
Abstract
Small intestinal neuroendocrine tumors (siNETs) are rare bowel tumors arising from malignant enteroendocrine cells, which normally regulate digestion throughout the intestine. Though infrequent, their incidence is rising through better diagnosis, fostering research into their origin and treatment. To date, siNETs are considered to be a single entity and are clinically treated as such. Here, by performing a multi-omics analysis of siNETs, we unveil four distinct molecular groups with strong clinical relevance and provide a resource to study their origin and clinical features. Transcriptomic, genetic and DNA methylation profiles identify two groups linked to distinct enteroendocrine differentiation patterns, another with a strong immune phenotype, and the last with mesenchymal properties. This latter subtype displays the worst prognosis and resistance to treatments in line with infiltration of cancer-associated fibroblasts. These data provide insights into the origin and diversity of these rare diseases, in the hope of improving clinical research into their management.
Similar content being viewed by others


Introduction
Neuroendocrine cells are present throughout the body where they secrete hormones that trigger long- and short-range cellular responses. In the intestinal tract, they are known as enteroendocrine cells (EECs). EECs are involved in digestion and nutrient uptake, as well as in intestinal motility1,2. Various EEC subtypes have been described and were recently analyzed at the single-cell level1,3,4,5. The traditional nomenclature categorizes EECs according to their localization and the hormone(s) they secrete: A cells (secrete grelin and nestatin), G cells (gastrin), enterochromaffin-like-cells (histamine) and P cells (leptin) were described in the stomach; N cells (neurotensin), M cells (motilin), I cells (cholecystokinin), S cells (secretin), and K cells (glucose-dependent insulinotropic peptide) in the small intestine; L cells (glucagon-like peptides 1 and 2, peptide YY, oxyntomodulin) in the colon; and D cells (somatostatin) and enterochromaffin cells (serotonin) are expressed throughout the gastro-intestinal tract6.
Neuroendocrine tumors of the small intestine arise from EECs and are classified by the WHO as neuroendocrine tumors (NETs) or neuroendocrine carcinomas (NECs) of the digestive system. The former category comprises three groups based on histological analysis of cell proliferation: NET-G1 (Ki67 index <3%), NET-G2 (3%-20%), and NET-G3 ( > 20%) constituted by well-differentiated neoplasms. Conversely, NECs are poorly differentiated, clinically very aggressive and associated with poor patient survival. SiNETs present indolent growth but patients frequently relapse or present metastases at diagnosis, with a 5-year survival rate of 69% when metastatic, even following multiple lines of treatment7. SiNETs also secrete neurotransmitters, like serotonin, leading to the development of a carcinoid syndrome and carcinoid heart disease (CHD), which can result in patient death if no anti-secretory treatment and rapid surgical replacement of the heart valves is conducted8. Recently, omics-based analyses of NECs identified two subgroups: a ductal and an acinar type, with a clear difference in neuroendocrine origin in the same organ9. Conversely, siNETs are still considered as a unique pathology both at the molecular and clinical levels, and are thus treated as such. SiNETs are reported to be poorly mutated and there is an unusual absence of somatic driver mutations and of infiltration by immune cells10,11,12. However, the section of the gut between the duodenum, jejunum and ileum has scarcely been analyzed, despite the fact that (i) EECs are spatially different, for instance gastrin-secreting cells are only located in the duodenum, and that (ii) 30–50% of patients present multiple forms of the pathology13 from 2 to over 100 tumors, which may imply differences between these tumors and their cell-of-origin. We thus wondered whether different siNET subtypes could be identified.
We herein conduct a large-scale analysis of a cohort of human siNETs from 122 patients by integrating data from (i) RNA sequencing of 206 separate intestinal samples or metastases, (ii) DNA methylation arrays of 200 samples, (iii) genotyping arrays of 183 samples and (iv) whole genome sequencing (WGS) of 37 samples. We unveil several relevant subgroups of the pathology in terms of clinical response by depicting molecular portraits at different omics levels. More specifically, we identify a group of well-differentiated tumors correlated with good patient outcome, and a mesenchymal profile predictive of poor prognosis and weak response to treatments linked with strong cancer-associated fibroblast (CAF) infiltrations that we demonstrate as pro-tumoral. This study argues in favor of considering siNETs as a group of tumors comprising different subtypes, useful for their clinical management, and provides a valuable resource to study human tumoral EECs and siNETs, offering practical insights for the research community.
Results
A valuable resource for studying siNETs
In order to analyze the clinical and biological diversity of siNETs, we collected 219 samples including primary tumors (from the duodenum, jejunum or ileum; n = 170), invaded mesenteric lymph nodes (n = 22), liver metastases (n = 8), and others (blood, adjacent normal tissues, adenomas, normal lymph node; n = 19), from a cohort of 122 patients including cases with multi- or unifocal forms (Fig. 1a; Supplementary Figs 1, 2). All clinical parameters, including overall survival (OS) and progression-free survival (PFS), were documented and samples were verified by pathologists for the expression of typical NET markers, such as Chromogranin A (CgA), to confirm diagnosis (Fig. 1b, c). To analyze the clinical diversity of siNETs, we first performed a multiple correspondence analysis (MCA) based on 20 clinical parameters of the cohort such as age, sex, number of metastases and their location (Supplementary Figs. 1a–c). This clearly showed that (i) many clinical parameters were not discriminative in the two first axes of the MCA (age, gender, tumor size, proliferative status…) (Supplementary Figs. 1a–c), albeit (ii) patients with a functional tumor (secreting hormones) displayed an uncontrolled carcinoid syndrome (p < 0.001) associated with the detection of CgA in blood at the time of surgery (p < 0.001) and the presence of metastases (p = 0.015) (Fig. 1d). Moreover, these parameters were also strongly correlated with worse OS and PFS, highlighting the capacity of the database to link clinical and biological features (Fig. 1d).

A valuable resource for the study of siNETs. a Schematic representation of the siNET cohort. SNP (Single Nucleotide Polymorphism); FFPE (Formalin-Fixed Paraffin-Embedded). Created with BioRender.com. b Representative hematoxylin and eosin (H&E) and Chromogranin-A immunohistochemistry (IHC) staining for pT1, pT2 and pT3 statuses. All 122 cases have undergone H&E staining and Chromogranin-A IHC for diagnosis. Scale bar 1 mm. c Table of the clinical characteristics of the siNET cohort. WHO (World Health Organization); G (grade); ENETS (European Neuroendocrine Tumor Society); UICC (Unio Internationalis Contra Cancrum); LN (Lymph Node); SRI (Somatostatin Receptor Imaging); CI (Confidence Interval); NR (Not Reached) d left panels: representation of four out twenty clinical variables used for the multiple correspondence analysis (MCA) in the first two planes (functionality, uncontrolled carcinoid syndrome, Chromogranin A (CgA) dosage at surgery time and number of metastatic sites variables) (see Supplementary Fig.1). Right panels: Kaplan-Meier, log-rank test and Cox proportional hazards regression model methods were used to study overall (OS) and progression-free (PFS) survival for the four selected clinical features. The shaded areas represent 95% confidence intervals around the curves; p-values are indicated below the graphs.
Human siNETs comprise 4 different molecular traits
The disparity in clinical parameters and correlations obtained with patient survival led us to speculate that siNETs may comprise different subtypes. To test this hypothesis, we initially performed adequate quality controls (QCs), before subjecting samples to mRNA sequencing. Of note, there was no correlation between how long specimens had been frozen and their quality. To limit bias due to the presence of several samples from the same patient (in multifocal forms), we only analyzed one sample per patient (n = 111, discovery cohort), namely the largest tumor, which would have been used at diagnosis to characterize tumor stage. We thus performed an unsupervised clustering analysis by selecting the top 10% variant protein-coding genes among the whole discovery cohort. Consensus clustering analysis (CCP) indicated that the optimal number of gene clusters was four. These gene clusters were then identified by Leiden clustering, projected into the UMAP first plan, and were submitted to pathway enrichment analysis (Fig. 2a–c; Supplementary Data 3). In order to link tumors with these four biological phenotypes, a ssGSEA scoring system was used to label all samples as either positive or negative for the corresponding representative gene cluster (Fig. 2d bottom tracks; Supplementary Figs. 2a–d). Differential expression analyses were then carried out between positive and negative samples regarding the four gene clusters, in order to comprehensively characterize biological functions underlying each of them. The first cluster (Epithelial Cluster or Epi, green) included 431 genes and was characterized by an enrichment in intestinal epithelial brush border cells displaying enterocyte features (Fig. 2c, d; Supplementary Figs. 2a, 3a; Supplementary Data 4). The second cluster (Vesicular Cluster or Vesi, red) comprised 913 genes and displayed an enrichment in vesicular transport pathways, as well as in synaptic and neural features, and co-occurred with classical EEC markers, like neurotransmitters (Fig. 2c, d; Supplementary Figs 2b, 3b; Supplementary Data 5). The third cluster (Immune, blue) was enriched in immune cell markers (124 genes) (Fig. 2c, d; Supplementary Figs. 2c, 3c; Supplementary Data 6). The last cluster (Mesenchymal Cluster or Mes, gold) encompassed 515 genes and displayed an enrichment in pathways related to extracellular matrix remodeling and epithelial to mesenchymal transition (EMT), and also exhibited fewer neuroendocrine markers (Fig. 2c, d; Supplementary Figs. 2d, 3d; Supplementary Data 7). Survival analysis showed that a positive Mes status was associated with the worst OS (p = 0.002). Conversely, a positive Vesi status was predictive of a better OS (p = 0.055) (Fig. 2e). No difference in OS was observed in the Epi and Immune signatures. We confirmed these findings in the entire cohort (Supplementary Fig. 2e). Hence, our analyses highlight four transcriptomic signatures in siNETs that relate to specific clinical outcomes, expanding current knowledge in this area.

Exploring the transcriptional landscape of siNETs by RNA-sequencing. a Determination of the optimal number of gene clusters in the siNETs cohort with consensus clustering based on the 10% most variant genes among the discovery cohort of siNETs. CDF (Cumulative Distribution Function) b Leiden clustering analysis (w/ 4 clusters) projected in UMAP-2D plan. c Pathway enrichment analysis of the 4 siNETs cluster described in (a, b) (epithelial in green, vesicular in red, immune in blue and mesenchymal in gold). Adjusted two-tailed p-values (hypergeometric test) are shown on each bar (BH: Benjamini-Hochberg). d Hierarchical clustering (clustering method: Ward’s; distance: Euclidean): genes (row) are split according to the Leiden gene expression clusters: epithelial (green), vesicular (red), immune (blue), mesenchymal (gold). Recapitulative ssGSEA score of each cluster is indicated as bottom annotation (see Supplementary Fig. 2). TPM (Transcripts Per Million). e Kaplan-Meier, log-rank test and Cox proportional hazards regression model methods were used to study overall (OS) survival for epithelial, vesicular, immune, mesenchymal statuses respectively in green, red, blue and gold. The shaded areas represent 95% confidence intervals around the curves; the median survival rate is represented by dotted line; p-values are indicated below the graphs.
A cell-of-origin phenotype for tumor enteroendocrine cells?
We were initially able to determine that tumors of the duodenum were transcriptionally different from those of the ileum, implying that the localization of EECs is important, and reinforcing the hypothesis that siNETs are not a single and homogeneous population (Supplementary Fig. 3e). We then investigated whether siNET subtypes and representative signatures arose from a single type of EECs using ‘exclusive’ signatures previously obtained by single cell RNA sequencing analysis of normal cells of the intestine14,15. Some groups have shown that the different types of EECs derived from LGR5+ stem cells at the bottom of the crypts. Two differentiations could then take place, separating enterochromaffin and L, M, N, D, P, I and K type EECs5,6,16. Patients expressing the Epi signature (i.e., depicted by ssGSEA scores- bottom lines) displayed an enrichment in enterocyte/goblet cell precursor signatures, whereas a positive Vesi status was associated with enterochromaffin properties (Fig. 3a). The Mes signature was correlated with different types of progenitors with a more “stem-like” phenotype (Fig. 3a). We analyzed the expression of transcription factors previously described in the literature as important in intestinal differentiation. We identified that Vesi cells could be characterized as LMX1A+; ATOH1- which confirms their advanced enteroendocrine state of differentiation. Epi cells were defined as ATOH1+ which seems to confirm the differentiation pathway that exists for normal EECs. Mes cells seemed to correspond more to progenitor cells, defined as DCLK1+. Duodenal cells formed a specific cluster defined by the ARX+, highlighting the specificity of this subgroup compared to other EECs (Fig. 3b). Moreover, we analyzed the transcriptional expression of hormones associated with EEC functions, as this was previously used as a basis to dichotomize two normal types of EECs6,16. We unveiled a clear distinction between the Epi and Vesi signatures. The former was associated with neurotensin, pro-glucagon, PYY, cholecystokinin and somatostatin expression, likely corresponding to an EEC origin, whereas the latter was associated with the enzyme that generates serotonin TPH1, PCSK1 and NUCB2, suggesting that these tumors develop from enterochromaffin cells (Fig. 3c). Interestingly, the single expression of PCSK1, pro-glucagon (GCG) or their absence was sufficient to discriminate between the Epi, Vesi and Mes signatures (Fig. 3d), as the latter was associated with the complete absence of hormone expression, which may suggest a loss of neuroendocrine characters consistent with a more stem/dedifferentiated phenotype (Fig. 3c, d). Overall, these results suggest that parallel differentiations that exist in normal EECs persist during tumorigenesis. The overall clinical management of these groups is therefore questionable, since only those least involved in differentiation showed a poor OS (i.e., the Mes group that does not express hormones).

siNETs tumors are defined by different precursors and/or types of differentiation. a Unsupervised clustering of ssGSEA scores for fetal small intestine single-cell RNASeq-based cell type signatures in the full siNETs cohort (n = 206). b Unsupervised clustering of gene expression of transcription factors, described as essential for intestinal differentiation, across the siNETs cohort. ATOH1 (Atonal BHLH Transcription Factor 1); ARX (Aristaless Related Homeobox); DCLK1 (Doublecortin Like Kinase 1); LMX1A (LIM Homeobox Transcription Factor 1 Alpha); ssGSEA (Single Sample Gene Set Enrichment Analysis). c, d Unsupervised clusterings of gene expression of endocrine hormones or converting enzymes: full list (c) or short list (d) across the entire siNETs cohort. CCK (Cholecystokinin); GAST (Gastrin); GCG (Pro-glucagon: Glucagon-like peptide 1 et 2, Oxyntomodulin); GHRL (Ghrelin); GIP (Gastric inhibitory polypeptide); HDC (Histidine decarboxylase); LEP (Leptin); MLN (Motilin); NTS (Neurotensin); NUCB2 (Nesfatin-1); PCSK1 (prohormone convertase 1 for glucagon-like peptide 1 et 2, oxyntomodulin); PYY (Peptide YY); SCTR (Secretin); SI (Small Intestine); SST (Somatostatin); TPH1 (Tryptophan Hydroxylase 1, biosynthesis of serotonin). Clustering method: Ward’s; distance: Spearman. Recapitulative ssGSEA score of each gene cluster (epithelial, vesicular, mesenchymal and immune) is indicated as bottom annotation. LN (Lymph Node); MN (Mesenteric Node); T (Tumor); TPM (Transcripts Per Million).
SiNETs are poorly mutated but specific chromosomal rearrangements predict outcome
SiNETs were reported to display a low tumor mutational burden and no obvious driver mutations12,17. We performed WGS analyses of normal tissue and tumor tissue from multi- or unifocal patients, metastatic or not (n = 37). We identified a few genes mutated in at least 2 samples, including BCOR, FAT1, MUC5AC and MCAM, and detected previously described genes like CDKN1B (Fig. 4). Overall, we found no correlation between driver mutations or clinical indications, supporting the uniqueness of these pathologies. In addition, we took advantage of WGS analyses to investigate chromosomal rearrangements that are recurrent in siNETs18,19,20, and revealed an enrichment in chr18 deletion and chr4, 5, 7, 10, 14 and 20 gains among WGS samples (Fig. 4). We thus conducted an extensive copy number alteration (CNA) detection analysis on 183 samples including 99 samples from the discovery cohort, from which we established a global CNA landscape of siNETs using genotyping arrays (Fig. 5a). We observed chromosomal rearrangements, including some previously reported, and identified a peak of deletion in the RB1 locus, a key suppressor gene in other NET pathologies21 but undescribed in siNETs (Fig. 5a). We then performed a hierarchical clustering of CNAs of the discovery cohort and reported their status for all tumors. Based solely on chromosome rearrangements, four phenotypes were observed, the first completely lacked rearrangement, the second encompassed siNETs with exclusive loss of chromosome 18, the third with gain in chromosomes 4, 5, 7, 10, 14 and 20, and the last, in which loss of chromosome 18 was accompanied by other deletions (Fig. 5b). The first group likely presented very few cancer cells, as detected by the ASCAT model and putative existing rearrangements were not detectable. Nevertheless, we maintained this group in order to conduct unbiased analyses of our patients. Survival was neither correlated with the level of global chromosome rearrangement (Fraction genome altered - FGA), nor with loss of chromosome 18, a major event associated with the disease20 (Fig. 5c; Supplementary Fig. 4a). Nonetheless, gain of chromosomes 4, 10 and 14 were predictive of poor outcome (p = 0.013, p = 0.025, p = 0.0085, respectively), and seemed to be associated with the Mes signature, especially the amplification of chromosomes 10 and 14, but not with other clusters (Fig. 5d; Supplementary Figs. 4b–e). In line with these data, we created a chromosome amplification score based on the concomitant gain of chromosomes 4, 10 and 14. This score was predictive of a worse prognosis in OS and PFS (p < 0.001; p = 0.026 respectively) (Fig. 5d), making this score a potential tool for patient diagnosis using Fluorescence In Situ Hybridization (FISH). Regarding our 4 representative gene signatures, no rearrangement, loss or gain was significantly associated with a given siNET signature, except for the Mes signature which was associated with chromosome 10 and 14 rearrangements, highlighting the key role of these events in siNETs of poor prognosis (Supplementary Fig. 4f).

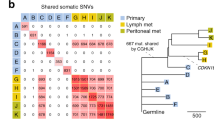
Human siNETs are poorly mutated without clear genetic drivers. OncoPrint of DNA somatic alterations from 24 tumors sequenced in whole genome. The percentage on the right indicates the mutation frequency of each gene across samples. OncoPrint is split in 3 panels: frequently altered genes across WGS cohort (top), frequently altered genes among TCGA cancer pathways (middle), frequently altered whole chromosomes (bottom). Mutation counts (substitutions and small indels) and COSMIC mutational signatures distribution per sample were inferred from whole genome data. Phylogenetic relationships between primary and metastasis samples are indicated with a solid line above the tissue annotation track. Tumor purity, ploidy and fraction of genome altered (FGA) were estimated from FACETS analysis. LOH (Loss Of Heterozygosity); MN (Mesenteric Node); T (Tumor).

siNETs can be defined by specific chromosomic rearrangements. a Frequency plots of DNA gains (yellow) and losses (blue) in the discovery cohort. RB1 (Retinoblastoma 1). b Unsupervised hierarchical clustering of copy number alterations in the ileum discovery cohort. A 20260 genes x 99 samples matrix encoded with −1 (loss), 0 (no alteration), +1 (gain) was used (see Methods). Tumor purity, ploidy and fraction of genome altered (FGA) were estimated from ASCAT analysis. Recapitulative ssGSEA score of each gene cluster (epithelial, vesicular, mesenchymal and immune) is indicated as bottom annotation. CNA (Copy Number Alterations). Clustering method: Ward’s; distance: binary. c, d Kaplan-Meier, log-rank test and Cox proportional hazards regression model methods were used to study overall (OS) and progression-free (PFS) survival for chr18.del and chr4.10.14.gains statuses. chr4.10.14.gains was set to YES if at least one of chromosomes 4, 10 or 14 was gained, NO otherwise. The shaded areas represent 95% confidence intervals around the curves; p-values are indicated below the graphs.
DNA methylation landscape of siNETs
As described above, unsupervised analysis of siNETs was conducted in the discovery cohort (n = 98) using the top 1000 most variable CpG probes. This resulted in the identification of four patient DNA methylation (DNAm) consensus clusters, designated as the FGA-enriched, epithelial-enriched, hypomethylated, and unifocal-enriched clusters, containing 18, 23, 36, and 21 samples, respectively (Fig. 6a, Supplementary Data 8). Clustering also resulted in 4 groups of probes, designated DNAm A-D. The patient DNAm consensus clusters exhibited significantly different mean levels of DNA methylation (Fig. 6b, c). Notably, samples in the unifocal-enriched cluster exhibited significantly lower average DNAm levels on CpGs in probe cluster B compared to the other consensus clusters, suggesting that DNA methylation levels on these CpGs may allow the identification of multifocal disease (Fig. 6a, c).

Methylation refines the siNET groups. a Unsupervised k-means consensus clustering was found to be optimal with 4 groups for the top 1,000 most variable CpGs methylation probes (in row: A, B, C and D) and samples (in column, Sample.Methyl.Clust: epithelial-enriched, hypomethylated, unifocal-enriched and FGA-enriched). Clustering method: Ward’s; distance: Euclidean. Recapitulative ssGSEA score of each gene cluster (epithelial, vesicular, mesenchymal and immune), chr18.del and chr4.10.14.gains statuses together with methylation subtypes are indicated as bottom annotations. b Boxplots of the methylation levels (mean beta value) of the 1000 most variants probes (left) and each of the four probe clusters (right: A-D) across the four k-means based methylation clusters (hypomethylated, n = 41; epithelial-enriched, n = 18; unifocal-enriched, n = 26 and FGA-enriched, n = 22). Two-tailed exact p-values were determined by Kruskal-Wallis test. Boxplots: center line = median, box range 25th–75th percentile, minimum/maximum denoted by whiskers. c Barplots for co-occurrence analysis of methylation clusters vs expression clusters, FGA levels, chr18 deletion, chr4.10.14 gains and tumor type (unifocal or multifocal). Significance was determined by two-tailed Fisher’s exact test.
The patient DNAm consensus clusters were significantly associated with unifocality, deletions in chromosome 18, gains in chromosomes 4, 10, and 14, as well as three of the four siNET groups identified above based on unsupervised clustering of transcriptomics results (Fig. 6b, c). The hypomethylated and unifocal-enriched clusters overlapped extensively with the Vesi-cluster (26/36 of the hypomethylated and 20/21 of the unifocal-enriched clusters were also classified as Vesi-positive), while the epithelial-enriched (18/23) and FGA-enriched clusters (12/18) were primarily comprised of patients classified as immune/epithelial-positive (Fig. 6a). Chr18 deletions were frequently observed in the hypomethylated cluster (26/36). While it may be tempting to postulate that deletions in Chr18 may be responsible for the observed lower levels of DNA methylation in this DNAm consensus, we noted that the CpG mapping to chromosome 18 made up only 26 of the 1000 most variable probes used in this consensus clustering. Thus, it is unlikely that chromosome deletions are directly responsible for the extensive variation in DNA methylation in the siNET primary cohort. While gains in either of the tested chromosomes (4, 5, 7, 10, 14, and 20) were not associated with any particular siNET subtype, at least one of the above listed chromosome gains was observed in 14/18 patients clustered into the FGA-enriched cluster (Fig. 6b).
Integrated Multi-Omics analysis highlight key findings in tumors
In order to integrate all omics data types into a low-dimensional representation, we performed an unsupervised decomposition of siNET molecular heterogeneity using Multi-Omics Factor Analysis (MOFA)22, with transcriptomic, genomic and epigenetic layers of data (Supplementary Fig. 5a). We identified seven independent latent factors individually explaining more than 10% of variation in at least one molecular layer and some factors displayed strong correlation with clinical features (Supplementary Fig. 5b, c). We focused specifically on factor 1 that globally summarized 63.7% of the variation and which was associated with both OS (HR = 1.95; CI = [1.354, 2.810]; p = 0.000328) and PFS (HR = 1.36; CI = [1.098, 1.68]; p = 0.005) (Supplementary Fig. 5d, e). Enrichment analysis of gene features supporting factor 1, both in terms of gene expression and methylation (promoter, enhancer region and gene body) exhibited an enrichment of enterocyte precursor genes for up-regulated or hyper methylated gene body or hypo-methylated promoter or enhancer features and an enrichment of enteroendocrine/enterochromaffin markers for down-regulated or hypo-methylated gene body or hyper-methylated promoter or enhancer features (Supplementary Fig. 5f, g). This highlighted that a lesser neuroendocrine differentiation is correlated with a poor prognosis, confirming our previous observations.
Multiple tumor forms are not linked with a transcriptional subtype, but express specific markers and are characterized by distinct methylation groups
One of the most intriguing features of siNETs is the formation of multiple forms of tumors. Elias and colleagues elegantly showed that they all arose from independent tumors12. We first confirmed this result after WGS analysis of tumors within the same patient (Fig. 7a). We analyzed the transcriptomic divergences between unifocal and multifocal forms, and identified 239 differentially expressed coding genes (p adj < 0.05). We then established a signature containing 9 genes that clearly discriminated between uni- and multifocal tumors (p adj < 0.01 and |log2(FoldChange)| >1), namely SEMA3E, CAMKK1, NWD2, PLA2G2C, LIPF, mainly expressed in multifocal forms, and NCR1, ATP8B3, PI3KC2G and HOXC10 mainly expressed in unifocal forms (Fig. 7b, c). We were unable to determine any association with the four siNET Epi, Vesi, Immune and Mes signatures, though a slight association was observed between the Vesi signature and unifocal tumors (Fig. 7c). Differential methylation analyses revealed that 836 individual CpG sites and 1115 genomic regions were significantly differentially methylated when comparing unifocal tumors with multifocal forms (Fig. 7d, e; Supplementary Data 9, 10). Genomic regions hypermethylated in the multifocal tumors were associated with 68 unique genes, while hypomethylated regions were associated with 841 unique genes. As highlighted in the heatmap representation, multifocal tumors displayed lower levels of DNA methylation in the DMPs (differentially methylated probes) and DMRs (differentially methylated regions) compared to unifocal tumors (Figs. 6a, 7d, e). Interestingly, the transcriptional expression of several hormones, such as GIP and SST, also discriminated unifocal vs multifocal forms implying that they could be used as a diagnostic test (Fig. 7f).

Omics can define unifocal vs multifocal forms of siNETs a Venn-diagram of somatic point mutations and small indels on the whole-genome scale for available WGS samples of multifocal patients P101, P17, P41 and P68 (patient P68 was analyzed using a tumor as a paired sample). b Volcano plot for multifocal (n = 45) vs unifocal (n = 57) differential gene expression (DE) analysis; p-values are indicated on the y-axis (two-tailed Wald test with Benjamini-Hochberg correction). c Hierarchical clustering based on the 9 highly significantly differentially expressed coding genes ( | log2(FoldChange)|>1 & pval.adj < 1e-2). ATP8B3 (ATPase Phospholipid Transporting 8B3); CAMKK1 (Calcium/Calmodulin Dependent Protein Kinase Kinase 1); HOXC10 (Homeobox C10); LIPF (Lipase F, Gastric Type); NCR1 (Natural Cytotoxicity Triggering Receptor 1); NWD2 (NACHT And WD Repeat Domain Containing 2); PIK3C2G (Phosphatidylinositol-4-Phosphate 3-Kinase Catalytic Subunit Type 2 Gamma); PLA2G2C (Phospholipase A2 Group IIC); SEMA3E (Semaphorin 3E). d Volcano plot for multifocal (n = 44) vs unifocal (n = 54) differential methylation (DM) analysis; p-values are indicated on the y-axis (two-tailed T-test with Benjamini-Hochberg correction). e Hierarchical clustering based on the 836 significantly differentially methylated probes (delta(Beta)>10%, pval.adj < 5e-2). f Multifocal (n = 45) vs unifocal (n = 57) boxplots of gene expression for endocrine hormones or converting enzymes (log2TPM). GAST (Gastrin); GCG (Pro-glucagon: Glucagon-like peptide 1 et 2, Oxyntomodulin); GIP (Gastric inhibitory polypeptide); HDC (Histidine decarboxylase); NTS (Neurotensin); SST (Somatostatin). Boxplots: center line = median, box range 25th–75th percentile, minimum/maximum denoted by whiskers. Significance was determined by Mann-Whitney U tests. Clustering method: Ward’s; distance: Spearman. Recapitulative ssGSEA score of each gene cluster (epithelial, vesicular, mesenchymal and immune), chr18.del and chr4.10.14.gains statuses together with methylation subtypes are indicated as bottom annotations. FGA (Fraction genome altered); TPM (Transcripts Per Million).
The tumor microenvironment influences siNET subgroups
The tumor microenvironment (TME) plays a major role in tumor growth, including infiltrating immune cells and cancer-associated fibroblasts. To investigate the role of the TME in siNETs, we conducted several deconvolution methods to analyze the subpopulations within the TME23,24,25,26. Three different profiles could be isolated, and remarkably they segregated with the 4 molecular signatures (Fig. 8a). A first profile was closely correlated with the Vesi signature, the second myeloid profile segregated with the Epi signature, and the last profile containing few immune cells, abundant CAFs and endothelial cells was close to the Mes signature (Fig. 8a). We also observed a group constituted of about 10% of patients who highly expressed immune checkpoint markers (ICM), strongly correlated with the Immune signature and who would be the most likely to respond to these therapies (Supplementary Fig.6). Of note, Epi and Vesi did not express the same ICM, which also underlines the difference between these molecular groups.

CAFs favor severe forms of siNETs. a Unsupervised clustering of MCPcounter scores estimating the abundance of immune and stromal cells infiltrate across the full siNETs cohort (n = 206). Clustering method: Ward’s; distance: Spearman. X-cell derived Immune and Stromal Scores, recapitulative ssGSEA score of each gene cluster (epithelial, vesicular, mesenchymal and immune), chr18.del and chr4.10.14.gains statuses together with methylation subtypes are indicated as bottom annotations. LN (Lymph Node); MN (Mesenteric Node); T (Tumor). Clustering method: Ward’s; distance: Spearman. b Representative anti-alpha-smooth muscle actin (αSMA) immunohistochemistry (IHC) staining illustrating CAF infiltration of siNETs (low CAF score vs high CAF score). Twenty-one tumors have undergone αSMA IHC. Scale bar 100 µm. c Prognostic value for OS (Overall Survival) and PFS (Progression Free Survival) of the CAFs score in siNETs tumors. A cut-off in EPIC-related CAF scores distribution was applied (score=0.025) to label samples either as low (below cut-off) or high (above cut-off) for CAF infiltration. Kaplan-Meier, log-rank test and Cox proportional hazards regression model methods were used to study overall (left) and progression-free (right) survival. The shaded areas represent 95% confidence intervals around the curves; the median survival rate is represented by dotted line; p-values are indicated below the graphs. d Schematic representation of purification, culture and production of conditioned media from patients’ siNETs associated fibroblasts and transfer to the GOT-1 cell line. Created with BioRender.com. e Analysis of 2-months proliferation of GOT-1 cells treated with CAF conditioned medium (p = 0.0286). Two-tailed exact p-value was calculated using a Mann-Whitney U test. Error bars represent means ± SEM, n = 4. Source data are provided as a Source Data file. RLU (Relative Light Units).
Next, we focused on the most aggressive Mes signature. We speculated that it was linked either to EMT activation or to the presence of mesenchymal tissue, and studied the genes contributing to this signature like ZEB1, a widely recognized transcription factor with a key role in EMT27. By IHC we observed the presence of nuclear ZEB1 in fibroblasts but not cancer cells, indicating that EMT is likely not involved in this particular phenotype (Supplementary Fig. 7a). The mesenchymal cluster was also defined by the expression of smooth muscle-related genes. Estimation of cancer cell purity in the tumors with a positive Mes status indicated that this was not due to contamination of healthy tissue. CAFs are known to express these markers, in particular the smooth muscle actin gene. Interestingly, the CAF signature was strongly correlated with the Mes signature, which displayed abundant CAF infiltration observed in bioinformatics analysis and confirmed by IHC on patient tumor samples (Fig. 8b; Supplementary Figs. 7b, c), and this was predictive of a strong decrease in long-term survival and PFS, reflecting treatment resistance (Fig. 8c). To analyze this finding functionally, we isolated CAFs from 4 different siNETs from patients and cultured them (Fig. 8d). Given that CAF secretions are considered to be at the origin of tumor progression, we cultured the only available siNET cell line, GOT-1, in conditioned medium from these CAFs and analyzed tumor proliferation at 2 months. CAF secretions significantly increased siNET proliferation (Fig. 8e), unveiling the relevance of our resource dataset both in terms of diagnosis and therapy, as anti-drugs are already under development to target CAFs and could thus constitute siNETs treatments.
Discussion
In the present paper, we classified a cancer so far considered as a single pathology in four major molecular subgroups with high clinical significance. We identified a so-called Vesicular group, with the most differentiated enteroendocrine cells phenotype that had retained strong neuroendocrine features with typical enterochromaffin marker expression (i.e., serotonin). This group displayed an overall signature related to vesicle transport and secretion/synapse and had the best OS. The Epithelial-group displayed a brush border phenotype which may be an intermediate state of neuroendocrine differentiation, arising from a distinct differentiation pathway as attested by the different hormones expressed. The Immune group was characterized by an immune cell infiltrate in the tumor bulk, suggesting that this group is the most likely to respond to immune checkpoint inhibitors in future clinical treatments28. This group was highly correlated with the Epi-cluster, from which it derives. The Mesenchymal group had the worst prognosis. This cluster was characterized by a very low hormonal secretion with a more “stem-like” phenotype associated with progenitor markers. This phenotype implies that resistance to chemotherapies is genetically intrinsic to the primary tumor and not acquired following treatment cycles -i.e., the gene signature of the removed primary tumor was predictive of death and patient-free survival 10 years after surgery. Of note, the Mesenchymal subtype was not associated with any of the tested clinical parameters. The Mes tumor microenvironment was characterized by the presence of CAFs and by an overall absence of immune infiltrate. We were able to determine by RNA sequencing and IHC analyses that this CAFs infiltration was correlated with a very poor patient prognosis. We trust that this parameter could become a relatively simple criterion for pathologists to predict severe siNET cases at the time of surgery. Indeed, specific markers of CAFs have been widely described29,30, however the description of these CAF subtypes was beyond the scope of our study. Therapies targeting CAFs also seem promising in this group of patients31, as we were able to functionally show by isolating patient CAFs that they promote the growth of siNETs, making them therapeutic targets for future studies. Further analyses to improve our understanding of tumor heterogeneity, for example using single cell/spatial transcriptomic technologies, will be necessary to clarify these mechanisms in mixed groups, to understand if EMT or CAFs are activated/infiltrated only in some spatially-defined compartments of the tumors. These same studies will be important to define specific markers of differentiation of the different tumor subtypes, such as specific expression of transcription factors, which we determined within bulk RNAseq and may therefore be non-specific to tumor cells. The results of our current study did not yield statistically significant findings regarding metastatic lesions. This outcome is likely attributable to the limited sample size, which constrained the statistical power of our analysis. The modest number of samples in our study may have prevented the detection of meaningful patterns or correlations that could elucidate the characteristics and behaviors of metastatic lesions. Given these limitations, it is imperative to conduct further research with larger cohorts to obtain more robust and generalizable data. Increased sample sizes will enhance the ability to discern subtle differences and provide a more comprehensive understanding of the metastatic process.
Our study raises several puzzling issues, including the differences observed in other NETs in terms of etiology and therapies32,33. The absence of recurrent driver mutations specific to siNETs implies that the role of chromosome rearrangements in the pathology must be decisive. These did not segregate randomly (chr18 del; mixed or gain chromosome 4, 5, 10, 14, 20) but were not related to expression clusters. These rearrangements affect a very large number of genes, making it very complicated to understand the reasons for these modifications. Furthermore, the DNA methylome analysis of siNETs also demonstrated the existence of four distinct subtypes of siNETs which reflected unifocal tumors (mainly Vesi), the four observed transcriptomic clusters and the occurrence of chromosomal 4, 5, 7, 10, 14, and 20 gain and probably explains previous work in the field34. Differentially methylated sites and unsupervised analysis of methylation status indicated that unifocal vs multifocal tumors can be isolated at the molecular level, suggesting two different diseases. Multifocal forms showed characteristic hypomethylation, suggesting that significant global perturbations in the epigenetic regulation of siNETs are involved in the emergence of multifocal tumors.
Overall, our study provides sheds light on the heterogeneity of siNETs. The identification of a subpopulation of patients with a poor prognosis highlights an aspect that contrasts with the perceived homogeneity of these tumors from a pathologist’s point of view. CAFs thus seem to play a major role in the aggressiveness of siNETs, and this will certainly be very important to study in the future. There are, however, some shortcomings, such as the need to place EEC differentiation in a more general context of the intestine with other epithelial cell subtypes35,36. Issues such as inflammation in the small intestine37 and, more generally, the immune system, could not be addressed in our study, nor could intestinal metabolism and the microbiota38,39. Given the comprehensive nature of the dataset we generated, our analysis represents only an initial exploration, and future studies should investigate the role of these events in siNET pathology.
Methods
Statement of ethics
This study follows the laws set by the 1975 WMA Declaration of Helsinki and was approved by both the Medical Ethics Research Committee of Hospices Civils de Lyon (June 14, 2021, No 21_390) and the National Data Protection Commission (November 6, 2015, No. 15-111 of the Commission nationale de l’informatique et des libertés, CNIL). Written information was given and signed by each patient included in the study. Their consent is not required by French law, but patients were informed about their right to withdraw their data from the cohort (Reference Methodology MR-004 according to 2016–41 law dated 26 January 2016 on the modernisation of the French health system).
Human tumor samples
Human samples were collected from a cohort of 122 consecutive patients with small-intestinal neuroendocrine tumors (siNETs). Inclusion criteria were patients who underwent resection of at least one primary siNET (poorly differentiated neuroendocrine carcinoma and non-neuroendocrine tumor were not included), by the same surgeon (GP) between 10/09/1998 and 31/07/2019, in Lyon EURACAN/ ENETS Center of Excellence, according to the French regulations on the protection of persons (French Ethics Committee). Exclusion criteria were patient refusal and insufficient material for molecular analysis. Briefly, based on the previously described operative protocol40, median laparotomy was performed to explore the full length of the small intestine visually and by digital palpation and compression in order to find multiple siNETs. The clinico-morphological and pathological data are summarized in Supplementary Fig. 1a. All participants were included without financial compensation. Only the sex of the participants (not the gender) was presented in this study. The sex of the participants was self-reported.
siNET surgical specimens were reviewed by VH, TF and JYS, three members of the national French TENpath pathological network (Reseau national d’expertise pour le diagnostic anatomopathologique des tumeurs neuroendocrines de l’adulte, familiales et sporadiques). Samples were stored within the “Tissu-Tumorothèque Est” (CRB-HCL, Hospices Civils de Lyon Biobank, BB-0033-00046). The biobank works in strict accordance with French ethical and regulatory issues.
Following patient agreement, primary tumors were obtained after surgery, and were directly frozen before storage. Fresh tissue samples were stored in liquid nitrogen until use and grinded to a fine powder using a cryogenic mill. All Prep DNA/RNA Mini kit (Qiagen, 80204) was then used for a simultaneous purification of genomic DNA and total RNA from a maximum of 30 mg of the same tissue sample, following the manufacturer’s instructions.
Tissue samples for diagnostic purposes were fixed in 10% buffered formalin and embedded in paraffin wax. 4-µm-thick sections were then prepared according to conventional procedures.
Immunohistochemistry
Immunohistochemistry was performed on Benchmark and Discovery systems (Roche). Tissue sections were first subjected to antigen retrieval. The proliferation index of the tumors was determined by counting the number of Ki67-positive nuclei among 1,000 cells. The following antibodies were used: anti-Chromogranin A (Dilution 1/1000; DAK-A3, Dako, M0869, lot 41257389), anti-Zeb-1 (Dilution 1/300; Bethyl IHC-00419, lot #2), anti-Smooth Muscle Actin (Dilution 1/1500 ;1A4, Dako M0851, lot 41415169).
Statistical analyses
All analyses and statistical tests were carried out with the R software (v4.1.0) (R Core Team, 2021). Plots were generated either with R base functions or with ggplot2 R package (v3.3.6). Figures were assembled with GraphPad Prism 9 (San Diego, California), drawings were made using BioRender (BioRender.com) and Servier Medical Art (smart.servier.com). All statistical tests were two-tailed and p-values were corrected, when indicated (“p-adjust”), with the Benjamini-Hochberg method41. Venn diagrams were performed using ggVennDiagram R package (v1.2.2). Multiple correspondence analysis (MCA) was carried out on twenty clinical variables for all 122 patients with ade4 R package (v 1.7-19). Co-occurrence and mutual exclusivity were tested using Fisher’s exact test. Survival analyses, Kaplan-Meier curves and log-rank tests were conducted using survival (v3.4-0) and survminer (v0.4.9) R packages. OS (overall survival) corresponds to the length of time between diagnosis and death (censored by last follow-up date in case of survival), while PFS (progression-free survival) represents the duration between surgery treatment and progression or relapse event (censored by last follow-up date in case of absence of progression or relapse).
Sample selection for discovery cohorts
In order to analyze gene expression, DNA copy-number alterations and DNA methylation across siNET samples in an unbiased manner, we built discovery cohorts composed exclusively of tumor samples, from which we retained only one sample from each patient (in case of multifocal pathology or multi-sampled unifocal patients). The selection was based on (i) the biggest tumor size in case of multifocal disease, (ii) RNAseq, SNP array and methylation array data availability for unifocal patients associated with more than one sample and multifocal cases with equivalent tumor sizes and (iii) the highest SNP array-based tumor purity estimation (through ASCAT analysis) in case of equivalent tumor sample size and data availability. It is of note that lesion size did not differ significantly between unifocal and multiple tumors. The discovery cohorts for each sequencing and array data were thus composed of 111 samples for RNAseq, 99 for SNP array and 107 for methylation (the initial complete cohorts were composed of 206, 183 and 200 samples, respectively, for a total of 219 processed samples. Healthy tissue samples for WGS analysis, adenoma and lymph node were also included in the total number of samples. All these sample-by-sample data, together with the Omics performed, are compiled in Supplementary Data 2.
RNAseq and Whole Genome sequencing (WGS)
RNA quality was assessed on a TapeStation system (Agilent) in the Centre Léon Bérard Genomic Facility in 206 samples. Libraries were then prepared using a TruSeq Stranded mRNA kit (Illumina) following the manufacturer’s recommendations. The key steps include PolyA mRNA capture with oligo dT beads to isolate 1 μg total RNA, cDNA double strand synthesis, and ligation of adaptors, library amplification and sequencing. Sequencing was performed using the NovaSeq6000 Illumina sequencer in 75 bp paired-end in the CRCL genomic facility (Lyon, France). All WGS were generated in Centre National de Recherche en Génomique Humaine - CNRGH (Evry, France) on NovaSeq6000 machines with a 2x151bp paired-end protocol.
RNAseq data processing
Sequencing control metrics were computed using FastQC (v4.0.0)42. For careful QC and batch effect analyses, raw data were aligned on the human genome (GRCh38) with STAR (v2.7.0 f), and RNA control metrics were evaluated using RSeQC (v4.0.0)42,43. Gene expression was then quantified with Salmon (1.4.0) from the raw sequencing reads, using gencode v37 comprehensive annotation set44. Further gene expression analyses were restricted to protein-coding genes.
Gene expression analysis
Starting from log2 transformed TPM normalized data, unsupervised analyses were conducted by selecting the top 10% most variable protein-coding genes as input data. The optimal number of clusters (k = 4) was identified through consensus clustering with the ConsensusClusterPlus R package. Selected genes were then clustered with a Leiden clustering from the leiden R package using “modularity” objective function and setting the resolution parameter to 0.3 to produce k = 4 gene clusters. Principal component analysis (PCA) was completed with the R package ade445. Differential expression analyses were performed on ileal tumor samples (n = 102) using the R package DESeq2 (v1.32.0)46, with the Wald test, sequencing batches correction and apeglm shrinkage estimator (v1.14.0)47 All heatmaps were generated with the Ward.D clustering method, using the R package ComplexHeatmap (v2.8.0)48.
Microenvironment analysis
Microenvironment analysis was conducted using MCPcounter25, Xcell23 and EPICS26 methods through Immunedeconv R package24. Immune checkpoint genes list was obtained from literature review49.
SNP array data processing and analysis
Genotyping of the cohort was performed at the CNRGH. Before genotyping, a QC was systematically performed on the samples, including a quantification in duplicate (Quant-It kits, Thermofisher) and an assessment of the quality of the DNAs (10% of the samples). After QC, DNAs were aliquoted in 96-well plates (JANUS liquid handling robot, Perkin Elmer) for genotyping; sample tracking was ensured by a systematic barcode scanning for each sample. Two DNA positive controls were systematically inserted in a random fashion into the plates. Genotyping was performed on a high throughput Illumina automated platform, using the GSA-MD v3 array and standard automated protocols from Illumina ® (Illumina ®, San Diego, USA). Reading of the chips was performed on iScan+ scanners (Illumina®, San Diego, USA) and primary analysis of the results was done using the GenomeStudio software (Illumina®, San Diego, USA). The analysis of the internal controls provided by Illumina and the randomly distributed positive controls allowed the validation of the technological process. The QC of the genotypes was performed for each marker by measuring the deviation from the expected Hardy-Weinberg genotypic proportions for each individual, by measuring the success rate, the average heterozygosity and detecting duplications and outliers. Low quality probes were removed from the analysis as well as probes localized on chromosome X and Y, resulting in 652339 final probes for analysis. Log-ratio (LR) and B-allele frequency (BAF) signals for each sample were then extracted from the Genome Studio processed files and analyzed through ASCAT R package (v3.0.0, https://github.com/VanLoo-lab/ascat) using hg37 reference files. Tumor purity and tumor mean ploidy estimation for each sample were derived from ASCAT model. Gains and losses were determined upon ASCAT segmentation using a rounded value of tumor mean ploidy as a reference. Reference ploidy was defined as follows: if mean ploidy was lower than 3 then reference ploidy was set to 2. For mean ploidy greater than 3, reference ploidy is the closest integer to mean ploidy.
WGS data processing
Raw sequencing data (FASTQ files) were aligned to the GRCh38 primary assembly (downloaded from GENCODE and PAR regions masked on chrY) of the human reference genome with bwa (v0.7.17) aligner. Duplicate alignments were subsequently marked using biobambam (v2.0.89). To check for putative sample swaps and to confirm relatedness of sequenced DNA samples, identity monitoring was performed with NGSCheckMate (commit 8ea2c04 from https://github.com/parklab/NGSCheckMate). Coverage quality metrics were obtained using mosdepth (v0.2.9).
Point mutations and mutational signatures analysis
Somatic point mutations and small indels ( < 100 bp) were called with Mutect2 from GATK software suite (v4.1.2.0) in a tumor/normal setting. A panel of “normal tissues” of 75 patients sequenced in the same conditions was used to remove systematic sequencing artifacts and variants were annotated with ensembl-vep (v98.3) using its internal cache to identify and account for recurrent technical artifacts as recommended by the Broad Institute (https://gatk.broadinstitute.org/hc/en-us/articles/360035890631-Panel-of-Normals-PON). Per sample mutational signatures deconvolutions were obtained using SigProfilerSingleSample (v0.0.0.27) and COSMIC (v3.3) signatures repertoire.
Copy number alterations analysis (WGS)
Somatic copy number alterations (CNA) from WGS were detected with Facets (0.5.14). Due to the absence of heterozygous SNPs on chrX for male patients, a manual curation was applied in some cases to correct erroneously called homozygous deletions. One copy of chrX for male (normal status) was labelled “hemizygous chrX” in Fig. 5. Reference ploidy was determined as the most represented copy number level. Gains and losses were then called relatively to this reference ploidy.
DNA methylation profiling
DNA methylation profiling was conducted on 200 samples using the Infinium MethylationEPIC BeadChip arrays (HM850K, Illumina) by Diagenode (Seraing, Belgium) (genomic DNA extraction, purity and quantity determination, conversion and processing by microarray). Data were analyzed using the methylkey R package (v1.0, accessible at https://github.com/IARCbioinfo/methylkey). Briefly, raw intensity data files were preprocessed and normalized using the minfi Bioconductor package (v.3.17). Quality checks on all samples profiled were conducted. Beta-values, which measure the proportion of methylated alleles at any given locus, were generated for each CpG site. CpG sites for which detected signals were not significantly different from background levels (p > 0.02) and for which data were missing in >20% of the samples were excluded. Cross-reactive probes, probes located on the X and Y chromosomes and at chromosome locations associated with known SNPs were also excluded, resulting in a final dataset of 792,079 CpG probes.
Methylation consensus clustering
Unsupervised clustering of the top 1000 most variable CpGs by standard deviation in beta-values across the primary cohort was conducted using the Consensus Cluster Plus R package (v. 1.62.0) with Euclidean distance and k-means clustering. The association between clinical features across clusters was tested using Fisher’s exact test, while the difference in DNA methylation levels between clusters was compared using Kruskal-Wallis test.
Differential methylation analysis
Beta-values were transformed to M-values for differential methylation analysis, which was conducted using robust linear regression as implemented in the limma R package (v.3.57.6). Differential methylation analysis between multifocal vs unifocal tumors was conducted on a sub-cohort of 98 ileum samples which excluded normal tissue samples and included only one representative sample for each patient with multifocal tumors. CpG sites were considered to be significantly differentially methylated probes (DMPs) if the difference between multi- and unifocal tumors was significant after false discovery rate (FDR) correction (q < 0.05) and the average between-group difference in beta values was above 0.10. Regional analyses were conducted using the DMRcate package.
Multi‐omics factor analysis (MOFA)
MOFA models were performed using the r/bioconductor package mofa2 version 1.10.0 on 3 omics data: (i) for SNP data, gains and losses matrices (derived from ASCAT) were summarized into chromosome gain and loss ratio (number of probes gained (resp. lost) relative to total number of probes by chromosome); (ii) for RNA expression data, the 5% most variant genes were selected from the log2-abundance matrix; (iii) for methylation profiling, the M-value matrix was split according to 3 modalities (promoter, enhancer and gene body methylation) from IlluminaHumanMethylationEPICanno.ilm10b2.hg19 version 0.6.0 annotations. A probe associated with several modalities was assigned a promoter, enhancer or gene body label in this order of priority. For each modality, the 5 % most variant methylation probes were selected from the input matrix (log2-scale).
Default parameters for model training (number of factors = 15, convergence mode = “slow”, maxiter = “2000”, seed = “42”) were used. We investigated whether any of the inferred latent factors were related to clinico-morphological and pathological data by using Pearson correlation with Benjamini-Hochberg correction.
Survival analysis has been performed using Cox’s proportional hazard model from which the significance of the hazard ratio between the reference and the other levels has been evaluated using Wald tests. We assessed the global significance of the model using the logrank test statistic (R package survival version 3.5-5) and drew Kaplan–Meier and forest plots using the R package survminer (version 0.4.9).
After we ran feature set enrichment analysis with the MOFA version slightly modified of the pcgse function. It computes the statistical association between GO C8 gene sets (msigdbr version 7.5.1) and positive or negative RNA Factors.
Then to have an overview of implication of each modality Gene ontology over-representation test between GO C8 gene sets and gene associated with weight factor 1: > 0.2 RNA expression, > 0.2 methylation gene body, < −0.2 methylation promoter gene, < -0.2 methylation enhancer gene are done.
Pathway enrichment analysis and score computation
Enrichment of lists of genes in specific biological pathways were tested using clusterProfiler (v4.0.2) and msgidbr (v7.4.1) R packages50,51. All tested pathway lists originated from MSigDB (Molecular Signatures Database): Hallmark (H) gene sets52, C5 gene sets (encompassing GO pathways) and C8 gene sets (relative to cell type signatures identified in single-cell RNASeq data). Single sample GSEA (ssGSEA) scores were computed on TPM normalized data through gsva R package53. Gaussian finite models were performed with Rmixmod R package (v2.1.8).
CAFs isolation and conditioned media
Patients’ CAFs were obtained by tumor mincing and subsequent digestion with DNase I and Collagenase I. Fibroblasts were isolated by consecutive trypsinizations and cultured in 10% fetal bovine serum containing RPMI media for at least 3 passages before preparation of the conditioned media.
CAFs were then seeded in Petri dishes, and the culture medium was replaced with 2% FBS medium containing RPMI when the cells reached 80% confluence. Conditioned media were collected 72 hours later, filtered through 0.45 µm pore membranes, aliquoted, and stored at −80 °C until use.
Cell culture and viability assay
GOT1 cells were a kind gift from Ola Nilsson (Sahlgrenska Cancer Center, University of Gothenburg, Sweden) and are usually cultured in 10% fetal bovine serum containing RPMI media with 5 µg/mL of both insulin and transferrin54,55. In our experiment, GOT1 cells were cultured for at least 60 days in our CAFs conditioned media or in a control medium (2% FBS medium containing RPMI) supplemented with 8% FBS, insulin and transferrin.
Ten thousand cells per well were plated in white 96-well plates. Cell viability was evaluated as previously described56 after three days using the CellTiter-Glo® kit (Promega, Charbonnières-les-Bains, France) according to manufacturer’s instructions.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The methylation arrays data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under accession code GSE229203. The sequencing data that support the findings in the article are deposited and available at the European Genome–Phenome Archive (EGA; https://ega-archive.org), which is hosted by the European Bioinformatics Institute (EBI) and the Centre for Genomic Regulation (CRG), through the primary accession study code EGAS50000000642. The transcriptomic [https://ega-archive.org/datasets/EGAD50000000906], WGS [https://ega-archive.org/datasets/EGAD50000000907] and genotyping arrays [https://ega-archive.org/datasets/EGAD50000000905] data are available under restricted access due to ethical and legal reasons. Data use conditions attached to this EGA dataset limits its use to approved users at a specific institution for a specific a health/medical/biomedical project and dictates that useful results should be made available to the wider scientific community. All data are available after agreement of the scientific committee as required by the French law on data protection. Source data are provided with this paper.
Code availability
The code used for analysis and figure generation is available on request. Other requests can be addressed to Dr. Benjamin Gibert (benjamin.gibert@lyon.unicancer.fr).
References
Beumer, J. et al. High-Resolution mRNA and Secretome Atlas of Human Enteroendocrine Cells. Cell 181, 1291–1306.e19 (2020).
Worthington, J. J., Reimann, F. & Gribble, F. M. Enteroendocrine cells-sensory sentinels of the intestinal environment and orchestrators of mucosal immunity. Mucosal Immunol. 11, 3–20 (2018).
Haber, A. L. et al. A single-cell survey of the small intestinal epithelium. Nature 551, 333–339 (2017).
Yan, K. S. et al. Intestinal Enteroendocrine Lineage Cells Possess Homeostatic and Injury-Inducible Stem Cell Activity. Cell Stem Cell 21, 78–90.e6 (2017).
Lin, L. et al. Unbiased transcription factor CRISPR screen identifies ZNF800 as master repressor of enteroendocrine differentiation. Science 382, 451–458 (2023).
Gehart, H. et al. Identification of Enteroendocrine Regulators by Real-Time Single-Cell Differentiation Mapping. Cell 176, 1158–1173.e16 (2019).
Dasari, A. et al. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 3, 1335 (2017).
Hassan, S. A. et al. Carcinoid heart disease. Heart 103, 1488–1495 (2017).
Yachida, S. et al. Comprehensive Genomic Profiling of Neuroendocrine Carcinomas of the Gastrointestinal System. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-21-0669 (2021).
Banck, M. S. et al. The genomic landscape of small intestine neuroendocrine tumors. J. Clin. Invest. 123, 2502–2508 (2013).
Scarpa, A. The landscape of molecular alterations in pancreatic and small intestinal neuroendocrine tumours. Annales d.’Endocrinologie 80, 153–158 (2019).
Elias, E. et al. Independent somatic evolution underlies clustered neuroendocrine tumors in the human small intestine. Nat. Commun. 12, 6367 (2021).
de Mestier, L. et al. Digestive Neuroendocrine Neoplasms (NEN): French Intergroup clinical practice guidelines for diagnosis, treatment and follow-up (SNFGE, GTE, RENATEN, TENPATH, FFCD, GERCOR, UNICANCER, SFCD, SFED, SFRO, SFR). Digestive Liver Dis. 52, 473–492 (2020).
Gao, S. et al. Tracing the temporal-spatial transcriptome landscapes of the human fetal digestive tract using single-cell RNA-sequencing. Nat. Cell Biol. 20, 721–734 (2018).
Cao, J. et al. A human cell atlas of fetal gene expression. Science 370, eaba7721 (2020).
Egerod, K. L. et al. A Major Lineage of Enteroendocrine Cells Coexpress CCK, Secretin, GIP, GLP-1, PYY, and Neurotensin but Not Somatostatin. Endocrinology 153, 5782–5795 (2012).
Kawasaki, K. et al. An Organoid Biobank of Neuroendocrine Neoplasms Enables Genotype-Phenotype Mapping. Cell 183, 1420–1435.e21 (2020).
Andersson, E., Swärd, C., Stenman, G., Ahlman, H. & Nilsson, O. High-resolution genomic profiling reveals gain of chromosome 14 as a predictor of poor outcome in ileal carcinoids. Endocr.-Relat. Cancer 16, 953–966 (2009).
Zhang, Z. et al. Patterns of chromosome 18 loss of heterozygosity in multifocal ileal neuroendocrine tumors. Genes Chromosomes Cancer 59, 535–539 (2020).
Nieser, M. et al. Loss of Chromosome 18 in Neuroendocrine Tumors of the Small Intestine: The Enigma Remains. Neuroendocrinology 104, 302–312 (2017).
George, J. et al. Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53 (2015).
Argelaguet, R. et al. Multi‐Omics Factor Analysis—a framework for unsupervised integration of multi‐omics data sets. Mol. Syst. Biol. 14, e8124 (2018).
Aran, D., Hu, Z. & Butte, A. J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220 (2017).
Finotello, F. et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med 11, 34 (2019).
Becht, E. et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 17, 218 (2016).
Racle, J., de Jonge, K., Baumgaertner, P., Speiser, D. E. & Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife 6, e26476 (2017).
Chaffer, C. L., San Juan, B. P., Lim, E. & Weinberg, R. A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 35, 645–654 (2016).
Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018).
Lavie, D., Ben-Shmuel, A., Erez, N. & Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 3, 793–807 (2022).
Luo, H. et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat. Commun. 13, 6619 (2022).
Kieffer, Y. et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 10, 1330–1351 (2020).
van Riet, J. et al. The genomic landscape of 85 advanced neuroendocrine neoplasms reveals subtype-heterogeneity and potential therapeutic targets. Nat. Commun. 12, 4612 (2021).
Alvarez, M. J. et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet 50, 979–989 (2018).
Karpathakis, A. et al. Prognostic Impact of Novel Molecular Subtypes of Small Intestinal Neuroendocrine Tumor. Clin. Cancer Res. 22, 250–258 (2016).
Wallaeys, C., Garcia‐Gonzalez, N. & Libert, C. Paneth cells as the cornerstones of intestinal and organismal health: a primer. EMBO Mol. Med 15, e16427 (2023).
Yavitt, F. M. et al. In situ modulation of intestinal organoid epithelial curvature through photoinduced viscoelasticity directs crypt morphogenesis. Sci. Adv. 9, eadd5668 (2023).
Tisch, N. et al. Caspase‐8 in endothelial cells maintains gut homeostasis and prevents small bowel inflammation in mice. EMBO Mol. Med 14, e14121 (2022).
Marcelino, V. R. et al. Disease-specific loss of microbial cross-feeding interactions in the human gut. Nat. Commun. 14, 6546 (2023).
Wit, M. et al. When fat meets the gut—focus on intestinal lipid handling in metabolic health and disease. EMBO Mol. Med 14, e14742 (2022).
Pasquer, A., Walter, T., Milot, L., Hervieu, V. & Poncet, G. Principles of Surgical Management of Small Intestinal NET. Cancers 13, 5473 (2021).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc.: Ser. B (Methodol.) 57, 289–300 (1995).
Wang, L., Wang, S. & Li, W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Dray, S. & Dufour, A.-B. The ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat. Soft. 22, 1–20 (2007).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Zhu, A., Ibrahim, J. G. & Love, M. I. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092 (2019).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Hu, F.-F., Liu, C.-J., Liu, L.-L., Zhang, Q. & Guo, A.-Y. Expression profile of immune checkpoint genes and their roles in predicting immunotherapy response. Brief. Bioinforma. 22, bbaa176 (2021).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS: A J. Integr. Biol. 16, 284–287 (2012).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. 102, 15545–15550 (2005).
Liberzon, A. et al. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 1, 417–425 (2015).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinforma. 14, 7 (2013).
Hofving, T. et al. The neuroendocrine phenotype, genomic profile and therapeutic sensitivity of GEPNET cell lines. Endocr.-Relat. Cancer 25, 367–380 (2018).
Kölby, L. et al. A Transplantable Human Carcinoid as Model for Somatostatin Receptor-Mediated and Amine Transporter-Mediated Radionuclide Uptake. Am. J. Pathol. 158, 745–755 (2001).
Jiang, S. et al. Targeting netrin‐3 in small cell lung cancer and neuroblastoma. EMBO Mol. Med 13, e12878 (2021).
Acknowledgements
We thank Brigitte Manship for proofreading the manuscript. This work was supported by institutional grants from INCa Lyrican+ (BG, TW), Centre Léon Bérard (BG), ANR JCJC (BG), and industrial Chair ANR (BG). This work was also supported by grants from Groupement d’étude des Tumeurs Endocrines GTE (BG-TW-MO).
Author information
Authors and Affiliations
Contributions
C.P. coordinated the study logistics, collected the samples, performed sample preparation and RNA/DNA isolation, performed the CAF-conditioned media experiment and wrote and revised the manuscript, R.M.P. curated the metadata, conducted the RNA sequencing data analyses and prepared the corresponding figures, wrote and revised the manuscript, A.F. analyzed the genotyping and WGS data, prepared the corresponding figures and wrote and revised the manuscript, F.C. and M.O. analyzed the methylation data, prepared the corresponding figures and wrote and revised the manuscript, P.Mo. curated the metadata, draw the diagrams and performed MOFA, M.R., M.H., S.B. and M.A. performed sample preparation and RNA/DNA isolation, L.G. collected the clinical information and contributed with clinical expertise, V.H., T.F. and J-Y.S. provided the samples and conducted pathological evaluation, G.P. performed the surgeries, V.C. and R.K. contributed with methylation data analysis expertise, A-S.S, A.V. and N.R. contributed with bioinformatics expertise, A.B. and D.B-D performed WGS, B.D. performed sample preparation, J.M. contributed with scientific expertise, C.R. collected the samples and contributed with scientific expertise, P.Me., A.V. and J-F.D. supervised the study, T.W. contributed with clinical expertise and created the clinical database, B.G. designed and supervised the project, wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest. Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy, or views of the International Agency for Research on Cancer/World Health Organization.
Peer review
Peer review information
Nature Communications thanks Annemiek Walenkamp and the other anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Patte, C., Pommier, R.M., Ferrari, A. et al. Comprehensive molecular portrait reveals genetic diversity and distinct molecular subtypes of small intestinal neuroendocrine tumors. Nat Commun 16, 2197 (2025). https://doi.org/10.1038/s41467-025-57305-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57305-8
This article is cited by
-
Novel concepts of cell-of origin in neuroendocrine neoplasms
Virchows Archiv (2026)
-
Pituitary Neuroendocrine Tumor or Pituitary Adenoma? Let’s Ask the Epigenome!
Endocrine Pathology (2025)
