
Abstract
Surgical removal of primary tumors reverses tumor-mediated immune suppression in pre-clinical models with metastatic disease. However, how cytoreductive surgery in the metastatic setting modulates the immune responses in patients, especially in the context of immune checkpoint therapy (ICT), is not understood. We report the first prospective, pilot, non-comparative clinical trial (NCT02210117) to evaluate the feasibility, clinical benefits, and immunologic changes of combining three different ICT-containing strategies with cytoreductive surgery or biopsy for patients with metastatic clear cell renal cell carcinoma. Primary safety endpoint of this trial has been met, with 43 patients completing cytoreductive surgery, 36 patients undergoing post-ICT biopsy, and 25 patients without either procedure due to progressive disease or toxicities or withdrawal of consent (total N = 104). Patients receiving ICT with cytoreductive surgery or biopsy, did not experience additional ICT- or procedure-related toxicities. The median overall survival was 54.7 months for patients who received ICT plus cytoreductive surgery. Immune-monitoring studies demonstrated that cytoreductive surgery increased antigen-presenting dendritic cell population and decreased KDM6B-expressing immune-suppressive myeloid cells in the peripheral blood. This study highlighted the feasibility of combining ICT with cytoreductive surgery in a metastatic setting and demonstrated the potential enhancement of immune responses following ICT plus cytoreductive surgery.
Similar content being viewed by others
Introduction
Patients diagnosed with stage IV solid tumors, characterized by multiple metastatic lesions throughout the body, have traditionally been considered ineligible candidates for surgical interventions targeting the primary tumor or any metastatic sites1,2. Nonetheless, in pre-clinical murine models bearing metastatic disease, removing the primary tumor resulted in the reversal of tumor-mediated immune suppression3, highlighting the possibility of improved response to immune-based therapy in the remaining metastatic disease. Additionally, a retrospective study of patients with metastatic melanoma treated with immune checkpoint therapy (ICT) demonstrated that patients who subsequently underwent complete surgical resection of metastases had improved survival compared to those with incomplete resection, suggesting that surgery may provide a survival benefit in the setting of ICT4. However, the concept of debulking surgery to remove a single lesion (the primary tumor-bearing organ or a metastatic lesion) to enhance anti-tumor response with continued post-surgery ICT to treat other metastatic sites of disease in the same patient has not previously been investigated in a prospective study.
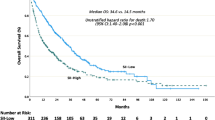
To investigate the safety as well as potential clinical and biological outcomes of ICT plus cytoreductive surgery in the metastatic disease setting, we designed an open-label, pilot, non-comparative clinical trial (NCT02210117) with three different ICT-containing regimens plus cytoreductive surgery, or biopsy if a patient is not eligible for surgery, in patients with mccRCC (N = 104). Each patient on this trial was given 6 weeks of systemic therapy consisting of nivolumab, nivolumab plus bevacizumab, or nivolumab plus ipilimumab, before surgery or biopsy, followed by nivolumab maintenance therapy for up to 2 years until disease progression, toxicity, or withdrawal from the protocol. The primary endpoint of the study was safety for all patients, and the secondary endpoints included best overall response, progression-free survival (PFS), overall survival (OS), and correlative immunological responses. We noted that a combination of ICT plus cytoreductive surgery or biopsy in the metastatic setting is safe. The median OS was 54.7 months for patients who received ICT plus surgery (n = 43). Additionally, an ad hoc non-comparative analysis showed a median OS of 23.5 months for patients who received ICT without surgery (n = 61).
Baseline and post-treatment tissue samples and peripheral blood samples were analyzed to assess immune-genomic markers correlating with clinical benefit. Tumor interferon-gamma (IFN-γ) gene signature and tertiary lymphoid structure (TLS) gene signature correlated with improved clinical response. However, we observed a cohort of patients with high IFN-γ and TLS gene signatures who did not respond to ICT. Analyzes of spatial conformation of tumor immune cell subsets in patients with high IFN-γ and high TLS gene signatures including those without clinical response to ICT delineated distinct cellular distribution and enrichment of certain cellular neighborhoods that accurately correlated with clinical response to ICT. Importantly, longitudinal analyzes of matched peripheral blood samples at a single-cell level by sc-RNA-seq demonstrated increased conventional dendritic cell population with a concurrent reduction in KDM6B expressing immune-suppressive myeloid cells in patients who underwent surgical removal of the tumor compared to patients who did not.
Overall, we report herein the safety and immune correlative data from this prospective pilot trial. Our data demonstrated the feasibility of combining ICT with cytoreductive surgery in mccRCC, highlighted the importance of the spatial distribution of immune cell subsets in determining response to ICT, and demonstrated the effect of cytoreductive surgery in modulation of the immune responses in the metastatic disease setting.
Results
Combination of ICT plus surgery or biopsy is safe and feasible in the metastatic setting
Between July 2015 and March 2018, 105 patients were randomly assigned to receive nivolumab (n = 30), nivolumab+ bevacizumab (n = 45), or nivolumab + ipilimumab (n = 30) for a total of 6 weeks, then underwent cytoreductive surgery or tumor biopsy, followed by maintenance nivolumab therapy for up to 2 years. Baseline and post-treatment tissue samples and peripheral blood samples were collected for immunologic and genomic analyzes (Fig. 1a, Supplementary Fig. 1a, b). One patient was not treated in the nivolumab arm due to inadvertent randomization prior to completing screening and ultimately was not eligible for enrollment in the trial (and thus N = 104 were used for clinical outcome analysis). After enrollment into the trial, patients were concurrently evaluated by medical oncologists and urologists specialized in RCC, at baseline and after completing 6 weeks of systemic therapy, to assess the suitability of debulking surgery or biopsy. Based upon baseline evaluation and surgical eligibility after 6 weeks of ICT treatment, 43 patients on this trial proceeded with cytoreductive surgery, while 36 patients who had medical comorbidities preventing surgery or did not have a lesion amenable for surgical resection underwent post-ICT biopsy as specified in the clinical trial protocol, and 25 patients who discontinued study participation due to progressive disease (n = 17) or toxicities (n = 3) or withdrawal of consent (n = 5) did not receive either procedure (Supplementary Fig. 1a, b). All (N = 104) patients were evaluated for safety and clinical responses with a median follow-up of 76.1 months. Patient characteristics including age, gender, performance status, and international metastatic RCC database consortium (IMDC) prognostic risk group were described in Supplementary Table 1. Overall, the ICT-related toxicity profile for this study (Table 1 and Supplementary Table 2) was expected and comparable to previously published data on ICT monotherapy or combination therapy in mccRCC5,6. For the patients who underwent cytoreductive surgery (n = 43), treatment with ICT did not result in any delays in surgery or wound complications. Specifically, the 90-day surgical complications among patients who received combination treatment with ICT plus surgery in this trial was 14% (6/43) as compared to historical data for cytoreductive surgical complications which ranged between 12-57%7 (Table 2 and Supplementary Table 3).

a Schema for clinical trial NCT02210117. Patients with mccRCC underwent baseline tumor biopsy and blood sample collection before being randomly assigned to receive nivolumab (n = 30), nivolumab + bevacizumab (n = 45), or nivolumab + ipilimumab (n = 30) for a total of 6 weeks. Four to six weeks after the ICT treatment, based upon evaluation by medical oncologists and urologists specialized on RCC, patients underwent either cytoreductive surgery or tumor biopsy. Two to four weeks after cytoreductive surgery or biopsy, nivolumab was given as maintenance therapy to each patient for up to 2 years or until disease progression or intolerable toxicities or withdrawal from the protocol. Tissue and blood samples were collected at pre-ICT treatment and at the time of surgery or biopsy (4–6 weeks after the initial 6 weeks of ICT treatment) for correlative studies. b Overall survival (OS) in Arm A (nivolumab). c OS in Arm B (nivolumab + bevacizumab). d OS in Arm C (nivolumab + ipilimumab).
Clinical responses per RECISTv1.1 criteria were assessed at 12 weeks after treatment initiation as best overall response. We assessed BOR in patients who still had metastatic lesions that could be followed by imaging studies. The BOR at 12 weeks (excluding the surgery effect) was 34% for all patients, 45% in the nivolumab arm, 36% in the nivolumab + bevacizumab arm, and 30% in nivolumab + ipilumimab arm (Supplementary Table 4). Additionally, we performed ad hoc non-comparative analyzes examining outcomes for patients separately for those who received surgery and those who did not. Patients with ICT plus surgery had BOR of 79% in all ICT arms combined (Supplementary Table 4). For individual ICT arms, the BOR in the group of patients who were treated with ICT plus surgery was 86% in the nivolumab arm, 81% in the nivolumab + bevacizumab arm, and 69% in the nivolumab + ipilimumab arm (Supplementary Table 4). These unusually high response rates are partly because cytoreductive surgery removed target lesions in some patients. After adjusting for the effect of surgical resection of targeted lesions, the BOR was 57% in the nivolumab arm, 56% in the nivolumab + bevacizumab arm, 38% in the nivolumab + ipilimumab arm, and 51% in all arms combined in patients who received ICT plus surgery (Supplementary Table 4). For patients who received ICT without surgery (n = 61), the BOR was 33% in the nivolumab arm, 24% in the nivolumab + bevacizumab arm, 24% in the nivolumab + ipilimumab arm, and 26% in all arms combined (Supplementary Table 4). Since this study is a non-comparative clinical trial with primary endpoint of safety, no statistical comparisons were made between different treatment arms of ICT or between the surgery group and non-surgery group.
With a median follow-up time of 76.1 months, the median OS was 46.6 months (95% confidence interval [CI] 24.0, 69.2) in the nivolumab arm, 35.5 months (95% CI 20.1, 46.3) in the nivolumab + bevacizumab arm, and 30 months (95% CI 18.3, not reached [NR]) in the nivolumab + ipilimumab arm (Fig. 1b–d). The median PFS was 12.4 months (95% CI 5.5, 16.8) in the nivolumab arm, 7.6 months (95% CI 4.8, 9.1) in the nivolumab + bevacizumab arm, and 8.6 months (95% CI 2.1, 16.8) in the nivolumab + ipilimumab arm (Supplementary Fig. 2a–c). Continuing the ad hoc non-comparative analyzes of patients treated with ICT plus surgery, the median OS was 54.7 months (95% CI 40.2, NR) (Supplementary Fig. 3a); and the median PFS was 12.4 months (95% CI 8.0, 18.3) (Supplementary Fig. 3b). For all patients who received ICT without surgery (n = 61), the median OS was 23.5 months (95% CI 14.3, 35.5) (Supplementary Fig. 3c) and the median PFS was 4.7 months (95% CI 2.0, 7.8) (Supplementary Fig. 3d). Representative images of the clinical response to ICT plus surgery are shown in Supplementary Fig. 3e.
Together, these data demonstrated the safety and feasibility of combining ICT with cytoreductive surgery or biopsy in mccRCC. Interestingly, we noted that patients who could undergo surgery appeared to have durable clinical outcomes, although this is based upon an ad hoc evaluation instead of pre-conceived analysis of randomized data and thus may be, at least in part, a reflection of selection of “fit” patients for surgery.
Patients with PBRM-1 and SETD-2 mutations demonstrate improved clinical response
An important secondary objective for this pilot trial was to perform immune monitoring analyzes. to assess genomic and immunologic changes that correlate with clinical response, which was defined as partial response (PR), stable disease (SD) or progressive disease (PD). Among a total of 104 patients on this trial, we were able to obtain tissue samples from 94 patients for immune monitoring studies. The treatment allocation and the number of patients who had tissue available for analysis are shown in the CONSORT diagram in Supplementary Fig. 4a.
Whole exome sequencing (WES) of available tumor samples (n = 58) with paired peripheral blood mononuclear cells (PBMC), was performed to identify tumor-specific mutations correlating with clinical benefit. WES demonstrated VHL, PBRM1, and SETD2 to be the three most commonly mutated genes (Fig. 2a and Supplementary Fig. 4b). Most of the PBRM1 mutations were frameshift deletions and nonsense mutations, with few missense mutations whereas the majority of SETD2 mutations were nonsense mutations (Fig. 2a). Importantly, mutations in either PBRM1 or SETD2 were enriched among patients with PR or SD (Fig. 2b). Analysis of copy number alterations (CNA) of top 20 most commonly mutated genes in ccRCC did not show any correlation of CNA with clinical response (Supplementary Fig. 4c). Next, we calculated tumor mutational burden (TMB) based on counts of somatic mutations per megabase (Mb) of the captured region. We did not note any correlation of TMB with clinical response (Supplementary Fig.4d). Additionally, we did not detect any correlation of predicted neoantigen load with clinical benefit (Supplementary Fig. 4e).

a Oncoplot showing the somatic mutation landscape of the top 5 mutated genes [from TCGA. Kidney Renal Clear Cell Carcinoma (KIRC)]. A total of 58 tumor tissue samples were. analyzed and each column represents a patient. The color bar at the bottom shows response for. each patient (PD=progressive disease, SD=stable disease, PR=partial response). The genes are listed on the left and their respective frequencies are listed on the right of the heatmap. The colored rectangles indicate different types of somatic mutations and the key identifying each mutation type is shown at the bottom of the heatmap. The bar plot on the top shows the somatic mutation count for each patient. The bar plot on the right side shows the counts of mutations for each gene and the colors in the bar plots correspond to the colors showing mutation types in the body of the heatmap. b Stacked bar plot showing a positive association of genomic signature (mutations in PBRM1 or SETD2 genes) with clinical responses. Patients (n = 58) were stratified into Mut (patients with mutations in SETD2 or PBRM1, n = 25) and WT (patients with wild-type SETD2 and PBRM1. genes, n = 33) groups (p = 0.03). c–d Box plots showing association of IFN-γ signature (c) and. TLS signature (d) with clinical responses (n = 83). Box plots represent the median, interquartile. range and the whiskers represent 1.5 x the upper and lower interquartile range values. Welch’s. ANOVA test across the 3 groups for IFN-γ signature (p = 0.025) and TLS signature (p = 0.039). Stacked bar plot showing a positive association of IFN-γ signature (e) and TLS signature (f). with response. Patients (83) were stratified into IFNGhigh (n = 41) and IFNGlow (n = 42) groups. (p = 0.02) (e) or into TLShigh (n = 41) and TLSlow (n = 42) (f). Statistical significance was calculated using two-sided Welch’s ANOVA, Welch’s t-test and Fisher’s exact test for comparing z scores in 3 or more unpaired groups, comparing z scores in 2 unpaired groups and comparing group counts, respectively. p < 0.05 was considered statistically significant. The following color scheme is used in all figures showing biological response groups; PD=blue, SD=yellow, and PR=red.
IFN-γ gene signature is often correlated with clinical response in various tumor types, thus multiple studies are currently investigating the use of IFN-γ gene signature as a predictive biomarker8,9,10. Therefore, we performed NanoString gene expression analysis to evaluate IFN-γ gene signature in available pre-treatment (n = 83) samples. Pre-treatment samples were pooled from all patients since the tumor tissues were not exposed to ICTs. Analysis of pre-treatment samples demonstrated a correlation of IFN-γ gene signature with clinical responses in patients receiving ICT with nivolumab as a common backbone across the three treatment groups (Fig. 2c). Similarly, patients with PR had higher TLS-gene expression scores as compared to patients with PD (Fig. 2d). Next, sequential analyzes of gene expression in matched pre- and post-treatment samples (n = 60) also showed higher IFN-γ gene signature and TLS-gene signature in the post-treatment samples (Supplementary Fig. 5a–b) and differential gene expression (DEG) analysis showed upregulation of genes including CD8A, Granzyme K (GZMK), CXCL13, CCL19, CCR7, and PDCD1 (Supplementary Fig. 5c) suggest a pro-inflammatory change in the tumor immune microenvironment following ICT-based therapy. Although IFN-γ and TLS gene signatures correlated with improved response, we observed a cohort of patients with high IFN- γ and TLS gene signatures who did not respond to ICT (Fig. 2e, f).
Spatial distribution of immune cell subsets is a critical determinant of response to ICT
We hypothesized that the lack of correlation between IFN-γ and TLS gene signatures and clinical responses in a subset of patients may be due to the fact that gene expression data do not provide information on cellular spatial distribution; therefore, we performed co-detection by indexing (CODEX) analyzes to evaluate cellular phenotype and distribution pattern in patients who had high IFN-γ and TLS gene signature associated with partial clinical response (PR, n = 4) as compared to patients who had high IFN-γ and TLS gene signature associated with progressive disease (PD, n = 4). CODEX, a multiplexed cytometric imaging approach11,12 allows spatial analyzes of single cells and their distribution within the cellular neighborhood. We assessed the cell subsets assigned by Leiden-based clustering on a staining panel consisting of 25 markers and the annotated clusters were further validated by manual inspection of multiplexed immunostains on images. This led to the identification of 15 unique cell clusters comprising of CD8 and CD4 T cells, neutrophils, tumor/epithelial cells, blood vessels, DC/APCs, macrophages, and other immune clusters. (Fig. 3a–c). We noted a distinct pattern of cell subsets in patients with high IFN-γ and TLS gene signatures who responded to ICT compared to patients who had high IFN-γ and TLS gene signatures but did not respond to ICT. While the percentage for B cells, CD8 T cells, and dendritic cells (DCs) was higher in PR cases, more tumor/epithelial cells were noted in the PD cases (Fig. 3d). To understand the spatial organization of the cell clusters, we assessed cell neighborhoods based on the 15 annotated cell clusters and identified 6 distinct cellular neighborhoods (CNs) (Fig. 3e). Enrichment of CD8 T cell neighborhood (CN1), B cell neighborhood (CN3) and DCs/APC cell neighborhood (CN5) were identified in PR while tumor cell neighborhood (CN0) was more dominant in PD (Fig. 3f). To investigate cell-cell interactions, we conducted a preliminary analysis using a single pair of PR and PD cases. This analysis identified and validated 13 clusters (Supplementary Fig. 6a). Among these cell types, we observed stronger intercellular interactions between CD4, CD8 T cells, B cells, and DCs in the PR case compared to the PD case within the 3–100-micron range13 (Supplementary Fig. 6b). Next, we expanded our initial cell-cell interaction analysis to include 25 regions of interest across all 8 patient samples (PD = 4, PR = 4), and a spatial cellular graph was constructed for each cell to its 10 closest neighbors. The volcano plots showed interactions between tumor cells and CD4 T cells as well as DCs with B cells in PR while interactions between CD8 and Tregs as well as B cells and macrophages were seen in PD (Supplementary Fig. 6c). Additionally, we computed the average minimal cell distance between different pairs of cellular subsets in PR and PD cases. Our analysis showed that B cells were closely aggregated with CD8, CD4, and DCs in PR compared to PD cases (Fig. 3g, Supplementary Fig. 7). Together, this data demonstrated that although IFN-γ response is required to mount an immune response, the spatial organization and distribution of immune cell subsets finally dictate the outcome to ICT.

a Multiplex IF of immune cell aggregate for a representative partial response (PR) and progressive disease (PD) case showing staining for CD4, CD8, CD20, CD68, Ki-67, Pan CK and CD31 (PR, n = 4; PD, n = 4) (b) Heatmap of the average expression of 25 markers in the different cell clusters is shown. (c) The UMAP plots for PR and PD cases shows a total of 15 cell clusters that were validated by manual inspection of multiplexed immunostains from a 25 markers panel staining using CODEX. Color- code correspond to those for each cluster in the fig. (b). d Pie graph shows percentages of most abundant cell clusters in PR and PD cases. (e, f) Neighborhood (CN) are defined based on the presence of the 15 validated clusters. A total of six CN are identified. The stacked bar graph shows distribution of each cell neighborhood between PR (n = 4) and PD (n = 4) cases. The statistical test used is two-sided pairwise t-tests on the transformed proportions, comparing PR and PD cohorts. The resulting p-values were corrected for multiple testing by the Bonferroni method. g The bar plots show average cell distance to nearest B cells from different cell subsets when comparing 25 regions of interest from a total of eight cases (PR, n = 4; PD, n = 4), each dots represent each region of interest from all PR and PD cases. Box plots represent the median, interquartile range and the whiskers represent 1.5 x the upper and lower interquartile range values. The statistical test used is two-sided Welch Two Sample t-test on the average minimum distance metric between our two patient cohorts.
Cytoreductive surgery is associated with the pro-inflammatory skewing of the peripheral immune status in patients with mccRCC
Based on our adhoc analyzes, we noted that patients with mccRCC who underwent surgery had a median OS of 54.7 months. Further, metastatic models in preclinical settings previously showed that removal of the primary tumor reverts tumor-mediated immune suppression3. Therefore, to garner insight into how cytoreductive surgery in mccRCC might modulate the immune system and enhance response to ICT, we performed sc-RNA-seq analyzes on matched peripheral blood samples from mccRCC patients (n = 38). We assessed the changes in the immune cell subsets in patients who underwent surgery (n = 20, baseline=10, post-surgery=10) and compared them to patients who underwent biopsy (n = 18, baseline=9, post-biopsy=9). Using graph-based clustering of uniform manifold approximation and projection (UMAP), we identified 4 major immune cell clusters (T cell, NK cell, myeloid cell, and B cell) based on the expression of canonical genes (Supplementary Fig. 8a, b), Cluster frequency of the major immune cell subsets showed no significant difference between post-surgery or post-biopsy group when compared to their corresponding baseline (Supplementary Fig. 8c). To identify changes in T or NK cell subsets, we further sub-clustered them into 15 clusters (Supplementary Fig. 9a, b) and myeloid cell subsets into 8 clusters (Fig. 4a, b). Although there were no significant differences in T or NK cell subsets (Supplementary Fig. 9c), we identified significant changes in the frequency of immune-stimulatory and immune-inhibitory myeloid cell subsets in the peripheral blood of patients who underwent cytoreductive surgery (Fig. 4c–e & Supplementary Fig. 10). We noted the presence of S100A12hi monocytes (C1), HLA-DRhi monocytes (C2), KDM6B+HIF1A+ monocytes (C3), non-classical monocytes (C4 and C6), conventional dendritic cells (cDCs) (C5), plasmacytoid DCs (pDCs) (C7) and neutrophils (C8) (Fig. 4a, b). Further evaluation of the myeloid cell subsets revealed that the abundance of the antigen-presenting cells including cDCs (C5) and HLA-DRhi monocytes (C2) increased significantly in post-surgery samples but not in post-biopsy samples compared to their matched baseline (Fig. 4c, d). Importantly, we noted a concurrent decrease in a KDM6B+HIF1A+ immune-suppressive myeloid cell subset following cytoreductive surgery but not in patients who underwent biopsy compared to the matched baseline samples (Fig. 4e). cDCs are known antigen-presenting cells, which are critical in mounting T cell-mediated immune response. Differential gene expression (DEG) analyzes further showed higher expression of HLA molecules in the cDC subsets (C5), confirming their antigen-presenting capacity (Fig. 4f). We recently characterized the immune-suppressive nature of KDM6B expressing myeloid cells which inhibit T cell-mediated antitumor immunity14. DEG analyzes also demonstrated higher expression of other immune-suppressive genes such as HIF1A in this subset (Fig. 4g, Supplementary Data 1). Cumulatively, this data suggests that ICT treatment followed by cytoreductive surgery is associated with a pro-inflammatory skewing of the peripheral immune signature with increasing cDC population and HLA-DRhi monocytes and a concurrent reduction in KDM6B expressing immune-suppressive myeloid cells in the peripheral blood. Thus, highlighting the potential reversion of tumor-mediated immune-suppression in patients with mccRCC following cytoreductive surgery which is possibly linked to 2-year overall survival of 84% of cases seen in this cohort of patients.

a UMAP projection of myeloid cell subclusters in peripheral blood. b Dot plot indicating the. average expression of indicated genes as well as the percentage of cells expressing the gene to define myeloid subclusters. c–e Box plots of frequency of cDCs (c), HLA-DRhi monocytes (d), and KDM6B+HIF1A+ monocytes (e), from patients who underwent surgery (n = 10) or biopsy (n = 9) comparing post-surgery or post-biopsy samples to baseline (BL). Box plots represent the median, interquartile range and the whiskers represent 1.5 x the upper and lower interquartile range values. P values indicated were analyzed using two-sided Wilcoxon signed-rank test. f, g Identity of differentially expressed genes (DEGs) in cDCs (f) and KDM6B+HIF1A+ monocytes (g) from PBMCs. P values indicated were analyzed using two-sided Wilcoxon rank-sum test and Bonferroni adjustment was performed for multiple comparisons.
Discussion
Surgery for patients with mccRCC has been explored as a treatment approach through surgical removal of the primary tumor-bearing kidney (nephrectomy) and/or metastasis (metastasectomy), which has been referred to as cytoreductive or “debulking” surgery. The combination of systemic therapy, such as cytokine therapy or targeted therapy, with cytoreductive surgery in mccRCC patients remains controversial due to the limited and/or conflicting evidence demonstrating a therapeutic benefit. ICT15,16,17,18,19 enhances anti-tumor T cell responses and provides durable clinical benefits in patients with mccRCC5,6. However, the value of combining ICT with debulking surgery in mccRCC is not known and currently, there is no data to prospectively evaluate the feasibility and benefits of ICT combined with surgery for patients with metastatic disease. Therefore, we designed the pilot clinical trial to test the safety and feasibility, as well as the clinical and biological outcomes of ICT with cytoreductive surgery or biopsy for patients with mccRCC.
The treatment landscape of mccRCC has changed dramatically since the initiation of this trial. While ICT agents such as nivolumab and ipilimumab were not FDA approved before initiation of this trial, bevacizumab was used in clinical practice. This trial enrolled the first patient in July 2015 before nivolumab and ipilimumab were approved by the FDA for metastatic RCC. Therefore, this trial offered patients with mccRCC the promising clinical benefits of nivolumab, bevacizumab, and ipilimumab. Although nivolumab plus ipilimumab is now FDA approved as front line and nivolumab monotherapy is approved for subsequent lines of therapy, none of this has been tested in a pre-surgical setting in combination with cytoreductive surgery in mccRCC. Therefore, this trial allowed to test the safety and feasibility of combining cytoreductive surgery (or biopsy) plus ICT with nivolumab as a common backbone across the three treatment groups. This non-comparative trial was designed to describe rather than compare clinical efficacy between different treatment arms of ICT. However, it is worth noting that in our trial the nivolumab arm has higher RR and PFS as compared to the nivolumab plus bevacizumab arm and the nivolumab plus ipilimumab arm likely due to due to the nivolumab arm has 1) younger patients; 2) more untreated patients; 3) fewer metastatic sites; and 4) less bone metastasis and more lung metastasis (Supplementary Table 1). Further, it is important to highlight that clinical response was assessed by 12 weeks in our trial, whereas other studies e.g. IMMotion 15120 and JAVELIN Renal 10121 assessed clinical response at maximal response time, which could explain the difference in the CR rate between our trial and other studies.
Our data demonstrated that ICT can be safely combined with cytoreductive surgery for the treatment of patients with mccRCC who have multiple metastatic lesions. In addition, this trial was not statistically designed to compare clinical outcomes for patients who received ICT plus surgery versus patients who received ICT without surgery. The ad hoc non-comparative analyzes examining outcomes for patients separately for those who received surgery and those who did not receive surgery. We noted that a median OS of 54.7 months for patients who received ICT plus surgery. For those patients who received ICT only without surgery, the median OS was 23.5 months. This is likely due to the fact that the ICT plus surgery group enriched clinically “fit” patients who were candidates for surgery. Similarly, we noted a generally noted a high BOR rates (38-57%, Supplementary Table 4) for patients who received ICT plus surgery. For those patients who received ICT only without surgery, the BOR rates were 24-33% (Supplementary Table 4). This likely due to the fact the ICT without surgery group had a higher proportion of patients with >3 metastatic sites and with previous localized and systemic therapies (Supplementary Table 5).
Prior to the ICT era, cytoreductive surgery has been combined with either cytokine therapy5,6,8 or targeted therapy7,22 in clinical trials for patients with metastatic ccRCC, the reported median OS in these studies was generally less than 17 months (Supplementary Table 6). The prolonged survival in patients treated with ICT plus surgery in our study could be due to multiple factors including ICT as a superior therapy compared to cytokine therapy and targeted therapy, improved synergy between ICT and cytoreductive surgery, and selection bias for patients who were “fit” for surgery. Therefore, future prospective, randomized, controlled trials will need to test the hypothesis of whether cytoreductive surgery adds efficacy with ICT.
Over the past few years, much effort has been undertaken to identify biomarkers to predict clinical response to ICT in mccRCC22,23,24,25,26,27,28. In our study, we identified both tumor intrinsic (PBRM1/SETD2 mutations) and extrinsic (IFN-γ and TLS gene signature) components correlating with response to ICT in mccRCC. Based on our TCGA analysis, no correlation of OS with TLS and IFN-G gene signatures was found in patients with mccRCC (data not shown), suggesting these biomarkers are more likely to be predictive than prognostic. However, further analyzes might be required to distinguish the predictive vs prognostic value of these biomarkers with matched samples. Importantly, we demonstrated that although IFN-γ response is required to drive an immune response, the spatial distribution of immune cell subsets finally regulate the outcome to ICT. Thus, this study provided insight demonstrating the spatial organization of immune cell subsets within the tumor microenvironment as a critical factor dictating response to ICT. Factors regulating differential spatial conformation of cellular neighborhoods within the tumor immune microenvironment will require further investigations.
The clinical data, coupled with increased peripheral antigen-presenting cells and reduction in KDM6B expressing immune-suppressive myeloid cell subsets in patients who underwent surgical removal of the tumor provided the potential mechanistic insight into immune modulation following cytoreductive surgery in a metastatic setting. This data led to the hypothesis that antigen release during cytoreductive surgery could potentially enhance anti-tumor immunity in patients, which will need to be further interrogated. Overall, our clinical and translational data may serve as a foundation to guide larger randomized, comparative clinical trials for further investigation of ICT plus cytoreductive surgery for patients with mccRCC and other tumor types as a combinatorial strategy to enhance response to ICT.
Methods
Study population and trial design
This study complies with all relevant ethical regulations and was approved by the Institutional Review Board (IRB) at the University of Texas MD Anderson Cancer Center. This trial was registered on 08-04-2024 at: https://clinicaltrials.gov/study/NCT02210117 with a trial identification number of NCT02210117. The trial protocol is included in the Supplemental Data. The study design and conduct complied with all relevant regulations regarding the use of human study participants and was conducted in accordance with the criteria set by the Declaration of Helsinki. This trial was designed prior to SAGER guidelines and did not include sex, gender, or age in the trial design. The patients in this study included adults with histologically confirmed metastatic clear cell RCC with measurable disease who were eligible for cytoreductive nephrectomy, metastasectomy or post-treatment biopsy. In addition, patients needed to have good performance status and adequate organ functions. Patients with organ allografts, serious autoimmune diseases, active human immunodeficiency virus (HIV), acquired immunodeficiency syndrome (AIDS), hepatitis B virus (HBV), hepatitis C virus (HCV), uncontrolled hypertension, or grade 2 or higher proteinuria were excluded from this study. In addition, patients who were previously treated with anti-CTLA-4, anti-PD1, or bevacizumab were excluded from this trial. Furthermore, patients on systemic immune suppression medications such as high-dose steroids (e.g., >10 mg prednisone daily or equivalent) or infliximab were also excluded from this study. This trial was a pilot, non-comparative, randomized study with a combination of three different ICT-containing strategies with cytoreductive cytoreductive surgery or biopsy for the treatment of patients with mccRCC (Fig. 1a). Written informed consent was obtained from all patients for participation in the trial. In addition, all patients provided informed consent for the IRB-approved laboratory protocol MDACC PA13-0291, and all blood and tumor samples used for correlative studies were collected under this protocol. Patients on this trial underwent baseline tumor biopsy and blood sample collection for research use.
Patients were randomized in a 2:3:2 fashion onto this trial to receive nivolumab (n = 30) [intravascular (IV) 3 mg/kg every 2 weeks x3 doses], or nivolumab + bevacizumab (n = 45) (IV 10 mg/kg every 2 weeks x3 doses), or nivolumab + ipilimumab (n = 30) (IV 1 mg/kg every 3 weeks x2 doses) for a total of 6 weeks. The stratified randomization, including randomly sized blocks, was set up in the Clinical Trial Conduct website by the trial statistician. The website is housed on a secure server maintained by the MDACC Department of Biostatistics, allowing online access through usernames and passwords for personnel responsible for enrolling patients while maintaining allocation concealment. Of note, when this trial was designed, there were other studies with nivolumab, or nivolumab plus ipilimumab, but no combination of nivolumab plus bevacizumab at that time. We decided to allocate more patients in the nivolumab plus bevacizumab arm to gather more clinical and biological data on this combination (nivolumab plus bevacizumab) based upon the decision from our group and the sponsoring company (for clinical convenience and financial practicality). Patients were concurrently evaluated by medical oncologists and urologists specialized on RCC, at baseline and after completing 6 weeks of ICT therapy, to assess the suitability of debulking surgery or biopsy. For this purpose, imaging studies such as CT scans were performed at 12 weeks (after 6 weeks of ICT treatment) and read by a collaborating radiologist to assess clinical responses and assist the decision for cytoreductive surgery or biopsy for each patient. Two to four weeks post cytoreductive surgery or biopsy, nivolumab maintenance therapy was given to each patient for up to 2 years or until disease progression or intolerable toxicities or withdrawal from the protocol (Fig. 1a). The trial underwent 2 major revisions. The initial design included only patients planning nephrectomy. To increase accrual, patients with planned metastasectomy or biopsy were added, and stratification was added to the randomization. One the original trial was complete, additional patients were added to each arm to gather more precise estimates of the endpoints. The analysis plan and methods remained the same.
Of note, cytoreductive surgery was decided based upon the following criteria (in addition to consent from the patient): 1) Patients should have a resectable primary tumor; 2) ECOG performance score of 0 or 1; 3) Low surgical risk, i.e. absence of significant co-morbid illnesses; 4) Absence of uncontrolled CNS or uncontrolled spine metastasis; 5) Absence of multiple liver metastasis; 6) Absence of multiple bone metastasis; 7) Patients should be candidates for planned systemic therapy; 8) Patients should not have dominant sarcomatoid, transitional cell, or collecting duct carcinoma histology; 9) No active infection, i.e. negative culture for any previously active infection; 10) Patients should have a predicted adequate renal function after nephrectomy; 11) Patients should not have more than 2 organs involved with metastases.
Ninety-day surgical complications were defined according to the Clavien-Dindo classification system29. Complications were considered intraoperative if they occurred between the time from the induction of anesthesia to the time when the patient left the post-anesthesia recovery unit.
Surgical approach was not mandated by the trial design and was at the discretion of the primary surgeon.
The clinical outcomes of patients were reported but the trial was not designed or powered for comparison between the treatment arms. The primary endpoint of this trial was safety and the secondary endpoints include best overall response, progression free survival (PFS), and overall survival (OS), and correlative immunologic changes. Safety stopping rules were implemented separately for each arm according to the methos of Thall30. The safety stopping endpoint was grade 3 or higher adverse events that were related to protocol therapy occurring in the first 6 weeks of therapy with 2 exceptions that only counted as events if they did not improve to grade 1 or better within 2 weeks: 1) immune-related adverse events that were treatable with steroids; or 2) any bevacizumab related events that were amenable to medical management. The proportion of patients with these events was controlled at 30% assuming an uninformative prior of beta(0.6, 1.4) which is the equivalent of adding 2 patients with a 30% safety event rate. Clinical responses were defined per RECISTv1.1 criteria and assessed at 12 weeks as complete response (CR), partial response (PR), stable disease (SD), or progression of disease (PD). BOR was defined as response status at 12 weeks. These responses were confirmed by another restaging studies about 3 months later (except in patients with apparent rapid disease progression or death or withdrawal from the trial). A response was defined as a CR or PR, and no response was defined as SD or PD. For the clinical response rate and survival analyzes, all eligible patients (N = 104) were included in the analyzes. One patient was randomized prior to enrollment based upon “human error” and was not followed medically, so we cannot include all randomized patients (N = 105)), but this did not change our analysis plan. Of the 104 patients who received ICT, 94 patients had available tissue samples for correlation of biomarkers with clinical response (Supplementary Fig. 4a).
We defined a modified BOR excluding surgery effect to correlate potential biomarkers to true biological responses without interference from cytoreductive surgery that removed target lesion(s) from some patients (Supplementary Table 4). For patients (n = 12) who had surgery removing target lesion(s), BOR excluding surgery effect was based on assessment of the remaining target lesions with exclusion of resected target lesion(s). For those patients (n = 4) with only one metastatic lesion resected by metastasectomy, BOR was based upon the clinical response before surgery. Disease control was defined as a CR, PR or SD. Overall survival was defined as number of months from randomization to death (event) or last contact for patients who were alive at the final data collection. PFS was defined as the time from randomization until progression or death, whichever came first (event), or last follow-up for disease assessment among patients who were alive and free of disease at the last assessment. Estimates of OS and PFS (including those in ad hoc analyzes of patients who received ICT plus surgery and patients who received only ICT without surgery) were calculated and graphed by Kaplan-Meier methods. Patient treatment allocation and tissue analysis is shown in Supplementary Fig. 1.
Whole exome sequencing
Tumor tissue samples and matched peripheral blood mononuclear cells (PBMCs, controls) from 42 patients were processed for whole exome sequencing. DNA from FFPE tissues and PBMCs was extracted using the QiaAmp DNA FFPE Tissue Kit and QiaAmp DNA Mini kit, respectively (Qiagen). Library construction was performed using manufacturer’s instructions. Briefly, ~250 ng genomic DNA was sheared using the Covaris S2 sonicator. KAPA Hyper Prep Kit with Agilent SureSelect XT Target Enrichment System was used for end repair, A-base addition, adapter ligation, and library enrichment PCR. Sample concentrations were measured following library construction using the Agilent Tapestation. Hybridization reaction was then performed for exon capture using the manufacturer’s guidelines (Agilent SureSelect-XT Human All Exon v4). The libraries were normalized to equal concentrations using a QuantStudio 6 Flex instrument and pooled to equimolar amounts. Libraries were quantified using the Agilent Tapestation and sequenced using the Illumina HiSeq 2500 platform at a coverage of ~200X for tumor samples and ~100X for normal samples. The BWA aligner 40 was used for sequence alignment to the human reference genome, GRCh37 (UCSC genome browser: genome.ucsc.edu). The average exome- wide coverage ranges in 93.6-302.3-fold (median 200.7) in tumor samples and 54.1-294.3-fold (median 99.5) in the matched PBMC samples. SNV and indel calls were made with Mutect31 and Pindel32 respectively. The mutations were annotated by ANNOVAR33. Germline variants were filtered using germline DNA from paired blood samples. The resulting variants were filtered further by the following criteria to get the final variants: (a) dbSNPs that were “novel” and the ones already existing in COSMIC were included; (b) each variant had a coverage of at least 20x for tumor and 10x for normal samples; (c) SNVs with a VAF ≥ 0.05 and <0.02 for tumor and normal samples, respectively and at least 3 reads to support the call at SNV in tumor sample were included; (d) for exclusion of common variants, only variants with AF < 0.01 in Exome Aggregation Consortium (EXAC), ESP600 and 1000 Genome (1KG) were included; (e) only variants with LOD score ≥ 6.3 (Mutect default) for tumor samples were included; (f) silent mutations, 5’UTR and 3’UTR mutations were excluded. Tumor mutational burden (TMB) was calculated based on counts of somatic mutations per Mb of captured region. Neoantigen prediction was performed using NetMHCpan (http://www.cbs.dtu.dk/services/NetMHCpan/). Briefly, all possible 8 to 12-mer peptides containing the mutated amino acid were used in the prediction. A binding affinity of less than 500 nM was used as cut off of predicted neoantigen for each nonsynonymous mutation. Statistical significance was calculated using Wilcoxon rank sum test to compare TMB or neoantigen load between groups. P < 0.05 was considered statistically significant. Copy number alterations (CNAs) were identified using in-house algorithm as previously described34. In brief, the copy number log2 ratios of tumor versus matched normal were calculated across the entire capture regions and then subjected to segmentation using circular binary segmentation (CBS)35. A cutoff of log2 ratio <= -0.325 was applied to identify copy number loss and log2 ratio ≥ 0.325 was applied for copy number gain. An oncoplot plot was generated using maftools36.
NanoString analysis
Pre-treatment tumor tissue samples from 83 patients were processed for RNA isolation. FFPE tissues were subjected to de-waxing using deparaffinization solution (Qiagen, Valencia, CA) prior to RNA isolation. Total RNA was extracted using the RecoverALL™ Total Nucleic Acid Isolation kit (Ambion, Austin, TX) for FFPE tissues and RiboPure™ RNA Purification Kit (Thermo Fisher Scientific) for fresh-frozen tissues according to the manufacturer’s instructions. Extracted RNA was quantified by ND Nanodrop1000 spectrometer (Thermo Scientific, Wilmington, MA, USA). For NanoString assay, 100 ng of RNA was used to detect immune gene expression using nCounter PanCancer Immune Profiling panel along with custom CodeSet. Counts of the reporter probes were tabulated for each sample by the nCounter Digital Analyzer and raw data output was imported into nSolver (http://www.nanostring.com/products/nSolver, v4.0) for normalization. Negative controls were subtracted as a background correction. Positive controls and housekeeping genes were used for normalization with the default parameters. Batch effect was corrected using the R sva package37. A 24-gene TLS signature was derived using a candidate gene approach and genes were selected based on two criteria: (i) biological relevance and (ii) gene sets from published studies of TLS38,39,40,41,42. Z scores were computed for TLS signature, IFN-γ signature10. Briefly, for gene expression signature analysis, the z score standardized values of each member gene in the gene set was averaged into a combined z score by using the square root of the number of member genes as the denominator to stabilize the variance of the mean. The list of genes for the TLS and IFN-γ signatures can be found in Supplementary Table 7. Statistical significance was calculated using Welch’s t-test to compare Z scores between two groups and using Welch’s ANOVA to compare Z scores across three or more groups. Pairwise t-test was used to compare matched pre- treatment and post-treatment samples for signature Z scores and gene expression. Benjamini- Hochberg correction was applied for multiple tests. Patients were segregated into TLShigh and TLSlow group based on the median of TLS z score. Similarly, patients were segregated into IFNGhigh and IFNGlow group based on the median of IFN-γ z score. Statistical significance was calculated using Fisher’s exact test to compare patient counts in different groups. P < 0.05 was considered statistically significant.
Codex
Codex staining assays were carried out according to the manufactured protocol. Briefly, 4 µm FFPE tissue section of were placed on poly-L-lysine coated coverslip (22 mm×22 mm). Section were stored at 4 °C until use. Purified antibodies were obtained from the listed vendors (Supplementary Table 8). Barcode and reporters were purchased form Akoya Bioscience (Supplementary Table 8). Antibodies conjugated for CD107A, CD11C, CD20, CD21, CD31, CD44, CD45RO, CD68, CD8, E-CADHERIN, KI67 and PANCYTOKERATIN were tagged with CODEX® Barcodes at purchase (Akoya Biosciences). For CD15, CD23, CD3, CD4, CD47, EOMES, FOXP3, HLA-DR, ICOS, LAG3, MMP9, PD-1, and T-BET, 50ug of antibody (purified and free of BSA and glycerol) was conjugated in house following manufacture recommendations. Briefly, partially reduced antibodies were incubated with a unique DNA oligonucleotide (barcode), then barcode-conjugated antibody were purified using a 50-kDa centrifugal filter and collected with antibody storage solution. Purified barcoded-conjugated antibodies were stored at 4 °C and used within 6 months of conjugation. The antibody conjugation reactions were validated via protein gel electrophoresis. Codex staining were validated using human tonsil tissue. Antigen retrieval Tris–EDTA pH 9.0 was used during the staining protocol. Antibodies were incubated either O/N at 4 °C or 3 hours at room temperature, depending on the optimization protocol (Table S8).
Codex imaging
The regions of interest were determined using IHC and mIF staining and were representative of the microenvironment of the tissue sample. Stained sections were capture using a Keyence BZ-X810 inverted microscope with filter cubes 4900-UF1 Dapi, 49011-UF1 Alexa Fluor 488, 49004-UF1 Cy3 and 49006-UF1 Cy5 for the detection the corresponding fluorescent reporter. Exposure times for each antibody is shown in Supplementary Table 8. A region of interest of 2.754 × 2.065 mm (5 × 5 tiles) was capture using at 20x of magnification and Z-stack of pitch of 1.5 µm with 9 slices.
Image processing, segmentation and analysis
Alignment of images across cycles, stitching of tiles and subtraction of auto-fluorescence was performed using CODEX® Processor application. A neural network-based cell segmentation tool DeepCell10 was applied to pre-processed images on DAPI channels to identify nuclei, and these nuclear masks were dilated to obtain whole-cell segmented cells. Nuclear segmentation masks were stochastically dilated by flipping pixels with a probability equal to the fraction of already-assigned neighboring pixels, an algorithm that resembles a diffusion process: for rounds 1 to 9: for each nuclear mask “M”, for each pixel on the border of “M”, count the number of its neighbor pixels that are already assigned, compute the “p”, the fraction of neighbor pixels already assigned to cells with probability “p” add the pixel to mask “M”. The dilation was done 9 times to obtain cell masks approximating cell shapes.
CODEX data analysis
All mIF analyzes were run in R-4.0.5 unless otherwise indicated. R functions are specified using the following notation: “<package_name>::<function_name>”
Cell clustering
For cell clustering and cell neighborhood analysis, data from 8 cases (PD = 4, PR = 4) were combined using a total of twenty-five ROIs to include associated tissue heterogeneity. Possible batch effects were addressed by performing an inverse hyperbolic sine transform (“base::asinh”) on cell expression values for every marker, in every ROI. The normalized values were z-scaled across both cells and markers. To cluster cells, dimensionality reduction was first performed on scaled expression values using principal component analysis with 20 components (“stats::prcomp”). Next, a k-nearest neighbor graph was constructed to build a similarity network between cells in principal component space (“dbscan::kNN”, k = 30). Finally, cells were clustered using the Leiden graph clustering algorithm (“igraph::cluster_leiden”, cluster_resolution = 1.0). To label clusters, a heatmap showing the average normalized marker expression in each cluster was plotted. Clusters were annotated using their average expression to identify cell types, and these annotations were validated by manual inspection of multiplexed immunostains on images.
Construction of a spatial cellular graph
To perform cell to cell interaction, a spatial graph of nearest neighbor was first constructed. Cell coordinates were derived by taking the centroid of each segmented cell nucleus relative to the corner of the ROI. A k-nearest neighbor algorithm was next used on these coordinates (“dbscan::kNN”, k = 10). This graph thus represents, for each cell, its 10 closest neighbors in 2D space.
Cell distance analysis
An algorithm was computed to determine cell distance for a given cell type pair and the average minimum distance was calculated. The algorithm proceeds as follows: For each cell i of cell type A, compute distances to all cells of type B (rdist::cdist); (2) for each i, compute the shortest distance to a cell of type B (the minimum distance); and compute the average of all cells i in each ROI. Finally, to compare this metric between patient cohorts, we performed a Welch Two Sample t-test on the average minimum distance metric between our two patient cohorts.
Cell neighborhood analysis
To define cellular neighborhoods (CNs), the number of neighbors of each cell type was counted, resulting in a matrix of cells by cell clusters, with each row representing a cell, each column representing a cell annotation (cell type) from the clustering above, and each value representing the count of neighbors of the given annotation. The neighbor cell proportion was computed for each row. The resulting matrix was clustered using k-means clustering (“stats::kmeans”), where the optimal k was determined empirically by maximizing the silhouette score metric (“cluster::silhouette”). Each cluster was defined as a CN. Thus, each cell was given both a cell type annotation, which depends only on the cell’s own marker expression, and a cell neighborhood annotation, which depends on the cell’s type and the identities of its nearest neighbors. To compare CNs between patient cohorts, we determined the proportion of cells in each ROI belonging to each CN. Proportions were transformed using the inverse hyperbolic tangent (“base::asinh”) and split by cohort. We then performed pairwise t-tests (“stat::t.test”) on the transformed proportions, comparing each CN between PR and PD cohorts. The resulting p - values were corrected for multiple testing by the Bonferroni method (“stat::p.adjust”, method = “Bonferroni”).
Single-cell RNA sequencing
sc-RNA-seq was performed using the 10x Genomics Chromium Single Cell Controller. Briefly, single-cell suspensions were prepared from PBMCs. Cells were resuspended in freezing media containing 90% AB serum (derived from donors with AB blood type) and 10% dimethyl sulfoxide (DMSO) and stored in liquid nitrogen until analysis. For sc-RNA-seq analysis, cells were thawed, washed, and droplet-separated using the Chromium Single Cell 5′ v.2 Reagent Kit (10X Genomics) with the 10x Genomics microfluidic system creating cDNA library with individual barcodes for individual cells. Barcoded cDNA transcripts from patients were pooled and sequenced using the NovaSeq 6000 Sequencing System (Illumina).
Single-Cell RNA sequencing analysis
Raw sc-RNA-seq reads generated by Illumina sequencer were demultiplexed into FASTQ and aligned to GRCh38 reference genome to generate count matrices using Cell Ranger v7.1.0 analysis pipelines (10x Genomics). Potential doublets were removed with the DoubleFinder R package (v2.0.3)43. The Seurat R package (v4.0.3)44 was used to perform the analysis including filtering out low-quality cells, normalizing the data and clustering the cells. Briefly, genes presented in less 10 cells and cells with less than 500 genes or more than 5000 genes, or with more than 20% mitochondrial gene counts were excluded from downstream analysis. A global-scaling normalization method “LogNormalize” was applied to the raw expression (“Seurat::NormalizeData”) with the default scale factor (10000). The top 2000 highly variable genes were found with the “vst” method (“Seurat::FindVariableFeatures”) and their normalized expression was scaled (“Seurat::ScaleData”) with regression out library size and cell cycle effects. Principal component analysis (PCA) was performed with the highly variable genes (“Seurat::RunPCA”) and Harmony R package (v0.1.0)45 was used to integrate the data sets with the first 50 PCA components (“harmony::RunHarmony”). Then the first 30 components from harmony were used for constructing KNN (K-nearest neighbor) and SNN (shared nearest neighbor) graphs (“Seurat::FindNeighbors”). Cells were clustered with Louvain algorithm based on the SNN graph (“Seurat::FindClusters”) at resolution 0.4. UMAP projection was performed (“Seurat::RunUMAP”) with parameters (reduction = “harmony”, dims = 1:30, n.neighbors = 20, min.dist = 0.2, spread = 1). For T and NK cells subset analysis, we selected the T and NK cells and performed the similar data analysis described above with slightly different parameters (top 1000 variable genes, first 20 harmony components for building the graphs and UMAP projection, resolution 0.8 for clustering, min. dist 0.05 and spread 2 for UMAP projection) Principal component analysis (PCA) was applied to the top 2000 highly variable genes and Harmony R package (v0.1.0)45 was used to integrate the data sets with the first 50 PCA components. Then the first 30 components from harmony were used for constructing a KNN graph, clustering and UMAP projection.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All whole exome sequencing and sc-RNA sequencing that support the findings of this study have been deposited in European Genome-phenome Archive (EGA) and are accessible through the EGA accession number EGAS00001005667. CODEX data has been deposited in Zenodo (https://doi.org/10.5281/zenodo.14531275). All other relevant data related to the current study are available in the article and its Supplementary files or from the corresponding author (Padmanee Sharma) upon request which does not include confidential patient information. Source data are provided in this paper. Source data are provided with this paper.
Code availability
No custom codes and algorithms were used in this study. The whole exome sequencing, sc- RNA sequencing, and gene expression data (NanoString) analyzes presented in the manuscript were performed with open-source algorithms as described in Methods. Further details will be made available by the authors on request.
References
Badwe, R. et al. Locoregional treatment versus no treatment of the primary tumour in metastatic breast cancer: an open-label randomised controlled trial. Lancet Oncol. 16, 1380–1388 (2015).
Metcalfe, M. J., Smaldone, M. C., Lin, D. W., Aparicio, A. M. & Chapin, B. F. Role of radical prostatectomy in metastatic prostate cancer: A review. Urol. Oncol. 35, 125–134 (2017).
Danna, E. A. et al. Surgical removal of primary tumor reverses tumor-induced immunosuppression despite the presence of metastatic disease. Cancer Res 64, 2205–2211 (2004).
Bello, D. M. et al. Survival outcomes after metastasectomy in melanoma patients categorized by response to checkpoint blockade. Ann. Surg. Oncol. 27, 1180–1188 (2020).
Motzer, R. J. et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med 373, 1803–1813 (2015).
Motzer, R. J. et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N, Engl, J, Med, 378, 1277–1290 (2018).
Bhindi, B. et al. Systematic review of the role of cytoreductive nephrectomy in the targeted therapy era and beyond: an individualized approach to metastatic renal cell carcinoma. Eur. Urol. 75, 111–128 (2019).
Ayers, M. et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest 127, 2930–2940 (2017).
Cristescu, R. et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362, eaar3593 (2018).
Sharma, P. et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 18, 312–322 (2017).
Goltsev, Y. et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174, 968–981 e915 (2018).
Schurch, C. M. et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 183, 838 (2020).
Steinhart, B. et al. The spatial context of tumor-infiltrating immune cells associates with improved ovarian cancer survival. Mol. Cancer Res. 19, 1973–1979 (2021).
Goswami, S. et al. Myeloid-specific KDM6B inhibition sensitizes glioblastoma to PD1 blockade. Nat. Cancer 10, 1455–1473 (2023).
Bex, A. et al. Comparison of immediate vs deferred cytoreductive nephrectomy in patients with synchronous metastatic renal cell carcinoma receiving sunitinib: The SURTIME randomized clinical trial. JAMA Oncol. 5, 164–170 (2019).
Flanigan, R. C. et al. Cytoreductive nephrectomy in patients with metastatic renal cancer: a combined analysis. J. Urol. 171, 1071–1076 (2004).
Flanigan, R. C. et al. Nephrectomy followed by interferon alfa-2b compared with interferon alfa-2b alone for metastatic renal-cell cancer. N. Engl. J. Med 345, 1655–1659 (2001).
Mejean, A. et al. Sunitinib alone or after nephrectomy in metastatic renal-cell carcinoma. N. Engl. J. Med 379, 417–427 (2018).
Mickisch, G. H. et al. Radical nephrectomy plus interferon-alfa-based immunotherapy compared with interferon alfa alone in metastatic renal-cell carcinoma: a randomised trial. Lancet 358, 966–970 (2001).
Rini, B. I. et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet 393, 2404–2415 (2019).
Motzer, R. J. et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med 380, 1103–1115 (2019).
Au, L. et al. Determinants of anti-PD-1 response and resistance in clear cell renal cell carcinoma. Cancer Cell 39, 1497–1518 (2021).
Bi, K. et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 39, 649–661 (2021).
Krishna, C. et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 39, 662–677.e666 (2021).
McDermott, D. F. et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med 24, 749–757 (2018).
Miao, D. et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806 (2018).
Motzer, R. J. et al. Molecular subsets in renal cancer determine outcome to checkpoint and angiogenesis blockade. Cancer Cell 38, 803–817.e804 (2020).
Pan, D. et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775 (2018).
Dindo, D., Demartines, N. & Clavien, P. A. Classification of surgical complications: a new proposal with evaluation in a cohort of 6336 patients and results of a survey. Ann. Surg. 240, 205–213 (2004).
Thall, P. F., Simon, R. M. & Estey, E. H. Bayesian sequential monitoring designs for single-arm clinical trials with multiple outcomes. Stat. Med 14, 357–379 (1995).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Ye, K., Schulz, M. H., Long, Q., Apweiler, R. & Ning, Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871 (2009).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Gopalakrishnan, V. et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018).
Olshen, A. B., Venkatraman, E. S., Lucito, R. & Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572 (2004).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res 28, 1747–1756 (2018).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Becht, E. et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 17, 218 (2016).
Cabrita, R. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020).
Coppola, D. et al. Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am. J. Pathol. 179, 37–45 (2011).
Helmink, B. A. et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020).
Petitprez, F. et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560 (2020).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e324 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat. Methods 16, 1289–1296 (2019).
Acknowledgements
This research is supported in part through an alliance between BMS and the MD Anderson Cancer Center Immunotherapy Platform, the MD Anderson Physician Scientist Award (J.G., S.G), Khalifa Physician Scientist Award (J.G., S.G), Andrew Sabin Family Foundation Fellows Award (J.G., S.G), NCI/NIH 1R37CA279192-01 (S.G), NCI/NIH R01 CA254988-01A1 Award (J.G.), the David H. Koch Center for Applied Research of Genitourinary Cancers (J.G.), the Kidney Cancer Association Advanced Discovery Award (J.G.), and Cancer Research Institute- CLIP-sward (S.G). Dr. Sharma is a member of the Parker Institute for Cancer Immunotherapy, which supports their research work at MD Anderson Cancer Center. Dr. Goswami and Dr. Sharma are members of James P. Allison Institute at MD Anderson Cancer Center. We would like to thank the Immunotherapy Platform for performing immune monitoring assays, Lisa Pruitt (Data Coordinator), Jianfeng Chen, and the Advanced Technology Genomics Core at MD Anderson for performing sequencing. We would also like to acknowledge Dr. Christopher Wood, a legendary surgeon, researcher and mentor, who played a key role in the development of this manuscript but unfortunately passed away before its submission.
Author information
Authors and Affiliations
Contributions
P.S. conceptualized clinical trial and design and supervision of immune monitoring studies. P.S. served as Principal Investigator of the clinical trial, J.G. and C.W. served as Co-PIs and supervised the clinical trial. R.S.T. and Y.S. performed statistical analysis. P.S., J.G., S.G., S.B., and S.J. contributed to the writing, reviewing, and/or revision the manuscript. D.S., J.A.K., K.A., M.T.C., A.E.T, A.T.M., A.E., C.S., M.D.M., Y.C., W.L., Z.H., S.S.Y., Y.W., P.R., L.Z., J.Z., N.M.T., A.F., L.W. provided administrative, technical or material support (reporting or organizing data and constructing databases).
Corresponding author
Ethics declarations
Competing interests
Dr. Padmanee Sharma’s COI disclosures are as follows as a SAB member for these companies and is not related to any of the work in this paper: Achelois, Adaptive Biotechnologies, Affini-T Akoya Biosciences, Apricity, Asher Bio, BioAtla LLC, BioNTech, Candel Therapeutics, Catalio, C-Reveal Therapeutics, Dragonfly Therapeutics, Earli Inc, Enable Medicine, Glympse, Henlius/Hengenix, Hummingbird, ImaginAb, InterVenn Biosciences, JSL Health, LAVA Therapeutics, Lytix Biopharma, Marker Therapeutics, Matrisome, Oncolytics, Osteologic, PBM Capital, Phenomic AI, Polaris Pharma, Soley Therpeutics, Sporos, Spotlight, Time Bioventures, Trained Therapeutix Discovery, Two Bear Capital, Xilis, Inc. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ziad Bakouny and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Goswami, S., Gao, J., Basu, S. et al. Immune checkpoint inhibitors plus debulking surgery for patients with metastatic renal cell carcinoma: clinical outcomes and immunological correlates of a prospective pilot trial. Nat Commun 16, 1846 (2025). https://doi.org/10.1038/s41467-025-57009-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57009-z
