
Abstract
Lipid nanoparticle (LNP)-mRNA vaccines have demonstrated protective capability in combating SARS-CoV-2. Their extensive deployment across the global population leads to the broad presence of T-cell immunity against the SARS-CoV-2 spike protein, presenting an opportunity to harness this immunological response as a universal antigen target for cancer treatment. Herein, we design and synthesize a series of amino alcohol- or amino acid-derived ionizable lipids (AA lipids) and develop an LNP-RNA-based antigen presentation platform to redirect spike-specific T-cell immunity against cancer in mouse models. First, in a prime-boost regimen, AA2 LNP encapsulating spike mRNA elicit stronger T-cell immunity against the spike epitopes compared to FDA-approved LNPs (ALC-0315 and SM-102), highlighting the superior delivery efficiency of AA2 LNP. Next, AA15V LNP efficiently delivers self-amplifying RNAs (saRNAs) encoding spike epitope-loaded single-chain trimer (sSE-SCT) MHC I molecules into tumor tissues, thereby inducing the presentation of spike epitopes. Our results show that a single intratumoral (i.t.) treatment of AA15V LNP-sSE-SCTs suppresses tumor growth and extends the survival of B16F10 melanoma and A20 lymphoma tumor-bearing mice vaccinated with AA2 LNP-spike mRNA. Additionally, AA15V LNP-sSE-SCTs enable SE-SCT expression in ex vivo human glioblastoma and lung cancer samples, suggesting its potential in clinical translation.
Similar content being viewed by others


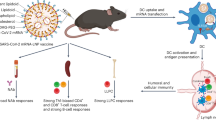
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a highly transmissible RNA virus, has posed a significant threat to global public health since the outbreak in late 20191,2. To counteract its morbidity and mortality, a variety of vaccines, such as virus-based vaccines and lipid nanoparticle (LNP)-mRNA vaccines, have been developed3,4. These vaccines have demonstrated abilities to elicit protective immune responses against the SARS-CoV-2 virus5,6. Among them, Pfizer/BioNTech’s BNT162b2 and Moderna’s mRNA-1273, both LNP-mRNA vaccines, encapsulate mRNA encoding a full-length spike protein, inducing protective spike-specific antibody and T-cell immunity7,8,9,10,11. Since the deployment of these vaccines, a significant portion of the global population has been immunized to limit the transmission of SARS-CoV-212. This widespread anti-spike T-cell immunity offers an opportunity for reprogramming toward cancer immunotherapy.
Immunotherapy has highlighted the benefit of harnessing the immune system for cancer treatment13,14. Effective antitumor immunity requires the priming of T cells against tumor-associated antigens (TAAs) presented by the major histocompatibility complex (MHC)15,16. However, due to tumor heterogeneity and individual differences, it is challenging to identify TAAs that display high universality, specificity, and immunogenicity17,18,19. To tackle this challenge, redirecting pre-existent immunity against cancer cells has emerged as a feasible and promising strategy20,21,22. For instance, a recent study leveraged virus-specific T cells to control tumors through intratumoral (i.t.) injection of viral peptides, demonstrating the ability of viral antigenic reprogramming21. In another study, cytomegalovirus (CMV) epitopes were conjugated with tumor-targeting antibodies, which facilitated their accumulation in tumors and subsequent presentation on tumor cell surface by MHC I molecules22. This approach enabled CMV-specific cytotoxic T lymphocytes (CTLs), which were already primed to attack CMV, to recognize and destroy cancer cells. Given the prevalence and efficacy of LNP-mRNA vaccine-induced anti-spike T-cell immunity, the spike-derived epitopes may serve as a new universal antigen that can be leveraged to redirect spike-specific T-cell immunity against tumors. However, stable presentation of desired epitopes by tumor MHC I is challenging23,24,25,26. Some tumors show impaired antigen presentation because of downregulation or even loss of MHC I23. Moreover, the competition between exogenously introduced and endogenously generated epitopes may compromise the presentation efficiency of MHC I27. This may reduce the immunostimulatory effect of viral peptides in these tumors.
The single-chain trimer (SCT) MHC I molecule, which consists of a polymorphic heavy chain, a β2 microglobulin (β2m) light chain, and a covalently attached epitope connected by a linker, has been developed to induce antigen-specific immune responses28,29. The covalent binding of an epitope in the SCT allows stable expression of defined epitope-loaded MHC I molecules on cell surface. Herein, we hypothesize that i.t. delivery of RNA encoding spike epitope-loaded SCTs (SE-SCTs) may retarget the anti-spike immunity against tumors in mice immunized by SARS-CoV-2 vaccines (Fig. 1a). To examine this hypothesis, we first designed a series of mRNAs and self-amplifying RNAs (saRNAs) encoding SE-SCTs. To effectively deliver RNA in vivo, we synthesized a library of amino alcohol- or amino acid-derived ionizable lipid materials (AA lipids) containing various headgroup structures (differ in types and quantities of hydrogen-donors) and types of hydrophobic tails (vary in length, biodegradability, and location of biodegradable groups). Through comprehensive explorations, we identified two lead LNP formulations, AA2 and AA15V. The FDA-approved LNP-mRNA vaccines, Moderna’s mRNA-1273 and Pfizer/BioNTech’s BNT162b2, utilize SM-102 and ALC-0315 as ionizable lipids, respectively30. Compared to the SM-102 and ALC-0315 LNPs, AA2 LNP showed higher spike mRNA delivery efficiency post intramuscular (i.m.) injection, leading to a stronger vaccination efficacy against SARS-CoV-2 as exhibited by the robust generation of spike-specific CD8+ T cells. Moreover, AA15V LNP demonstrated more potent RNA delivery efficiency to mouse cancer cells than the FDA-approved LNPs. As a result, a single i.t. treatment with AA15V LNP-saRNA encoding SE-SCTs (sSE-SCTs) significantly repressed tumor progression and extended the overall survival of the tumor-bearing mice that were immunized by AA2 LNP-spike mRNA vaccines. Additionally, AA15V LNP-sSE-SCTs facilitated SE-SCT expression in ex vivo human cancer samples, suggesting the potential for future clinical applications.

a Illustration of LNP-RNA-mediated antigen presentation platform. E spike epitope, TM transmembrane domain. Created in BioRender. Xue, Y. (2025) https://BioRender.com/g18v799. b Design and structures of AA lipids. c General synthetic routes of AA lipids.
Results
Ionizable lipids play a pivotal role in LNPs for the encapsulation of mRNA molecules30,31. Through electrostatic interactions, their positive head groups engage with the negatively charged phosphate groups of the mRNA, facilitating the complexation of the lipids with the mRNA and the formation of nanoparticles. Furthermore, it has been observed that the establishment of hydrogen bonds between the hydroxyl or amide groups present in these lipids and mRNA molecules can significantly enhance the entrapment of mRNA, thereby leading to improved mRNA translation32. The state-of-the-art ionizable lipids developed for COVID-19 vaccines, ALC-0315 and SM-102, incorporate hydroxyl groups in their structures, demonstrating effective mRNA delivery efficacy. In this work, we designed a library of amino alcohol- or amino acid-derived ionizable lipids, which contain hydroxylic or carboxylic moieties as the hydrogen bond donor. To enhance the chemical diversity of AA lipids, we first fine-tuned the quantities of hydrogen bond donors in the head groups, adjusted the length of carbon spacer between hydrogen bond donors and head group, and varied the types of hydrophobic tails (Fig. 1b). Particularly, AA1-AA26 lipids are amino alcohol-derived lipids featuring a hydroxyl group connected with the amino head groups via varying carbon spacers. AA27-AA45 lipids are amino acid-derived lipids incorporating a carboxylic acid group. In AA46-AA50 lipids, two hydroxyl groups were introduced to the head group via an ethylidene linker. To further augment the chemical diversity of the lipid library, four classes of hydrophobic tails were attached to the amino head groups, including non-biodegradable hydrocarbon tails (tail R1), biodegradable tails containing ester (tail R2), carbonate ester (tail R3-R6), and acid-labile acetal groups (tail R7-R9). The presence of these diverse hydrophobic tails in lipids can greatly affect their biodegradability, particle formation, and interactions with biological membranes33. The general synthesis routes of AA lipids are shown in Fig. 1c.
To assess AA lipids, AA LNPs were formulated with 1,2-dioleoyl-sn-glycerol-3-phosphoethanolamine (DOPE), cholesterol, and DMG-PEG2k with firefly luciferase (FLuc) mRNA. The AA LNP-FLuc mRNA displayed particle sizes between 86.4 ± 4.6 nm to 154.0 ± 4.5 nm, with a polydispersity index (PDI) < 0.3 (Supplementary Fig. 1a). These LNPs were slightly positively charged and obtained mRNA encapsulation efficiencies ranging from 29.7 ± 5.3% to 89.8 ± 1.9% (Supplementary Fig. 1b). Then, we treated C2C12 myoblasts and JAWSII dendritic cells (DCs) with AA LNPs to evaluate their mRNA delivery efficiency. These two cell lines were selected because i.m. vaccination tends to target muscle cells and antigen-presenting cells (APCs), including DCs for antigen presentation and immune stimulation. Among all these AA lipids, lipids with branched ester tails (tail R2), especially lipid AA2, exhibited superior mRNA delivery efficiency compared to other AA lipids and clinically approved SM-102 and ALC-0315 LNPs at an equivalent mRNA dose (Fig. 2a, b). This enhanced performance may be attributed to the hydrogen bond-forming capability of the hydroxyl group in the amino head groups of AA lipids. Moreover, the increased delivery efficiency of lipids with branched tails R2 may be attributed to their potential to adopt a more cone-shaped structure, facilitating improved endosomal escape compared to AA lipids with linear tails (such as hydrocarbonic, carbonated, and acetal tails)34,35. Conversely, the reduced mRNA delivery efficiency observed among AA46-AA50 lipids could be explained by the fact that their two-tailed core structure is less likely to adopt a cone-shaped structure compared to the three-tailed lipids34,35. Additionally, the diminished mRNA delivery efficiency of AA27-AA50 lipids may be associated with the deprotonation of their carboxylic acid group under physiological pH conditions, leading to decreased hydrogen bond donation capacity of lipids and limited mRNA entrapment (Supplementary Fig. 1b)33. Based on the in vitro results, we selected 18 lead AA LNPs with over 2-fold higher mRNA delivery efficiency than the SM-102 LNP in both cell lines for in vivo evaluations. These AA LNPs encapsulating FLuc mRNA were i.m. administered into mice, and the luminescence intensity at the injection site was measured after 6 h. Among all the AA LNPs tested, AA2 LNP displayed the most potent mRNA delivery efficiency with 5.4-fold and 2.4-fold higher luminescence intensity than ALC-0315 LNP and SM-102 LNP, respectively (Fig. 2c). Meanwhile, AA2 LNP showed lower off-target delivery to liver and spleen compared to ALC-0315 LNP and SM-102 LNP (Supplementary Fig. 2b–d). Notably, the luminescence intensity of AA2 LNP could still be observed at the injection site after 24 h (Fig. 2d; Supplementary Fig. 2a). Based on the above results, we selected AA2 LNP for detailed characterizations. The AA2 LNP exhibited a particle size of 108.6 ± 3.7 nm and an mRNA encapsulation efficiency of 89.0 ± 1.4% (Supplementary Fig. 2e, f). The particle was slightly positively charged and displayed a spherical morphology in Cryo-TEM images (Supplementary Fig. 2f, g).

a The luminescence intensity in C2C12 cells. b The luminescence intensity in JAWSII cells. c The luminescence intensity of the muscle in the lead AA LNP-FLuc mRNA-treated C57BL/6 mice. The intensity was normalized to the ALC-0315 LNP group. d Representative images of C57BL/6 mice i.m. treated with AA2 LNP-FLuc mRNA, ALC-0315 LNP-FLuc mRNA, and SM-102 LNP-FLuc mRNA. e Spike-specific IgG titer in blood drawn from C57BL/6 mice vaccinated with AA2 LNP-spike mRNA, ALC-0315 LNP-spike mRNA or SM-102 LNP-spike mRNA. Data in (a–d) are from n = 3 biologically independent samples. Data in (e) are from n = 6 biologically independent samples. Data are presented as mean ± SD. Statistical significance was determined by one-way ANOVA followed by Dunnett’s multiple comparison test. *P < 0.05, **P < 0.01, ****P < 0.0001.
To further evaluate the vaccination efficacy of AA2 LNP, we synthesized the mRNA encoding the full-length spike protein from SARS-CoV-2 and vaccinated mice via a prime-boost regimen. AA2 LNP generated 4.7-fold and 3.4-fold higher anti-spike IgG antibody titer than ALC-0315 LNP and SM-102 LNP, respectively (Fig. 2e). Importantly, activation-induced marker (AIM) assays revealed that AA2 LNP vaccination elicited spike-specific T cell populations in the blood and spleen (Fig. 3a). Particularly, to measure spike-specific CD4+ T cells in the vaccinated mice, we stimulated peripheral blood mononuclear cells (PBMCs) with a mixture of spike epitopes (SE; SGWTFGAGAALQIPF and VTWFHAIHVSGTNGT)36,37. The random peptide sequence AAAAFAAL was used as a negative control epitope (CE)38. Compared to treatment with CE, spike epitopes significantly increased activation markers of CD4+ T cells from blood (Fig. 3b; Supplementary Fig. 3a). Similarly, spike epitopes elicited higher percentages of CD137+CD134+ and CD154+CD134+ in CD4+ T cells from the spleen (Fig. 3d; Supplementary Fig. 3b). The CD4+ T cells isolated from the blood and spleen of mice vaccinated with SM-102 LNP also showed increased activation markers following stimulation with spike epitopes (Fig. 3b, d). However, the activation efficiency was lower than those observed with AA2 LNP. These results demonstrated the effective development of spike-specific CD4+ T cells induced by AA2 LNP-spike mRNA vaccinations. To detect spike-specific CD8+ T cells, we included one additional methodology, intracellular cytokine staining (ICS). Four spike-derived epitopes (SE; VNFNFNGL, SIIAYTMSL, VVFLHVTYV, VVLSFELL) were selected for CD8+ T cell activation based on their algorithm-predicted MHC I binding affinity and reported applications39,40,41,42,43. In both blood and spleen, AIM and ICS detected obvious CD8+ T cell responses to spike epitopes, with AA2 LNP-spike mRNA vaccination inducing increased levels of CD69+, CD69+CD137+, IFN-γ+, TNF-α+, and granzyme B+ compared to SM102 LNP (Fig. 3c, e; Supplementary Fig. 3). These results are consistent with the in vivo mRNA delivery efficiency and spike-specific IgG titers, indicating that vaccinations with AA2 LNP-spike mRNA generate stronger spike-specific T-cell immunity compared to SM-102 LNP-spike mRNA. This provides a solid foundation for the utilization of SE-SCTs to redirect spike-specific T-cell immune responses against tumors.

a Schematic depicting activation-induced marker (AIM) assay of T cells isolated from spleen and blood of mice vaccinated with AA2 LNP-spike mRNA or SM-102 LNP-spike mRNA. Created in BioRender. Xue, Y. (2025) https://BioRender.com/z43s143. b, d Spike epitope-specific activation of CD4+ T cells isolated from the blood (b) and spleen (d) of the mice vaccinated with AA2 LNP-spike mRNA or SM-102 LNP-spike mRNA. CE control epitope, SE spike epitope. c, e Spike epitope-specific activation of CD8+ T cells isolated from the blood (c) and spleen (e) of the mice vaccinated with AA2 LNP-spike mRNA or SM-102 LNP-spike mRNA. Gzmb granzyme B, CE control epitope, SE spike epitope. Data are from n = 5 biologically independent samples and are presented as mean ± SD. Statistical significance was determined by the two-tailed Student’s t-test. n.s. not significant P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Next, we evaluated the delivery efficiency of AA LNP-FLuc mRNA in B16F10 melanoma cells. Similarly, AA lipids with branched ester tails (tail R2) exhibited higher mRNA delivery efficiency than other tail types of AA lipids in B16F10 cells. The lead material, AA15 LNP, induced the highest luminescence intensity, which was over 25.6-fold and 5.3-fold greater than ALC-0315 LNP and SM-102 LNP, respectively (Fig. 4a). To further increase mRNA delivery capacity of AA15 LNP, we fine-tuned the molar ratios for each formulated lipid using an L16 (4)4 orthogonal table (Supplementary Fig. 4a). We also optimized the mass ratio of AA15 lipid to mRNA in the formulation. The lead formulation AA15V LNP demonstrated 1.5-fold and 3.1-fold higher luminescence intensity compared to the 1st-round top orthogonal formulation AA15I and the original formulation AA15, respectively (Supplementary Fig. 4b, c). The hydrodynamic diameter of AA15V LNP was 102.3 ± 4.1 nm with a PDI < 0.15. Over 85% mRNA was encapsulated in AA15V LNP, and the particle exhibited a slightly positive charge and spherical morphology (Supplementary Fig. 4d–f). Accordingly, AA15V LNP was chosen for mRNA delivery to cancer cells in the following studies.

a Screening of AA LNPs in B16F10 cells. b Construction of saRNA encoding MHC I single-chain trimer (SCT) containing spike epitopes. E spike epitope, TM transmembrane domain. Created in BioRender. Xue, Y. (2025) https://BioRender.com/k91i086. c Dynamic expression of H-2Kb+β2m+ in B16F10 cells. d Expression of OVA-H-2Kb+ in B16F10 cells after a single i.t. injection of AA15V LNP-sOP-SCTs. OP, OVA257-264 peptide (SIINFEKL). e Percentage of apoptotic B16F10 cells in the T cell-mediated killing assay. f Percentage of dead B16F10 cells in the T cell-mediated killing assay. g, h Spike epitope-specific activation of CD4+ T cells (g) and CD8+ T cells (h) isolated from B16F10 tumors in mice vaccinated with AA2 LNP-spike mRNA. CE control epitope, SE spike epitope. Data in (a) and (c) are from n = 3 biologically independent samples. Data in (d) are from n = 4 biologically independent samples. Data in (e–h) are from n = 5 biologically independent samples. Data are presented as mean ± SD. Statistical significance in (a, c) and (e, f) was determined by one-way ANOVA followed by Dunnett’s multiple comparison test. Statistical significance in (d) and (g, h) was determined by the two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The covalent binding of an epitope in a single-chain trimer (SCT) MHC I molecule enables stable expression of defined epitope-loaded MHC I molecules on the cell surface28,29. To express spike epitope (SE)-loaded single-chain trimer (SE-SCT) MHC I molecule on B16F10 cells, we constructed mRNAs and saRNAs encoding four distinct SE-SCTs with spike epitopes (VNFNFNGL, SIIAYTMSL, VVFLHVTYV, VVLSFELL, Fig. 4b). We refer to mRNAs and saRNAs encoding SE-SCTs as mSE-SCTs and sSE-SCTs, respectively. The saRNA encoding SCT with AAAAFAAL, the control epitope, was named sCE-SCTs. Both mRNAs encoding SE-SCTs (mSE-SCTs) and saRNAs encoding SE-SCTs (sSE-SCTs) delivered by AA15V enabled over 80% B16F10 cells to express H-2Kb+β2m+ with individual epitopes at 24 h (Supplementary Fig. 5a). To evaluate dynamic expression of SE-SCTs, B16F10 cells were treated with AA15V containing the four-mixed mSE-SCTs or sSE-SCTs. Although both treatments induced over 90% expression of SE-SCTs at 24 h (Fig. 4c; Supplementary Fig. 5b), the sSE-SCTs group retained 69.0 ± 2.9% H-2Kb+β2m+ B16F10 cells on day 3 while the mSE-SCTs group showed a sharply decreased level to 59.1 ± 2.5% on day 2 and further to 5.8 ± 1.8% on day 3. To assess the expression of antigen-specific H-2Kb molecules on tumor cells following i.t. injection of AA15V LNP encapsulating SCT-saRNAs in vivo, we constructed saRNAs encoding OVA257-264 peptide-SCTs (OP-SCTs), enabling the quantification of OVA-H-2Kb+ live tumor cells using established antibodies. We established the tumor model using B16F10-FLuc cells and found a single i.t. injection of AA15V LNP-sOP-SCTs resulted in 27.8 ± 7.7% of OVA-H-2Kb+ live B16F10 cells within tumor tissues (Fig. 4d; Supplementary Fig. 6). This result indicated our AA15V LNP-SCT saRNAs could effectively induce expression of antigen-specific H-2Kb molecules on cancer cells in vivo.
Cytotoxicity of spike-specific CD8+ T cells was evaluated by isolating CD8+ T cells from the spleens of vaccinated mice, which were then co-cultured with B16F10 cells that had been pretreated in vitro with PBS, AA15V LNP-sCE-SCTs, or AA15V LNP-sSE-SCTs. These CD8+ T cells exhibited cytotoxicity against B16F10 cells pretreated with AA15V LNP-sSE-SCTs (Fig. 4e, f; Supplementary Fig. 5c). In contrast, no notable T cell-mediating cell killing was observed in the AA15V LNP-sCE-SCTs group compared to PBS control. To assess whether spike-specific T cells could infiltrate tumors, C57BL/6 mice vaccinated with AA2 LNP-spike mRNA were inoculated with B16F10 cells, after which T cells were isolated from melanoma tissues and later co-cultured with CD4- and CD8-specific spike epitopes (Supplementary Fig. 5d). AIM assays revealed that specific epitopes targeting MHC II and MHC I stimulated tumor-infiltrating T cells, resulting in increased levels of CD69+CD134+ in CD4+ T cells and CD69+CD137+ in CD8+ T cells, respectively (Fig. 4g, h). These findings indicate the potential of AA15V LNP-sSE-SCTs to retarget spike-specific T-cell immunity against tumors in vaccinated mice.
To evaluate the antitumor effects of spike-specific T-cell immunity against cancer cells expressing SE-SCT, we performed a single i.t. treatment in a B16F10 melanoma mouse model, where the mice received prime-boost immunizations of AA2 LNP-spike mRNA before tumor inoculation (Fig. 5a). Compared to PBS, AA15V LNP-mSE-SCTs, and AA15V LNP-sCE-SCTs treatment, AA15V LNP-sSE-SCTs more effectively suppressed the tumor growth by 22.0-fold, 18.2-fold, and 7.5-fold (14 d post-inoculation), respectively, and substantially extended the overall survival time (Fig. 5b, c; Supplementary Fig. 7a–d). To evaluate the function of CD4+ and CD8+ T cells in AA15V LNP-sSE-SCTs treatment, tumor-bearing mice were treated with isotype control, anti-CD4, or anti-CD8 antibodies prior to receiving AA15V LNP-sSE-SCTs (Supplementary Fig. 8a). Depletion of CD8+ T cells markedly diminished the therapeutic effects of AA15V LNP-sSE-SCTs, as evidenced by reduced tumor regression and lower survival rates (Supplementary Fig. 8b–g). In contrast, while CD4+ T cell depletion moderately compromised tumor suppression, it did not lead to significant differences in overall survival between mice pretreated with isotype control or anti-CD4 antibodies (Supplementary Fig. 8b–g). These results indicate that CD8+ T cells play a more crucial role than CD4+ T cells in the antitumor effects of AA15V LNP-sSE-SCTs treatment.

a Schematic of the treatment regimen in the B16F10 tumor model. Created in BioRender. Xue, Y. (2025) https://BioRender.com/b49g753. b Tumor volumes in different groups. c Survival rates of the mice in the B16F10 tumor model. d Expression of cytokines and chemokines in tumor samples at 24 h post-treatment. e Expression level of representative cytokines and chemokines in tumor samples from (d). f Expression of cytokines and chemokines in blood samples at 24 h post-treatment. g Expression level of representative cytokines and chemokines in blood samples from (f). h–l Immune cell populations in tumor tissues. Populations of i activated DCs, j activated macrophages, k primed CD4+ T cells, and l primed CD8+ T cells in tumor tissues. Data in (b, c) and (h–l) are from n = 5 biologically independent samples. Data in (d–g) are from n = 3 biologically independent samples. Data are presented as mean ± SD. Statistical significance in (b) was determined by two-tailed Student’s t-test. Statistical significance in (c) was determined by the log-rank (Mantel–Cox) test. Statistical significance in (e–l) was determined by one-way ANOVA followed by Dunnett’s multiple comparison test. n.s. not significant P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To profile the changes in the tumor microenvironment (TME) after a single treatment, we examined the expression of cytokines and chemokines in tumor tissues and blood. Relative to PBS treatment, AA15V LNP-mSE-SCTs, AA15V LNP-sCE-SCTs, and AA15V LNP-sSE-SCTs all stimulated immune responses, as indicated by elevated levels of pro-inflammatory cytokines and chemokines in both tumor and blood samples (Fig. 5d–g). Notably, AA15V LNP-sCE-SCTs induced comparable or even higher levels of multiple pro-inflammatory cytokines and chemokines compared to AA15V LNP-mSE-SCTs. This may be attributed to the immunogenicity of saRNA which can form double-stranded RNA intermediates during translation, triggering innate immune responses44. Compared to these treatments, AA15V LNP-sSE-SCTs significantly increased levels of pro-inflammatory cytokines and chemokines, including IL-1α, IL-6, IL-7, IL-12p70, IL-15, IFN-γ, TNF-α, and CXCL9, in both tumor tissues and blood 24 h post-administration (Fig. 5d–g). Next, we detected the infiltration and activation of immune cells in tumors including conventional dendritic cells (cDC1 and cDC2), macrophages, CD4+, and CD8+ T cells (Fig. 5h–l; Supplementary Fig. 9). Compared to AA15V LNP-mSE-SCTs, AA15V LNP-sCE-SCTs demonstrated an equal or greater capacity of recruiting and activating immune cells within tumor tissues including macrophages and CD80+ cDC1s (Fig. 5h–l). Consistent with the trends observed in cytokine and chemokine levels, AA15V LNP-sSE-SCTs treatment recruited more immune cells to the tumor tissues and achieved stronger immune cell activation than other treatments. Specifically, AA15V LNP-sSE-SCTs treatment increased the populations of activated APCs, including CD80+/86+ DCs and CD80+/86+ macrophages (Fig. 5i–j), as well as activated CD4+ T cells expressing CD137+CD134+ and CD154+CD134+ within tumor tissues (Fig. 5k). Importantly, the treatment also upregulated the expression of Ki-67, IFN-γ, TNF-α, and granzyme B in CD8+ T cells compared to other groups, indicating activation of cytotoxic responses (Fig. 5l). These findings suggest that both saRNA-mediated innate immune responses and spike-specific T-cell immunity contribute to the antitumor effects of AA15V LNP-sSE-SCTs.
Although AA15V LNP-sSE-SCTs treatment showed obvious antitumor effects, the incomplete eradication of the tumor necessitates boosting the efficacy of primed T-cell responses. Immune checkpoint inhibitors (ICIs), including antibodies that target PD-1 and CTLA-4, enable enhanced T lymphocyte survival and efficacy in cancer treatments45. To further improve the antitumor effects of AA15V LNP-sSE-SCTs, we incorporate the ICI combination, anti-PD-1 and anti-CTLA-4 Abs, in our strategy. We intraperitoneally injected one dose of ICI prior to the AA15V LNP-sSE-SCTs treatment, followed by three subsequent doses given at three-day intervals throughout the treatment (Fig. 6a). The combination of AA15V LNP-sSE-SCTs (single dose) and ICI (four doses) dramatically inhibited tumor growth and prolonged survival of the tumor-bearing vaccinated mice in comparison to those treated with AA15V LNP-sSE-SCTs alone or AA15V LNP-sCE-SCTs (Fig. 6b, c; Supplementary Fig. 10a–d). Moreover, 12.5% of the mice treated with the AA15V LNP-sSE-SCTs + ICI showed complete regression (Fig. 6c). To explore the applicability of AA15V LNP-sSE-SCTs, we assessed the antitumor effects on the A20 tumor model in BALB/C mice. H-2Kd-based SE-SCTs were constructed to match the MHC I haplotype of BALB/C mice (Fig. 6d). Additionally, we incorporated control groups of unvaccinated mice to assess the role of spike-specific T-cell immunity in the antitumor efficacy of our strategy. The AA15V LNP-sSE-SCTs + ICI treatment induced completed tumor rejection and long-term survival in over 28.6% of the vaccinated mice (Fig. 6e, f; Supplementary Fig. 11a–e). However, such treatment in the unvaccinated mice failed to achieve a complete response, and all the mice reached the end point criteria before day 31, indicating the importance of spike-specific T-cell immunity in suppressing tumors expressing SE-SCTs.

a Schematic of the treatment regimen in B16F10 tumor model treated with the combination of AA15V LNP-sSE-SCTs and ICI. b Tumor volumes over time. c Survival rates of mice bearing B16F10 tumor. d Schematic of the treatment regimen in A20 tumor model. e Tumor volumes over time. f Survival rates of the mice bearing A20 tumor. g Schematic depicting ex vivo AA15V LNP-sSE-SCTs delivery in human tumor tissues. Created in BioRender. Xue, Y. (2025) https://BioRender.com/q02v567. h Expression of H-2Kb+β2m+ expression in CD45− cells from pediatric glioma dissections after ex vivo treatment with AA15V LNP-sSE-SCTs. i, j H-2Kb+β2m+ in CD45− cells from two separate lung left lower lobe (LLL) adenocarcinoma specimens after ex vivo treatment with AA15V LNP-sSE-SCTs. Data in (b, c) are from n = 6 (PBS group) and 8 (SCT groups) biologically independent samples, respectively. Data in (e, f) are from n = 6 (PBS group and UnVac groups) and 7 (Vac groups) biologically independent samples, respectively. Data in (h) and (j) are from n = 3 individual tissue slices. Data in (i) are from n = 4 individual tissue slices. Data are presented as mean ± SD. Statistical significance in (b, e) and (h–j) was determined by the two-tailed Student’s t-test. Statistical significance in (c) and (f) was determined by the log-rank (Mantel–Cox) test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To further explore the clinical translatability of AA15V LNP-sSE-SCTs, we examined AA15V LNP for the delivery of sSE-SCTs to human tumor tissues ex vivo (Fig. 6g). Glioma samples from one patient were treated with AA15V LNP-sSE-SCTs. Quantitative analysis revealed H-2Kb+β2m+ expression in 7.1 ± 0.2% of CD45− cells in the glioma samples while the untreated slices exhibited no detectable expression (Fig. 6h; Supplementary Fig. 12). Furthermore, in one lung left lower lobe (LLL) adenocarcinoma specimen from a patient, AA15V LNP-sSE-SCTs treatment resulted in 5.8 ± 2.2% of CD45− cells expressing H-2Kb+β2m+ (Fig. 6i). In the LLL adenocarcinoma sample from another adult human subject, AA15V LNP-sSE-SCTs treatment induced H-2Kb+β2m+ expression in 8.7 ± 2.0% of CD45− cells (Fig. 6j). Given the cellular heterogeneity within the tumor microenvironment of human tumors, the expression of H-2Kb+β2m+ could be present in both cancer cells and tumor stromal cells. These results demonstrate the ability of AA15V LNP for delivery of sSE-SCTs to primary human tumor samples.
Humans have encountered a multitude of viral infections. Following recovery, the body harbors memory T cells dispersed throughout its entirety46,47. These antiviral memory T cells exhibit notable characteristics: heightened vigilance, rapid response, and cytotoxic abilities. Upon encountering virus-specific epitopes, indicating potential reinfection, these memory T cells become primed, efficiently orchestrating immune defenses at the reinfection site. In contrast to human tumor antigens, which may be patient-specific and non-immunogenic, the epitopes recognized by virus-specific T cells are renowned for their universality and immunogenicity21. Therefore, redirecting established antiviral immunity may offer a therapeutic avenue for cancer immunotherapy, although more studies need to determine if the antitumor activity of vaccine-induced T cells mirrors that of T cells generated through natural viral infections.
The widespread use of mRNA vaccines against SARS-CoV-2 has elicited anti-spike T-cell immunity on a global scale, establishing spike epitopes as a viable antigenic target. Thus, we constructed RNA-encoding spike epitope-loaded SCTs (SE-SCT) and explored the therapeutic potential of leveraging LNP-RNA formulation-mediated antigen presentation to redirect spike-specific T-cell immunity against cancer. We synthesized and evaluated a library of amino alcohol- or amino acid-derived (AA) ionizable lipid materials, identifying AA2, the optimal LNP formulation for spike mRNA vaccination, and AA15V, the optimal LNP formulation for RNA delivery in cancer cells. AA15V LNP-sSE-SCTs enabled the presentation of SE-SCT on cancer cell surface, expediting the recognition by spike-specific T cells induced through AA2 LNP-spike mRNA vaccination. Such redirection of spike-specific T-cell immunity facilitated cancer cell death induced by T-cell-mediated cytotoxicity. Moreover, i.t. administration of AA15V LNP-sSE-SCTs reprogrammed the TME by eliciting antitumor phenotypes of immune cells and pro-inflammatory cytokines as well as chemokines. As a result, a single treatment significantly suppressed the growth of melanoma and lymphoma tumors and extended the survival of AA2 LNP-spike mRNA-vaccinated mice, particularly when in combination with ICI treatments.
Compared to AA15V LNP-mSE-SCTs, the enhanced antitumor effects observed with AA15V LNP-sSE-SCTs are likely due to the prolonged translation and increased immunogenicity of saRNAs. First, treatment with AA15V LNP-sSE-SCTs resulted in approximately 69.0% of B16F10 cells expressing SE-SCTs by day 3, in contrast to just 5.8% expression observed with AA15V LNP-mSE-SCTs (Fig. 4c), highlighting the extended translation capacity of saRNA. Second, AA15V LNP-sCE-SCTs encoding control epitopes demonstrated measurable antitumor efficacy (Fig. 5b, c), potentially due to the immunogenic properties of saRNA. During translation, saRNA forms double-stranded RNA intermediates that activate innate immune responses48. As shown in Fig. 5d, e, AA15V LNP-sCE-SCTs induced higher levels of IL-1α, IFN-γ, and TNF-α in tumor tissues compared to AA15V LNP-mSE-SCTs. Such nonspecific immune responses benefiting cancer treatment have also been reported in previous studies, where i.t. injection of LNP-saRNA encoding mCherry as a reporter gene showed significant antitumor efficacy in mouse models44. However, the antitumor effects of LNP-sCE-SCTs were limited relative to AA15V LNP-sSE-SCTs, likely due to the absence of spike-specific CD8+ T cell responses. As shown in Fig. 4h, spike-specific CD8+ T cells were detected in tumor tissues following vaccination. Moreover, i.t. injection of AA15V LNP-sSE-SCTs led to significantly elevated levels of pro-inflammatory cytokines and chemokines, such as IL-1α, IL-6, IL-7, IL-12p70, IL-15, IFN-γ, TNF-α, and CXCL9 (Fig. 5e, g) compared to AA15V LNP-sCE-SCTs. Consistently, AA15V LNP-sSE-SCTs recruited more immune cells to the TME and enhanced immune cell activation, evidenced by increased levels of CD80+/86+ DCs as well as IFN-γ+, TNF-α+, and granzyme B+ CD8+ T cells (Fig. 5i–l). Furthermore, depletion of CD8+ T cells significantly diminished the therapeutic effects of AA15V LNP-sSE-SCTs (Supplementary Fig. 8). All these results underscore the crucial roles of both saRNA-mediated innate immune responses and spike-specific T-cell immunity in the antitumor efficacy of AA15V LNP-sSE-SCTs.
Recent studies have explored the feasibility of using i.t. injection of viral epitopes or intravenous injection of antibody-conjugated viral epitopes to repurpose anti-virus T-cell immunity for cancer treatment21,22. However, certain tumors exhibit impaired antigen presentation due to the downregulation or complete loss of MHC I23. Additionally, competition between exogenously introduced epitopes and those generated endogenously can further reduce the efficiency of MHC I presentation27. The LNP-sSE-SCTs treatment restores MHC I presentation in cancer cells, potentially enhancing the immunogenicity of cancer cells. Moreover, the expression of sSE-SCTs can facilitate epitope loading onto MHC I molecules, circumventing competition with endogenous tumor epitopes with low immunogenicity.
One potential challenge to clinical translation may be the transitory duration of SARS-CoV-2-specific T-cell memory. Clinical trials have shown that booster immunizations with mRNA vaccines can enhance the persistence of anti-spike T-cell memory49. However, another key hurdle is the extensive diversity of human leukocyte antigens (HLA), which complicates the ‘one-size-fits-all’ approach of our platform. A single universal epitope may not be effective across individuals with different HLA alleles50. Recently, next-generation sequencing (NGS) has emerged as the preferred method for precise HLA typing in transplantation, offering high throughput and rapid processing51. Moreover, the production of mRNA or saRNA encoding target immunogens is rapid and scalable. The synergy between NGS and LNP-mRNA technologies has already led to several personalized cancer vaccines in clinical trials52,53, underscoring the potential of this platform as a personalized treatment strategy. Additionally, we recognize that i.t administration may not be suitable for all tumor types. Recent advancements in image-guided administration have the potential to improve the precision and safety of i.t. delivery, particularly for deep-seated tumors54,55,56,57. Some mRNA-based therapeutics are under evaluation in clinical trials using i.t. injections58,59,60. For eligible patients, i.t. delivery offers significant advantages, such as enhanced biodistribution within tumor tissues and reduced systemic side effects55,56.
In summary, we have developed an LNP-RNA-based antigen presentation platform designed to redirect spike-specific T-cell immunity against cancer. The clinical potential of this platform is highlighted by two key aspects: first, AA2 LNP demonstrates superior efficacy for mRNA vaccine delivery in vivo compared to FDA-approved LNPs; second, AA15V LNP facilitates the delivery of sSE-SCTs to human cancer samples. Considering that a significant portion of the global population has already developed SARS-CoV-2 T cell memory, this strategy provides a new avenue for cancer immunotherapy.
Methods
Reagents
1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), cholesterol, and 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG2K) were purchased from Avanti Polar Lipids. ALC-0315, ALC-0159 and SM-102 were obtained from MedKoo Biosciences. The purification of AA lipids was achieved via column chromatography using a RediSep Gold Resolution silica column (Teledyne Isco) with a CombiFlash Rf system employing gradient elution. ¹H NMR spectral analyses were conducted by a Bruker Avance 400 MHz device. Mass spectrometry analyses were executed using Acquity SQD UPLC/MS (Waters), LTQ Orbitrap XL mass spectrometer (Thermo Scientific), and the ultrafleXtreme MALDI-TOF mass spectrometer (Bruker) at The Ohio State University.
Cell culture
Murine myoblast C2C12 cell line (CRL-1772), murine JAWSII dendritic cell line (CRL-3612), murine melanoma B16F10 cell line (CRL-6475), and murine B cell lymphoma A20 cell line (TIB-208) were all obtained from American Type Culture Collection (ATCC). C2C12 cells and B16F10 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin (50 U/mL). JAWSII cells were cultured in RPMI-1640 medium with L-Glutamine, 20% FBS, penicillin-streptomycin (50 U/mL), and 5 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF).
RNA synthesis
The linear dsDNA of firefly luciferase (FLuc), the spike protein of SARS-CoV-2 Delta variant, and spike epitope single-chain trimer MHC class I molecules (SE-SCTs) were obtained from Integrated DNA Technologies. Corresponding plasmids were generated from pUC19 vector via NEBuilder® HiFi DNA assembly. mRNA and saRNA synthesis were conducted as previously reported61. All mRNAs used in this manuscript are chemically modified with N1-methylpseudouridine.
LNP formulation and characterization
LNPs were formulated using Rapid Nanomedicine System INano L+ (Micro&Nano biologics Technology Ltd.) by mixing an ethanol solution comprising ionizable lipids, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), cholesterol, and 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG2K), with aqueous citrate solution containing the mRNAs (10 mM, pH 3.0). For the preliminary screening phase, LNPs were formulated with AA lipids, DOPE, cholesterol, and DMG-PEG2K at the molar ratios of 20:30:40:0.75, respectively. Concurrently, FLuc mRNA was formulated at the mass ratio of AA lipid: mRNA set at 10:1. Subsequently, an L16 (4)4 orthogonal array was constructed to optimize the LNP formulations. The FDA-approved LNPs were formulated using previously established lipid ratios and lipid-to-mRNA mass ratios62. For in vitro screening, LNP-mRNA formulation was added to cells at the dose of 50 ng mRNA per 2 × 104 cells. mRNA delivery efficiency was evaluated 18 h after treatment by exposing cells to Bright-Glo luciferase substrate (Promega), followed by quantification of luminescence intensity using Cytation 5 cell imaging multi-mode reader (Biotek). Hydrodynamic diameter, zeta potential, and polydispersity index (PDI) were assessed by NanoZS Zetasizer (Malvern, USA). Encapsulation efficiency was evaluated using Ribogreen assay, while the morphology of LNPs was visualized by Glacios Cryo-TEM (Thermo Scientific, USA).
Intramuscular injection and vaccination
For in vivo screening, mice were intramuscularly injected with AA LNP-FLuc mRNA at 1.5 μg mRNA per mouse. At 6- and 24-h time points, 150 μL of D-luciferin substrate (30 mg/mL) was intraperitoneally injected into the mice. Following an 8-min interval, live mice were imaged using a Xenogen IVIS imaging system to quantify the luminescence signals at muscles. To investigate the biodistribution of AA2 LNP, two parallel groups of mice were sacrificed to dissect major organs for quantifying luminescence signals at 6- and 24-h time points, respectively.
For spike mRNA vaccinations, mice were intramuscularly immunized by AA2 LNP-spike mRNA at the mRNA dose of 0.3 mg/kg on day 0. Booster immunization with the same dose was conducted on day 21. Five days after the booster dose, mouse blood was collected with anticoagulant (3.8% Sodium citrate) and centrifugated at 1500×g for 10 min at 4 °C to obtain plasma before being stored at −80 °C until subsequent analysis. ELISA assay was used to detect the plasma titer of Delta SARS-CoV-2 spike-specific antibodies. Specifically, 250 ng of Delta SARS-CoV-2 spike S1 subunit peptide was applied to coat a 96-well plate, after which diluted mouse plasma was added to the wells. Goat anti-mouse IgG linked to HRP (Abcam, ab7068, 1:5000 dilution) was then added, and the mean optical density at 492 nm (OD492) was recorded using a Cytation 5 cell imaging multi-mode reader (Biotek). The anti-S1 antibody titer in the mouse plasma was quantified against a standard curve employing a recombinant anti-spike monoclonal antibody (Sino Biological, 40591-MM43).
Luminex analysis of cytokines and chemokines
Tumor tissues and blood samples from mice were collected at 24 h post-i.t. injection of PBS, AA15V LNP-mSE-SCTs, AA15V LNP-sCE-SCTs, or AA15V LNP-sSE-SCTs (10 μg RNA per mouse). Tumor tissues were promptly flash-frozen in liquid nitrogen, followed by pulverization and extraction using RIPA lysis buffer (Thermo Scientific, 89900) supplemented with protease inhibitors (Thermo Scientific, 87785). Murine whole blood was collected in sodium citrate-containing tubes, followed by centrifugation to isolate the plasma. Both tumor lysate and plasma samples were kept at −80 °C until further analysis. Mouse cytokine and chemokine levels were evaluated using mouse cytokine/chemokine discovery assay (Eve Technologies).
Expression of activation-induced markers (AIM) of T cells from SARS-CoV-2 vaccinated mice
Mouse spleens and blood samples were collected 5 days post-boost vaccination. Single-cell suspension was prepared from mouse spleen, and pan T cells were isolated according to the manufacturer’s instructions (T cell isolation kit, Miltenyi Biotec, Catalog # 130-096-130). Peripheral blood mononuclear cells (PMBCs) from the whole blood were isolated by centrifugation and resuspended in T cell medium for 6 h. B16F10 tumors from vaccinated mice were harvested when the largest tumor diameter exceeded 0.8 cm and dissociated into a single-cell suspension for T-cell isolation (Miltenyi Biotec, Catalog #130-096-730). The isolated pan T cells were resuspended in T cell medium and rested for 6 h. Following this, the T cells were stimulated with control, CD8, or CD4 epitopes. After 10–12 h of culture, AIM expression was then assessed by flow cytometry9,63. For CD8+ T cell killing assay, B16F10 cells receiving AA15V LNP-sSE-SCTs or controls were co-cultured with CD8+ T cells isolated from the spleen of the mice vaccinated with AA2 LNP-spike mRNA. The apoptosis of B16F10 cells was detected by dead cell apoptosis kit (V13242 Invitrogen™).
Intratumoral delivery of AA15V LNP-SCT saRNAs in vivo
C57BL/6 mice were subcutaneously (s.c.) inoculated with 1 × 105 B16F10-FLuc cells on the right flank. When the largest tumor diameter exceeded 0.8 cm, the mice were i.t. injected with AA15V LNP-sOP SCTs at 10 μg RNA dose. After 24 h, the tumor tissues were dissected and dissociated into a single-cell suspension using tumor dissociation kit (Miltenyi Biotec, Catalog # 130-096-730). Erythrocytes were removed with Red Blood Cell Lysis Solution (10×, Miltenyi Biotec, Catalog # 130-094-183). The cells were then stained with LIVE/DEAD™ Fixable Violet Dead Cell Stain Kit (Invitrogen™, L34955). After washing with PBS, the cells were stained with antibodies followed by flow cytometry analysis.
Tumor models and treatment regimens
C57BL/6 and BALB/c mice (male and female, 6–10 weeks) were purchased from the Jackson Laboratory and housed in the Icahn School of Medicine at Mount Sinai. All mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC) at The Ohio State University (2014A00000106) and Icahn School of Medicine at Mount Sinai (IPROTO202200000134), and complied with local, state, and federal regulations. In this study, a maximum of five mice were accommodated in each cage within a barrier environment, maintaining conditions of approximately 20 °C, 45% humidity, and a 12-h light/12-h dark cycle.
The mice received bi-dose vaccinations of AA2 LNP-spike mRNA at 0.3 mg/kg mRNA dose. Five days after the booster doses, around 1 × 105 B16F10 cells or 5 × 106 A20 cells were s.c. inoculated on the right flank of the mice. The A20 tumor model included unvaccinated mice as control groups. On day 6 post-tumor inoculation, mice with tumor size about or over 0.5 cm of the largest diameter were randomly separated into different groups. For T cell depletion tumor model, 1 × 105 B16F10 cells s.c. inoculated on the right flank of the mice. The tumor-bearing mice were intraperitoneally injected with anti-mouse CD8α, anti-mouse CD4, or anti-rat IgG2b isotype antibodies, respectively, on day 6 post-tumor inoculation. Each antibody was administered every 3 days. One day after the first antibody depletion treatment (Day 7), the tumor-bearing mice received a single i.t. dose of AA15V LNP-sSE-SCTs. For the B16F10 tumor model groups without combinatorial therapy of ICIs (anti-PD1+ anti-CTLA4 antibodies), a single dose of various treatments (10 μg RNA dose) was i.t. administered to the tumor-bearing mice on day 6 following tumor inoculation. For the combination therapy involving ICIs, starting from day 6, mice were injected intraperitoneally with anti-PD1 (100 µg/mouse, clone: RMP1-14, BioXcell) and anti-CTLA4 antibodies (100 µg/mouse, clone: 9D9, BioXcell). Each antibody was administered every 3 days for four doses. One day after the first ICI treatment (Day 7), the tumor-bearing mice received a single i.t. dose of various treatments.
Ex vivo saRNA delivery in human tumor tissues
Lung tumor samples were obtained from surgical specimens of patients undergoing resection at Mount Sinai Hospital (New York, NY) after obtaining informed consent in accordance with a protocol reviewed and approved by the Institutional Review Board at the Icahn School of Medicine at Mount Sinai (IRB Human Subjects Electronic Research Application 10-00472 A) and in collaboration with the Biorepository and Department of Pathology. All glioma tissue was banked, de-identified under approved institutional IRB protocol (STUDY-18-00983), and informed consent was secured from all participants. The obtained human tumor tissue biopsies were promptly placed on ice in PBS and sectioned into 600 μm thick slices using Leica VT 1200S microtome within 30 min. The slices were then cultured in RPMI-1640 medium containing 20% FBS for 1 h before being treated with 1 µg AA15V LNP-sSE-SCTs. After 24 h, the slices were dissociated into single-cell suspension and prepared for immunofluorescence staining with specific cell markers.
Statistics and reproducibility
For the comparison of multiple data sets, one-way ANOVA with Dunnett’s multiple comparison test was used, whereas the two-tailed Student’s t-test was used for comparing two groups. In the mouse tumor model, tumor sizes were compared using two-way ANOVA with Dunnett’s multiple comparison, and survival rates were analyzed with the log-rank (Mantel–Cox) test. P-values of <0.05 were considered statistically significant. All data adhered to the assumptions of the statistical tests applied, including normal distribution and equal variances, and underwent formal testing to validate the statistical analysis. The sample size was not predetermined by any statistical method.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and Supplementary Information files. Source data is available for Figs. 2–6 and Supplementary Figs. 1, 2, 4, 8, 10 and 11 in the associated source data file. Source data are provided with this paper.
References
Guan, W. J. et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 382, 1708–1720 (2020).
Merad, M., Blish, C. A., Sallusto, F. & Iwasaki, A. The immunology and immunopathology of COVID-19. Science 375, 1122–1127 (2022).
Krammer, F. SARS-CoV-2 vaccines in development. Nature 586, 516–527 (2020).
Barbier, A. J., Jiang, A. Y., Zhang, P., Wooster, R. & Anderson, D. G. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 40, 840–854 (2022).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2021).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Goel, R. R. et al. mRNA vaccines induce durable immune memory to SARS-CoV-2 and variants of concern. Science 374, abm0829 (2021).
Wang, L. et al. T cell immune memory after covid-19 and vaccination. BMJ Med. 2, e000468 (2023).
Gao, F. et al. Spheromers reveal robust T cell responses to the Pfizer/BioNTech vaccine and attenuated peripheral CD8+ T cell responses post SARS-CoV-2 infection. Immunity 56, 864–878.e864 (2023).
Collier, A.-R. Y. et al. Differential kinetics of immune responses elicited by Covid-19 vaccines. N. Engl. J. Med. 385, 2010–2012 (2021).
Liu, J. et al. Vaccines elicit highly conserved cellular immunity to SARS-CoV-2 Omicron. Nature 603, 493–496 (2022).
Lauring, A. S. et al. Clinical severity of, and effectiveness of mRNA vaccines against, covid-19 from omicron, delta, and alpha SARS-CoV-2 variants in the United States: prospective observational study. BMJ 376, e069761 (2022).
Waldman, A. D., Fritz, J. M. & Lenardo, M. J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 20, 651–668 (2020).
Demaria, O. et al. Harnessing innate immunity in cancer therapy. Nature 574, 45–56 (2019).
Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015).
Yarchoan, M. et al. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 17, 209–222 (2017).
Blass, E. & Ott, P. A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 18, 215–229 (2021).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013).
Hu, Z., Ott, P. A. & Wu, C. J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 18, 168–182 (2018).
Çuburu, N. et al. Harnessing anti-cytomegalovirus immunity for local immunotherapy against solid tumors. Proc. Natl Acad. Sci. USA 119, e2116738119 (2022).
Rosato, P. C. et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat. Commun. 10, 567 (2019).
Millar, D. G. et al. Antibody-mediated delivery of viral epitopes to tumors harnesses CMV-specific T cells for cancer therapy. Nat. Biotechnol. 38, 420–425 (2020).
Garrido, F., Aptsiauri, N., Doorduijn, E. M., Lora, A. M. G. & Van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 39, 44–51 (2016).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017).
Sade-Feldman, M. et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 8, 1136 (2017).
Neefjes, J., Jongsma, M. L. M., Paul, P. & Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 11, 823–836 (2011).
Cruz, F. M., Colbert, J. D., Merino, E., Kriegsman, B. A. & Rock, K. L. The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu. Rev. Immunol. 35, 149–176 (2017).
Yu, Y. Y., Netuschil, N., Lybarger, L., Connolly, J. M. & Hansen, T. H. Cutting edge: single-chain trimers of MHC class I molecules form stable structures that potently stimulate antigen-specific T cells and B cells. J. Immunol. 168, 3145–3149 (2002).
Huang, C. et al. Cancer immunotherapy using a DNA vaccine encoding a single-chain trimer of MHC class I linked to an HPV-16 E6 immunodominant CTL epitope. Gene Ther. 12, 1180–1186 (2005).
Hou, X., Zaks, T., Langer, R. & Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 6, 1078–1094 (2021).
Chen, S. et al. Nanotechnology-based mRNA vaccines. Nat. Rev. Methods Prim. 3, 63 (2023).
Tilstra, G. et al. Iterative design of ionizable lipids for intramuscular mRNA delivery. J. Am. Chem. Soc. 145, 2294–2304 (2023).
Zhang, Y., Sun, C., Wang, C., Jankovic, K. E. & Dong, Y. Lipids and lipid derivatives for RNA delivery. Chem. Rev. 121, 12181–12277 (2021).
Han, X. et al. An ionizable lipid toolbox for RNA delivery. Nat. Commun. 12, 7233 (2021).
Hajj, K. A. et al. Branched-tail lipid nanoparticles potently deliver mRNA in vivo due to enhanced ionization at endosomal pH. Small 15, e1805097 (2019).
Rakib, A. et al. Immunoinformatics-guided design of an epitope-based vaccine against severe acute respiratory syndrome coronavirus 2 spike glycoprotein. Comput. Biol. Med. 124, 103967 (2020).
Mao, T. et al. Unadjuvanted intranasal spike vaccine elicits protective mucosal immunity against sarbecoviruses. Science 378, eabo2523 (2022).
Son, E. T. et al. The self-peptide repertoire plays a critical role in transplant tolerance induction. J. Clin. Invest. 131, e146771 (2021).
Dangi, T., Class, J., Palacio, N., Richner, J. M. & Penaloza MacMaster, P. Combining spike- and nucleocapsid-based vaccines improves distal control of SARS-CoV-2. Cell Rep. 36, 109664 (2021).
Poluektov, Y., George, M., Daftarian, P. & Delcommenne, M. C. Assessment of SARS-CoV-2 specific CD4+ and CD8+ T cell responses using MHC class I and II tetramers. Vaccine 39, 2110–2116 (2021).
Ahmed, S. F., Quadeer, A. A. & McKay, M. R. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 12, 254 (2020).
Davenport, B. J., Morrison, T. E., Kedl, R. M. & Klarquist, J. Conserved and novel mouse CD8 T cell epitopes within SARS-CoV-2 spike receptor binding domain protein identified following subunit vaccination. J. Immunol. 206, 2503–2507 (2021).
Parn, S., Savsani, K. & Dakshanamurthy, S. SARS-CoV-2 Omicron (BA.1 and BA.2) specific novel CD8+ and CD4+ T cell epitopes targeting spike protein. ImmunoInformatics 8, 100020 (2022).
Li, Y. et al. Multifunctional oncolytic nanoparticles deliver self-replicating IL-12 RNA to eliminate established tumors and prime systemic immunity. Nat. Cancer 1, 882–893 (2020).
Darvin, P., Toor, S. M., Sasidharan Nair, V. & Elkord, E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp. Mol. Med. 50, 1–11 (2018).
Masopust, D., Vezys, V., Marzo, A. L. & Lefrançois, L. Preferential localization of effector memory cells in nonlymphoid tissue. Science 291, 2413–2417 (2001).
Thome, J. J. et al. Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat. Med. 22, 72–77 (2016).
Rappaport, A. R. et al. Low-dose self-amplifying mRNA COVID-19 vaccine drives strong protective immunity in non-human primates against SARS-CoV-2 infection. Nat. Commun. 13, 3289 (2022).
Arunachalam, P. S. et al. Durability of immune responses to mRNA booster vaccination against COVID-19. J. Clin. Invest. 133, e167955 (2023).
Mentzer, A. J. et al. Human leukocyte antigen alleles associate with COVID-19 vaccine immunogenicity and risk of breakthrough infection. Nat. Med. 29, 147–157 (2023).
Hosomichi, K., Shiina, T., Tajima, A. & Inoue, I. The impact of next-generation sequencing technologies on HLA research. J. Hum. Genet. 60, 665–673 (2015).
Yarchoan, M. et al. Personalized neoantigen vaccine and pembrolizumab in advanced hepatocellular carcinoma: a phase 1/2 trial. Nat. Med. 30, 1044–1053 (2024).
Rojas, L. A. et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 618, 144–150 (2023).
Sheth, R. A. et al. Assessment of image-guided intratumoral delivery of immunotherapeutics in patients with cancer. JAMA Netw. Open 3, e207911 (2020).
Som, A., Rosenboom, J. G., Chandler, A., Sheth, R. A. & Wehrenberg-Klee, E. Image-guided intratumoral immunotherapy: developing a clinically practical technology. Adv. Drug Deliv. Rev. 189, 114505 (2022).
Melero, I., Castanon, E., Alvarez, M., Champiat, S. & Marabelle, A. Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat. Rev. Clin. Oncol. 18, 558–576 (2021).
Sheth, R. A. et al. Intratumoral injection of immunotherapeutics: state of the art and future directions. Radiology 312, e232654 (2024).
Patel, M. et al. 539 Phase 1 study of mRNA-2752, a lipid nanoparticle encapsulating mRNAs encoding human OX40L/IL-23/IL-36γ, for intratumoral (ITu) injection +/- durvalumab in advanced solid tumors and lymphoma. J. Immunother. Cancer 9, A569 (2021).
Bechter, O. et al. 391 A first-in-human study of intratumoral SAR441000, an mRNA mixture encoding IL-12sc, interferon alpha2b, GM-CSF and IL-15sushi as monotherapy and in combination with cemiplimab in advanced solid tumors. J. Immunother. Cancer 8, A237 (2020).
Hamid, O. et al. 19O Preliminary safety, antitumor activity and pharmacodynamics results of HIT-IT MEDI1191 (mRNA IL-12) in patients with advanced solid tumours and superficial lesions. Ann. Oncol. 32, S9 (2021).
Xue, Y. et al. LNP-RNA-engineered adipose stem cells for accelerated diabetic wound healing. Nat. Commun. 15, 739 (2024).
Schoenmaker, L. et al. mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int. J. Pharm. 601, 120586 (2021).
Zhang, Z. et al. Humoral and cellular immune memory to four COVID-19 vaccines. Cell 185, 2434–2451.e2417 (2022).
Acknowledgements
The authors acknowledge the use of the core facility provided by the Campus Microscopy & Imaging Facility at Ohio State University. Electron microscopy was performed at the Center for Electron Microscopy and Analysis (CEMAS) at The Ohio State University. The authors thank Biorepository and Pathology CoRE Laboratory at the Icahn School of Medicine at Mount Sinai for support. Y.D. acknowledges the support from the Maximizing Investigators’ Research Award R35GM119679 and R35GM144117 from the National Institute of General Medical Sciences as well as funds from the Icahn School of Medicine at Mount Sinai. B.D.B. acknowledges the support (R01DK138025) from the National Institute of Diabetes and Digestive and Kidney Diseases. D.J.I. is an investigator of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Contributions
Y.X., X.H., Y.Zhong, and Y.Zhang conceived the work, performed the experiments, analyzed the data and wrote the paper. S.D., H.L., S.W., Z.L., M.T. and K.G. contributed to animal studies. L.W. and C.W. contributed to lipid synthesis. D.D.K. and D.C. contributed to mRNA synthesis. B.D. and D.W.M. contributed to Cryo-EM imaging. E.P. and J.D. contributed to the vibratome. P.H. and N.M.T. contributed to the collection of human brain tissues. B.D.B. and M.M. contributed to data analysis and manuscript editing. D.J.I. and R.W. contributed to the saRNA plasmid. Y.D. conceived and supervised the project and wrote the paper. The final paper was edited and approved by all authors.
Corresponding authors
Ethics declarations
Competing interests
Y.D. is a scientific advisor in Arbor Biotechnologies. Y.D. is a co-founder and holds equity in Immunanoengineering Therapeutics. D.J.I. and R.W. are scientific advisory board members and hold equity in Strand Therapeutics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dan Peer and Thorbald Van Hall for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xue, Y., Hou, X., Zhong, Y. et al. LNP-RNA-mediated antigen presentation leverages SARS-CoV-2-specific immunity for cancer treatment. Nat Commun 16, 2198 (2025). https://doi.org/10.1038/s41467-025-57149-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57149-2
This article is cited by
-
Nanoparticle–Host Interactions: The Impact of Physiological and Pathological Factors on Biodistribution, Immune Processes, and Translational Challenges
Cellular and Molecular Bioengineering (2026)
-
Technological advancements in antibody-based therapeutics for treatment of diseases
Journal of Biomedical Science (2025)
-
Antimicrobial peptide delivery to lung as peptibody mRNA in anti-inflammatory lipids treats multidrug-resistant bacterial pneumonia
Nature Biotechnology (2025)
