
Abstract
Covalent drug discovery efforts are growing rapidly but have major unaddressed limitations. These include high false positive rates during hit-to-lead identification; the inherent uncoupling of covalent drug concentration and effect [i.e., uncoupling of pharmacokinetics (PK) and pharmacodynamics (PD)]; and a lack of bioanalytical and modeling methods for determining PK and PD parameters. We present a covalent drug discovery workflow that addresses these limitations. Our bioanalytical methods are based upon a mass spectrometry (MS) assay that can measure the percentage of drug-target protein conjugation (% target engagement) in biological matrices. Further we develop an intact protein PK/PD model (iPK/PD) that outputs PK parameters (absorption and distribution) as well as PD parameters (mechanism of action, protein metabolic half-lives, dose, regimen, effect) based on time-dependent target engagement data. Notably, the iPK/PD model is applicable to any measurement (e.g., bottom-up MS and other drug binding studies) that yields % of target engaged. A Decision Tree is presented to guide researchers through the covalent drug development process. Our bioanalytical methods and the Decision Tree are applied to two approved drugs (ibrutinib and sotorasib); the most common plasma off-target, human serum albumin; three protein targets (KRAS, BTK, SOD1), and to a promising SOD1-targeting ALS drug candidates.
Similar content being viewed by others
Introduction
Traditionally, rational drug discovery focused on molecules that bind reversibly. This was due in part to the pharmaceutical industry’s aversion to covalent (i.e., irreversible) interactions1,2, which tend to raise safety concerns due to off-target binding that can result in allergies and severe idiosyncratic drug reactions, e.g., from haptenization and enzyme inhibition2. Nonetheless, meta-analyses show that 30% of marketed drugs, including blockbusters such as aspirin, omeprazole, and penicillin, employ covalent bonds2,3. Most of these were discovered using phenotypic assays (through serendipity) due to a lack of relevant methods for covalent inhibitors.
There is currently a surge in rational covalent drug development2,3. This began with an affinity-first approach, i.e., append to a high-affinity ligand, an electrophilic warhead positioned to bind a non-conserved cysteine. This approach led to at least ten marketed drugs3, e.g., ibrutinib targeting Bruton’s tyrosine kinase (BTK) and afatinib targeting epidermal growth factor receptor (EGFR). Covalent drug discovery recently adopted an electrophile-first approach, wherein relatively selective electrophiles, discovered by chemoproteomics, serve as a scaffold for further drug development. This approach is co-evolving with libraries of diverse electrophiles that expand the number of potential targets, and whole-proteome chemoproteomics technologies that identify—and assess the selectivity for—new targets4,5,6. The electrophile-first approach led to the discovery of SARS-CoV-2 inhibitor, nirmatrelvir (Paxlovid)7, and has been credited with making undruggable targets, such as KRAS, druggable (KRASG12C inhibitor, sotorasib [Lumakras])8,9.
Bioanalytical methods for traditional drugs (i.e., small molecule, non-covalent) are well-established10 and are predicated upon a basic tenet of pharmacology that states drug concentration at the site of action is related to effect. Non-covalent drugs are in equilibrium with their targets. As a result, if the equilibrium binding constant (Ki) is known and the free drug concentration is determined using a conventional liquid chromatography tandem mass spectrometry (LC-MS/MS) assay, the percent inhibition can be extrapolated using the Cheng Prusoff equation11,12. The time dependence of analyte concentration in blood is used to derive traditional pharmacokinetics (PK) parameters e.g., area under the curve (AUC), maximum concentration (Cmax), volume of distribution (Vd), clearance (CL), elimination half-life (t1/2), and to derive distribution and absorption coefficients. PK, biochemical (Ki), and phenotypic parameters are then input into a PK/PD model to predict, for example, effect and dose regimen.
For all but the highest affinity reversible ligands, PK findings connect to PD through the direct relationship of concentration to enzyme modulation, but with covalent ligands this direct relationship is lost. Drugs that form covalent bonds to their protein targets are pharmacologically unique because they bind irreversibly, which results in non-equilibrium kinetics and the uncoupling of PK and PD13. Therefore, with covalent drugs the free drug concentration at the site of action is not indicative of the effect, which limits the predictive power of traditional bioanalysis with respect to pharmacodynamics2,3. Moreover, the low concentration of free covalent drugs can impede the determination of an accurate AUC, and by extension absorption, distribution, metabolism, and excretion (ADME) parameters13. Additional PK parameters are also required for covalent drugs because in addition to the clearance of the free drug, clearance of the drug-target complex and its relationship to protein synthesis (i.e., turn-over rate) must be considered. In pioneering work to overcome these limitations, empirical relationships between PK and PD AUCs were established for a number of drugs14 including aspirin15, pantoprazole16, penicillin17,18, and anticancer drugs19,20,21.
Clearly, covalent drug discovery would benefit from bioanalytical methods and PK/PD models that complement their traditional counterparts. Intact protein MS is increasingly used for quality control of biotherapeutics22; in early covalent drug development23; and for quantitation, PK, biotransformation of biotherapeutics23,24. Intact protein MS methods combined with immunoprecipitation have been used to determine drug to antibody ratio (DAR) for antibody drug conjugates25,26, in a mass range ( > 100 kDa) that includes common covalent drug targets. Highly pertinent to the current study is the application of intact mass (using purified protein) to the determination of kinact/KI, which can be used to interpret structure-activity relationships (SARs) and measure selectivity. kinact/KI has been described as a critical in vitro parameter27,28 for selecting covalent inhibitors and may supplant IC50 (which is only appropriate for reversible inhibitors28) as the standard for predicting the in vivo occupancy of covalent drugs3,27,29,30,31. To our knowledge, intact protein MS has not been applied to in vivo PK/PD studies for covalent drug development.
Here, we show, by taking advantage of the property that covalent drugs and drug candidates (hereafter referred to as drugs for simplicity) remain conjugated to their targets, an intact protein MS assay that can analyze the drug-target complex, and assess both drug mechanism of action (MoA) and % target engagement (%TE). This bioanalytical method is applicable from screening to in vivo studies, and therefore throughout covalent drug development and clinical studies. We then derive a model to interpret time-dependent % TE data, which we term “intact protein PK/PD” (iPK/PD), that outputs both PK and PD parameters (absorption, distribution, and drug-target complex half-life, exposure, etc.). We present a workflow and create a results-based Decision Tree to guide researchers through drug candidate selection. We apply these to leading preclinical familial ALS (fALS) drug candidates targeting SOD1 and approved drugs for popular targets KRAS and BTK.
Results
Kinetic stabilization is employed by tafamidis to stabilize transthyretin32 and is a leading therapeutic strategy for stabilizing the quaternary structure of fALS-associated SOD1 variants33,34,35,36,37. Covalent approaches to kinetic stabilization of fALS SOD1 variants tend to target a Cys residue (Cys111) within the SOD1 dimer interface, and have employed cyclic disulfide derivatives38,39,40, disulfiram41, cisplatin42 and ebselen43. Our early screening efforts identified cyclic disulfide-based kinetic stabilizers using an infusion-ESI intact protein assay that could assess MoA44. Notably, this assay excluded 91 bond-forming compounds (compared to 9 true positive hits) that would have been false-positive hits in conventional assays (e.g., size exclusion-based and 35S-labeling). This illustrates the power of MS to improve covalent drug high-throughput screening efforts45. Our previous infusion-based MS assay, although capable of rapid sample analysis (<1 min/sample) is not compatible with biological matrices. All the abbreviations used throughout the manuscript are summarized in Supplementary Table 6.
Development of an LC-MS assay for determining target engagement within a biological matrix
We sought an intact protein liquid chromatography mass spectrometry (LC-MS) assay that is compatible with any phase of covalent drug development. Based upon previous intact protein LC-MS methods standardization efforts46 an LC-MS assay was developed to accommodate a variety of soluble proteins (16 proteins with diverse functions and molecular weights, Supplementary Fig. 1, Supplementary Table 1). To accommodate the complexity of biological matrices, an enrichment step is highly recommended prior to LC-MS. For SOD1 we employed a fast (~10 min) chloroform/ethanol partitioning technique that is applicable to multiple biological matrices (e.g., whole blood, red blood cells [RBCs], and brain) and when coupled to LC-MS requires less than a drop (20 µl) of blood for high S/N spectra. The output of this bioanalytical assay, %TE, serves as the organizing principle of this work. Moreover, accurate mass measurement provides an important quality control step on target protein preparations. Figure 1 illustrates the detection and chromatographic separation for unmodified SOD1 (fALS SOD1G93A) and SOD1 modified by a covalent kinetic stabilizer (cross-linker 1,2-dithiane-1-oxide, hereafter “S-XL6” to denote Sulfur mediated-crosslinking [XL] 6-membered cyclic thiosulfinate) and related sample preparation controls.

Whole blood from a transgenic fALS mouse model (SOD1G93A) fractionated using chloroform-ethanol without (a–e) and with (b–f) spiked S-XL6 (200 µM). S-XL6 is a kinetic stabilizer that cross-links the SOD1 dimer via two cysteines (Cys111subunit A and Cys111subunit B). Cross-linking forms two disulfide bonds that may be susceptible to cleavage by thiol-disulfide exchange with endogenous thiols (e.g., glutathione)79. To address this, we compared target engagement without (a, b, green) and with (c, d, purple; e, f, light blue) thiol alkylating (i.e., endogenous thiol blocking) agents iodoacetamide (IAA) and N-ethyl maleimide (NEM). The inset panels show the deconvoluted spectra. Ubiquitin peak eluted at 23.5 min. Alkylating agents did not improve cross-linking yield, indicating that a thiol blocking step was unnecessary in the final method. Mass spectra were obtained using reversed-phase liquid chromatography and quadrupole time-of-flight MS (Agilent 6560 LC-QToF-MS). % TE is defined as percentage of deconvoluted intensity of the drug-protein complex [drug-protein complex intensity / (drug-protein complex intensity + unbound protein intensity)]. Total ion current is represented as a.u. (arbitrary units) and mass is reported in kDa.
An MS-based decision tree for covalent drug development
For most covalent drugs, e.g., kinetic stabilizers or enzyme inhibitors, %TE is proportional to the on-target effect. Here, %TE from in vitro preparations is used for rapid, initial screening, and the time dependence of %TE in dosed animals is used to assess PK and PD parameters. Specifically, in this Decision Tree (Fig. 2) %TE is applied to preparations ranging from pure protein (Decision [D]1-2) to tissue extracts (D3-4) through dosed animals (D5-7).

Rectangles signify objectives and diamonds signify decisions. Blue arrows indicate advancement (go decisions), pink arrows indicate an unsuitable drug candidate, and black arrows indicate re-estimation. Once a covalent drug candidate reaches the green line, it can be advanced into safety pharmacology studies in higher animal species and Good Laboratory Practice (GLP) ADME studies. *Failures at these stages require input from traditional ADME assays. D# = Decision number, LD50 = lethal dose 50, EC50 = Half-maximal effective concentration, MoA = Mechanism of action, METE = Minimally effective target engagement.
Does the compound employ the correct MoA (D1) and sufficiently engage the purified target protein (D2)?
D1 depends upon confirmation of the proposed MoA in vitro by validation of the intended drug-protein molecular mass (Fig. 2). D2 depends upon the ability to reach a Minimally Effective Target Engagement (METE), as defined by disease biologists, following titration of the compounds into purified protein. For fALS 37% was defined as the METE to match the magnitude of the effect of the most effective fALS SOD1 modulating drug, tofersen47, an antisense oligonucleotide (ASO). The MoAs (D1 = go) and ability to reach METE (D2 = go) of leading fALS drug candidates were confirmed in purified proteins (Fig. 3) and these results were consistent with crystallographic studies of cisplatin, ebselen and S-XL6 (PDB IDs: 3RE042, 1UXM43, and 8CCX44, respectively).

Covalent drug candidates—disulfiram (green, column 1), cisplatin (light blue, column 2), ebselen (purple, column 3), and S-XL6 (pink, column 4) were chosen for screening as they target SOD1G93A, a fALS variant of SOD1 protein involved in ALS. (First row) The proposed mechanism of binding and the predicted mass shift for each covalent drug using intact protein LC-MS analysis. (Second row) Control sample for target protein SOD1G93A. Representative raw and deconvoluted spectra (inset) for each covalent drug. (Third row) Assessment of target engagement of covalent drug using purified SOD1G93A. a Disulfiram shows the predicted molecular mass at 16006 Da ( + 148 Da shift). *However, it also forms a SOD1G93A dimer via a non-native disulfide bond with an unknown mechanism, which can result in misfolding and aggregation41,80. b Cisplatin shows the predicted molecular mass at 16311 Da ( + 452 Da i.e., 226 × 2). c Ebselen shows a molecular mass of 16133 Da ( + 274 Da shift), which confirms its proposed MoA. d S-XL6 cross-links SOD1G93A to form a dimer at 31834 Da [15976 Da shift from (15858*2 + 136 – 18 = 118 Da)], that also confirms the proposed MoA45. (Fourth row). Specifically, the mechanism of S-XL6 involves (step 1) reversible thiolate disulfide interchange between a protein cysteine and S-XL6, which due to Brönstead characteristics results in ring-opening to expose a terminal sulfenic acid, which (step 2) forms a second disulfide (and crosslink) with a second cysteine via condensation. Evaluation of in vitro target engagement in target tissue homogenate (i.e., transgenic ALSG93A mouse brain homogenate). Both ebselen and S-XL6 show the predicted mass shifts at 16133 Da and 31834 Da, respectively. (Fifth row) In vivo target engagement (SC RoA for all candidates). Only S-XL6 in vivo dosing was feasible and resulted in successful detection of cross-linked dimer at 31834 Da in brain (1-hr post-dose via SC). A red “no” symbol indicates a “no-go” decision.
Can the target be sufficiently engaged in the biological matrix of interest (D3) at an acceptable dose (D4)?
D3 is intended for rapid screening (Fig. 2) and depends upon the ability to reach METE in a target tissue homogenate with the compound at a concentration that is higher than a reasonable therapeutic dose (e.g., 10 mM, which extrapolates to a 5 mg/ml for a 500 Da compound). If a compound does not succeed at this concentration, it is unlikely to be therapeutically relevant. Treating ALS requires a CNS-permeable compound, and so a mouse brain homogenate was chosen for the present study. A “no-go” for D3 eliminated cisplatin from further consideration as a fALS drug candidate because brain target engagement is unlikely (Fig. 3). D4 requires titration of compound in the homogenate of interest to extrapolate a minimally effective dose, and is “no-go” if the extrapolated dose is greater than the LD50 or above the established safety limit (Fig. 4). Considering the extrapolated doses were all below the LD50s and the traditional preclinical screening results were acceptable, ebselen, disulfiram, and S-XL6 were a “go” for D4.

S-XL6’s concentration-%TE relationship was established by titration into target tissue homogenate, a minimum effective dose was estimated and tested in vivo. a Titration of covalent drug in whole blood and target tissue homogenate followed by intact protein LC-MS analysis. S-XL6 was spiked at increasing concentrations in whole blood and brain homogenate (target tissue) prepared from fast-line B6SJL-Tg (SOD1*G93A)1Gur/J transgenic mouse model, incubated at 37°C for 1 h, extracted using chloroform-ethanol precipitation, and %TE was analyzed by LC-MS. Dose estimation of S-XL6 was based upon 37% METE. b In vivo administration of covalent binder S-XL6 to achieve effective target engagement in systemic (whole blood) and target organ (brain). Dosed ALS SOD1G93A mice achieved METE at all predicted doses (10 mg/kg intravenously, 12.5 mg/kg and 30 mg/kg subcutaneously). In vitro and in vivo experiments were performed in singlet and duplicate, respectively. c Intravenous (IV) administration of covalent drug S-XL6 at 10 mg/kg and (d) oral (PO) administration of S-XL6 at 150 mg/kg in transgenic SOD1G93A mice (n = 2 per RoA). Whole blood was collected at 1, 4, 8 24-, 48-, 72-, and 168-h post-dose (for IV), and up to 72-h for PO, and extracted and analyzed as described above. Area under the curves (AUCs) were estimated for IV and PO curves. Additionally, brain (target) tissue was collected 1-hr post IV dose and processed as described above. Kp[brain] was calculated using %TE values (%TE brain divided by %TE blood) was c.a. ~1. For c and d raw %TE data are shown as discrete data points, and the lines illustrate fits obtained to the two term iPK/PD model [\(C\left(t\right)=A{e}^{-\alpha t}+B{e}^{-\beta t}\)] derived in Supplementary Fig. 2 and described in Supplementary File. Data (c, d) are presented as Mean ± SD. RoA = route of administration, METE = Minimally effective target engagement. Source data are provided as a Source Data file.
The experiments proposed here for D1-D4 provide useful information for determining structure activity relationships, which may merit their inclusion in high-medium throughput screening efforts. D4 serves as a gatekeeper for humane animal studies and should therefore also consider traditional preclinical screening methods and their respective exclusion criteria44, as informed by therapeutic indication48, ideally with input from a drug development specialist. For example, preclinical screening for SXL-6 included physicochemical properties (e.g., logD) and in vitro ADMET assays (e.g., plasma protein binding, hERG channel inhibited, cytotoxicity, CYP profiling, etc.,) as potential “no-gos.” We suggest that screening drug candidates for HSA binding, which serves as a proxy for plasma binding, be used as an additional preclinical assay (Fig. 6).
Defining the iPK/PD model
To our knowledge, previous PK/PD studies of covalent drugs were based upon the empirical relationship of free drug concentration and effect. Here, we derive a model that accepts % target engagement as input. Compounds that pass D4 are administered at an acceptable dose (e.g., estimated 50% TE or <½ of the LD50) and %TE is measured as a function of time in the target tissue. Traditional PK/PD models link the dose, time, and drug concentration (PK) to effect (PD) and are not applicable to covalent drugs. That notwithstanding, drug concentration-time curves and %TE-time curves are indistinguishable in their graphical form. It stood to reason that %TE-time curves could be fit using identical mathematical operations as drug concentration-time curves, and that by considering the different input (%TE instead of drug concentration), the different outputs could be derived. The mathematical models used to treat traditional PK curves as well as methods for extrapolating these results to effect (PK/PD modeling) have been reviewed49,50,51. Based upon these studies, application of identical mathematical operations to a traditional PK curve or an iPK/PD curve yields analogous parameters, including Tmax, AUC, and t1/2. In either case (traditional PK or iPK/PD) the ratio of AUCs of a particular tissue to the central compartment is the permeability coefficient (Kp with respect to the drug molecule, and Kp [TE] with respect to the target, respectively) and the ratio of AUCs of different routes of administration (RoA) is the absorption coefficient (%F with respect to the drug molecule, and %F [TE] with respect to the target, respectively). With traditional PK analysis t1/2 is that of drug itself and with iPK/PD it is that of the drug-protein complex, which we note have no a priori relationship. The detailed mathematical derivation as well as the capabilities and limitations of iPK/PD compared to existing models are described in Supplementary Fig. 2. Notably, the %TE vs. time curve for drug-protein complex is analogous to a traditional concentration-time-effect (PK/PD curve) for small molecules. By analogy to well-established protein turnover measurements52, if the amount (e.g., intensity) of the labeled target-time is fit as shown in Fig. 4, the output will be the protein degradation half-life. If the %TE-time is modeled, the output will include protein synthesis, and yield the protein turnover number.
There are a few important differences in the biological implication of these analogous parameters, because iPK/PD analyzes target engagement over time and traditional PK analyzes drug concentration over time. First, iPK/PD has the disadvantage of being blind to any compound that is not conjugated to the target and therefore does not address the absorption, distribution, metabolism, or elimination of the total compound. These must be addressed by traditional PK studies. Second, compared to traditional PK/PD models, iPK/PD has the advantage of avoiding the assumptions used during extrapolation between drug concentration and effect. Third, whereas the half-life derived from traditional PK relates to the clearance of the compound itself, the half-life derived from iPK/PD relates to the turnover of the protein target. Fourth, although the half-lives of traditional PK, iPK/PD, and protein turnover are all derived by fitting curves where the numbers of Xe-kt terms49,52 equals the number of compartments, PK compartments relate to different tissues, and iPK/PD and protein turnover compartments relate to distinct pools of proteins.
Is the target sufficiently engaged in vivo at an acceptable dose (D5)?
At this point a dose has been estimated from the concentration-%TE relationship in target tissue homogenates. The required dose, however, will be subject to the combined effects of ADME. In particular, exposure will be affected by: absorption, which can be accounted for with an IV dose; distribution, which can be estimated based upon compound- and organ-related dependencies; and the disposition of the compound via biotransformation and elimination, whose cumulative effects will be determined for compounds that proceed to D6 and D7. With the objective of estimating the maximum value of %TE (%TEmax), animals are given the estimated dose, sacrificed at early time points (e.g., 15 min, 30 min, 1 h), and the target tissues are harvested, processed, and %TEs are determined. If the METE is reached the compound is “go” for D5; if not, the dose is re-estimated [New dose = (old dose) x (METE/ %TE old dose)] and %TE is reassessed as described above. The METE could not be reached for ebselen (at ~22% of LD50) and disulfiram (at a 20 mg/kg dose imposed by disulfiram’s solubility limit in our IV dose formulation [4 mg/mL]), eliminating these compounds from further consideration (D5 “no-go”). Interestingly, D5-related results indicated that ebselen, which improves survival in a SOD1G93A mouse model43, effected SOD1 in vivo by promoting cysteinylation (in brain) and glutathionylation (in blood) of SOD1G93A, consistent with target engagement followed by compound removal via thiol/selenosulfide exchange53. This finding illustrates that the metabolic products of the target-drug conjugate can be discovered using our methods. Exceptions to a D5 “no-go” can be made, for example, in indications where the target product profile (TPP) allows an alternative RoA that could increase exposure (e.g., intrathecal for neurodegeneration).
Is the predicted dose regimen acceptable (D6)?
The remaining protocols depend upon multiple variables (e.g., animal availability, drug accumulation, a Kp estimate that enables the analysis of blood rather than target tissue etc.) and can be adjusted. Animals are treated with a single dose and %TE-time is assessed in the target tissue at additional time points (e.g., with the dose and duration informed by D5 results). Notably, an iPK/PD study will require a longer duration than traditional PK studies (0-24 h) when the target-drug conjugate’s half-life is greater than 1 day (as illustrated below for the S-XL6-SOD1 complex). iPK/PD parameters are determined and from these parameters, in particular %TEmax and the one (TE = TE0 e−k[TE]*t) or two term [TE(t) = Ce-γt + De-δt] fits of %TE-time curve, the dose and dosing interval can be estimated using established criteria52,54 (detailed description in Supplementary Fig. 2). D6 is “go” if, given the TPP, the predicted dose interval is acceptable.
SOD1G93A is contained within RBCs, which permitted the determination of oral (%FPO) and subcutaneous (%FSC) absorption as well as blood-brain barrier (BBB) permeability estimation (%TEbrain/%TEblood). The ratio of AUCs (oral/IV) indicated %FPO = 7, which is consistent with the %FPO obtained using traditional PK bioanalytical analysis of total drug (15%) and the ratio of toxicity (LD50[IV]/ LD50[PO] x100 = 6%). For additional studies, animal usage was limited by acquiring snapshot (one timepoint at 1-h) measurements, which indicated high brain penetrance (Kp [TE]@1hour = 1.0) and acceptable SC permeability (%FSC, [TE]@1hour = 38). iPK/PD modeling of %TE-time in blood indicated two compartments with 11 and 87-h half-lives (60% and 40% of SOD1, respectively), and that over half of the initially engaged SOD1 remains at the trough in a 24-h dose interval, which indicated a “go” for D6. Guidance for minimizing animal usage is provided in Supplementary.
Can an acceptable dose regimen be achieved (D7)?
The design of optimal dose regimen has been described55 and depends here upon factors that affect the dosing intervals, e.g., dose at %TEmax, drug-target complex half-life, and METE. Using a time course and a dose frequency estimated from D6-related data, and at least two doses, %TE-time is measured. After sufficient administration this provides a steady state exposure-time relationship, which based upon prior biological knowledge, can be extrapolated to an exposure-effect relationship. If exposure is sufficient following multiple doses, D7 is a “go”, i.e., the compound has potential as a therapeutic agent. It can then progress to comparably more resource intensive studies including standard (e.g., 14 day toxicology, etc.) and covalent drug-related (chemo- and toxicoproteomics to identify off-targets) safety assessment, and the precise dose-finding needed for the development stage. These experiments are currently in progress for S-XL6 in the SOD1G93A fALS mouse model.
Broader application: considerations for screening different therapeutic targets
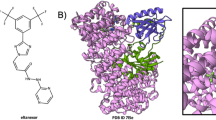
We explored the applicability of the workflow above for the most popular covalent drug targets, KRAS (23 kDa), BTK (77 kDa) (Fig. 5), and EGFR (69 kDa) and the most prevalent off-target, HSA (Fig. 6). KRAS and BTK recombinant purified proteins were incubated with covalent drugs sotorasib and ibrutinib respectively. The observed intact mass measurements provided a precise quality control step for these drug-target preparations and satisfied the MoA (D1 = go) and METE (D2 = go) criteria (Fig. 5). Additionally, for confirming the sotorasib mechanism of action, we used in house purified GNP (active) and GDP (inactive) bound forms of KRAS. As expected, sotorasib fully bound GDP-KRAS and minimially bound GNP-KRAS, effectively locking the protein in the inactive state that inhibits oncogenic signaling56. These results were consistent with crystallographic56 (PDB: 6OIM) as well as previously reported bottom-up mass spectrometry studies57.

Covalent inhibitors sotorasib and ibrutinib targeting the undruggable KRASG12C and BTK protein, correspondingly, were chosen to test the scope of the methods presented here. Representative raw and deconvoluted spectra (inset) with drug-related mass shift for each of the protein targets are shown. Together (a) and (b) confirm the expected sotorasib-related mass shift ( + 560.6 Da) using a commercially sourced KRASG12C (green). In addition, the mechanism of action (Decision#1, D1) and METE (D2) were confirmed using an in-house purified (c), (d) GNP (blue) and (e), (f) GDP-bound (purple) versions of KRAS, confirming sotorasib selectively binds the inactive GDP-KRASG12C, thereby trapping the protein in its inactive conformation. (g), (h) Ibrutinib covalently binds to BTK ( + 438.6 Da) (pink) confirming the MoA and METE criteria. The phosphorylated proteoforms of BTK are delineated with a bracket. Notably, this analysis shows that unmodified BTK as well as BTK phospoproteoforms with 1, 2, and 3 phosphorylation sites fully bind ibrutinib. The unbound mass of BTK (average mass: 77512.5 Da) corresponds to an unknown +88 Da modified proteoform of BTK that cannot bind ibrutinib. Total ion current is represented as a.u. (arbitrary units).

Covalent drugs were incubated with human serum albumin (HSA), the most abundant plasma protein, to test albumin-binding and to provide a go/no go decisions in the covalent discovery workflow. a Raw and deconvoluted (inset) spectra for control HSA (black). b Disulfiram ( + 147 Da) (purple), (c) cisplatin ( + 225 Da) (pink), and (d) ebselen ( + 274 Da) (blue) showed drug-related mass shifts with HSA, which affects their METE in vivo (D5 = No Go). As expected, rationally designed covalent drugs (e) afatinib (green), and (f) ibrutinib (orange), showed minimal binding to HSA. Additionally, (g) sotorasib (dark red) and (h) S-XL6 (red) showed negligible binding to HSA, which with S-XL6 is enabled by the unique ability to reversibly bind off-target lone cysteine residues, while covalently cross-linking closely spaced pairs of cysteines such as SOD1’s. *denotes cysteinylation (average mass: 66564.28 Da), which blocks the binding of cysteine-targeting drugs disulfiram and ebselen, but does not block the binding of cisplatin, presumably due to reaction with additional nucleophilic amino acids. Total ion current is represented as a.u. (arbitrary units).
The next step in the workflow requires screening for ability to reach METE in target tissue preparations. KRAS presented an analytical challenge at this step, because due to its instrinsically low abundance it could not be detected by IP-MS in the 10 mg human tissue samples available to us. The Kelleher group, however, showed that KRAS can be detected in IP-MS studies from 200 mg tissue samples, allowing studies with adequate tissue to progress through the entire iPK/PD workflow. To facilitate intact protein detection from tissue homogenates, we spiked tissues with KRAS and proceeded as describe above for SOD1 D3. Both immunoprecipitation (IP)-based and solvent extraction-based enrichment were successful in KRAS-spiked mouse lung homogenates and spiked patient lung samples (Supplementary Fig. 3), which then progressed into the iPK/PD workflow at D3. As expected, the above experiment confirmed that METE was reached with KRAS (D3 = Go). A summary of what is currently enabled for various targets, e.g., IP-MS and LC-MS methods, along with related references can be found in Supplemenatry Table 5. Briefly, SOD1 and KRAS can proceed through the entire workflow, BTK can proceed through D2 (go for ibrutinib), and EGFR could not be detected by intact protein LC-MS. At present, BTK has been immunopurified by others, and successful LC-MS studies have been performed by others, and a combined IP-MS method will need to be developed for steps beyond D2. EGFR has been immunopurified by others, but to our knowledge there are no published intact protein studies.
To overcome limitations related to immunopurification or intact protein detection, we derived the formula to convert chemoproteomics output, which has been successfully generated for BTK and EGFR, into %TE values (Supplementary Fig. 2) required for input into the iPK/PD model. To enable studies on new targets, sensitivity benchmarks and exemplar LOD, LOQ, and matrix effect calculations are provided for SOD1 in Supplementary Table 2, Supplementary Table 3, and Supplementary Table 4, respectively. Practically speaking, 1 picomol of the targets examined here (Fig. 5) should be sufficient for microflow (1-10 µL/min) LC-MS analysis. If additional sensitivity is required, more sample can be loaded or more sensitive nanoflow LC-MS analysis can be employed. For abundant the abundant target, SOD1, 1–10 mg of tissue is required, and for less abundant targets like KRAS, 200 mg of tissue is required58. For targets not considered here, the relative sample loading required can be estimated from the above considerations or the previously described relationship of intact MS sensitivity and protein size59.
Intact albumin screening reveals that more recent covalent drugs exhibit decreased off-target plasma binding
Serum albumin is the most abundant plasma protein (0.63 mM, constitutes ~60% total plasma protein) and a common off-target for covalent drugs that directly influences PK/PD60. As such, covalent compound binding to HSA can complement traditional preclinical plasma protein binding assays, by providing the MoA for HSA binding. The LC-MS assay described above was applied to HSA and a variety of covalent drugs (Fig. 6). Interestingly, the first-generation covalent drug (from phenotypic assay and not-rationally designed), disulfiram, converted 100% of HSA (at 20-fold molecular excess and equivalent to 20 mg/kg dose) to an HSA-disulfiram adduct by the mechanism shown in Fig. 3. This result is consistent with the previously described high plasma protein binding via albumin61. Second-generation covalent drugs (rationally designed by appending an electrophile to a high-affinity ligand) like afatinib and ibrutinib demonstrated limited, but detectable covalent binding to HSA. The third-generation covalent drug (electrophile-first with core electrophile discovered by chemoproteomics screening and rationally modified), soratosib, and fourth-generation electrophilic warhead (electrophile-first with electrophile rationally designed to minimize lone-cysteine binding), S-XL6, resulted in no detectable HSA binding.
Discussion
This work demonstrated how an intact protein MS assay, interpreted using our iPK/PD model, can advance covalent drug candidates throughout preclinical drug development. Provided that the tissues are available, the same techniques are applicable to clinical trials and personalized medicine. We tested the leading preclinical fALS SOD1 kinetic stabilizers and identified one compound, S-XL6, that engaged and kinetically stabilized its target and exhibited BBB penetration and bioavailability with different RoAs. We demonstrated the applicability of our workflow to assess target engagement of important targets like KRAS and BTK. For targets where intact mass measurements or protein fractionation haven’t been developed, we provide a means to port chemoproteomics data to the iPK/PD model. We extended our on-target analysis methods to off-target analysis, and demonstrated that the extent of HSA binding, which is a primary off-target for most RoAs, can be estimated. From these studies a fascinating and reassuring trend emerged—covalent drugs are evolving to exhibit less off-target binding to HSA. As we demonstrated previously for SOD1, sufficient enrichment62,63 can obviate the need for LC and prepare samples that are compatible with emerging native MS techniques64,65,66. Notably, iPK/PD analysis is applicable to any preparation where %TE can be measured or inferred, including non-covalent drugs analyzed by native MS techniques, endoproteinase digests where the unmodified and drug-modified peptides are detected67, and chemoproteomics methods that detect reactive cysteine abundance as a function of drug concentration. The iPK/PD model (i.e., % TE as input) applies to the target-bound drug, but does not directly apply to free drug absorption, distribution, metabolism, and excretion. The methods proposed here are therefore complementary to traditional bioanalytical methods for free drug analysis and chemoproteomics methods for off-target ID.
Methods
Ethical statement
All methods and animal manipulations were approved by the Northeastern University Institutional Animal Care and Use Committee (IACUC) under protocol #16-0303-R.
Chemicals
Cisplatin, ebselen, disulfiram, afatinib, sotorasib, and ibrutinib were purchased commercially from Sigma Aldrich (St Louis, MO). Human KRASG12C was purchased from Sino Biological (China) and Human Bruton’s Tyrosine kinase (BTK) was purchased from Abcam (Cambridge, UK). KRAS-GNP and KRAS-GDP were purified as per previously described68. Human Serum Albumin (HSA), Bovie Serum Albumin (BSA), NIST IgG1k mAb were purchased from Sigma-Aldrich (St Louis, MO). SOD1 was purified in-house as previously described44,62 from yeast.
Synthesis of cyclic thiosulfinate 1,2-dithiane-1-oxide (S-XL6)
Synthesis of 1,2-dithiane-1-oxide (S-XL6) was achieved according to the previously published literature procedure45. Briefly, commercially available 1,4-butanedithiol was reacted with bromine and silica gel to afford 1,2-dithiane. After crystallization from hexanes, 1,2-dithiane was then oxidized with sodium periodate and the final product 1,2-dithiane-1-oxide (S-XL6) was purified by flash column chromatography on silica gel.
Reaction monitoring of target engagement in vitro
Blood and brain were collected from a control hemizygous mouse expressing human SOD1G93A for titration experiment to determine the initial minimum concentration of compound to reach the target (e.g., hSOD1 protein). Brain homogenates were prepared using a Potter-Elvehjem homogenizer and 10 mM ammonium acetate, pH 7.8 (2:1, ammonium acetate to brain weight). 40 µL of both brain and blood were dispensed to separate vials and labeled accordingly. The test compounds for titration experiment were dispensed to both blood and brain vials according to the following final concentrations: 0.1, 0.5, 1, 5, 10, 50, 100, 500, 1000, 5000, 10000 µM ( < 2% DMSO). All samples were then fractionated using ethanol and chloroform extraction as previously described69.
Expression and purification of wild-type SOD1 and SOD1G93A
Expression and purification of SOD1 were conducted as previously described44,62. Briefly, EGy118ΔSOD1 yeast were transformed with a wild-type SOD1 and SOD1G93A YEp351 expression vector and grown at 30 °C for 44-hr. Cultures were centrifuged, lysed in a blender using 0.5 mm glass beads, and subjected to a 60% ammonium sulfate precipitation. Then the sample was centrifuged, and the resulting supernatant was diluted to 2.0 M ammonium sulfate. The diluted sample was passed through a phenyl-sepharose 6 fast flow (high sub) hydrophobic interaction chromatography column (Cytiva Life Sciences, Marlborough, MA, USA) using a linearly decreasing salt gradient from high salt buffer (2.0 M ammonium sulfate, 50 mM potassium phosphate dibasic, 150 mM sodium chloride, 0.1 mM EDTA, 0.25 mM DTT, pH 7.0) to low salt buffer (50 mM potassium phosphate dibasic, 150 mM sodium chloride, 0.1 mM EDTA, 0.25 mM DTT, pH 7.0) over 300 mL. Fractions containing SOD1 eluted between 1.6 and 1.1 M ammonium sulfate and were confirmed with SDS-PAGE. These fractions were pooled and exchanged into low salt buffer (10 mM Tris pH 8.0). Pooled fractions were then passed through a Mono Q 10/100 anion exchange column (Cytiva Life Sciences, Marlborough, MA, USA) using a linearly increasing salt gradient from low salt buffer to high salt buffer (10 mM Tris pH 8.0, 1 M sodium chloride) from 0 – 30%. SOD1 fractions were collected between 5 and 12% high salt buffer and were confirmed with SDS-PAGE, western blot, and Fourier Transform Ion Cyclotron Resonance Mass Spectrometry (FT-ICR-MS).
Confirmation of covalent drug-protein complex formation using LC-MS
In-house purified 40 µM wild-type SOD1 was diluted using 10 mM ammonium acetate, pH 7.4 and combined with 400 µM (10x, 0.25% final DMSO content) S-XL6 in equal volumes and incubated for 4 h at 37 °C and vortexed at 350 rpm. Cisplatin (1 mM) was incubated in hemizygous B6SJL mice expressing human SOD1G93A blood for 24-h at 37 °C. Complete cross-linking of cyclic thiosulfinates (S-XL6) and other reactions (e.g., drug-protein complex, post-IP (immunoprecipitation) samples, disulfiram, cisplatin, ebselen, afatinib, sotorasib and ibrutinib) were confirmed by either an Agilent 6560 QToF mass spectrometer (Santa Clara, CA) or by Bruker TIMS-ToF Flex mass spectrometer (Billerica, MA). Thiol alkylating agents – iodoacetamide (IAA, Fig. 1c, d) at 100 mM and N-ethylmaleimide (NEM, Fig. 1e, f) at 10 mM were incubated for 1 h with or without S-XL6 during method development. All in vitro assays were performed in duplicates and repeated on different MS instruments with similar results. All the samples (total: 218 and control: 6) were analyzed in duplicates by either an Agilent 1290 Infinity II-series UHPLC (Ultra-High Performance Liquid Chromatography) system coupled to an Agilent 6560 drift-tube ion mobility Q-ToF (quadrupole time of flight) mass spectrometer (IM-MS) (Santa Clara, CA) or a Bruker nanoElute2 (nanoLC) liquid chromatography system coupled to TIMS-ToF (Trapped Ion Mobility quadrupole time of flight) Flex as previously described44,46,68,70,71. The n/LC system was equipped with reversed phase Acquity UPLC Protein BEH C4 (300 Å pore size, 1.7 µm particle size, 100 mm bed length, 2.1 mm ID x 100 mm) column at 60 °C with a flow rate of 0.3 mL/min or Acquity UPLC Protein BEH C4 (300 Å pore size, 1.7 µm particle size, 100 mm bed length, 300 µm ID x 100 mm) column at 60 °C with a flow rate of 3 µL/min. The mobile phase consisted of a mixture of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B) and combined a gradient: 0-2 min: 95% A; 2-70 min: 30% A; 72-75 min: 5% A; 78-80 min: return to initial conditions. The sample was introduced in 10% formic acid and 5 or 1 µL was injected for analysis. The mass spectrometer was calibrated prior to all the samples acquisition and the instrument was operated in positive ion mode. The source temperature was set to 150 °C and 200 °C, with drying temperatures of 150 °C and 210 °C, drying gas flow rates of 10 L/min and 9 L/min, and nebulizer pressures of 30 psi and 26 psi for the Agilent 6560 QToF and Bruker TIMS-ToF Flex mass spectrometers, respectively. Fragmentor voltages were maintained at 250 V and 300 V, while capillary voltages were set to 5000 V and 4500 V for each instrument, respectively. Agilent MassHunter Workstation Data Acquisition software v9.0 (Santa Clara, CA) was used for controlling the Agilent LC-MS and BioConfirm v10.0 software was used for data processing. Similarly, timsControl software v5.1.8 (Billerica, MA) was used for controlling the TIMS-ToF nanoLC-MS and Bruker Data Analysis v6.1 software was used for data processing.
The MS data were analyzed as previously described44. Briefly, the chromatographic window containing the proteins of interest were assigned using EICs (Extracted Ion Chromatogram), e.g., m/z 1586.25-1587.25 (SOD1G93A monomer) and m/z 1516.50-1517.50 (SOD1G93A dimer) and mass spectra from this region were summed. The summed-up mass spectra from each region were deconvoluted and average masses were calculated using the MaxEnt algorithm in either Agilent BioConfirm v10.0 or Bruker Data Analysis v6.1 software. The %TE was calculated as the ratio of dimer intensity (31836 Da) to the monomer (15858 Da) plus dimer intensity. The fit of the measured dimer values was performed and calculated by incorporating 2-compartment model equation using GraphPad Prism (version 10.1.2, San Diego, California).
In vivo dosing and tissue sample preparation for mass spectrometry analysis
All methods and animal manipulations conformed to the guidelines and were approved by the Northeastern University Institutional Animal Care and Use Committee (IACUC) under protocol #16-0303-R. In vivo dosing or ex vivo tissue sample preparation was performed using hemizygous B6SJL mice expressing human SOD1G93A (fast-line Jackson Laboratory; B6SJL-Tg(SOD1*G93A)1Gur/J, also known as SOD1-G93A stock- 002726)72. Animals in the age range of 15–30 weeks were used for in vivo testing; sex was not considered in this study. Animals were acclimatized with a 12 h dark/light cycle (24°C, 30% humidity). Test articles were prepared either in 1x PBS or water (depending upon the solubility, <2% final DMSO content) and mice/rat were dosed slowly at a specified dose level (mg/kg) via a suitable route of administration (intravenous, subcutaneous, or oral). Mice were kept in a restrainer or sacrificed to collect blood and brain at different time points (1, 4, 8, 24, 48, 72, and 168 h) for pharmacokinetic/pharmacodynamic assay or at a single time point tissue collection. Protein extraction was performed as previously described69. Briefly, homogenates were treated with an 8 equivalents of 10 mM ammonium acetate (pH 7.4), followed by 0.15 equivalents of cold chloroform and 0.25 equivalents of cold ethanol, and vortexed at 1800 rpm at 4 °C for 15 min and centrifuged at 17,700 g for 10 min. The supernatant was collected and stored at -80 °C for LC-MS analysis. The analyses (n = 2 for each RoA) herein are therefore of the supernatant resulting from a mixture of 0.25 equivalents of ethanol, 0.15 equivalents of chloroform, 0.6 equivalents of water. Ebselen was injected via SC to a hemizygous mouse expressing human SOD1G93A at 20 mg/kg followed by sacrificing the mouse and collecting both blood and brain after 1 h for further processing the samples prior mass spectrometry run as mentioned earlier.
Tissue homogenization before Immunoprecipitation (IP)
Mouse lung samples were homogenized using ceramic beads and T-PER Tissue Protein Extraction Reagent (Thermo Scientific) for extracting total proteins from tissues, using the manufacturer’s recommended method. Protease inhibitor (SIGMAFAST protease inhibitor, Sigma-Aldrich) was added to the sample homogenization procedure to avoid any proteolysis.
Immunoprecipitation Mass Spectrometry (IP-MS) sample preparation
The immunoprecipitation method optimization was done using Dynabeads Protein G IP kit (Invitrogen) and mouse lung tissue. KRAS protein was enriched using human KRAS protein-specific monoclonal antibody (Thermo Scientific). 50 µL (1.5 mg) of magnetic beads were used at 30 mg/mL in PBS, pH 7.4, with 0.01% Tween-20 and 0.09% sodium azide (preservative), for each IP experiment. The anti-KRAS antibody (10 µg) was diluted in 200 µL antibody binding and washing buffer before adding it to the prepared magnetic beads. After washing step, 250–500 µL tissue homogenate samples (containing KRAS protein) were added to the antigen binding step to form the immune complex. KRAS protein was eluted using 0.2% TFA, which enabled subsequent downstream intact protein MS analysis. Due to inherently low abundance of KRAS protein in the tissues of interest, the eluted protein was concentrated using SpeedVac (Thermo Scientific), followed by reconstitution in 100% water. With the optimized IP procedure, KRAS protein was detected with the intact protein LC-MS method described above. The experiments were repeated three times with similar results.
Reproducibility, sensitivity, accuracy, matrix effect, and precision experiments
Blood was collected from control B6SJL hemizygous mouse for the calibration curve experiments to perform reproducibility and sensitivity measurements. 40 µL of blood was dispensed in separate tubes. For monomer standard curve, human SOD1 was added to blood vials according to the following final concentrations: 0.001, 0.01, 0.1, 0.5, 1, 2.5, 5, 10, 15, 20, and 25 µM. For the dimer standard curve, hSOD1 was dispensed to blood vials containing 5 mM S-XL6 previously spiked into the preparation to form a dimer. All vials were incubated at 37 °C for 3 h. All samples were then fractionated using ethanol and chloroform extraction as described69. The internal standard 13C, 14N-labeled ubiquitin, purchased from Cambridge Isotope Labs (Tewksbury, MA), was spiked into both monomer and dimer standard curves. Quality control samples i.e., 1, 5 and 15 µM for monomer and 2.5, 5 and 20 µM for dimer, were prepared separately for each calibration run (Supplementary table 2 and 3). Additionally, matrix effect quality control samples i.e., 1, 5 and 10 µM for monomer and dimer were prepared separately and % matrix effect was calculated based on mean peak area ratio of mean peak area of the analytes spiked post-extraction to the mean-peak area of the same analyte standards (Supplementary table 4). Statistical analysis was performed using GraphPad Prism (version 10.1.2, San Diego, California). Data from three independent measurements were analyzed to calculate precision, accuracy and matrix effect.
Derivation and detailed description of the iPK/PD model
Analysis of the target tissue is required due to violation of the free drug hypothesis by covalent drugs, which results in drug concentration asymmetry. Notably, drug concentration asymmetry is pervasive, including for reversible binders, and difficult to model for the CNS, the focus of this study.
Assumptions in the iPK/PD model
-
The ionization efficiency of the protein with and without the drug remains unchanged. Note, this only applies to the measurement of %TE, and is not an assumption (i.e., is factored out) when ratios of %TE are taken [i.e., Kp(TE) and F(TE)].
-
When extrapolating in vitro (target tissue homogenates) results to in vivo, there are certain underlying assumptions such as –
-
If k is unknown, there is no barrier between target and site of measurement.
-
Equal partitioning among the organs.
-
The off-target binding is the same across the organism.
-
Rate of distribution is significantly higher than the rate of metabolism of both the small molecule and the complex.
-
There is a lack of feedback drug-related up or down regulation of the target (e.g., from downstream signaling, activate proteases), and no target manipulation (tolerance).
-
The tissue homogenate extrapolation to whole organ follows the same rules and recommendations as suggested by health authorities73. Target expression studies can be conducted while validating a target for a disease indication. The complexity and dependencies of target expressed within tissue/cell types will then be extrapolated from these studies to consider target tissue for analysis.
-
Drug-target complex half-lives are obtained by fitting the curve with one or more of the generalized terms Cie-kit (C=Normalization Constants), e.g., TE = TE0 e−kTE*t and TE(t) = Ce-γt + De-δt for one and two compartment models, respectively, by analogy to traditional PK methods.
Corollaries of the iPK/PD model
-
The k[TE] is the turnover constant for the drug-protein complex and the half-life obtained is the drug-protein complex degradation half-life (turnover of the target).
-
%F(TE) is the absorption coefficient with respect to (only) the drug-protein complex. When target is present in blood, absolute %F (TE) can be calculated as ratio of area under the curves (AUCs) for a specific route of administration compared to intravenous route. When target is not present in blood, a relative %F(TE) can be obtained by comparing AUCs in target tissue for two different routes of administration.
-
Kp, (TE) is the tissue partition coefficient with respect to the drug-protein complex. It can be calculated as the ratio of %TE(tissue) to %TE(blood), only when the target is present in the blood.
-
The compartments in traditional PK refer to different tissues whereas, in protein metabolism refer to different pools of proteins within a cell.
Traditional PK/PD model assumptions that do not apply to iPK/PD
-
Drug concentration (free/unbound) is the driving force of the pharmacological effect.
-
Unbound drug concentration in blood is same as that at the site of action (at steady state).
-
Drug and target are in equilibrium.
-
Drug (specifically the Drug-Target complex) is eliminated from the body through the central compartment (metabolism in liver and or excretion via kidney).
Relating the iPK/PD model to traditional PK, PK/PD, and protein metabolism models
The mathematical derivation of iPK/PD model is shown and is related to traditional PK, PK/PD, and protein metabolism models in Supplementary Fig. 2.
Consideration of target turnover in iPK/PD
In iPK/PD model presented, the overall covalent drug PK and PD outcomes depend upon the %TE measurements of the drug-target protein complex. %TE inform the drug-target binding process, that includes target turnover rate. Target half-life and de novo synthesis of target influences the duration of PD response. A potential approach is using %TE from intact covalent-drug protein complex measurements that outputs the half-life of the complex (i.e., target), enabling a direct correlation of PK to PD, which otherwise is not possible with traditional PK74,75.
Avoiding terminal experiments through the analysis of proxy tissues (D6 and D7)
Unfortunately, the analysis of the target tissue often requires a terminal experiment (e.g., neurodegenerative diseases and brain neoplasms). In these cases, certain terminal experiments can be avoided if the target is also present in an accessible tissue (ideally blood), so long as a target-specific Kp(TE) is determined (%TEtarget tissue/ %TEblood). This allows extrapolation of exposure in the target tissue through the following equation [%TEtarget tissue = %TEblood * Kp(TE)]. Likewise, if an alternative RoA (to IV) is to be considered and the target is present in blood, the ratio of the AUCs (%TE vs. time) in blood for oral vs. IV administration provides an absolute F(TE) (F with respect only to Target Engagement, for derivation, see Supplementary Fig. 2). The oral LD50 may then be determined to assure the predicted dose is within the safety margin. If the target is not present in an accessible tissue, a relative F(TE) can only be determined as the ratio of AUCs in the target tissue for two different routes of administration, as described in the main text.
Reproducibility, sensitivity, accuracy, precision, and matrix effect results
The linearity of the intact protein LC-MS assay was established over the concentration range of 0.5 µM – 25 µM for hSOD1 monomer, and with similar concentration range of hSOD1 incubated with a fixed amount of S-XL6 (dimer concentrations), spiked into mouse whole blood. The selected concentration range covers the therapeutically relevant concentrations for SOD1 in healthy and disease-state (fALS) patients. The internal standard used in the quantitation was 13C, 15N-labeled ubiquitin eluting at 22.6 min in the LC-MS method76. Regression analysis was performed using GraphPad Prism software (10.1.2) and correlations of R2 ~ 0.93 (monomer) and R2 ~ 0.95 (dimer) were obtained. The lower limit of quantitation (LLOQ) of 0.5 µM for both monomer and dimer curves was obtained using peak area ratios. The selectivity of the LC-MS method was established with the detection of intact SOD1 monomer peak at 24.2 min and S-XL6-mediated SOD1 dimer peak at 25.5 min in total ion chromatograms, without interferring with any endogenous components (Fig. 1). The data for accuracy (% bias), precision, and matrix effect for the intact protein LC-MS assay are summarized in Supplementary Table 2 (monomer), Supplementary Table 3 (dimer), and Supplementary Table 4, respectively. The intra-run bias was between -5.97 – 1.63% for monomer and -3.25 – 19.2% for dimer, with intra-run precision values ranged from 1.9 – 8.4 % and 0.6 – 0.9% for monomer and dimer, correspondingly.
Consideration of semi-physiological PK parameters
We derived the iPK/PD model from the anachronistic (simplest) PK/PD model. In the anachronistic model the number of compartments is equal to the number of exponential terms. When distribution or elimination occurs in multiple compartments (e.g., tissues), this can be expanded for a given drug and target to more customized physiologically or semi-physiologically based PK models77,78. For example, a simplistic semi-physiological PK model would include blood compartment, assumes similar perfusion across all compartments to reduce model complexity, and produces a quick snapshot of the covalent drug’s fate in vivo. A more complex semi-physiological PK model would include different rate constants for different tissues. Jones H. M. et al., reviews the use of multi-tissue compartments as either perfusion (rate of blood flow) or permeability (drug-specific)-limiting kinetics77. When the iPK/PD requires more than two terms for a fit, it is important to recognize (see Supplementary section derivation of the iPK/PD) that the terms likely involve protein degradation constant, i.e., they are compartments in the sense of protein metabolism/catabolism; not compartments in the physiological sense (e.g., organ). The derivation and meaning of such terms of different organ compartments can be found in the work of Burlingame52. In the three term models of Burlingame’s work, one compartment is the free amino acid pool, the second is the total protein pool, and the third is protein of interest pool.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry data generated in this study have been deposited in the ProteomeXchange Consortium via the PRIDE database under accession code PXD046903. All relevant data are within the paper and its Supporting Information files. Source data are provided with this paper.
References
Singh, J. The ascension of targeted covalent inhibitors. J. Med. Chem. 65, 5886–5901 (2022).
Singh, J., Petter, R. C., Baillie, T. A. & Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 10, 307–317 (2011).
Boike, L., Henning, N. J. & Nomura, D. K. Advances in covalent drug discovery. Nat. Rev. Drug Discov. 21, 881–898 (2022).
Pichler, C. M., Krysiak, J. & Breinbauer, R. Target identification of covalently binding drugs by activity-based protein profiling (ABPP). Bioorg. Med. Chem. 24, 3291–3303 (2016).
Deng, H., Lei, Q., Wu, Y., He, Y. & Li, W. Activity-based protein profiling: recent advances in medicinal chemistry. Eur. J. Med. Chem. 191, 112151 (2020).
Chan, W. C., Sharifzadeh, S., Buhrlage, S. J. & Marto, J. A. Chemoproteomic methods for covalent drug discovery. Chem. Soc. Rev. 50, 8361–8381 (2021).
Duveau, D. Y. & Thomas, C. J. The remarkable selectivity of nirmatrelvir. ACS Pharmacol. Transl. Sci 5, 445–447 (2022).
Kessler, D. et al. Drugging an undruggable pocket on KRAS. Proc. Natl Acad. Sci. USA. 116, 15823–15829 (2019).
Ghosh, A. K., Samanta, I., Mondal, A. & Liu, W. R. Covalent inhibition in drug discovery. ChemMedChem 14, 889–906 (2019).
Thakur, A. et al. Bioanalytical strategies in drug discovery and development. Drug Metab. Rev. 53, 434–458 (2021).
Summerfield, S. G., Yates, J. W. T. & Fairman, D. A. Free drug theory - no longer just a hypothesis? Pharm. Res. 39, 213–222 (2022).
Cheng, Y. & Prusoff, W. H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 (1973).
Zhang, D. et al. Drug concentration asymmetry in tissues and plasma for small molecule-related therapeutic modalities. Drug Metab. Dispos. 47, 1122–1135 (2019).
Swinney, D. C. The role of binding kinetics in therapeutically useful drug action. Curr. Opi.n Drug Discov. Devel. 12, 31–39 (2009).
Hong, Y., Gengo, F. M., Rainka, M. M., Bates, V. E. & Mager, D. E. Population pharmacodynamic modelling of aspirin- and Ibuprofen-induced inhibition of platelet aggregation in healthy subjects. Clin. Pharmacokinet 47, 129–137 (2008).
Ferron, G. M., McKeand, W. & Mayer, P. R. Pharmacodynamic modeling of pantoprazole’s irreversible effect on gastric acid secretion in humans and rats. J. Clin. Pharmacol. 41, 149–156 (2001).
Turnidge, J. D. The pharmacodynamics of beta-lactams. Clin. Infect. Dis. 27, 10–22 (1998).
M.E. Levison, J.H. Levison, Pharmacokinetics and pharmacodynamics of antibacterial agents. Infect. Dis. Clin. North Am. 23, 791–815 (2009).
Sutanto, F., Konstantinidou, M. & Dömling, A. Covalent inhibitors: a rational approach to drug discovery. RSC Med. Chem. 11, 876–884 (2020).
Bauer, R. A. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 20, 1061–1073 (2015).
Hong, D. S. et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 383, 1207–1217 (2020).
Carillo, S., Criscuolo, A., Füssl, F., Cook, K. & Bones, J. Intact multi-attribute method (iMAM): a flexible tool for the analysis of monoclonal antibodies. Eur. J. Pharm. Biopharm. 177, 241–248 (2022).
Kellie, J. F. et al. Intact protein mass spectrometry for therapeutic protein quantitation, pharmacokinetics, and biotransformation in preclinical and clinical studies: an industry perspective. J. Am. Soc. Mass Spectrom. 32, 1886–1900 (2021).
Vasicek, L. A., Spellman, D. S. & Bateman, K. P. Intact mass quantitation of therapeutic antibodies for pharmacokinetic studies using immuno-purification. Methods Mol. Biol. 2313, 259–268 (2022).
Xu, K. et al. Characterization of intact antibody-drug conjugates from plasma/serum in vivo by affinity capture capillary liquid chromatography-mass spectrometry. Anal. Biochem. 412, 56–66 (2011).
Zhu, X., Huo, S., Xue, C., An, B. & Qu, J. Current LC-MS-based strategies for characterization and quantification of antibody-drug conjugates. J. Pharm. Anal. 10, 209–220 (2020).
Li, K. S. et al. High-throughput kinetic characterization of irreversible covalent inhibitors of KRAS(G12C) by intact protein MS and targeted MRM. Anal. Chem. 94, 1230–1239 (2022).
Miyahisa, I., Sameshima, T. & Hixon, M. S. Rapid determination of the specificity constant of irreversible inhibitors (kinact/KI) by means of an endpoint competition assay. Angew. Chem. Int. Ed. Engl. 54, 14099–14102 (2015).
Mons, E., Roet, S., Kim, R. Q., Mulder, M. P. C. & Comprehensive Guide, A. for assessing covalent inhibition in enzymatic assays illustrated with kinetic simulations. Curr. Protoc. 2, e419 (2022).
Campuzano, I. D. et al. High-throughput mass spectrometric analysis of covalent protein-inhibitor adducts for the discovery of irreversible inhibitors: a complete workflow. J. Biomol. Screen. 21, 136–144 (2016).
Ozbal, C. C. et al. High throughput screening via mass spectrometry: a case study using acetylcholinesterase. Assay Drug Dev. Technol 2, 373–381 (2004).
Maurer, M. S. et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 379, 1007–1016 (2018).
Liu, H.-N. et al. Targeting of monomer/misfolded SOD1 as a therapeutic strategy for amyotrophic lateral sclerosis. J. Neurosci. 32, 8791–8799 (2012).
Ray, S. S. & Lansbury, P. T. A possible therapeutic target for Lou Gehrig’s disease. Proc. Natl Acad. Sci. USA 101, 5701–5702 (2004).
Rakhit, R. & Chakrabartty, A. Structure, folding, and misfolding of Cu,Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochimica. et Biophysica. Acta (BBA) Mol. Basis Dis. 1762, 1025–1037 (2006).
Hough, M. A. et al. Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc. Natl Acad. Sci. USA 101, 5976–5981 (2004).
Cropley, T. C. et al. Structure relaxation approximation (SRA) for elucidation of protein structures from ion mobility measurements (II). Protein complexes. J. Phys. Chem. B 127, 5553–5565 (2023).
Broom, H. R., Rumfeldt, J. A., Vassall, K. A. & Meiering, E. M. Destabilization of the dimer interface is a common consequence of diverse ALS-associated mutations in metal free SOD1. Protein Sci. 24, 2081–2089 (2015).
Anzai, I. et al. A misfolded dimer of Cu/Zn-superoxide dismutase leading to pathological oligomerization in amyotrophic lateral sclerosis. Protein Sci. 26, 484–496 (2017).
Doucette, P. A. et al. Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant: insights into the molecular basis for dimer stability. J. Biol. Chem. 279, 54558–54566 (2004).
Anzai, I. et al. Oxidative misfolding of Cu/Zn-superoxide dismutase triggered by non-canonical intramolecular disulfide formation. Free Radic. Biol. Med. 147, 187–199 (2020).
Banci, L. et al. Interaction of cisplatin with human superoxide dismutase. J. Am. Chem. Soc. 134, 7009–7014 (2012).
Amporndanai, K. et al. Novel Selenium-based compounds with therapeutic potential for SOD1-linked amyotrophic lateral sclerosis. EBioMedicine 59, 102980 (2020).
Hossain, M. A. et al. Evaluating protein cross-linking as a therapeutic strategy to stabilize SOD1 variants in a mouse model of familial ALS. PLoS Biol. 22, e3002462 (2024).
Donnelly, D. P. et al. Cyclic thiosulfinates and cyclic disulfides selectively cross-link thiols while avoiding modification of lone thiols. J. Am. Chem. Soc. 140, 7377–7380 (2018).
Donnelly, D. P. et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 16, 587–594 (2019).
Miller, T. M. et al. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N. Engl. J. Med. 387, 1099–1110 (2022).
Steinmetz, K. L. & Spack, E. G. The basics of preclinical drug development for neurodegenerative disease indications. BMC Neurol. 9, S2 (2009).
Greenblatt, D. J. & Abourjaily, P. N. Pharmacokinetics and pharmacodynamics for medical students: a proposed course outline. J. Clin. Pharmacol. 56, 1180–1195 (2016).
Jusko, W. J. Moving from basic toward systems pharmacodynamic models. J. Pharm. Sci. 102, 2930–2940 (2013).
Derendorf, H. & Meibohm, B. Modeling of pharmacokinetic/pharmacodynamic (PK/PD) relationships: concepts and perspectives. Pharm. Res. 16, 176–185 (1999).
Guan, S., Price, J. C., Ghaemmaghami, S., Prusiner, S. B. & Burlingame, A. L. Compartment modeling for mammalian protein turnover studies by stable isotope metabolic labeling. Anal. Chem. 84, 4014–4021 (2012).
Hondal, R. J., Marino, S. M. & Gladyshev, V. N. Selenocysteine in thiol/disulfide-like exchange reactions. Antioxid. Redox Signal. 18, 1675–1689 (2013).
Yang, Z. Achieving a low human dose for targeted covalent drugs: pharmacokinetic and pharmacodynamic considerations on target characteristics and drug attributes. Biopharm. Drug Dispos. 42, 150–159 (2021).
Sime, F. B., Roberts, M. S. & Roberts, J. A. Optimization of dosing regimens and dosing in special populations. Clin. Microbiol. Infect. 21, 886–893 (2015).
Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019).
Chambers, A. G. et al. Mass spectrometry quantifies target engagement for a KRASG12C inhibitor in FFPE tumor tissue. Clin. Proteom. 20, 47 (2023).
Adams, L. M., DeHart, C. J. & Kelleher, N. L. Precise characterization of KRAS4B proteoforms by combining immunoprecipitation with top-down mass spectrometry. Methods Mol. Biol. 2262, 47–64 (2021).
Compton, P. D., Zamdborg, L., Thomas, P. M. & Kelleher, N. L. On the scalability and requirements of whole protein mass spectrometry. Anal. Chem. 83, 6868–6874 (2011).
Yang, F., Zhang, Y. & Liang, H. Interactive association of drugs binding to human serum albumin. Int. J. Mol. Sci. 15, 3580–3595 (2014).
Johansson, B. Plasma protein binding of disulfiram and its metabolite diethylthiocarbamic acid methyl ester. J. Pharm. Pharmacol. 42, 806–807 (1990).
Auclair, J. R., Boggio, K. J., Petsko, G. A., Ringe, D. & Agar, J. N. Strategies for stabilizing superoxide dismutase (SOD1), the protein destabilized in the most common form of familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 107, 21394–21399 (2010).
Taylor, D. M. et al. Tryptophan 32 potentiates aggregation and cytotoxicity of a copper/zinc superoxide dismutase mutant associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 282, 16329–16335 (2007).
Taylor, R. M. & Jesaitis, A. J. Immunoaffinity purification of human phagocyte flavocytochrome b and analysis of conformational dynamics. Methods Mol. Biol. 412, 429–437 (2007).
Illes-Toth, E. et al. Mass spectrometry detection and imaging of a non-covalent protein-drug complex in tissue from orally dosed rats. Angew. Chem. Int. Ed. Engl. 61, e202202075 (2022).
Habeck, T. et al. Top-down mass spectrometry of native proteoforms and their complexes: a community study. Nat. Methods 21, 2388–2396 (2024).
Hansen, R. et al. An internally controlled quantitative target occupancy assay for covalent inhibitors. Sci. Rep. 8, 14312 (2018).
M.R. Packer. et al. Raf promotes dimerization of the Ras G-domain with increased allosteric connections. Proc. Natl. Acad. Sci. USA 118, e2015648118 (2021).
McCord, J. M. & Fridovich, I. Superoxide dismutase. Enzymic. Funnct. Erythrocuprein. (hemocuprein). J. Biol. Chem. 244, 6049–6055 (1969).
Aluri, K. C. et al. Cyclic thiosulfinates as a novel class of disulfide cleavable cross-linkers for rapid hydrogel synthesis. Bioconjug. Chem. 32, 584–594 (2021).
Miller, A. et al. Spatially resolved metabolomics and isotope tracing reveal dynamic metabolic responses of dentate granule neurons with acute stimulation. Nat. Metab 5, 1820–1835 (2023).
Gurney, M. E. et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775 (1994).
Timmerman, P. et al. Recommendations from the European. Bioanalysis Forum Method Establishment Tissue Homogenates. Bioanal. 6, 1647–1656 (2014).
An, G. Concept of pharmacologic target-mediated drug disposition in large-molecule and small-molecule compounds. J. Clin. Pharmacol. 60, 149–163 (2020).
Gabrielsson, J. & Hjorth, S. Turn on, tune in, turnover! target biology impacts in vivo potency, efficacy, and clearance. Pharmacol. Rev. 75, 416–462 (2023).
Bronsema, K. J., Bischoff, R. & van de Merbel, N. C. Internal standards in the quantitative determination of protein biopharmaceuticals using liquid chromatography coupled to mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 893-894, 1–14 (2012).
Jones, H. & Rowland-Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst. Pharmacol. 2, e63 (2013).
Zhao, P., Rowland, M. & Huang, S. M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin. Pharmacol. Ther. 92, 17–20 (2012).
Auclair, J. R. et al. Artifacts to avoid while taking advantage of top-down mass spectrometry based detection of protein S-thiolation. Proteomics 14, 1152–1157 (2014).
Stefani, M. Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world. Biochim. Biophys. Acta 5–25, 2004 (1739).
Acknowledgements
We want to thank Dr. Daniel B. Wall for his guidance. In the memory of Jakal Amin, we want to express our deepest gratitude and appreciation for their early contributions to the discussion and design of the PK/PD studies in this work, their insights, and dedication were instrumental in shaping the direction of our project, and their impact is profoundly felt and remembered. This work was supported by the National Institute of Neurological Disorders and Stroke of National Institute of Health (R01NS065263 to J.N.A.), the ALS Association (18-IIA-420 to J.N.A. and R.M.), Johnston Educational Ventures (#685162 to J.N.A.).
Author information
Authors and Affiliations
Contributions
J.N.A., M.A.H., and R.R.B. designed and initiated the study. M.A.H., R.R.B., B.C.M., J.A., M.B., and J.L.S. performed all the experiments and analyzed the data. J.N.A., M.A.H., and R.R.B. wrote and edited the manuscript with input from the coauthors J.R.A., C.M., Q.W., N.Y.R.A., D.J.G., and R.M. J.N.A. led the project.
Corresponding author
Ethics declarations
Competing interests
Authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Miaomiao Liu, Jing Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hossain, M.A., Brahme, R.R., Miller, B.C. et al. Mass spectrometry methods and mathematical PK/PD model for decision tree-guided covalent drug development. Nat Commun 16, 1777 (2025). https://doi.org/10.1038/s41467-025-56985-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56985-6
