits remarkable neuroprotective effects and ameliorating PD-like behavioral deficits.
Aspirin-promoted α-syn clearance is mediated by K63 ubiquitination in vivo
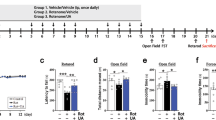
To further validate the mechanism of aspirin to enhance α-syn clearance through K63 ubiquitination, we generated adeno-associated virus (AAV) vectors expressing three ubiquitin variants (WT, K48R, K63R) in PD mice. Consistent with previous procedures, mouse α-syn PFFs were injected bilaterally into the striatum. Four weeks later, the three AAV vectors into the same region. After two weeks of AAV expression, mice were treated with aspirin at a dose of 6 mg/kg/d (Fig. 7a). Behavioral experiments were conducted eight weeks post-administration. While no significant differences were observed in open-field tests among groups (Fig. 7b), the pole test and rotarod test – key assessments of motor abilities – revealed significantly worse performance in mice expressing K63R-Ubiquitin compared to those expressing K48R-ubiquitin or WT ubiquitin (Fig. 7c, d).

a Experimental scheme for aspirin treatment in a PD mouse model overexpressing different ubiquitin variants. AAV vectors expressing three ubiquitin variants (WT, K48R, K63R) were introduced to PD mice. 6 mice were used for each of the PFF-WT-Ub, the PFF-K48R-Ub, and the PFF-K63R-Ub treatments, respectively. The figure was created using BioRender.com. Open field test (b), pole test (c) and rotarod test (d) for PD mice with different ubiquitin variants. Each dot represents a mouse (n = 6, One-way ANOVA with FDR correction for multiple comparisons, data presented as mean ± SEM). e The K63R ubiquitin significantly weakens the clearance effect of p-α-syn caused by aspirin in the striatum. Representative immunofluorescence images of the striatum (scale bar, 100 μm) are shown. Zoom-in merged images are shown on the right (scale bar, 10 μm). Quantitative results are shown in f–h, and the dot represents the mouse brain. Impact of different ubiquitin variants, when treated with aspirin, on p-α-syn intensity (f) and density (g), as well as DAT intensity (h) in the striatum (n = 5 or 6, One way ANOVA with FDR correction for multiple comparisons, data expressed as mean ± SEM). i The K63R ubiquitin significantly decreases the number of DA neurons in the substantia nigra. Shown are representative immunofluorescence images of the substantia nigra (scale bar, 200 μm). Zoom-in of the merged images are shown on the right (scale bar, 20 μm). Quantitative results are shown in j–m, and the dot represents the mouse brain. Impact of different ubiquitin variants, when treated with aspirin, on p-α-syn intensity (j) and density (k), as well as DAT intensity (l) and DA neuron number (m) in the substantia nigra. The number of DA neurons was determined by counting DAT-positive cells in the substantia nigra (n = 4, 5 or 6, One way ANOVA with FDR correction for multiple comparisons, data expressed as mean ± SEM). Source data are provided as a Source Data file.
In addition, immunofluorescence assays were employed to assess differences in p-α-syn aggregates and DA neuronal loss among mice expressing different ubiquitin variants. Although there was no difference in the density of p-α-syn aggregates in the striatum, the intensity of p-α-syn was notably elevated in the K63R group compared to the WT group, with no difference observed between the K48R and WT groups (Fig. 7e–h). In the substantia nigra, the number of DA neurons in the K63R group was significantly lower than in the WT and K48R groups (Fig. 7i–m).
Altogether, these findings indicate that aspirin promotes the degradation of α-syn through K63 ubiquitination, thereby alleviating PD-like symptoms in vivo.
Discussion
Protein degradation is crucial for maintaining proteomic and cellular homeostasis, and dysregulation of this process is closely linked to many human diseases. Using the developed rp-SILAC strategy, our study has revealed that aspirin, an “all-purpose” drug and strong acetyl donor, can affect the degradation of both soluble and insoluble proteins in a bidirectional manner. This finding provides a new dimension to our understanding of its pharmacology.
As demonstrated by our data, aspirin is not as potent as epoxomicin, which has the capability to completely inhibit proteasome activity. Aspirin-induced acetylation appears to act as a mild regulator or inhibitor, influencing the rates of activity. However, it should be noted that, considering acetylation is a covalent modification, a long-lasting effect caused by acetylation may persist even if its direct impact on proteasome activity is not as robust as that of epoxomicin.
One surprising discovery is that, despite lysine being a hub residue for multiple important PTMs, the two most prevalent lysine modifications, acetylation and ubiquitination, rarely compete directly. This is due in part to the fact that the transfer of acetyl or ubiquitin to substrates is catalyzed by different enzymes and pathways, which recognize substrates or regional structures with selectivity. Additionally, these modifications are low stoichiometric events16,35,36. As a consequence, finding a vacant lysine residue is for most modification occurrences not difficult.
Aspirin has been shown to have a beneficial effect for neurodegenerative diseases, which are typically characterized by pathological protein aggregates in the central nervous system of patients, such as Aβ in Alzheimer’s disease and α-syn in PD28,29,30,37,38,39,40,41. Its therapeutic effect becomes more pronounced at a higher dose. Taking 2 or more aspirin tablets per day has been associated with the potential to delay or prevent the occurrence of PD30. A recent report also highlighted a positive correlation between the dose of aspirin tablets taken per week and the duration of aspirin intake with the age of onset of PD37. Besides its neuroprotective effects through its anti-inflammatory action42,43,44, previous studies have found that aspirin can promote lysosome biogenesis by upregulating transcription factor EB (TFEB) and accelerating Aβ clearance45. In addition, aspirin can inhibit protein aggregation by reducing the phosphorylation of Aβ pathogenic proteins46. Furthermore, studies have revealed that USP8, which mediates the removal of K63-linked ubiquitin chains on α-syn, can reduce its lysosomal degradation in dopaminergic neurons, thus contributing to α-synuclein accumulation in Lewy body disease47. This finding is highly consistent with our conclusion, as our study demonstrates the importance of aspirin-induced K63 ubiquitination in decreasing α-syn aggregates. This finding strongly supports the theory that endo-lysosomal degradation is a primary contributor to age-related neurodegeneration and raises its potential as a therapeutic target48,49. Studies have shown that tau-positive inclusions that are K63 ubiquitinated can be recognized and degraded by the autophagy pathway32. In addition, K63 chains play a role in heterophagy, where extracellular or membrane proteins are internalized and modified with K63 ubiquitination, recognized by ubiquitin-binding domains (UBD)-containing endosomal sorting complexes required for transport (ESCRT) complexes on the endosome membrane and after internalization degraded in the lysosome13.
Moreover, we note that the concentration of aspirin used for cell experiments in this study may seem higher than the typical working concentration in humans. Actually, the highest concentration of free salicylic acid (SA), a metabolite of aspirin, can reach 0.4 mM, with an average plasma concentration of 0.2 mM within 10 h after ingestion50. Furthermore, the primary objective of our research was to investigate aspirin-induced acetylation, which is known to be more pronounced at the concentrations we chose. By employing a higher concentration with a shorter treatment duration, we aimed to simulate the acetylation effects comparable to a lower concentration with a longer treatment period. This selection allowed us to effectively analyze the aspirin-induced acetylation by integrating drug treatment with metabolic labeling. Notably, the treatment of PD mice with aspirin was conducted using a dosage consistent with daily clinical practice, further validating our findings in cell-based studies.
Unresolved questions from our sutdy include how aspirin induces an increase in K63-linked ubiquitination on α-syn. The increase of K48-linked ubiquitination can be explained by our finding that aspirin inhibits the proteasomal degradation pathway that relies on K48 ubiquitination, leading to an accumulation of undegradable K48 ubiquitin chains. Inhibition of proteasome activity has been identified as a mechanism of aspirin in inhibiting cancer cell proliferation51. However, the mechanisms underlying the interplay between aspirin treatment and upregulated K63 ubiquitination are still not understood. One possible mechanism involves key players in the UPS, such as ubiquitin, key E2 and E3 enzymes, and deubiquitinases, whose functions can be modulated by protein acetylation52,53. For instance, studies have reported that HDAC inhibitors can increase the levels of acetylated K6 and K48 ubiquitin, which can impact the extension of ubiquitin chains54. Furthermore, our data demonstrated that the ATN combo significantly increased the ubiquitination levels of E2 and E3 enzymes (Fig. 2j), which might also be critical for E3 ligase activity and substrate recognition55. Although we established that aspirin can induce K63 ubiquitination on α-syn aggregates to promote their clearance, further detailed mechanistic studies could provide deeper insights into aspirin’s remarkable therapeutic effects and hold significant implications for the development of new targets and treatment strategies for Parkinson’s disease and other conditions characterized by pathological protein aggregates.
Methods
Cell lines and cell culture
HeLa, BV2, and H4 cells were cultured in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin/streptomycin at 37 °C in a 5% CO2 atmosphere. For SILAC experiments, HeLa cells were cultured in SILAC DMEM (Silantes, 280001200), in which lysine and arginine amino acids were excluded, with 10% (v/v) dialyzed FBS (Silantes, 281001200), 1% (v/v) penicillin/streptomycin, and 2 mM L-glutamine. In addition, stable isotopes labeled L-arginine and L-lysine (Light); or M-arginine (13C6) and M-lysine (2H4) (Medium-heavy); or H-arginine (13C6, 15N4) and H-lysine (13C6, 15N2) (Heavy) were added to the SILAC DMEM.
Animal studies
Male C57BL/6 J mice (4-weeks-old) were from the animal facility (Shanghai Ling Chang Biotechnology Co.). The mice were group-housed and maintained under standard conditions with a light-dark (12 h-12 h) cycle, room temperature of 25 ± 2 °C, and ad libitum access to food and water.
The mice were anesthetized with 0.5 mL/min isoflurane delivered in 1 mL/min O2. α-syn PFFs (2 μg/μL) were stereotaxically injected into dorsolateral striatum (dlSTR) of both hemispheres at a dose of 0.2 μg/g (body weight). Sterile phosphate-buffered saline (PBS) was injected at a dose of 0.1 μL/g (body weight) in control mice. The injection was controlled at 0.5 μL/min for the first 0.2 μL and 0.2 μL/min for the remaining volume using a micro-syringe. The following coordination: +0.2 mm to bregma, ± 2.0 mm from the midline, and −2.6 mm from the dura, were used. By multiplying the scaling factor ζ \(\left({\zeta}=\frac{{{\rm{Measured}}\; {{\rm{distance}}}\; {{\rm{between}}}\; {{\rm{bregma}}}\; {{\rm{and}}}\; {{\rm{lambda}}}}\left({{\rm{mm}}}\right)}{4.21\,{{\rm{mm}}}}\right)\), the accurate and consistent injecting location coordinates of each mouse were obtained. The mice were allowed to recover from the anesthesia and transferred back to their home cages until the next experiments.
For PD mice injected additionally with AAV vectors carrying different ubiquitin variants, 1 μL of AAV (titer: 1 × 10¹² VG/mL) was bilaterally injected into the striatum four weeks after the initial α-syn PFF injection, following the same stereotaxic method. After a two-week recovery period, subsequent experiments were conducted.
Reagents
The following antibodies were used: α-syn monoclonal rabbit antibody (1:1000, Abcam, ab138501), LC3B polyclonal rabbit antibody (1:1000, Sigma, L7543), Acetyl lysine rabbit antibody (1:1000, ImmuneChem, ICP0380), linkage-specific K48 ubiqutin monoclonal rabbit antibody (1:1000, Abcam, ab140601), linkage-specific K63 ubiqutin monoclonal rabbit antibody (1:1000, Abcam, ab179434), β-actin monoclonal mouse antibody (1:1000, Sigma, A1978), GAPDH monoclonal mouse antibody (1:1000, Sigma, G8795), HRP-conjugated β-tubulin monoclonal mouse antibody (1:10000, abclonal, AC030), HRP-conjugated polyclonal goat anti-rabbit antibody (1:1000, Beyotime, A0208), and HRP-conjugated goat polyclonal anti-mouse antibody (1:1000, Beyotime, A0216). DAT monoclonal rat antibody (1:500, Abcam, ab5990); phospho-α-syn staining (S129) monoclonal rabbit antibody (1:250, Abcam, ab51253); Flag monoclonal mouse antibody (1:1000, Sigma, F1804); Rab5 monoclonal rabbit antibody (1:200, Cell Signaling Technology, 3547S); Rab7 monoclonal rabbit antibody (1:200, Cell Signaling Technology, 9367S); LAMP2 polyclonal rabbit antibody (1:200, Abcam, ab18528); goat anti-rabbit Alexa Fluor 568 (1:1,000, Abcam, ab175471), and goat anti-rat Alexa Fluor 647 (1:150, Abcam, ab150159), goat anti-mouse Alexa Fluor 647 (1:1000, Abcam, ab150115). All blots are provided in the source data file.
Reversed-pulsed-SILAC
HeLa cells were initially fully labeled in medium-heavy (MH) or heavy (H) DMEM. The medium was switched to normal light (L) when 1‰ DMSO or an acetylation inducer (3 mM aspirin or a combination of 3 mM aspirin, 10 μM TSA, and 10 mM NAM) was added to MH- or H-labeled cells separately (Fig. 1a). Cells were continued to grow in the L-DMEM for an additional 12 h. The cells were harvested, and the proteins were subjected to quantitative shotgun proteomic experiments. Four independent biological replicates were conducted.
Triple SILAC
HeLa cells were fully labeled in L-, MH-, or H-DMEM. The L-cells were treated with 1‰ DMSO for 12 h and used as controls. The MH- and H- cells were treated with ATN (3 mM aspirin, 10 μM TSA, 10 mM NAM) and 5 μM MG132 for 12 h, respectively. Cells were harvested, and the proteins were subjected to proteomic, or acetylomic or ubiquitinomic analyses. Three independent biological replicates were conducted.
Aspirin treatment in cultured cells with α-syn PFFs
BV2, HeLa, and H4 cells were incubated with 3 μM human α-syn PFFs for 4 h. For immunofluorescence experiments, H4 cells were incubated with 3 μM human Flag α-syn PFFs for 4 h. The cells were then washed twice with PBS, and a fresh medium containing different concentrations of aspirin was added for 18 h. The cells were then harvested and lysed in a loading buffer containing 2% SDS. The proteins were boiled at 95 °C for 30 min and separated by 10% SDS-PAGE and analyzed by western blotting.
Myc-tagged wild-type (WT) ubiquitin, as well as Myc-tagged mutant ubiquitin (K48R, K63R, or K63only), were subcloned into pcDNA 3.1 vector. The mutagenesis and subcloning products were verified by DNA sequencing. H4 cells were transiently transfected with plasmids encoding WT and mutant ubiquitin using the manufacturer’s protocol (Signagen, SL100688). The α-syn PFFs and aspirin were treated as previously described, and the levels of α-syn protein were analyzed by western blotting.
For experiments involving the addition of autophagy inhibitors NH4Cl and 3-MA, or proteasome inhibitor MG132, the following protocol was used. H4 cells were also incubated with 3 μM α-syn PFFs for 4 h and then the medium was replaced with fresh medium containing 3 mM aspirin for 18 h. 5 μM 3-MA was cotreated with aspirin for 18 h. 20 mM NH4Cl or 10 μM MG132 was added for 4 h before sample collection.
AP-MS analysis ubiquitinated α-syn
H4 cells expressing myc-tagged ubiquitin (WT, K48R, and K63R) were treated with 3 μM α-syn PFFs for 4 h, after which the medium was replaced with fresh medium containing 3 mM aspirin for 18 h, along with 1 μM USP8 inhibitor (DUB-IN-2) to inhibit α-syn deubiquitination for 18 h. 20 mM NH4Cl was added for 4 h prior to sample collection to inhibit α-syn lysosomal degradation. The cell lysates were subjected to a standard AP-MS protocol. Briefly, the cells were lysed using RIPA lysis buffer containing 2% (w/v) SDS, 1% (w/v) Triton X-100, 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 50 mM Tris−HCl (pH 8), and a protease inhibitor cocktail, with sonication. The lysates were then centrifuged at 18,000 g for 15 min, and the resulting supernatants were diluted 1:20 and incubated overnight at 4 °C with anti-c-Myc magnetic beads. The beads were washed three times with Tris-buffered saline containing 0.1% SDS, resuspended in a loading buffer containing 2% SDS, and heated at 95 °C for 15 min before loading onto the gel. Following the in-gel digestion protocol, the α-syn were analyzed by mass spectrometry.
Sample preparation for LC-MS analysis
Cells that received different treatments were washed three times with pre-chilled PBS on ice. They were then lysed using RIPA lysis buffer containing 0.1% (w/v) SDS, 1% (w/v) Triton X-100, 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 50 mM Tris−HCl (pH 8), and protease inhibitor cocktail, with sonication. The lysates were centrifuged at 18,000 g for 15 min, and the resulting supernatants were transferred into clean tubes for protein concentration measurements using the bicinchoninic acid (BCA) assay.
In SILAC or rp-SILAC experiments, 50 μg of cell lysate protein labeled with different isotopes were mixed at a 1:1 (w/w) ratio. For triple-SILAC-based acetylomic and ubiquitinomic experiments, 2 mg of protein from each isotopic channel were mixed at a 1:1:1 (w/w/w) ratio. Label-free quantitative ubiquitination analysis used 2 mg of protein for each condition.
Proteins were precipitated on ice by adding 4 volumes of cold acetone. The protein pellets were obtained by centrifugation at 14,000 g for 15 min and then redissolved in a buffer containing 8 M urea and 500 mM Tris−HCl (pH 8.5). Disulfide bridges were reduced by adding Tris (2-carboxyethyl) phosphine (TCEP) at a final concentration of 5 mM for 20 min. Reduced cysteine residues were then alkylated by adding 10 mM iodoacetamide (IAA) and incubating for 15 min in the dark at room temperature. The urea concentration was reduced to 2 M by adding 100 mM Tris−HCl (pH 8.5) and 1 mM CaCl2. The protein mixture was digested overnight at 37°C with trypsin (V5111, Promega) at an enzyme-to-substrate ratio of 1:100 (w/w). Tryptic peptides were collected by centrifugation at 18,000 g for 20 min and the supernatants were desalted using C18 Stage tips. The peptides were further fractionated by high-pH reverse-phase high-performance liquid chromatography (HPLC) or subjected to PTM enrichments.
The mouse brains were homogenized using a bead-beating homogenizer (Bertin Precellys 24 Dual) in a RIPA lysis buffer. The resulting lysates were centrifuged at 18,000 g for 20 min, and the supernatants were collected and processed using the same protocol as described above.
High-pH reverse-phase liquid chromatography fractionation
High-pH reverse-phase separation was performed on an Agilent 1260 HPLC system using an XBridge C18 column (3.5 μm, 1.0 × 150 mm). The mobile phases consisted of 20 mM ammonium formate, pH 10.0 (A) and 90% (v/v) acetonitrile and 20 mM ammonium formate, pH 10.0 (B), and were used to develop a gradient. The 88-min gradient profile was set as follows: 5% B at 1 min, 20% B at 15 min, 30% B at 35 min, 40% B at 50 min, 60% B at 60 min, 80% B at 70 min, 90% B at 80 min, and 5% B at 88 min. The tryptic peptides of HeLa cells for SILAC and rp-SILAC analyses were dissolved in phase A solution and separated at a flow rate of 60 μL/min. The eluent was collected in a 96-well plate every minute, which resulted in 88 initial fractions. The 88 fractions were combined into eight fractions in a noncontinuous manner, dried under vacuum, and then subjected to LC-MS/MS analysis.
Acetylated peptide enrichment
The peptide mixture was dissolved in 200 μL of immunoprecipitation (IP) buffer containing 100 mM NaCl, 1 mM EDTA, 20 mM Tris−HCl, 0.5% NP-40, pH 8.0. Then, a mixture of 20 μL pan anti-acetyl lysine antibody agarose conjugated beads from two different vendors (PTM-104 from PTM Biolabs and ICP0388 from ImmuneChem) was added at a 1:1 ratio for 2 mg peptide sample. The beads were washed twice with cold PBS, resuspended in 200 μL of IP buffer, and then loaded into home-made C8 Stage tips. The peptides were added to C8 Stage tips and flowed through the beads by centrifuging slowly at 50 g for about 1 h. The nonspecifically bound peptides were removed by washing the beads sequentially with 200 μL of IP buffer, wash buffer (100 mM NaCl, 1 mM EDTA, 20 mM Tris−HCl, pH 8.0), and water. The acetylated peptides were eluted with 200 μL of 0.1% (v/v) trifluoroacetic acid in water. The elute was dried by centrifugation under vacuum at 4 °C, and then resuspended in 0.1% (v/v) formic acid (FA) for LC−MS/MS analysis.
Ubiquitinated peptide enrichment
The peptide mixture was dissolved in 200 μL of IP buffer containing 50 mM MOPS, 10 mM Na2HPO4, 50 mM NaCl, 0.3% NP-40, pH 7.5. A total of 20 μL of pan anti-K-GG antibody agarose conjugated beads (PTM Biolabs, PTM-1104) were washed twice with cold PBS and resuspended in 200 μL of IP buffer, which was then loaded into home-made C8 Stage tips. 2 mg of peptides were added to the C8 Stage tips and flowed through the beads by centrifuging slowly at 50 g for approximately 1 h. The nonspecifically bound peptides were removed by washing the beads three times with 200 μL of IP buffer, wash buffer (50 mM MOPS, 10 mM Na2HPO4, 50 mM NaCl, pH 7.5), and water, sequentially. The ubiquitinated peptides were eluted with 200 μL of 0.15% (v/v) trifluoroacetic acid in water. The eluted peptides were dried by centrifugation under vacuum at 4 °C, and then resuspended in 0.1% (v/v) FA for LC−MS/MS analysis.
A sequential enrichment method was employed for label-free acetylomic and ubiquitinomic analyses. Briefly, acetylated peptides were first enriched using an anti-acetyl lysine antibody. The resulting flow-through fraction was dried, re-dissolved in IP buffer, and subsequently enriched for ubiquitinated peptides using an anti-K-GG antibody.
LC-MS/MS analysis
For analyzing proteome and degradome of HeLa cells, the peptide mixture was subjected to online analysis using EASY-nL-LC 1000 coupled with an Orbitrap Fusion mass spectrometer (Thermo Scientific). The sample was loaded directly onto a 15 cm home-made capillary column (C18-AQ, 1.9 μm resin, 100 μm I.D.). A gradient was developed using mobile phase A (0.1% FA, 2% ACN, and 98% H2O) and B (0.1% FA, 2% H2O, and 98% ACN), with a 90-min gradient set as 5% B at 3 min, 12% B at 35 min, 22% B at 67 min, 30% B at 83 min, 90% B at 85 min, and 90% B at 90 min. A static flow rate of 300 nL/ min was used. Mass spectra were acquired in a data-dependent mode with one full scan in the Orbitrap (m/z: 300–1800; resolution: 120,000; AGC target value: 400,000; maximal injection time: 50 ms; exclusion duration: 60 s; mass tolerance: 10 ppm), followed by MS2 scans in the linear trap (activation type: HCD; collision energy: 32; ion trap scan rate: rapid; AGC target value: 10,000; maximal injection time: 100 ms; isolation window: 1.6 m/z).
For acetylome and ubiquitinome profiling using a triple SILAC strategy, the peptide mixture was analyzed on an online EASY-nL-LC 1200 coupled with a Q Exactive HF-X mass spectrometer (Thermo Scientific). Mobile phase A (0.1% FA), and B (0.1% FA, 80% ACN and 20% H2O) were used to develop a gradient. A 240-min gradient was developed on a 30 cm column for in-depth analysis, and a short 60-min gradient was used for ubiquitin-centric analysis on a 15 cm column. Mass spectra were acquired in a data-dependent mode (top 10) with one full scan in the Orbitrap (m/z: 350–1500; resolution: 60,000; AGC target value: 3,000,000; maximal injection time: 20 ms), followed by MS2 scans in the Orbitrap (collision energy: 27; resolution: 15,000; AGC target value: 100,000; maximal injection time: 150 ms; isolation window: 1.6 m/z).
For acetylome and ubiquitinome profiling using a label-free strategy, the peptide mixture was analyzed on a nanoElute LC system coupled online to TIMS TOF Pro2 mass spectrometer (Bruker) via a CaptiveSpray nano-electrospray ion source. The peptides were separated on a 25 cm home-made analytical column (C18, 1.5 μm resin, 75 μm I.D.). A gradient was developed using mobile phase A (0.1% FA in water) and B (0.1% FA in ACN), with a 120-min gradient set as 5% B at 0 min, 22% B at 99 min, 35% B at 110 min, 80% B at 115 min, 80% B at 120 min. A static flow rate of 300 nL/ min was used. The column oven temperature was maintained at 50°C. All spectra were acquired within an m/z range of 100 to 1700 and an ion mobility range from 0.75 to 1.40 Vs/cm2. The dual TIMS analyzer was operated at a fixed duty cycle with a ramp time of 100 ms, and the total cycle time was 1.16 s. Data-dependent acquisition (DDA) was performed in PASEF mode, with 10 PASEF scans per topN acquisition cycle.
Proteasome activity assay
The experiment was performed according to a previously described method56. The three enzymatically active subunits in the 20S complex were assayed with fluorogenic peptide substrates (BACHEM, Z-LLE-AMC for β1, Boc-LSTR-AMC for β2, and Suc-LLVY-AMC for β5).
For purified human 26S proteasomes (Boston Biochem, E-365), the assay was carried out in a buffer containing 50 mM Tris-HCl, pH 7.5, 40 mM KCl, 5 mM MgCl2, 0.5 mM ATP, 1 mM dithiothreitol, and 50 μg/μL BSA. Aspirin was added and the mixture was incubated at 37 °C for 30 min. A positive control of 20 μM epoxomicin was used. Then, 0.5 nM purified proteasomes were incubated with 100 μL of substrate (100 μM Z-LLE-AMC, 100 μM Boc-LSTR-AMC, 100 μM Suc-LLVY-AMC) in a 96-well black fluorescence plate at 37 °C for 60 min. The fluorescence intensity at 460 nm was recorded with 380 nm excitation.
For assaying proteasome activity in crude cell extracts, HeLa cells or H4 cells were cultured in six-well plates until they reached 70-80% confluency. After treating cells with different doses of aspirin, or a combination of TSA and NAM, or a combination of aspirin, TSA, and NAM for 12 h, cells were washed three times with PBS and then resuspended in homogenization buffer (50 mM Tris-HCl, pH 7.5, 250 mM sucrose, 5 mM MgCl2, 2 mM ATP, 1 mM DTT, 0.5 mM EDTA, 0.025% digitonin). The cell lysates were obtained as supernatants by centrifuging at 20,000 g for 15 min at 4 °C and the protein concentration was determined using a Bradford assay. 20 μM epoxomicin was added and incubated for 30 min as a positive control. 20 μg of cell lysates were incubated with 200 μL of substrate (100 μM Z-LLE-AMC, 600 μM Boc-LSTR-AMC, 100 μM Suc-LLVY-AMC) in a 96-well black fluorescence plate at 37 °C for 30 min. The fluorescence intensity at 460 nm was recorded with 380 nm excitation.
Proteasome integrity analyzed by native gel electrophoresis
The experiment was conducted using purified 26S proteasome (Boston Biochem, E-365) in a buffer consisting of 50 mM Tris-HCl (pH 7.5), 40 mM KCl, 5 mM MgCl2, 0.5 mM ATP, 1 mM dithiothreitol, and 50 μg/μL BSA according to a previously described method57. 2 μg of purified 26S proteasomes were incubated with different concentrations of aspirin at 37 °C for 30 min. 20 μM MG132 was used as a negative control. The integrity of the 26S proteasome was assessed using a 6% native gel (Meilunbio, MA0172). The band positions were visualized using Coomassie blue staining.
Preparation of the recombinant mouse and human α-syn PFFs
The recombinant mouse and human α-syn were overexpressed and purified using the same protocol as described previously58. In brief, the gene encoding full-length α-syn of mouse or human was inserted into the pET22 vector, and then expressed in E. coli BL21 (DE3) cells along with yeast N-acetyltransferase complex B59. The α-syn proteins were purified using a strong anion exchange column (GE Healthcare, 17-5156-01) and preparative gel filtration chromatography Superdex 75 (GE Healthcare, 28-9893-33). To obtain mouse and human α-syn fibrils, 100 μM α-syn monomer was incubated in a buffer containing 50 mM Tris, pH 7.5, 150 mM KCl, 0.05% NaN3, in the presence of 0.5% (mol/mol) α-syn PFFs, at 37 °C with constant agitation (900 rpm) for 7 days.
Next, the fibrils were collected by centrifugation at 14,462 g for 45 min. The pellets of mouse or human α-syn fibrils were further washed with PBS and then suspended in PBS to a concentration of 2 μg/μL and 500 mM respectively. The α-syn PFFs were obtained by sonication at 20% power for 15 cycles (1 s on/off) on ice. Transmission electron microscopy (TEM) was used to examine the physical state of the fibrils.
Mouse primary neurons
Mouse primary cortical neurons were prepared from the cortex of embryonic day E16-18 C57BL/6 J mice purchased from animal facility (Shanghai Ling Chang Biotechnology Co.). The primary neurons were cultured in Neurobasal medium supplemented with 2% (v/v) B-27 supplement, 0.25% (v/v) glutamax, and 1% (v/v) penicillin/streptomycin at 37 °C in a 5% CO2 atmosphere for 7 days. Subsequently, the neurons were treated with PBS or 200 nM human α-syn PFFs for 7 days, followed by infection with lentiviral vectors expressing three ubiquitin variants (WT, K48R, K63R) with HA tag. The lentivirus-containing medium was removed 12 h later. Six days after infection, 1 mM or 3 mM aspirin was added and incubated for 18 h. For the group without lentivirus infection, the medium was changed in the same manner, and the cells were further cultured for an additional 6 days. Subsequently, the treatment with aspirin was consistent across all groups. 1‰ DMSO was used as a control. The primary neurons were harvested and lysed in a loading buffer containing 2% SDS. The proteins were boiled at 95 °C for 30 min and separated by 10% SDS-PAGE and analyzed by western blotting.
Oral administration of aspirin in mice
Aspirin was mixed into strawberry-flavored sweetened jelly and given to mice60. 15.2 mg of aspirin was dissolved in 20 mL of Splenda solution with 0.1% methyl cellulose. Jelly alone was used as a vehicle. As mice exhibited avoidance to new chow type, 3–4 days of pretraining was introduced until they could finish the jelly within 1 min. The PD mouse model was established by injecting α-syn PFFs in the striatum. Four weeks after the injection, mice started to consume the vehicle or aspirin jelly over an 8-week period (6 mg/kg (body weight), once per day). Mice injected with the AAV vector received the aspirin-containing jelly two weeks after the AAV injection. For precise dosing, the required jelly amount for each mouse was calculated carefully based on the individual mouse’s body weight. 10, 11, and 12 mice were used for the PBS, the PFF-control, and the PFF-aspirin treatments, respectively. 6 mice were used for each of the PFF-WT-Ub, the PFF-K48R-Ub, and the PFF-K63R-Ub conditions, respectively.
Behavioral tests
All behavioral trainings and tests were carried out between Zeitgeber time 4 to 8 (4 to 8 h post-light on). To ensure that the mice were fully acclimatized to the testing environment, they were transferred to the behavioral room at least 1 h before testing. For the open field test, a 40 × 40 cm chamber was placed in a light/sound-proof box with a camera hanging on the ceiling. The light in the chamber was kept at 30∼35 lx. After habituating to the chamber, mice were placed in the center of the chamber and allowed to explore for 10 min. The camera was used to monitor and record mice’s activities, which were then analyzed by EthoVision XT (Noldus 11.5). The distance explored by mice within the chamber is used to assess the locomotor activity. A shorter distance correlated with more severe movement impairment. The chamber was cleaned thoroughly with 75% alcohol and left to air dry between each test.
For the rotarod test, all mice were pre-trained on a rotarod (Sansbio, YLS-4C) for three consecutive days. The training consisted of two constant speed trials (4 rpm/min) and one accelerating speed trial (0-40 rpm/min) per day. Each trial lasted 2 min, with a 3-min interval between trials. After a day’s break, mice were tested twice consecutively on the rotarod with an accelerating speed from 0 to 40 rpm/min in 1.5 min, followed by maintenance at 40 rpm/min for an additional 3.5 min. The total duration on the rotarod was recorded and used for analysis. The results of the rotarod test were normalized to the batch-specific PBS mice in each independent experimental batch or PFF-WT-Ub mice. The rotarod was cleaned thoroughly with 75% alcohol and left to air dry between two tests.
For the pole test, a metal rod (length = 50 cm, diameter = 10 mm) was wrapped with anti-slip tape. After acclimating to the environment for half an hour, mice were placed head down on top of the rod. All mice were trained to climb down the rod once before being tested five times continuously to measure the climbing down duration. Each test was completed within 60 sec. Subsequently, mice were returned to their home cages for at least 30 min of rest before undergoing five consecutive tests for turn duration. Mice were positioned head-up on top of the rod, and the time taken for them to turn their heads downward was recorded. For data analysis, the four shortest durations from both tests were averaged to estimate climbing down and turn duration, respectively. The total duration was calculated as the sum of the climbing down duration and turn duration. A higher score correlated with more severe movement impairment.
Immunofluorescence staining and confocal imaging
For animal models, mice were anesthetized with isoflurane vapor, then perfused transcardially with 10 mL ice-cold PBS and 40 mL 4% paraformaldehyde (PFA). The whole brain was removed and kept in 4% PFA overnight. After fixation, the brain was placed in PBS containing 20% sucrose overnight to ensure the mice brains sank to the bottom and then transferred to 30% sucrose for 2 days. Subsequently, the brains were snap-frozen in optimal cutting temperature compound (OCT, Tissue Freezing Medium, Leica) and stored at −80 °C. Next, 25-μm coronal slices were sectioned using a Leica cryostat (model: CM3050s-1-1-1) and mounted on poly-lysine-coated glass slides. Slices were stored at −20 °C. Before staining, slices were rinsed with PBS and incubated for 2 h at room temperature with a blocking solution consisting of 5% normal goat serum (Sigma) and 0.1% Triton X-100 in PBS.
For primary neurons and H4 cells, cells cultured on coverslips were washed three times with PBS and fixed with fixation buffer (4% PFA and 4% sucrose in PBS) for 10 min at room temperature. The cells were then permeabilized with PBS containing 0.15% Triton X-100 for 15 min. Next, the cells were incubated with blocking solution (5% bovine serum albumin in PBS) for 30 min at room temperature.
Brain or cell slices were incubated with primary antibodies at 4 °C overnight, followed by washing three times with PBST buffer (0.1% Tween 20 in PBS). The slices were then incubated with secondary antibodies at room temperature for 2 h in the dark, followed by washing three times with PBST. Next, the slices were mounted with mounting medium (Invitrogen, ProLong Gold antifade reagent containing DAPI), and fluorescence was visualized with either the SP8 confocal microscope (Lesia) or spinning disk microscope (Ador). Image analysis was performed using ImageJ and Imaris (version 9.3.1) software. For labeling DA neurons and their neurites, DAT staining was used. α-syn aggregation was labeled with phospho-α-syn staining (S129, p-α-syn). Flag α-syn staining was used to label exogenous α-syn aggregates. Early endosomes were labeled with Rab5 staining, late endosomes were labeled with Rab7 staining, and LAMP2 staining was used to label late endosomes and lysosomes.
High content imaging (HCI)
H4-GFP-LC3 cells were treated with different concentrations of aspirin for 18 h in 96-well plates. A positive autophagy activator, 400 nM of rapamycin was used as a control. The cells were fixed with 4% PFA and then stained with DAPI. Images were acquired using a High Content Analysis System (PerkinElmer, Operetta CLS) and analyzed using Harmony software (version 4.6).
MS data analysis
MaxQuant software (version 1.5.3.30) was used for SILAC quantifications61. The MS data were searched against the SwissProt human protein database (downloaded in October 2018, 20,409 entries) and the built-in contaminant protein list. The “match between runs” option was applied. Trypsin was set as the enzyme, and the maximum missed cleavage was set to 1 and 4 for non-enriched and PTM-enriched data, respectively. The heavy stable isotopes were appropriately set in SILAC data processing. Carbamidomethyl of cysteine (+57.02 Da) was designated as a fixed modification, while oxidation of methionine (+15.99 Da), and acetylation of protein N termini (+42.01 Da) were set as variable modifications. For acetylome and ubiquitinome data analysis, acetylation of lysine (+42.01 Da) and diGly of lysine (+114.04 Da) were additionally set as variable modifications. The first-search peptide mass tolerance and main-search peptide tolerance were set to 20 and 4.5 ppm, respectively. The MS/MS match tolerance was set to 0.5 Da for ITMS and 20 ppm for FTMS. Only unique peptides and razor peptides were used for quantification, and the minimum ratio count for protein identification was 1. The re-quantify option was selected. A false discovery rate (FDR) of 1% was set at protein, peptide, and site levels. The acetylated and ubiquitinated sites were required to have a minimal localization probability of 0.75.
FragPipe software (version 20.0) was used for label-free acetylome and ubiquitinome data analyses62. Precursor and fragment mass tolerances were set to 20 ppm. Trypsin was set as the enzyme, and the maximum missed cleavage was set to 4. Carbamidomethyl of cysteine (+57.02 Da) was designated as a fixed modification, while oxidation of methionine (+15.99 Da), acetylation of protein N termini (+42.01 Da) were set as variable modifications. Additionally, acetylation of lysine (+42.01 Da) and diGly of lysine (+114.04 Da) were set as variable modifications for the acetylome and ubiquitinome data, respectively. A closed database search was performed with MSFragger (version 3.8). Percolator and ProteinProphet in Philosopher (version 5.0.0) were used to filter all peptide-spectrum matches (PSMs), peptides, and proteins to 1% FDR. PTM localization probabilities were provided by PTM-Prophet. Label-free quantification with FDR-controlled “match between runs” quantification analysis was performed with IonQuant (version 1.9.8). In IonQuant, mass tolerance was set to 10 ppm, retention time tolerance was set to 0.4 min, ion mobility tolerance was set to 0.05, normalization was enabled, minimum scans were set to 3, minimum isotope count was set to 2, and minimum site localization probability was set to 0.75. Missing values were imputed using the “replace missing values from normal distribution” method in Perseus63.
Image analysis
Six mice in each group were used for immunofluorescence imaging. 2 slices for each mouse and the dot represents the mouse brain. Images were batched and analyzed by Image J and Imaris x64 (Oxford Instruments, version 9.3.1). First, all images were projected in z-direction with max intensity followed by background subtraction. For the striatum, the whole view was analyzed for signal intensity for all channels. To better visualize and analyze SN, montage images were selected, and SN was manually selected by referring to the mouse brain atlas. With identical background signal subtraction and the same contrast transformation for each data set, the mean intensity of p-α-syn and DAT were measured. P-α-syn aggregates were analyzed automatically by using the particle analysis module in ImageJ (auto black and white contrast, smooth, particle size between 2 and 80 μm). Aggregate density (#/1 mm2) and size distribution were calculated and reported. DAT-positive DA neurons in SN were selected and counted by the Spot module of Imaris. Briefly, manually select the soma of one typical DAT-positive cell as a template. Imaris automatically selects all spherical structures with 12-μm diameter, which avoids most of the non-soma DAT+ neurites signal. Then, the “Quality” filter was applied. Finally, using “Edit” module to remove signals that are obviously not neurons or manually select somas that are missed by Imaris. The distribution and average of quality lower threshold used in each group were equal. To report the immunofluorescence results, the mean p-α-syn signal intensity and the mean DAT signal intensity were normalized to the PFF-control or PFF-WT-Ub group. For different batches of experiments, groups were normalized to their own control before combination. All datasets were presented as mean ± SEM.
Statistics
The statistical analysis was conducted using GraphPad Prism (version 8.0.2) or Perseus (version 1.5.3.3). The data were obtained from at least three biological replicates and presented as mean ± SEM or individual data points. The p-value was calculated using one-way ANOVA for pairwise multiple comparisons between groups and using a two-tailed Student’s t-test for two independent groups.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The MS data for this study have been deposited in the ProteomeXchange Consortium through its iProX partner repository PXD059866. Source data are provided with this paper.
References
Prasad, K. N., Wu, M. & Bondy, S. C. Telomere shortening during aging: Attenuation by antioxidants and anti-inflammatory agents. Mech. Ageing Dev. 164, 61–66 (2017).
Flossmann, E. & Rothwell, P. M. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet 369, 1603–1613 (2007).
Ayyadevara, S. et al. Aspirin inhibits oxidant stress, reduces age-associated functional declines, and extends lifespan of Caenorhabditis elegans. Antioxid. Redox Signal 18, 481–490 (2013).
Green, G. A. Understanding NSAIDs: from aspirin to COX-2. Clin. Cornerstone 3, 50–60 (2001).
Guo, L., Gao, J., Gao, Y., Zhu, Z. & Zhang, Y. Aspirin reshapes acetylomes in inflammatory and cancer cells via CoA-dependent and CoA-independent pathways. J. Proteome 19, 962–972 (2020).
Tatham, M. H. et al. A proteomic approach to analyze the Aspirin-mediated Lysine Acetylome. Mol. Cell. Proteom. 16, 310–326 (2017).
Liu, Z. et al. CPLM: a database of protein lysine modifications. Nucleic Acids Res. 42, D531–D536 (2014).
Zhang, Y. et al. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 113, 2343–2394 (2013).
Archer, S. Y. & Hodin, R. A. Histone acetylation and cancer. Curr. Opin. Genet. Dev. 9, 171–174 (1999).
Ropero, S. & Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 1, 19–25 (2007).
Harper, J. W. & Bennett, E. J. Proteome complexity and the forces that drive proteome imbalance. Nature 537, 328–338 (2016).
Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev. Biochem. 78, 477–513 (2009).
Erpapazoglou, Z., Walker, O. & Haguenauer-Tsapis, R. Versatile roles of k63-linked ubiquitin chains in trafficking. Cells 3, 1027–1088 (2014).
Al-Hakim, A. et al. The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair 9, 1229–1240 (2010).
Wang, G. et al. K63-linked ubiquitination in kinase activation and cancer. Front. Oncol. 2, 5 (2012).
Ordureau, A., Münch, C. & Harper, J. W. Quantifying ubiquitin signaling. Mol. Cell 58, 660–676 (2015).
Qiang, J. et al. Multi-omic analyses reveal minimal impact of the CRISPR-Cas9 nuclease on cultured human cells. J. Proteome 18, 1054–1063 (2019).
Schwanhäusser, B., Gossen, M., Dittmar, G. & Selbach, M. Global analysis of cellular protein translation by pulsed SILAC. Proteomics 9, 205–209 (2009).
Bantscheff, M. et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 29, 255–265 (2011).
Cambridge, S. B. et al. Systems-wide proteomic analysis in mammalian cells reveals conserved, functional protein turnover. J. Proteome Res. 10, 5275–5284 (2011).
Huang, J. X. et al. High throughput discovery of functional protein modifications by Hotspot Thermal Profiling. Nat. Methods 16, 894–901 (2019).
Choudhary, C. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 (2009).
Zhang, N. et al. Comprehensive profiling of lysine ubiquitome reveals diverse functions of lysine ubiquitination in common wheat. Sci. Rep. 7, 13601 (2017).
Pohl, C. & Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366, 818–822 (2019).
Nalepa, G., Rolfe, M. & Harper, J. W. Drug discovery in the ubiquitin-proteasome system. Nat. Rev. Drug Discov. 5, 596–613 (2006).
Sugeno, N. et al. Lys-63-linked ubiquitination by E3 ubiquitin ligase Nedd4-1 facilitates endosomal sequestration of internalized alpha-synuclein. J. Biol. Chem. 289, 18137–18151 (2014).
Tofaris, G. K. et al. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl Acad. Sci. USA 108, 17004–17009 (2011).
Kern, S., Skoog, I., Ostling, S., Kern, J. & Börjesson-Hanson, A. Does low-dose acetylsalicylic acid prevent cognitive decline in women with high cardiovascular risk? A 5-year follow-up of a non-demented population-based cohort of Swedish elderly women. BMJ Open 2, e001288 (2012).
Rangasamy, S. B., Dasarathi, S., Pahan, P., Jana, M. & Pahan, K. Low-dose Aspirin Upregulates Tyrosine Hydroxylase and increases dopamine production in dopaminergic neurons: implications for Parkinson’s disease. J. Neuroimmune Pharmacol. 14, 173–187 (2018).
Chen, H. et al. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch. Neurol. 60, 1059–1064 (2003).
Luk, K. C. et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953 (2012).
Tan, J. M. et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet 17, 431–439 (2008).
Long, H. et al. Wild-type α-synuclein inherits the structure and exacerbated neuropathology of E46K mutant fibril strain by cross-seeding. Proc. Natl Acad. Sci. USA 118, e2012435118 (2021).
Antony, P. M., Diederich, N. J., Krüger, R. & Balling, R. The hallmarks of Parkinson’s disease. FEBS J. 280, 5981–5993 (2013).
Hansen, B. K., Gupta, R., Baldus, L., Lyon, D. & Narita, T. Analysis of human acetylation stoichiometry defines mechanistic constraints on protein regulation. Nat. Commun. 10, 1055 (2019).
Li, Y. et al. A quantitative chemical proteomics approach for site-specific stoichiometry analysis of ubiquitination. Angew. Chem. Int Ed. Engl. 58, 537–541 (2019).
Gabbert, C. et al. Coffee, smoking and aspirin are associated with age at onset in idiopathic Parkinson’s disease. J. Neurol. 269, 4195–4203 (2022).
Nilsson, S. E. et al. Does aspirin protect against Alzheimer’s dementia? A study in a Swedish population-based sample aged ?80 years. Eur. J. Clin. Pharmacol. 59, 313–319 (2003).
Ton, T. G. et al. Nonsteroidal anti-inflammatory drugs and risk of Parkinson’s disease. Mov. Disord. 21, 964–969 (2006).
Wahner, A. D., Bronstein, J. M., Bordelon, Y. M. & Ritz, B. Nonsteroidal anti-inflammatory drugs may protect against Parkinson disease. Neurology 69, 1836–1842 (2007).
Zong, N. et al. Lysine ubiquitination and acetylation of human cardiac 20S proteasomes. Proteom. Clin. Appl 8, 590–594 (2014).
Pettit, L. K., Varsanyi, C., Tadros, J. & Vassiliou, E. Modulating the inflammatory properties of activated microglia with Docosahexaenoic acid and Aspirin. Lipids Health Dis. 12, 16 (2013).
Zandi, P. P. et al. Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology 59, 880–886 (2002).
Asanuma, M., Miyazaki, I. & Ogawa, N. Neuroprotective effects of nonsteroidal anti-inflammatory drugs on neurodegenerative diseases. Curr. Pharm. Des. 10, 695–700 (2004).
Chandra, S., Jana, M. & Pahan, K. Aspirin induces lysosomal biogenesis and attenuates amyloid plaque pathology in a mouse model of Alzheimer’s disease via PPARα. J. Neurosci. 38, 6682–6699 (2018).
Ayyadevara, S. et al. Aspirin-mediated acetylation protects against multiple neurodegenerative pathologies by impeding protein aggregation. Antioxid. Redox Signal. 27, 1383–1396 (2017).
Alexopoulou, Z. et al. Deubiquitinase Usp8 regulates α-synuclein clearance and modifies its toxicity in Lewy body disease. Proc. Natl Acad. Sci. USA 113, E4688–E4697 (2016).
Root, J., Merino, P., Nuckols, A., Johnson, M. & Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 154, 105360 (2021).
Mukherjee, A. B. et al. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol. Neurodegener. 14, 4 (2019).
Ijaz, A., Nawaz Bhatti, H., Rasheed, S., Sadaf, B. & Nawaz, R. Pharmacokinetic study of Aspirin in healthy female volunteers. Pak. J. Biol. Sci. 6, 1404–1407 (2003).
Dikshit, P., Chatterjee, M., Goswami, A., Mishra, A. & Jana, N. R. Aspirin induces apoptosis through the inhibition of proteasome function. J. Biol. Chem. 281, 29228–29235 (2006).
Swatek, K. N. & Komander, D. Ubiquitin modifications. Cell Res 26, 399–422 (2016).
Snyder, N. A. & Silva, G. M. Deubiquitinating enzymes (DUBs): Regulation, homeostasis, and oxidative stress response. J. Biol. Chem. 297, 101077 (2021).
Ohtake, F. et al. Ubiquitin acetylation inhibits polyubiquitin chain elongation. EMBO Rep. 16, 192–201 (2015).
De Bie, P. & Ciechanover, A. Ubiquitination of E3 ligases: self-regulation of the ubiquitin system via proteolytic and non-proteolytic mechanisms. Cell Death Differ. 18, 1393–1402 (2011).
Kisselev, A. F. & Goldberg, A. L. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol. 398, 364–378 (2005).
Yazgili, A. S. et al. In-gel proteasome assay to determine the activity, amount, and composition of proteasome complexes from mammalian cells or tissues. STAR Protoc. 2, 100526 (2021).
Li, Y. et al. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res. 28, 897–903 (2018).
Johnson, M., Coulton, A. T., Geeves, M. A. & Mulvihill, D. P. Targeted amino-terminal acetylation of recombinant proteins in E. coli. PLoS One 5, e15801 (2010).
Zhang, L. Method for voluntary oral administration of drugs in mice. STAR Protoc. 2, 100330 (2021).
Cox, J. et al. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 4, 698–705 (2009).
Yu, F. et al. Fast Quantitative Analysis of timsTOF PASEF Data with MSFragger and IonQuant. Mol. Cell. Proteom.: MCP 19, 1575–1585 (2020).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Acknowledgements
We would like to acknowledge the funding support from the National Natural Science Foundation of China (32071245 to Y.Z.), the Natural Science Foundation of Shanghai (20ZR1474400 to Y.Z.), the Shanghai Municipal Science and Technology Major Project, the Shanghai Basic Research Pioneer Project, and the Shanghai Key Laboratory of Aging Studies (19DZ2260400 to Y.Z).
Author information
Authors and Affiliations
Contributions
Y.Z. and K.H. designed and supervised the study. J.G. and Y.L. conducted the experiments and analyzed the data. C.S., X.L., H.L., S.H. and D.L. provided experimental materials and technical assistance. R.G. and D.X. did the HCI experiment. D.X., Z.Z-R., C.L., B.S., and C.W.T. contributed their expertise and provided constructive discussions. Y.Z., J.G., K.H., and C.W.T wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
Y.Z., J.G. and K.H. have filed a patent application related to the use of aspirin or its derivatives for treating diseases associated with abnormal aggregation of α-syn. All other authors declare no competing interests.
Ethics
All animal experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the Interdisciplinary Research Center on Biology and Chemistry, Chinese Academy of Sciences, and are in accordance with the Guide for the Care and Use of Laboratory Animals of the Chinese Academy of Sciences.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gao, J., Liu, Y., Si, C. et al. Aspirin inhibits proteasomal degradation and promotes α-synuclein aggregate clearance through K63 ubiquitination. Nat Commun 16, 1438 (2025). https://doi.org/10.1038/s41467-025-56737-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56737-6
