
Abstract
The type-1 ryanodine receptor (RyR1) is an intracellular calcium release channel for skeletal muscle excitation-contraction coupling. Previous structural studies showed that the RyR1 activity is modulated by the exogenous regulators including caffeine, ryanodine, PCB-95 and diamide. An additional transmembrane helix, located adjacent to S1 and S4, has been observed in some structures, although its function remains unclear. Here, we report that using a mild purification procedure, this helix is co-purified with RyR1 and is designated as S0. When RyR1 is coupled with S0, it can be activated by Ca2+ to an open state; however when decoupled from S0, it remains in primed state. S0 regulates the channel conformation by directly affecting the TM domain via the pVSD-S0-S4/S5 linker coupling, which facilitates the dilation of S6. Our results demonstrate that S0 is an essential component of RyR1 and plays a key role in the physiological regulation of RyR1 channel gating.
Similar content being viewed by others

Introduction
Ryanodine receptors (RyRs) are a class of calcium (Ca2+) release channels located in the sarcoplasmic reticulum (SR) membranes of excitable cells, which release Ca2+ from SR into the cytosol in response to stimuli. RyRs play a key role in Ca2+-induced biological processes, including neural excitation, signal transduction, and muscle contraction1,2,3,4. During excitation-contraction coupling (E-C coupling), the functions of RyRs are modulated by a variety of factors, including proteins (junctin, triadin, calsequestrin (CSQ), calmodulin, FK506-binding protein (FKBP), etc.)5,6,7,8,9, small molecules (ATP, ryanodine, caffeine, PCB-95, etc.)10,11,12, and ions (Ca2+, Mg2+, etc.)13.
Ca2+ and ATP are key endogenous regulators of RyRs. Functional studies demonstrated that Ca2+ alone is sufficient to activate RyR channels; the open probability of RyR channel follows a bell-shaped Ca2+ concentration dependence curve14,15,16,17,18. The critical Ca2+ activation concentration is 10−7 M, and the channel open probability increases with increasing Ca2+ concentration at a range of 10−7–10−4 M16,17. Within this concentration range, ATP enhances the open probability of the RyR channel. At a Ca2+ concentration of 10−5 M, ATP increases the channel open probability to almost 100%19.
The RyR structures have been studied exhaustively by cryo-electron microscopy, with structures of open and closed states captured20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39. The majority of the reported open-state structures were obtained with the assistance of exogenous pharmacological activators, such as caffeine, ryanodine, PCB-95, and diamide23,24,31. An exception is the structure reported by Nayak AR et al., which was obtained in a lipidic environment38. Contradictory to functional studies, the RyR1 channel can be activated only to a “primed” state by Ca2+ alone or even in the presence of ATP24,27,36,39, where the gate is not fully open for Ca2+ release. The discrepancy between structural and functional studies remains a mystery. The structural studies are based on purified RyR1 samples, whereas some functional studies are based on partially purified RyR1 samples or even SR vesicles, suggesting that some key factors might be absent in the purified RyR1 samples, leading to the loss of RyR1 full activation by Ca2+ and Ca2+/ATP in the structural studies.
Here, we report that by employing a mild purification process, which omits the multiple affinity chromatography elution steps, we obtained the sample of RyR1 with the S0 helix coupled. Our structural analyses reveal that the channel coupled with S0 is activated by nanomolar concentration of Ca2+, exhibiting both primed and open states, in contrast to the RyR1 channel decoupled from S0, which is only activated in the primed state. Specifically, when RyR1 is coupled with S0, the channel transitions to an open state under the conditions of 20 μM Ca2+ and 2 mM ATP, whereas it remains in the primed state when S0 is decoupled. The structures revealed that S0 spans the SR membrane adjacent to S1 and aligns parallel to it, making hydrophobic contacts with S1, S4, and the S4/S5 linker. These interactions enhance the coupling between the pVSD and S4/S5 linker. The synergistic actions of these interactions with Ca2+ or Ca2+/ATP generate a pulling force on the S6 of RyR1 within the channel pore. This force promotes the dilation of S6, thereby facilitating the opening of the RyR1 channel. Our findings underscore the critical role of S0 as a component of RyR1, highlighting its function in the channel gating mechanism under physiological conditions.
Results
S0 is a transmembrane helix of RyR1
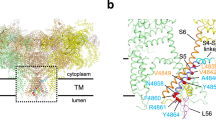
In the majority of RyR1 structures, six transmembrane helices are observed in each of the four subunits. However, the structures reported by Bai et al. (EMD-9521)23 and des Georges A, et al. (EMD-8378, EMD-8387, and EMD-8391)24 included an additional transmemebrane helix, which is modeled as RyR(4320-4345)24. This helix spans across the SR membrane, runs parallel to S1, and extends to the S4/S5 linker in the cytoplasm. Canonical purification of RyR1 includes multiple affinity chromatography elution steps40, which exclude the additional transmembrane helix (referred to as RyR1-sample 1, prepared with Protocol 1) (Fig. S1a–c and Table S1). In contrast, we prepared the RyR1-sample by sucrose gradient sedimentation41 after solubilization of the SR membrane, and the additional TM helix is evident in the structures (referred to as RyR1-sample 2, prepared with Protocol 2) (Fig. S1d–g and Table S1). To investigate the role of the additional helix, we first prepared RyR1-sample 2 with 5 mM Ca2+ and determined the structure of RyR1 at a global resolution of 3.3 Å (Fig. S2g, h). The resolution of the transmembrane helices was further improved to 3.0 Å by focused refinement using a mask that included only the transmembrane domain (Fig. S3a, b). ModelAngelo tool42,43 is utilized to predict the sequence of the transmembrane helix, resulting in four potential sequences: chain An (QLRRLSLKSAAEGLGALFFSLIAGRLGL), chain Aj (RNISSRSAAIALAALFFSLISRLL), chain BJ (KLRRINSRSAATALGAVFFSLLRRLGG) and chain AI (FRKVTLRSAATAIAALFYSLLLRLG). These sequences were then submitted to PSI-BLSAT to search against the proteome of Oryctolagus cuniculus (Rabbit), with RyR1 emerging as the top hit for chain BJ. RyR1 was also found in the research results for the other three sequences. Therefore, RyR1 4318-4344 (RLRRLTAREAATALAALLWAVVARAGA) was manually modeled based on our density map. This model of RyR1 4318-4344 also aligns well with the previously deposited map in EMDB 8391 (4.4 Å)24, despite the lower resolution. The segment 4323-4340 is modeled as a transmembrane helix and named S0 (Fig. S3c, d and Movie 1).
The structure shows that the transmembrane helix of S0 is located in the RyR1 transmembrane domain, adjacent to S1, and runs parallel to it (Fig. 1a, b and S1d–g, S4c). The interactions between S0 and RyR1 are mainly hydrophobic, where the cytoplasmic half of S0 (L4334-V4339) are surrounded by cluster of hydrophobic residues from TM helices S1(F4564, L4567, F4568, and F4571), S4(I4816, V4820), and residues V4838, M4839, V4846, and A4845 from S5(II) in the neighboring subunit (Fig. 1b, c, e and S4a, b). Furthermore, the hydrophobic interactions between S4/S5 linker (L4823, I4826) and S5(II) (L4843, M4839), along with the hydrogen bond between T4822S4/S5 linker and K4835S5(II) (Fig. 1c), also contribute to stabilizing the connection between S0 and the transmembrane helices. These observations are summarized as the coupling structure of the pVSD-S0-S4/S5 linker. On the luminal side, A4328S0 and A4332S0 interact hydrophobically with L4578S1 and F4575S1(Fig. 1d, f). R4325S0 interacts with the E4634S1-S2 loop and T4636S1-S2 loop by electrostatic interaction and hydrogen bond (Fig. 1b, d, f and S4c). R4321S0 also forms electrostatic interaction with E4908 in neighboring S6(III) (Fig. 1b, d), further enhancing the coupling between S0 and pVSD.

a The cryo-EM density map of S0/5 mM[Ca2+] shows an extra density, indicating an extra TM helix S0 with a small loop in lumen (dark green). Three RyR1 subunits (I, II, and III) are shown in the cutting section to allow a clear visualization of S0. The model of S0/5 mM[Ca2+] in S0 and luminal domain (red box in (a)) are illustrated in side-view (b), cytoplasmic view (c), and luminal view (d). e, f are enlarged regions from c (circle) and d (rectangle), respectively. I, II, III, and IV represent four subunits in the RyR1 tetramer. * indicates the mutagenesis in patients with a family history of congenital myopathy (CCD).
In previous reports, the structure of S0 has been observed and modeled in only a few cases, and its role in RyR1 activity remains unclear. To understand how S0 affects the channel gating, we performed a cryo-EM study of the RyR1-S0 sample in the presence of Ca2+ and Ca2+/ATP, to simulate the physiological conditions. Seven cryo-EM structures are obtained at six different conditions: (1) RyR1-sample 1 in EGTA (5 mM EGTA, denoted EGTA, resolution 4.06 Å (Fig. S2a, h); (2) RyR1-sample 1 in the presence of Ca2+ alone (RyR1 + 100 nM [Ca2+], denoted 100 nM[Ca2+], resolution 3.43 Å (Fig. S2b, h); (3) RyR1-sample 2 in the presence of Ca2+ alone (RyR1-S0 + 100 nM[Ca2+], two conformations were obtained and named as S0/100 nM[Ca2+] Class I (resolution 3.67 Å, (Fig. S2c, h)) and S0/100 nM[Ca2+] Class II (resolution 4.39 Å, (Fig. S2d, h)); (4) RyR1-sample 1 in the presence of Ca2+ and ATP (RyR1 + 20 μM [Ca2+] + 2 mM ATP, denoted 20 μM[Ca2+]/ATP, resolution 3.8 Å (Fig. S2e, h); (5) RyR1-sample 2 in the presence of Ca2+ and ATP (RyR1-S0 + 20 μM [Ca2+] + 2 mM ATP, denoted S0/20 μM[Ca2+]/ATP, resolution 3.94 Å (Fig. S2f, h)); (6) RyR1-sample 2 in the presence of 5 mM [Ca2+] (denoted S0/5 mM[Ca2+], resolution 3.32 Å (Fig. S2g, h). Condition 1 was used to capture the RyR1-sample 1 structure in its resting (closed) state (Fig. S1a, h, o); Condition 2 was used to obtain the structure of RyR1-sample 1 at critical Ca2+ activation concentration (Fig. S1b, i, o); Conditions 3 and 6 were conducted on RyR1-sample 2 to observe the effect of S0 on RyR1 gating by Ca2+ alone at critical Ca2+ activation (Fig. S1d, e, j, k, o) and inhibition concentrations (Fig. S1g, n, o), respectively; Condition 4 was applied to obtain the structure of RyR1-sample 1 activated by Ca2+ and ATP (Fig. S1c, l, o); and Condition 5 were conducted on RyR1-sample 2 to analysis the effect of S0 on RyR1 gating by Ca2+ and ATP (Fig. S1f, m, o). The data collection and image processing procedures are detailed in Fig. S5, and the data collection and model statistics are summarized in Table S2. In total, seven different structures were obtained (Fig. S5). According to the single-channel electrophysiological studies44,45, the channel is closed in the absence of Ca2+, termed the resting (closed) state. With the nM concentration of Ca2+, the cytoplasmic RyR is activated but the channel remains closed, named as primed (pre-open) state. With the μM Ca2+ and mM ATP, RyR is fully activated to facilitate Ca2+ release, which is termed the activated state. At mM concentration of Ca2+, the RyR1 is inactivated, and the channel is closed, termed the inhibited (closed) state. Generally, these structures can be categorized into four channel states: resting (closed), primed (pre-open), activated (open), and inhibited (closed) states (Fig. S1a–g).
S0 facilitates the RyR1 channel gating in response to Ca2+ and ATP
The mushroom-like cytoplasmic region of RyR exhibits a “breathing” motion as the channel transitions from the closed to the open state, with a noticeable expansion of the outer shell and a downward rotation of the core region. As shown in Fig. 2b, upon the Ca2+ loading at 100 nM[Ca2+] (condition 2), the shell region, comprising the NTD, Helical domain (HD1), and Handle domain, moves outward from the central axis and downward toward the SR membrane, while the central domain contracts, thereby expanding the entire cytoplasmic domain46,47. The CTD, VSD, and TM domains remain inactive, steering the structure toward the pre-open, or primed state (Fig. 2b and Movie 2). The domain symbols used in this manuscript are consistent with those in Yan Z et al., and the definitions of each domain are listed in Table S320,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39.

a Structural illustration of domain organization. b Conformational transitions of RyR1-sample 1 upon Ca2+ loading. Condition 1 (EGTA) is shown in gray; condition 2 (100 nM [Ca2+]) is colored by domains. HD1, green; NTD, yellow; Handle, cyan; central domain, purple; CTD, brown; pVSD, light blue; Pore, blue. c Conformational transitions from RyR1-sample 1 (condition 2, same color scheme as panel b) to RyR1-sample 2 in 100 nM Ca2+ (condition 3, brownish red). d Conformational transitions of RyR1-sample 1 from condition 2 (same color scheme as (b)) to condition 4 (20 μM[Ca2+]/ATP, pink). e Conformational transitions of RyR1-S0 from condition 4 (pink) to condition 5 (S0/20 μM[Ca2+]/ATP, teal). The insets show enlarged transmembrane structures and conformational change of the gate (I4937).
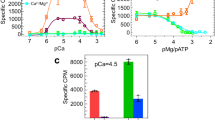
Notably, contrary to the previous reports, we observed two structures of RyR1 in the presence of 100 nM[Ca2+] (condition 3, RyR1-sample 2), which correspond to the primed and activated states. In the class I structure, the cytoplasmic domain is more extensive, yet the gate size (6.0 Å) (Fig. S1d, j) is nearly identical to that in RyR1-sample 1 structure in 100 nM[Ca2+] (6.2 Å) (Fig. S1b, i), indicating it is in the primed state. In the class II structure, there is an additional outward movement of the handle domain, central domain, and TM domain (S1, S2, S4, and S6) (Fig. 2c and Movie 2). The pore diameter expands from 6.2 to 13.8 Å, which indicates the channel is in the open state (Fig. 2c and S1e, k, o). The diagonal distance of the I4937 side chain in the gate is even larger than that observed in the RyR-Ca2+/ATP/caffeine structure reported by ref. 24 and in the RyR-Ca2+/ATP/caffeine/CHL structure reported by ref. 31, thus confirming that class II structure is activated. The class I was obtained from 60% of the particles (Figs. S5, S6). The coexistence of class I and class II structures in the S0/100 nM[Ca2+] sample indicates that S0, together with nM [Ca2+], can activate RyR1 to an open state, allowing the release of hydrated Ca2+, with an open probability of 40%.
When the Ca2+ concentration is increased to mM level for RyR1-sample 2 (condition 6, S0/5 mM[Ca2+]), the cytoplasmic domains move towards the central axis, and the gate diameter constricts to 5.0 Å (Fig. S1g, n, o). Although the additional Ca2+ binding site is not resolved in this structure, RyR1 remains in an inhibited state with a closed gate (Fig. S6). Generally, the conformational changes induced by the inhibitory concentration of Ca2+ is the reverse of those induced by the activation concentration of Ca2+. The open probability of RyR1 with S0 coupled (RyR1-sample 2) is consistent with the bell-shaped curve.
Since both Ca2+ and ATP are important modulators of RyR1, we further compared the structures of RyR1-sample 1 and RyR1-sample 2 under the condition of 20 μM Ca2+ and 2 mM ATP. In condition 4 (20 μM[Ca2+]/ATP), where S0 is decoupled, the presence of μM[Ca2+]/ATP induces significant conformational changes in the cytoplasmic domain, yet it is not sufficient for S6 dilation (Fig. 2d and Movie 3). ATP interacts with the CTD and the U motif, thereby stabilizing the calcium-activated central domain. However, the gate remains the same size (6.2 Å) (Fig. 2d and S1c, l, o) as in 100 nM[Ca2+](condition 2) (Fig. 2d and S1b, i, o) and the structure remains in the primed state. For the sample that S0 is coupled (condition 5, S0/20 μM[Ca2+]/ATP), the RyR1 adopts an open conformation characterized by a substantial outward shift of the CTD, U motif, and TM helix (Fig. 2e and Movie 3). The pore diameter expands to 15.1 Å (Fig. S1f, m, o), which is more extensive than the previously observed open states. We refer to this conformation as a fully open state. In this sample, only the fully open conformation is observed, indicating an open probability of 100% (Fig. S6).
S0 enhanced the conformational transitions of RyR1 in cooperation with Ca2+/ATP
To understand the synergistic regulations of S0, Ca2+, and ATP, we compared the conformational transitions of RyR1 from its resting state (EGTA) to its activated open state (S0/100 nM[Ca2+] Class II) (Fig. 3b–f). The global transition involves complex cooperation among various domains in the core region, such as the central domain, VSC, CTD, and TM domain. The interface among these four domains is called O ring23,28 (Fig. 3b, g and S7j). The high-affinity Ca2+ binding site48 is located at the interface between α10, α13, and α16 in the central domain and the H3-H4 loop in the CTD, which brings the central domain and CTD into closer proximity(Fig. 3b, g and S7k, m, n). The movement of the central domain causes an inward shift of α4 (Fig. 3b, c) and an outward shift of α22 and α21 (i.e., the U motif) (Fig. 3b–d). The interaction between α4 and C3’/C2 (Fig. S4e), and the interaction between α22 and S2’ of the VSC domain (Fig. S4f), enable an outward movement of VSC domain (Fig. 3b, c). The changes of S2’ and S3’ of VSC are transmitted to S2 and S3 in the TM domain(Fig. 3a), exerting a pushing force to pVSD (Fig. 3e). Given that the signal transduction occurs via a long-range allosteric gating mechanism from central domain to the TM domain24,26, we name this pathway as the ɑ4/U motif-VSC-pVSD-S4/S5 linker-gate path (path I) (Fig. 4g). On the other hand, the CTD is also clamped by the U motif through a hydrophobic chamber, directly transferring the movement to S6c of the TM domain (Fig. 3d, f). Since the four residues I4937 of S6 from RyR tetramers form the gate of Ca2+ release, the movement of CTD directly triggers channel opening. This signal transduction pathway is termed the U motif/CTD-S6c-gate path (path II) (Fig. 4g). S0, along with S1, S4, S4/S5 linker, and adjacent S5, forms a cyclic hydrophobic ring structure, (Fig. 1c–e and S4a, b), which helps to pull the S4/S5 linker away from the central axis. The interaction between S4/S5 linker (I4826, V4830, H4832, N4833) and S6 (E4942, F4940, D4938, and I4936) can facilitate the dilation of S6 (Fig. 3f and S4g)24.

a Structural illustration of domain organization: Central domain, purple; VSD, green; CTD, brown; pVSD, light blue; Pore, blue. b–f show the domain transitions from resting state (closed) (condition 1, gray) to activated state (open) (condition 3, colored). g–k show the domain transitions from the primed state (pre-open) (condition 4, pink) to the activated state (full open) (condition 5, colored).

a RyR1 in resting state without Ca2+ and the channel is closed. b In the presence of Ca2+, but S0 is decoupled to pVSD, RyR1 is in a primed state with a closed gate. c In the presence of Ca2+ and S0 is coupled to pVSD, RyR1 have two conformations (solid box), which are primed (gate is closed) and activated states (gate is open). d In the presence of Ca2+, ATP, and S0 is coupled to pVSD, RyR1 is in full open state. e In the presence of Ca2+ and ATP but S0 is decoupled to pVSD, RyR1 is in the primed state with a closed gate. f In the presence of Ca2+, ATP, and exogenous regulator (e.g., caffeine), RyR1 have two conformations (dashed box), which are primed (gate is closed) and open states (gate is open)24,36. g Schematic diagram of RyR1 channel gating mechanism. Ca2+/ATP/Caf regulates the channel through ɑ4/U motif-VSC-pVSD-S4/S5 linker-gate path (path I) and U motif/CTD-S6c path (path II), as indicated by the gray arrows. When S0 is coupled to pVSD, Ca2+/ATP regulates the channel through ɑ4/U motif-VSC-pVSD-S0-S4/S5 linker-gate path (path I) and path II. S0 enhances the coupling of pVSD with the S4/S5 linker, greatly facilitating the dilation of S6. h Color codes for the various domains and compounds. The positions of each domain within the full-length RyR1 are also marked. The dotted rectangular outline for S0 indicates that the density for S0 was not observed in the corresponding cryo-EM structures.
In previously reported open structures of RyR1, the fully activated conformations are predominantly obtained with the assistance of regulatory molecules, such as ATP/Caffeine, ryanodine, PCB-95, and diamide23,24,31,36. ATP interacts with H2 of CTD, α21 of U motif, and the cytoplasmic side of S6 (Fig. 2a and S7l, o, p). Caffeine interacts with α4, VSC, CTD, and α22, locking the central domain in a contracted state (Fig. 4f, g). The additional presence of ATP/Caffeine enhances the signal transmission along both the ɑ4/U motif-VSC-pVSD-S4/S5 linker-gate path and the U motif/CTD-S6c-gate path, leading to S6 dilation24,27,36. PCB-95 can also strengthen the contraction of the central domain23,28,29. The diamide insecticide (CHL) interacts with the pVSD of the TM domain, inducing a displacement of the S4/S5 linker, which causes movement of S5 and S6 to open the gate31. In our activated structure (S0/100 nM[Ca2+]/class II), S0 is coupled to pVSD, enhancing the interaction between pVSD and S4/S5 linker (Fig. 1c, e and S4a, b). This coupling significantly facilitates the dilation of S6, thereby promoting the opening of the RyR1 channel.
Although RyR1 can be fully activated by μM[Ca2+]/mM ATP in the membrane environment, our structure (condition 4, 20 μM[Ca2+]/ATP) shows that RyR1-sample 1, where S0 is decoupled, is in a primed state even in presence of μM[Ca2+]/mM ATP (Figs S1c, l, o). However, the structure transitions to a fully open state when S0 is coupled (condition 5, S0/20 μM[Ca2+]/ATP) (Fig. S6). The overall structures of the upper part of the O ring at pre-open and full-open state are very similar, with ɑ4, ɑ10, ɑ13, ɑ16, ɑ19 (central domain), and H3-H4 loop (CTD) showing minimal shift (Fig. 2e). The regions interacting with TM domain exhibits a large outward movement, such as the U motif (ɑ21n, ɑ21, ɑ22), CTD (H2, H3), and VSC (S2’, C2, C3) (Figs. 2e, 3g–i). As expected, the TM domains S1, S2, and the S4/S5 linker move outward due to the coupling of S0 with pVSD (Fig. 3j, k). We noticed that the structure of Ca2+ and ATP binding sites are similar to the pre-open state, with the exception of a global movement of these subregions. We speculate that S0 is the key element in the pVSD-S0-S4/S5 linker coupling structure that directly pulls S6 outward (Fig. 1c, e and S4a, b). An additional mM concentration of ATP can increase the open probability by locking the interface between CTD and S6.
Discussion
S0 is critical for the channel to open in the native membrane environment
Single-channel recording experiments have demonstrated that in membrane environment, RyR1 can be activated by Ca2+/ATP in 10−7–10−5 M Ca2+ range. The open probability increases with Ca2+ concentration16,17,19, and it reaches nearly 100% with the additional mM concentration of ATP19. However, RyR1, purified through multiple affinity chromatography elution steps, such as in our RyR1-sample 1 with 20 μM[Ca2+]/ATP and as previously reported structures with 50 μM Ca2+ and 7 mM ATP27, are in a primed state with a closed gate under buffer conditions of μM Ca2+ and mM ATP. This discrepancy between structural and functional studies raises questions about whether there might be elements missing during the chromatography purification process. In our RyR1-sample 2, where the density of S0 is consistently observed, the channel open probability correlates well with the observations from single-channel recording experiments. RyR1 can be activated by a low Ca2+ concentration (nanomolar range), although a substantial 60% of the protein population remains with a closed gate (Fig. S6). With μM[Ca2+]/ATP, all channels are in full open state, corresponding to the open probability 100%. We speculated that during the elution of multi-step affinity chromatography, the hydrophobic interactions between S0 and the pVSD might be disrupted. This disruption could lead to the displacement of S0 by the detergent, causing it to move flexibly within the SR membrane or even out of it. In either case, the density will not be observed in cryo-EM maps due to the increased mobility of S0. These results indicate that, in addition to exogenous regulators such as Caffeine, PCB-95, and CHL, endogenous element plays a critical role in facilitating pore opening in the native SR membrane.
The structural analysis revealed that S0 runs parallel to the transmembrane helix S1, making hydrophobic contacts with S1, S4, and S4/S5 linker (Fig. 1b, c, e and S4a, b). We have illustrated the differences between the S0-coupled and S0-decoupled RyR1 structures under 100 nM Ca2+ conditions in Fig. S4h, i. Figure S4h presents the primed state, where the conformations of the pVSD and the S4-S5 linker are nearly identical, with a subtle overall displacement when S0 is coupled. In contrast, Fig. S4i shows the activated state, a distinct conformational change in the pVSD and S4-S5 linker is evident with S0 coupled, leading to channel opening. In addition, R4325S0 interacts electrostatically with E4634 and T4636 from the S1-S2 loop on the luminal side (Fig. 1b, d, f and S4c). Our earlier study indicated that patients with a family history of congenital myopathy (CCD) possess mutations at positions 4634 (E4634G, case 11) and 4636 (T4636A, case 13), resulting in varying degrees of motor muscle dysfunction49. We speculate that through these interactions, S0 may enhance the coupling of pVSD and S4/S5 linker, greatly facilitating the dilation of S6. However, in the disease model, the interactions between R4325S0 and E4634/T4636 are disrupted, thereby affecting channel opening. Mutagenesis in the disease model also implies that S0 plays a critical role in regulating channel gating.
RyR1 activation is regulated by different allosteric communication pathway
The modulation of RyR1 is crucial for channel activities and calcium homeostasis in cells. While various modulators, such as proteins, small molecules, and ions, can synergistically regulate the channel kinetics in situ8,10,50. The stability of RyR1 could potentially be affected by different purification methods, as certain domains, such as the corner domain and FKBP, may not be visible in the structures. We are also interested in the possibility that the method we use to prepare the RyR1-sample could affect its stability, particularly in relation to the role of S0. In our structural analysis, HD2 is prominently visible under 100 nM Ca2+ conditions, irrespective of whether the purification process was harsh or mild, as shown in Fig. S1b,e. In the harsh purification protocol, the channel is in a primed state, with S0 absent from the structure; conversely, in the mild purification protocol, the channel is activated, and S0 is coupled in the structure. When HD2 is not visible (20 µM Ca/ATP, Figs. S1c, S2e) and S0 is decoupled from pVSD (invisible in the EM map, Fig. S1c), the RyR1 channel can only be activated to the primed state (Fig. S1c). However, when S0 is coupled to pVSD, 20µMCa/ATP can activate the RyR1 channel to a fully open state (Figs. S1f, S2f). These observations suggests that S0 is essential for channel activation under activation concentrations of Ca2+/ATP, regardless of whether the RyR1 is sufficiently stabilized or not.
On the other hand, FKBP12 is known for its ability to stabilize the RyR1 channel and has been utilized for the purification of the receptor22. It is essential to consider the role of FKBP12, when discussing the regulation of channel activity. The structures reported by des Georges et al. were prepared using FKBP affinity chromatography, and FKBP is present in all the structures24. The channel is in the primed state at 30 µM Ca2+, in which S0 is missing in the structure; the channel is activated with additional ATP and caffeine. In fact, S0 (they termed as TMx) also exists in the activated structure. Among our seven structures, the density of FKBP is blurry in most cases, except for RyR1-sample 2 in 20 µM Ca2+ and 2 mM ATP, where all RyR1 particles are in the open state. In the case where FKBP is absent, such as at 100 nM Ca2+, the channel is in the activated state only when S0 is observed in the structure. These observations again suggest that S0 is essential for channel activation, regardless of the existence or absence of FKBP.
The channel open probability is primarily determined by the cytoplasmic calcium concentration, following a bell-shaped curve15,16,18. RyR1 is in a closed state when the Ca2+ concentration is below the nanomolar level, such as in the presence of EGTA (Fig. 4a and S1a, h). Upon activation by Ca2+ at concentration between nanomolar and micro-molar range, Ca2+ binds to the high-affinity binding site in the central domain, leading to the conformational changes of the neighboring domains, such as α4, CTD and U motif. Consequently, the channel is activated to a primed state, but the gate remains closed (Fig. 4b and S1b, i). However, Ca2+ alone, even with the addition of mM ATP, is not sufficient to drive the channel to an open state (Fig. 4e and S1c, l). ATP interacts with the CTD, U motif, and cytoplasmic S6, regulating the channel open probability by directly pulling S6 outward. In our structure under condition 4, the gate has the same size as in the primed state (Fig. S1c, l). Additional caffeine can lock the α4 in the central domain to a more contracted conformation, thus to enhance the pulling force from ATP to S6 (Fig. 4f)24,27,36. ATP/Caffeine can active the RyR1 channel through ɑ4/U motif-VSC-pVSD-S4/S5 linker-gate path and U motif/CTD-S6c-gate path (Fig. 4g). Besides, 37% of the particles of RyR1 in Ca2+/ATP/Caffeine were activated, while the rest had a closed gate, corresponding to the state (condition 3, S0/100 nM[Ca2+]) we reported here (Fig. 4c and S5, S6).
In our study, RyR1 can be activated by a mechanism without the help of ATP/Caf. S0 spans across the SR membrane, adjacent to the RyR1 S1-S4 helix bundle, enhances the coupling between pVSD and S4/S5 linker and exerting a pulling force on S6 to facilitate pore open. In our structure model, Ca2+ regulating the channel opening through α4/U motif-VSC following with pVSD-S0-S4/S5 linker, which is consistent with the mechanism modulated by insecticide CHL31. The coupling of the pVSD-S0-S4/S5 linker facilitates S6 dilation, promoting 40% of the channel to an open state (Fig. 4c and S6). Additional ATP increases the open probability and further stabilizes the channel in the open state (Fig. 4d and S6).
Methods
Ethical statement
The source of tissue for RyR1 purification is a New Zealand white rabbit, purchased from the Animal Centre at Peking University. The sacrifice of the New Zealand white rabbit was carried out by venous air embolism, and no further ethics oversight was required. The Animal center follows all applicable local laws.
RyR1 purification
RyR1 was purified from CHAPS-solubilized skeletal heavy SR essentially as described previously with modifications (Table S1)26,40,41,51. Briefly, Rabbit skeletal heavy SR vesicles were suspended in buffer A (0.2 M NaCl, 20 mM Na-HEPES (pH 7.4), 2 mM DTT, 0.2 mM PMSF, 1:1 000 diluted protease inhibitor cocktail (Sigma-Aldrich, P8340)) and 1.2% CHAPS (AMRESCO)/0.6% soybean lecithin (Sigma-Aldrich, P3644); the CHAPS/protein ratio was 12:1 (wt/wt)). The sample was centrifuged for 1 h in a Beckman Type 45Ti rotor at 110 000 × g after incubation on ice for 30 min with shaking. The supernatant was loaded onto a 5-ml hydroxyapatite ceramic (Bio-Rad) column equilibrated with buffer B (200 mM NaCl, 20 mM Na-HEPES (pH 7.4), 0.5% CHAPS/0.25% soybean lecithin, 2 mM DTT, 0.2 mM PMSF, 1:1 000 diluted protease inhibitors cocktail). The column was washed with buffer B containing 10 mM K2HPO4, followed by washing with buffer B containing 50 mM K2HPO4. Proteins were then eluted with 15 ml of buffer B containing 200 mM K2HPO4. The elution samples containing RyR1 were loaded onto the top of a 5–20% (w/v) linear sucrose gradient in buffer B. After centrifugation for 16 h in a Beckman SW28 rotor at 110,000 × g, the gradient was fractionated into 1.5-ml fractions. The fractions containing RyR1 were further purified with the second HA column, using the same elution buffer as before without soybean lecithin. The eluate was collected and concentrated by centrifugation at 1000 × g in a 100-kDa cut-off Amicon centrifugal filter (Millipore) and stored in small aliquots at −80 °C. RyR1-sample 1 is prepared with the above protocol, while RyR1-sample 2 was prepared using a 5–25% (w/v) linear sucrose gradient following the solubilization of the SR membrane (Protocol 2 in Supplementary Table S1). The buffer in the sucrose gradient for Protocol 2 is prepared the same as above without soybean lecithin. The fractions containing RyR1 were collected for cryo-EM analysis.
RyR1-sample preparation for cryo-EM and data collection
The RyR1 samples prepared in two protocols are incubated with the corresponding buffers (EGTA, Ca2+, ATP) conditions 1-6 for one hour before plunge freezing. Ca2+ was buffered using ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) and free Ca2+ concentration ([Ca2+]free) was calculated by WinMaxc32 version 2.5052 (https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/downloads.htm). Aliquots of 3 μl of RyR1 at a concentration of ~25 nM is applied to glow-discharged Lacey carbon grids (400-mesh Cu TED PELLA). Grids were then blotted for 4.5 s at blot force 1, with a wait time of 1 s and no drain time, prior to vitrification by plunge freezing into liquid ethane chilled with liquid nitrogen with a Vitrobot Mark IV (Thermo Fisher) operated at 4 oC with 100% relative humidity53,54. Ashless filter paper (Whatman) was used to limit Ca2+ contamination. Grids were transferred to the FEI Titan Krios electron microscope (Thermo Fisher) that was operating at 300 kV. Images were collected automatically using software EPU and recorded in movie mode using a FEI Falcon-III detector at a nominal magnification of 75,000×, corresponding to a pixel size of 1.09 Å. A dose fractioned data collection was used to take each micrograph with 30 frames, resulting in a total dose of 50 e− per Å2. A total number of 2265/5364/6454/7665/6633/6046 movie stacks for RyR1 samples in conditions 1-6 were obtained with the defocus range 1.2–2.2 μm (Fig. S5 and Table S2).
Cryo-EM data analysis
For all of the datasets, the movie frames were further processed with MotionCorr 2 for motion correction and dose weighting55. The contrast transfer function parameters were estimated by Gctf56. Similar image processing procedures were performed on all the datasets, and the details are shown in Fig. S5. With regard to the dataset of condition 2 (100 nM[Ca2+]), 5364 micrographs were obtained after evaluation with CTF parameters. About 657,243 particles were picked automatically with the Laplacian-of-Gaussian approach and extracted in Relion 3.0/3.157 with a binned pixel size of 2.18 Å. After two rounds of 2D classification and two rounds of 3D classification, the particles were cleaned up and re-centered, re-extracted with the original pixel size of 1.09 Å. The remaining 183,859 good particles were imported into CryoSPARC v3.2.058, and subjected to a non-uniform refinement, which yield a reconstruction at 3.43 Å (C4 symmetry). The resolution estimation was reported according to the gold-standard Fourier shell correlation (FSC) using the 0.143 criterion. The local resolution map was calculated by Blocres59. For the datasets of condition 1 (EGTA), condition 4 (20 μM[Ca2+]/ATP), condition 5 (S0/20 μM[Ca2+]/ATP) and condition 6 (S0/5 mM[Ca2+]), the individual particles stacks with the number of 60,110/114,423/103,688/279,704 were reconstructed into 4.06, 3.8, 3.94, and 3.32 Å, respectively (Fig. S5 and Table S2).
For the dataset of condition 3 (S0/100 nM[Ca2+]), an ab initio reconstruction and one round of heterogeneous refinement were performed after imported into CryoSPARC and resulted in two different classes. After the non-uniform refinement, one class of 132,916 good particles yield a reconstruction of RyR1 in closed state at 3.67 Å and another class of 78,533 particles with a reconstruction of RyR1 in open state at 4.39 Å (Fig. S5 and Table S2).
Model building, refinement, analysis
To build the structural models, a recently published structure (PDB ID: 6WOT) was used as the initial model30. This structure was modified and docked into the density maps by CHIMERA60. The atomic models were then refined against maps iteratively by cycles of real space refinement in PHENIX with secondary structure and geometry restrained61. The refined models were further manually improved in Coot62. Extensive reports on evaluation were provided by the PHENIX comprehensive validation program. The analysis of pore dimensions was conducted using HOLE and illustrated in VMD63.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support this study are available from the corresponding authors upon request. The cryo-EM maps and correlated models of RyR1-sample 1 in 5 mM EGTA, 100 nM Ca2+, and 20 μM Ca2+/2 mM ATP have been deposited in the Electron Microscopy Databank (EMDB) and Protein Data Bank (PDB) with the following codes: EMD-38042/8X48 (EGTA), EMD-38043/8X49 (100 nM[Ca2+]) and EMD-38046/8X4C (20 μM[Ca2+]/ATP), respectively. The cryo-EM maps and correlated models of RyR1-sample 2 in 100 nM Ca2+, 20 μM Ca2+/2 mM ATP, and 5 mM [Ca2+] have been deposited in the Electron Microscopy Databank (EMDB) and Protein Data Bank (PDB) with the following codes: EMD-38044/8X4A (S0/100 nM[Ca2+]/Class I), EMD-38045/8X4B (S0/100 nM[Ca2+]/Class II), EMD-38047/8X4D (S0/20 μM[Ca2+]/ATP) and EMD-38048/8X4E (S0/5 mM[Ca2+]), respectively.
References
Pessah, I. N., Waterhouse, A. L. & Casida, J. E. The calcium-ryanodine receptor complex of skeletal and cardiac muscle. Biochem. Biophys. Res. Commun. 128, 449–456 (1985).
Agnew, W. S. Excitation contraction coupling - proteins that bridge the gap. Nature 334, 299–300 (1988).
Lanner, J. T., Georgiou, D. K., Joshi, A. D. & Hamilton, S. L. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2, a003996 (2010).
Takekura, H., Nishi, M., Noda, T., Takeshima, H. & Franzini-Armstrong, C. Abnormal junctions between surface membrane and sarcoplasmic reticulum in skeletal muscle with a mutation targeted to the ryanodine receptor. Proc. Natl Acad. Sci. USA 92, 3381–3385 (1995).
Tripathy, A., Xu, L., Mann, G. & Meissner, G. Calmodulin activation and inhibition of skeletal muscle Ca2+ release channel (ryanodine receptor). Biophys. J. 69, 106–119 (1995).
Jeyakumar, L. H. et al. FKBP binding characteristics of cardiac microsomes from diverse vertebrates. Biochem. Biophys. Res. Commun. 281, 979–986 (2001).
Beard, N. A. & Dulhunty, A. F. C-terminal residues of skeletal muscle calsequestrin are essential for calcium binding and for skeletal ryanodine receptor inhibition. Skelet. Muscle 5, 6 (2015).
Zhang, L., Kelley, J., Schmeisser, G., Kobayashi, Y. M. & Jones, L. R. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 272, 23389–23397 (1997).
Gyorke, I., Hester, N., Jones, L. R. & Gyorke, S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 86, 2121–2128 (2004).
Kong, H. et al. Caffeine induces Ca2+ release by reducing the threshold for luminal Ca2+ activation of the ryanodine receptor. Biochem. J. 414, 441–452 (2008).
Gong, D. S., Yan, N. E. & Ledford, H. A. Structural basis for the modulation of ryanodine receptors. Trends Biochem. Sci. 46, 489–501 (2021).
Xu, L., Mann, G. & Meissner, G. Regulation of cardiac Ca2+ release channel (ryanodine receptor) by Ca2+, H+, Mg2+, and adenine nucleotides under normal and simulated ischemic conditions. Circ. Res. 79, 1100–1109 (1996).
Meissner, G. & Henderson, J. S. Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. J. Biol. Chem. 262, 3065–3073 (1987).
Balshaw, D., Gao, L. & Meissner, G. Luminal loop of the ryanodine receptor: a pore-forming segment? Proc. Natl Acad. Sci. USA 96, 3345–3347 (1999).
Quinn, K. E., Castellani, L., Ondrias, K. & Ehrlich, B. E. Characterization of the ryanodine receptor/channel of invertebrate muscle. Am. J. Physiol. 274, R494–502, (1998).
Ahern, G. P., Junankar, P. R. & Dulhunty, A. F. Subconductance states in single-channel activity of skeletal muscle ryanodine receptors after removal of FKBP12. Biophys. J. 72, 146–162 (1997).
Chen, S. R. et al. Single-channel properties of the recombinant skeletal muscle Ca2+ release channel (ryanodine receptor). Biophys. J. 73, 1904–1912 (1997).
Bezprozvanny, I., Watras, J. & Ehrlich, B. E. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351, 751–754 (1991).
Smith, J. S. et al. Purified ryanodine receptor from rabbit skeletal muscle is the calcium-release channel of sarcoplasmic reticulum. J. Gen. Physiol. 92, 1–26 (1988).
Efremov, R. G., Leitner, A., Aebersold, R. & Raunser, S. Architecture and conformational switch mechanism of the ryanodine receptor. Nature 517, 39–43 (2015).
Yan, Z. et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 517, 50–55 (2015).
Zalk, R. et al. Structure of a mammalian ryanodine receptor. Nature 517, 44–49 (2015).
Bai, X. C., Yan, Z., Wu, J., Li, Z. & Yan, N. The Central domain of RyR1 is the transducer for long-range allosteric gating of channel opening. Cell Res. 26, 995–1006 (2016).
des Georges, A. et al. Structural basis for gating and activation of RyR1. Cell 167, 145–157.e117 (2016).
Peng, W. et al. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 354, aah5324 (2016).
Wei, R. et al. Structural insights into Ca(2+)-activated long-range allosteric channel gating of RyR1. Cell Res. 26, 977–994 (2016).
Willegems, K. & Efremov, R. G. Influence of lipid mimetics on gating of ryanodine receptor. Structure 26, 1303–1313.e1304 (2018).
Chi, X. et al. Molecular basis for allosteric regulation of the type 2 ryanodine receptor channel gating by key modulators. Proc. Natl Acad. Sci. USA 116, 25575–25582 (2019).
Gong, D. et al. Modulation of cardiac ryanodine receptor 2 by calmodulin. Nature 572, 347–351 (2019).
Iyer, K. A. et al. Structural mechanism of two gain-of-function cardiac and skeletal RyR mutations at an equivalent site by cryo-EM. Sci. Adv. 6, eabb2964 (2020).
Ma, R. et al. Structural basis for diamide modulation of ryanodine receptor. Nat. Chem. Biol. 16, 1246–1254 (2020).
Woll, K. A., Haji-Ghassemi, O. & Van Petegem, F. Pathological conformations of disease mutant ryanodine receptors revealed by cryo-EM. Nat. Commun. 12, 807 (2021).
Iyer, K. A., Hu, Y., Klose, T., Murayama, T. & Samso, M. Molecular mechanism of the severe MH/CCD mutation Y522S in skeletal ryanodine receptor (RyR1) by cryo-EM. Proc. Natl Acad. Sci. USA 119, e2122140119 (2022).
Kobayashi, T. et al. Molecular basis for gating of cardiac ryanodine receptor explains the mechanisms for gain- and loss-of function mutations. Nat. Commun. 13, 2821 (2022).
Melville, Z. et al. A drug and ATP binding site in type 1 ryanodine receptor. Structure 30, 1025–1034.e1024 (2022).
Melville, Z., Kim, K., Clarke, O. B. & Marks, A. R. High-resolution structure of the membrane-embedded skeletal muscle ryanodine receptor. Structure 30, 172–180.e173 (2022).
Miotto, M. C. et al. Structural analyses of human ryanodine receptor type 2 channels reveal the mechanisms for sudden cardiac death and treatment. Sci. Adv. 8, eabo1272 (2022).
Nayak, A. R. & Samso, M. Ca(2+) inactivation of the mammalian ryanodine receptor type 1 in a lipidic environment revealed by cryo-EM. eLife 11, e75568 (2022).
Cholak, S. et al. Allosteric modulation of ryanodine receptor RyR1 by nucleotide derivatives. Structure 31, 790–800.e794 (2023).
Inui, M., Saito, A. & Fleischer, S. Purification of the ryanodine receptor and identity with feet structures of junctional terminal cisternae of sarcoplasmic reticulum from fast skeletal muscle. J. Biol. Chem. 262, 1740–1747 (1987).
Lai, F. A., Erickson, H. P., Rousseau, E., Liu, Q. Y. & Meissner, G. Purification and reconstitution of the calcium release channel from skeletal muscle. Nature 331, 315–319 (1988).
Jamali, K. et al. Automated model building and protein identification in cryo-EM maps.Nature 628, 450–457 (2024).
Healy, M. D. et al. Structure of the endosomal Commander complex linked to Ritscher-Schinzel syndrome. Cell 186, 2219–2237.e2229 (2023).
Aldrich, R. W., Corey, D. P. & Stevens, C. F. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature 306, 436–441 (1983).
Fill, M. & Copello, J. A. Ryanodine receptor calcium release channels. Physiol. Rev. 82, 893–922 (2002).
Samso, M. A guide to the 3D structure of the ryanodine receptor type 1 by cryoEM. Protein Sci. 26, 52–68 (2017).
Van Petegem, F. How to open a ryanodine receptor. Cell Res. 26, 1073–1074 (2016).
Laver, D. R. Regulation of the RyR channel gating by Ca(2+) and Mg(2). Biophys. Rev. 10, 1087–1095 (2018).
Chang, X. et al. Correlation of phenotype-genotype and protein structure in RYR1-related myopathy. Front. Neurol. 13, 870285 (2022).
Li, L. W. et al. A new cytoplasmic interaction between junctin and ryanodine receptor Ca2+ release channels. J. Cell Sci. 128, 951–963 (2015).
Yin, C. C. & Lai, F. A. Intrinsic lattice formation by the ryanodine receptor calcium-release channel. Nat. Cell Biol. 2, 669–671 (2000).
Gaburjakova, J. & Gaburjakova, M. Coupled gating modifies the regulation of cardiac ryanodine receptors by luminal Ca(2+). Biochim. Biophys. Acta 1838, 867–873 (2014).
Dubochet, J. et al. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 21, 129–228 (1988).
Wagenknecht, T., Grassucci, R. & Frank, J. Electron microscopy and computer image averaging of ice-embedded large ribosomal subunits from Escherichia coli. J. Mol. Biol. 199, 137–147 (1988).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Heymann, J. B. & Belnap, D. M. Bsoft: image processing and molecular modeling for electron microscopy. J. Struct. Biol. 157, 3–18 (2007).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Adams, P. D. et al. The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 (2011).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Smart, O. S., Neduvelil, J. G., Wang, X., Wallace, B. A. & Sansom, M. S. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14, 354–360 (1996). 376.
Acknowledgements
This work was in part supported by the National Key Research and Development Program of China (Grant No. 2017YFA0504700 to C.-C.Y.) and the National Natural Science Foundation of China (Grant No. 31770785 to C.-C.Y.). This work was also supported by the National Natural Science Foundation of China, Youth Science Fund (Project No. 31900868 to C.L. and 32100963 to H.H.). This work was supported by the Shenzhen Science and Technology Innovation Committee (Projects No. JCYJ20210324131802008 to H.H.) and Kobilka Institute of Innovative Drug Discovery and Presidential Fellowship and University Development Fund at the Chinese University of Hong Kong, Shenzhen (H.H. and Q.C.). We thank the Cryo-EM Centre and Department of Biology, Southern University of Science and Technology, Shenzhen, for cryo-EM data collection.
Author information
Authors and Affiliations
Contributions
R.W. performed the protein purification; L.Z., C.L., Congcong L., and Q.C. performed the EM analysis, collected the cryo-EM data, performed image processing and 3D reconstruction; R.W., H.H., and C.-C.Y. analyzed the data; R.W., C.L., C.-C.Y., and H.H. initiated the project, supervised the research, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Deshun Gong and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wei, R., Chen, Q., Zhang, L. et al. Structural insights into transmembrane helix S0 facilitated RyR1 channel gating by Ca2+/ATP. Nat Commun 16, 1936 (2025). https://doi.org/10.1038/s41467-025-57074-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57074-4
This article is cited by
-
Statins, skeletal muscle, and ryanodine receptor activation: resolving a 30-year mystery behind statin myotoxicity
Cardiovascular Diabetology – Endocrinology Reports (2025)
