
Abstract
The presence of all five of the virulence-associated genes iucA, iroB, peg-344, rmpA, and rmpA2 is presently the most accurate genomic means for predicting hypervirulent Klebsiella pneumoniae (hvKp-p). With this longitudinal cohort study, we firstly provide novel insights into the clinical and genomic characteristics of hvKp-p in high-risk regions. Through propensity score matching, we show that hvKp-p is less likely to acquire antimicrobial resistance but develops more severe disease and result in increased mortality. HvKp-p are predominantly isolated from hospital settings and caused pneumonia in majority of the cases. ST23 and KL1 are the most common types in the hvKp-p cohort. Community-acquired and healthcare-associated infections are also identified as independent risk factors for hvKp-p. This genomic definition, albeit imperfect, offers a practical and efficient alternative to murine models, allowing for early identification and timely intervention in clinical settings.
Similar content being viewed by others


Introduction
Klebsiella pneumoniae (Kp) is an important Gram-negative pathogen, which is responsible for various infections1. Notably, Kp is highly prevalent in China2 and has evolved into two pathotypes: hypervirulent Klebsiella pneumoniae (hvKp) and classical Klebsiella pneumoniae (cKp)3. cKp is an important member of critical threats reported by the World Health Organization (WHO), is typically associated with hospital-acquired infections (HAI), and is known for its ability to acquire multiple antimicrobial resistance (AMR) genes2,4,5. By contrast, hvKp was initially associated with community-acquired infections (CAI), is known for its ability to cause pyogenic liver abscess (PLA), endophthalmitis6 and metastatic infection. A recent WHO warning7 was issued to heighten awareness of the capability of hvKp to cause life-threatening conditions, and significant morbidity and mortality rates in healthy individuals, thereby posing a threat to the public health. Moreover, hvKp has recently been reported as an emerging and concerning global pathogen, particularly for isolates that have acquired carbapenem resistance8,9,10. The epicenter for hvKp infections still appears to be centered in the Asian Pacific rim and has imposed a significant health threat in China due to its rapidly increasing prevalence in recent years1,9. Therefore, the ability to accurately identify hvKp for surveillance and clinical management is of great importance.
Since the first report of hvKp emergence in Taiwan in the 1980s, the definition of hvKp is evolving3,6,11. Initial studies identified a hypermucoviscous phenotype with positive string test results (> 5 mm) as the definition of hvKp12. However, this diagnostic test is not optimally accurate3. Similarly, the Galleria mellonella infection model is also unable to differentiate hvKp from cKp13. The current gold standard for hvKp identification is murine infection models. However, this modality has a pragmatic limitation due to high costs, ethical concerns, and operational complexity, making it impractical for clinical studies, surveillance of large strain collections, and patient care. Albeit imperfect, recent data support the presence of all five of the hvKp virulence plasmid-associated genes peg-344, rmpA, rmpA2, iroB, and iucA as being the most accurate means to identify hvKp14. The possession of these five biomarkers does not fully reflect the overall hypervirulent phenotype due to the complexity of pathogenesis15. However, assays targeting these markers can be efficiently developed and validated for use in clinical settings. This approach provides a practical and effective solution for clinical utility, enabling early detection and timely interventions to improve patient outcomes. Unfortunately, many studies in the literature have not defined hvKp by either murine studies or the presence of all 5 of peg-344, rmpA, rmpA2, iroB, and iucA.
Therefore, we performed a study in a large cohort to systematically investigate the clinical and genomic characteristics of hvKp-p (as defined by possessing all 5 of these biomarkers) compared to cKp-p (0−4 of these biomarkers). Our findings revealed that hvKp-p was associated with significantly lower AMR carriage but increased severity of disease and increased mortality compared to matched cKp-p cohorts. These data highlight the importance of accurately identifying hvKp, which may lead to improved surveillance, management, and outcomes.
Results
Comparisons between the hvKp-p and cKp-p cohorts revealed different clinical characteristics and outcomes
From 2017 to 2023, a total of 1179 Kp infection cases with information on 14, 28, and 90-day outcomes were enrolled in the study (Fig. 1A and Table 1). Detailed clinical characteristics of the cohort are described in Supplementary Information (Supplementary Fig. 1 and Fig. 1B).

A Annual isolate counts from 2017 to 2023 (total n = 1179), colored by specimen type. The blue line shows the number of hvKp-p isolates per year according to the right-hand y-axis. Data are presented as mean values ± SEM (central point indicates frequency, error bars indicate standard error of the mean). B Charlson Comorbidity Index (CCI) distribution colored by specimen type: Respiratory Tract (n = 605), Urogenital Tract (n = 276), Bloodstream (n = 141), Abdominal Cavity (n = 64), Skin and Soft Tissue (n = 53), Others (n = 40). Each point represents an individual patient. The half-violin plots show the density of CCI. The half-box plots indicate the median, interquartile range (IQR) including 25th and 75th percentiles, and the whiskers extend up to 1.5 × IQR. A two-tailed Kruskal-Wallis test was performed, followed by Dunn’s post-test with Bonferroni correction for multiple comparisons. P-values from left to right: < 0.001, 0.020, < 0.001. *: P-value between 0.01 and 0.05, **: P-value between 0.001 and 0.01, ***: P-value < 0.001. C Survival up to days 14, 28, and 90 for the hvKp-p (n = 118) and cKp-p cohort 2 (n = 118) after PSM. The corresponding table shows the number at risk over time. A two-tailed log-rank test was performed, and p-values are indicated in the figures. D Risk factors associated with hvKp-p infection (n = 127). Horizontal lines indicate 95% confidence intervals (CI), and squares represent odds ratios (ORs) calculated for each predictor. In the univariate logistic regression model, ORs were calculated for each listed variable. In the multivariable regression, variables with p < 0.1 in the univariate analysis were included to calculate adjusted ORs. A two-sided Wald test was performed. Source data are provided as a Source Data file.
Among them, Kp isolates from 127 cases (10.7%, 127/1179) that harbored the biomarkers iucA, rmpA, rmpA2, iroB, and peg-344 comprised the hvKp-p cohort, while the remaining 1052 cases comprised the cKp-p cohort. For the initial analysis, these cohorts were not propensity-matched. In the hvKp-p and cKp-p cohorts, over half of the patients were male (58.3% and 55.9%; P = 0.610), and the mean age of the hvKp-p cohort was slightly lower than that of the cKp-p cohort (63.3 ± 18.0 vs 65.2 ± 23.5; P = 0.275). No significant differences were observed among various socio-economic factors. The mean CCI in the hvKp-p cohort was 2.7 ± 2.5 (Supplementary Fig. 1B). The hvKp-p cohort had fewer comorbid conditions with a significantly lower percentage of high CCI (CCI ≥ 3) compared to the cKp-p cohort (38.6% vs 51.7%; P = 0.005). Fewer patients in the hvKp-p cohort suffered from digestive disorders (27.6% vs 37.3%; P = 0.032) and urinary diseases (20.5% vs 33.4%; P = 0.003, Table 1).
Patients in the hvKp-p cohort were numerically more likely to acquire infection in the ambulatory setting compared to the cKp-p cohort (24.4% vs 17.7%; P = 0.065), but this difference was not statistically significant. Surprisingly, the majority of infections in the hvKp-p cohort were either HAI or HCAI (45.7% and 29.9%), and as expected the majority of infections in the cKp-p cohort were also either HAI or HCAI (60.3% and 22.1%); HAI infections were significantly more common in cKp-p cohort (P = 0.002) and HCAI infections were significantly more common in hvKp-p cohort (P = 0.046). However, the hvKp-p cohort was more likely to acquire infections outside of the hospital (CAI and HCAI) (54.3% vs 39.8%, P = 0.002). In the hvKp-p cohort, pneumonia was the most common infection, which accounted for 64.5% (20/31) in CAI, 67.2% (39/58) in HAI, and 57.9% (22/38) in HCAI respectively (Supplementary Fig. 1C). Pneumonia was also significantly more common than in the cKp-p cohort (49.9%, P = 0.003). By contrast, urinary tract infection was significantly more common in the cKp-p cohort (16.7% vs 7.1%; P = 0.005) (Table 1).
Interestingly, the hvKp-p cohort had a significantly lower ICU admission rate than cKp-p cohort (19.7% vs 30.4%; P = 0.012), where most of the ICU-admitted hvKp-p cohort presented with respiratory tract infection (Supplementary Fig. 1C). In addition, patients with respiratory tract infections also exhibited the highest mortality rate (Supplementary Fig. 1D). However, in the unmatched comparison, no statistical differences were observed between the hvKp-p and cKp-p cohorts for 14, 28, and 90-day poor prognosis and mortality (Table 1).
Increased mortality occurred at 14 days in hvKp-p cohort compared to cKp-p cohort
To more accurately investigate the impact of hvKp-p infection on both disease severity and outcomes, a PSM case-control analysis was performed. The propensity score was calculated using a multivariate logistic regression model that included all of the imbalanced factors listed in Table 1, generating 126 pairs of fully matched cases (Supplementary Table 1). The baseline characteristics were balanced between matched cohorts with SMDs < 0.1.
The results showed that the hvKp-p cohort experienced significantly more severe infections and poor prognosis compared to the cKp-p cohort (Table 2). The incidence of sepsis (62.7% vs 29.4%; P < 0.001) and septic shock (23.0% vs 9.5%; P = 0.004) was notably higher in the hvKp-p cohort. This increased infection severity was also reflected in greater treatment needs, as the hvKp-p cohort required more oxygen therapy (40.5% vs 27.0%; P = 0.024) and invasive mechanical ventilation (22.2% vs 12.7%; P = 0.046). Interestingly, drainage was performed more often in the hvKp-p cohort (18.3% vs 7.1%; P = 0.008), suggesting a greater need for source control. In terms of clinical outcomes, the poor prognosis was significantly higher in the hvKp-p cohort compared to the cKp-p cohort at 14 days (19.0% vs 10.3%, P = 0.050) and at 28 days (23.0% vs 12.7%; P = 0.032). The rate at 90 days (31.0% vs 20.6%; P = 0.061) demonstrated a similar trend, but statistical significance was not achieved. Mortality was significantly higher in the hvKp-p cohort at 14 days (17.0% vs 7.4%; P = 0.032). This trend was also exhibited at 28 days (18.9% vs 10.2%; P = 0.071) and 90 days (19.8% vs 16.7%; P = 0.551), but statistical significance was not achieved. The Kaplan–Meier curves showed the impact of the number of biomarkers. The cKp-p cohort had a significant 14-day survival benefit compared to the hvKp-p cohort (P = 0.037) (Supplementary Fig. 1E).
Initial treatment failure (20.6% vs 19.8%; P = 0.875), and the mean length of hospital stay (12.4 ± 18.6 days vs 11.0 ± 10.8 days; P = 0.224) were similar between two cohorts. Notably, the cKp-p cohort had significantly higher rates of re-admission (19.0% vs 4.0%; P < 0.001) and re-infection (19.8% vs 4.8%; P < 0.001), indicating potentially worse long-term outcomes despite the less severe acute presentation. Additionally, there was a trend for greater alcohol use in the hvKp-p cohort, which was not statistically significant (23.8% vs 15.1%; P = 0.080). No statistically significant difference was observed in the immunodeficiency status (21.2% vs 18.6%; P = 0.625) (Table 2).
Increased mortality occurred in hvKp-p cohort compared to different cKp-p sub-cohorts
Next, we explored whether there was also a difference in disease severity and outcomes between the hvKp-p cohort (all 5 biomarkers), cKp-p sub-cohort 1 (1-4 biomarkers) and cKp-p sub-cohort 2 (0 biomarkers). The hvKp-p cohort was then subjected to 1:1 matching with these sub-cohorts, resulting in two additional sets of outcome comparisons (Supplementary Table 2, 3).
In the propensity match analysis comparing hvKp-p with cKp-p sub-cohort 1, the hvKp-p cohort was significantly more likely to develop sepsis compared to the cKp-p sub-cohort 1 (61.7% vs 40.9%; P = 0.002) but not septic shock (23.5% vs 18.3%; P = 0.330). Further, the hvKp-p cohort showed higher rates of poor prognosis and mortality at 14, 28, and 90 days compared to the cKp-p sub-cohort 1, although no statistical significance was observed (Supplementary Table 4 and Supplementary Fig. 1F).
Increased disease severity was also seen when comparing the matched hvKp-p cohort to the cKp-p sub-cohort 2 (Table 3), with significantly higher rates of sepsis (61.9% vs 26.3%; P < 0.001) and septic shock (23.7% vs 7.6%; P < 0.001). In addition, the poor prognosis was significantly greater in the hvKp-p cohort compared to the cKp-p sub-cohort 2 at 14, 28, and 90 days (19.5%, 23.7%, 31.4% vs 8.5%, 9.3%, 18.6%; P = 0.015, 0.003, 0.024). Similarly, mortality was significantly increased at 14 days (17.2% vs 7.3%; P = 0.029) and 28 days (19.2% vs 8.3%; P = 0.021). The cKp-p sub-cohort 2 had a higher 14-day (P = 0.055) and 28-day (P = 0.042) survival benefit compared to the hvKp-p cohort (Fig. 1C). However, no significant difference was observed at 90 days (P = 0.810). In addition, the hvKp-p cohort showed a greater need for oxygen therapy or respiratory support compared to the cKp-p sub-cohorts, and the hvKp-p cohort had a longer mean hospital stay compared to cKp-p sub-cohort 2 (12.7 ± 19.1 vs 9.0 ± 8.7 days; P = 0.058).
Taken together, these data strongly support that the severity of disease and poor prognosis is greater in the hvKp-p cohort compared to the cKp-p sub-cohorts.
Risk factors of infection by hvKp-p isolates
Univariate analysis revealed that patients with HCAI (P = 0.047, OR = 1.228) and CAI (P = 0.066, OR = 1.503) were more likely to have hvKp-p infection compared to the cKp-p cohort. Whereas, ICU admission (OR = 0.561), CCI ≥ 3 (OR = 0.587), HAI (OR = 0.821), the presence of digestive (OR = 0.641), or urologic diseases (OR = 0.514) were statistically more likely to occur in the cKp-p cohort (Fig. 1D). With multivariate analysis, CAI (P = 0.035, OR = 1.664) and HCAI (P = 0.021, OR = 1.296) were identified as being more common in the hvKp-p cohort, and conversely, CCI ≥ 3 (P = 0.023, OR = 0.640) was identified as being more common in the cKp-p cohort (Fig. 1D). Taken together, although these data suggest that hvKp-p infection is more likely than cKp-p infection with CAI or HCAI caused by Kp in the patients with fewer underlying diseases. However, it should be noted that 45.7% of infections in the hvKp-p cohort were hospital-acquired.
Molecular and phylogenetic characteristics of the hvKp-p and cKp-p isolates
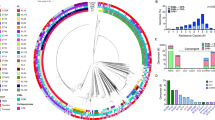
By WGS and analysis, we generated a phylogenetic map of the hvKp-p and cKp-p isolates (Fig. 2A). In line with the relatedness revealed by MLST (Fig. 2B), genomes of the same clonal groups (CGs) and STs were clustered together, and different CGs and STs were separated on the tree. CGs and STs, including hvKp-p isolates, were also dispersed. The isolates belonging to the clinically more common CGs and STs were closely clustered into different clades, including CG11, CG23, CG15, CG65, CG412, ST307, and ST86, supporting the ongoing spread of these STs and CGs within the hospital.

A Circular phylogenetic tree of the 1179 isolates reconstructed from whole genome data by the maximum likelihood method. From the inner to outer circles, the first circle adjacent to the isolate names shows whether the isolates carry all of the five virulence genes, whether the patients infected by the isolate present sepsis, septic shock, or poor prognosis (N: negative; P: positive). B Minimum spanning tree constructed based on the allelic profiles of the associated STs by the goeBURST algorithm. The circles represent the STs, and the red fans represent the proportion of isolates (n = 127) carrying all of the five virulence genes among each ST. The numbers on the lines represent the numbers of different alleles between two STs. Source data are provided as a Source Data file.
Among the total 1179 Kp isolates, 127 (10.8%) were categorized as hvKp-p with 23 distinct STs (Supplementary Fig. 2A), including common CGs and STs like CG23, CG65, CG412 and ST86, but no hvKp-p isolates were present in other common CGs and STs such as CG11, CG15 and ST307. ST23 was the most common ST in the hvKp-p cohort (30.7%, 39/127); although significantly less common, 9.3% (98/1052) of the cKp-p cohort was also ST23 (P < 0.001). Other common STs in the hvKp-p cohort were ST412 (n = 24, 18.9%), ST86 (n = 10, 7.9%), and ST65 (n = 7, 5.5%) (Supplementary Table 5), including ST23 accounted for 63.0% of the hvKp-p isolates. ST11 was the most common ST in the cKp-p cohort (22.9%, 241/1052); none of the isolates in the hvKp-p cohort were ST11. Notably, up to 18 different STs were detected in the remaining hvKp-p isolates, showing high genetic diversity within this cohort.
In total, 101 distinct K locus (KL) types were identified among the Kp isolates. The most common was KL1 (156/1179,13.2%), which was mainly associated with pneumonia (92/156), followed by bloodstream (27/156) and abdominal infections (20/156) (Fig. 3A). Furthermore, the diversity of KL types was also exhibited across different cohorts (Fig. 3B). Among them, KL1 (35.4% vs 10.6%; P < 0.001), KL2 (24.4% vs 8.2%; P < 0.001), KL57 (27.6% vs 2.7%; P < 0.001) were significantly more common in the hvKp-p isolates compared to the cKp-p isolates. By contrast, KL47 (0.0% vs 11.0%; P < 0.001) and KL102 (0.0% vs 7.8%; P = 0.001) were only found in the cKp-p isolates. Further, not all hvKp-p KL1 isolates possessed ST23. Among the 45 hvKp-p KL1 isolates, 86.7% were ST23 (39/45), and 8.9% (4/45) belonged to three other STs in the CG23 group including ST1265 (2/45, 4.4%), ST1769 (1/45, 2.2%) and ST23-1LV (1/45, 2.2%). Two isolates (2/45, 4.4%) belonged to ST2159 but were not in the CG23 group. Fourteen O-antigen types were detected in this study (Fig. 3C). The O1 (61.4% vs 37.1%; P < 0.001) and O3b (19.7% vs 8.4%; P < 0.001) were more likely to be identified in hvKp-p cohort compared to the cKp-p (Fig. 3D), suggesting a potential target for the vaccine (Supplementary Table 5).

A, B) Cumulative prevalence of K-loci ordered by mean prevalence across specimen type and virulence factors (VFs). Lines in Fig. 3A are colored by specimen type as per panel A of Fig. 1. Lines in Fig. 3B are colored by VFs. 5VF: isolates harboring all five key virulence factors, including iucA, iroB, peg-344, rmpA, and rmpA2. 1-4VF: isolates harboring any combination of fewer than five key virulence factors. 0VF: isolates harboring none of the five key virulence factors. C, D Cumulative prevalence of predicted O-types ordered by mean prevalence across specimen type and VFs. E Distribution of the number of acquired antimicrobial resistance (AMR) genes between the hvKp-p and cKp-p isolates, stratified by specimen type. F Distribution of the number of AMR genes across four groups: cKp-p cohort without ICU admission (n = 732), cKp-p cohort with ICU admission (n = 320), hvKp-p cohort without ICU admission (n = 102) and hvKp-p cohort with ICU admission (n = 25). Bars are colored by ICU admission status (blue = not admitted, red = admitted). The center line represents the median, the box bounds the 25th and 75th percentiles, and the whiskers extend up to 1.5 × IQR. A two-tailed Kruskal-Wallis test was performed, followed by Dunn’s post-test with Bonferroni correction for multiple comparisons. P-values from left to right: < 0.001, < 0.001, < 0.001. *: P-value between 0.01 and 0.05, **: P-value between 0.001 and 0.01, ***: P-value < 0.001. G Frequency of genomes with different VFs, shown by ESBL, CRKP, and KPC gene status. Bars are colored by specimen type as shown in Panel A of Fig. 1. Source data are provided as a Source Data file.
Possession of genetic elements associated with virulence and resistance among the hvKp-p and cKp-p isolates
The canonical hvKp capsule types KL1 and KL2 were present in isolates categorized as cKp-p that variably possessed iucA, iroB, rmpA, rmpA2, and peg-344, suggesting the presence of incomplete pVir plasmids. The virulence-associated factors possessed by the three cohorts are delineated in the Supplementary Information (Supplementary Fig. 2B, C).
The possession of various gene combinations in cKp-p isolates showed high diversity (Supplementary Table 6). The majority (616/1052, 58.6%) harbored none of the five virulence genes, 19.8% (n = 208) with four genes, 9.7% (n = 102) with three genes, 4.4% (n = 46) with two genes, and 11.9% (n = 80) with one gene (Supplementary Table 7). In addition to these five genes, another gene, peg-589, that is present on the hvKp-associated virulence plasmid was significantly more commonly detected in the hvKp-p isolates compared to the cKp-p isolates (99.2% (126/127) vs 33.2% (349/1052); P < 0.001) (Supplementary Table 5). In addition, the IncHI1B plasmid replication was significantly more commonly present in hvKp-p isolates compared to the cKp-p isolates (63.0% vs 30.4%; P < 0.001) (Supplementary Table 5), whereas AMR-associated plasmid replications were more common in cKp-p isolates. The overall characteristics of antimicrobial resistance were described in the Supplementary Information (Supplementary Fig. 2D, E and Supplementary Table 8, 9). Interestingly, hvKp-p isolates were more likely to be antimicrobial sensitive. The cKp-p cohort exhibited a significantly higher AMR gene carriage compared to the hvKp-p cohort (4.8 ± 5.2 vs 0.2 ± 0.9; P < 0.001). In the cKp-p cohort, skin and soft tissue infections showed the highest AMR gene carriage (5.8 ± 5.9), whereas in the hvKp-p cohort, pneumonia had the highest AMR gene carriage (0.2 ± 1.0) (Fig. 3E). In the cKp-p cohort, ICU-admitted patients were further found to have a significantly higher AMR gene carriage (6.3 ± 5.1 vs 4.1 ± 5.1; P < 0.001) (Fig. 3F). Additionally, only two hvKp-p isolates were identified as ESBL-producing (Fig. 3G). The detailed clinical and molecular characteristics of these two isolates were shown in Supplementary Table 9.
Discussion
One of the challenges with studies on hvKp is the lack of an accurate consensus definition. Murine studies are the gold standard but are less pragmatic when assessing a large number of strains. Presently, the best alternative is using the co-presence of all five of the genes rmpA, rmpA2, iroB, iucA, and peg-344, which are located on the hvKp-associated virulence plasmid. The presence of all of these biomarkers increases the likelihood that a complete virulence plasmid is present. Therefore, in this report, we assembled a large, contemporary cohort infected with Kp from 2017 to 2023. In total, we collected over 1000 infection cases from a hvKp high-risk region. Cases were divided into a cohort that was putatively infected with hvKp based on the presence of all 5 biomarkers (hvKp-p) and a cohort that was putatively infected with cKp based on having less than 5 of these markers (cKp-p). Further for some analyses, the cKp-p cohort was divided into 2 sub-cohorts, one that was infected with isolates variably possessed 1–4 of these biomarkers (cKp-p sub-cohort 1) and one that was infected with isolates that possessed none of these biomarkers (cKp-p sub-cohort 2). To fully characterize the predicted hypervirulent phenotype, the hvKp-p cohort was compared to each of the three cKp-p cohorts using PSM. It ensured that the differences in early-onset mortality were attributable to the hypervirulent phenotype. These patient cohorts were compared to assess for severity of disease, management, and outcomes. The isolates from these cohorts also underwent comparison.
The strain comparison revealed that strains with a predicted hypervirulent phenotype possessed remarkably low AMR, only 2/127 possessed ESBL and none carbapenemases. Although the impact of acquired antimicrobial resistance on the hvKp phenotype has not been completely resolved, hvKp isolates that possess ESBLs and/or carbapenemases have been appropriately validated via murine studies14. Other reports that have described a higher prevalence of drug-resistant hvKp isolates but have not always used rigorous definitions for the hvKp phenotype6,11,16. The Galleria model, serum resistance, a positive string test, or the presence of 1-2 biomarkers are often used but have been shown not to be able to differentiate between the cKp and hvKp phenotype13,17,18. Short of murine studies, which is the gold standard but not always pragmatic due to their high costs, time demands, and availability, the genomic definition used in this study is the most accurate means to identify hvKp14 and provides a more practical and efficient way to identify hvKp strains, enabling surveillance, early diagnosis, and timely intervention. Interestingly, not all of the hvKp-p isolates that possessed KL1 were ST23. This capsule locus was also identified in ST1265, ST1769, and ST23-1LV. Future studies assessing the relative virulence of these isolates compared to ST23/KL1 would be of interest, and ongoing surveillance would be important.
Clinical comparisons of these cohorts revealed that infection with hvKp-p isolates generally resulted in more severe disease, as measured by sepsis, septic shock, and mortality as compared to the cKp-p, cKp-p sub-cohort 1, and cKp-p sub-cohort 2. Some9, but not all studies13 have had similar findings. One explanation for studies that did not demonstrate more severe disease with hvKp infection could be that attribution of infection being due to an hvKp isolate may not have used rigorous criteria and, as a result, the isolate(s), in reality, possessed the cKp phenotype as discussed above. Another potential explanation is that hvKp infection can occur in younger, healthier patients than cKp infection. Therefore, propensity-matching controls for this potential variable, as was done in this study, is important.
An interesting finding in this study was that pneumonia was more common in the hvKp-p cohort than the cKp-p cohort and accounted for 63.8% of hvKp infections. Although prior studies have recognized that hvKp is a common cause of community-acquired pneumonia in the Asian Pacific Rim, and in some studies a similar prevalence as Streptococcus pneumoniae19, this study further strengthens that observation and has important clinical management implications since the management of infection caused by these two agents would differ such as searching for occult abscesses, vigilance for endophthalmitis, and infection control for hvKp infection. This study also reinforces earlier work that although hvKp infection can occur in individuals from the community (24.4% in this study), HCAI (29.9% in this study) and HAI (45.7% in this study) are becoming increasing common.
This study has some limitations. Since the cases are primarily from the Beijing municipality, this study reflects a relatively narrow geographic region and may not be representative of all locales. In addition, differentiating infection from colonization can be challenging, particularly for isolates obtained from non-sterile sites. While it is not entirely possible to exclude the possibility that some Kp isolates were colonizers, we employed a rigorous definition and carefully designed inclusion process, independently assessed by two physicians, to minimize this likelihood. Further, we used a genomic definition for putative hvKp isolates, which was defined by possessing all 5 of the biomarkers rmpA, rmpA2, iroB, iucA, and peg-344 (hvKp-p cohort). Although presently, this is the most accurate and efficient means to genomically define hvKp, it is imperfect. Some strains can harbor all 5 of these markers but not possess the hvKp pathotype14. Likewise, it is possible that strains in this study that were categorized as being putatively cKp that possessed 3-4 of these biomarkers could possess the hvKp pathotype. However, miscategorization of strains would blunt, and not inappropriately magnify, the identified differences between cohorts. Therefore, this limitation will not invalidate the results and conclusions.
The use of more rigorous criteria to define hvKp and propensity matching of patient cohorts in this study demonstrated that putative hvKp strains possessed less antimicrobial resistance than expected. Infection due to putative hvKp strains resulted in more severe disease and worse outcomes compared to putative cKp strains. Further, this genomic definition, albeit imperfect, provided an efficient and pragmatic alternative to murine models and can enable prompt identification for use in surveillance and clinical management.
Methods
Enrolled patients and demographic and clinical data collection
We conducted a systematic retrospective investigation on a large cohort infected with K. pneumoniae from 2017 to 2023 in Beijing, China4. Demographic information was obtained from electronic medical records. Social-economical characteristics, including the permanent residence area and medical insurance, were also recorded. Residence in either a village or a city was determined by the detailed address recorded in the medical record. We also collected the medical insurance types of the patients, which were categorized into superior and non-superior groups. Clinical characteristics, including underlying diseases, the Charlson Comorbidity Index (CCI), ICU admission, and specimen types, were also collected. Infection was defined as: (1) signs, symptoms, and/or laboratory and radiographic findings were consistent with infection from the site where Kp was cultured; (2) antibiotic therapy initiation following the positive culture result; (3) Kp was cultured from a sterile site. The inclusion of infection cases was first assessed by a junior physician and then reviewed by a senior physician. The process was independently conducted to ensure accuracy and reliability. Meeting any of the above criteria was defined as infection. Hospital-acquired infection (HAI) was defined as patients who developed the infection after being admitted for more than 48 h. Community-acquired infection (CAI) was defined as cases without any prior exposure to healthcare facilities, and healthcare-associated infection (HCAI) was defined as an infection in a patient who had prior interaction with the healthcare system before developing their infection. Sepsis and septic shock were defined as previous described9. Poor prognosis, measured at 14, 28, and 90 days, was defined as death or withdrawal of life-sustaining therapy. Patients lost to follow-up who do not have the 90-day outcome were excluded from the mortality analysis.
In addition, alcohol usage, immunodeficiency state, life-support strategy, treatment failure, length of stay, re-admission, and re-infection were also collected and assessed for the propensity score-matched analysis. Life-support interventions included supplemental oxygen, mechanical ventilation, usage of vasoactive agents, extracorporeal membrane oxygenation (ECMO), renal replacement, debridement, and abscess drainage. Treatment failure was defined as infection-attributable death or the need to re-treat. The immunodeficiency state was determined as previously described9. Re-infection was defined as Kp infection in a patient with a previous Kp infection and re-admission was defined as an admission for a Kp infection after being discharged for a previous Kp infection. The protocol for this study was approved by the Peking University Third Hospital Medical Science Research Ethics Committee (M2021545). Consent waiver granted due to the retrospective nature of the study, no additional health risks beyond standard care, no personally identifiable information, and no commercial interests.
Clinical isolates identification and classification
All strains were stored at − 80 °C. Strain identification was performed by the Vitek Compact 2 system and MALDI-TOF mass spectrometry. The Klebsiella species were further characterized through whole-genome sequencing (WGS), analyzed using the Kleborate software v2.3.2 as well as additional bioinformatic analyses. K. pneumoniae isolates that harbored all five of the biomarkers rmpA, rmpA2, iroB, icuA, and peg-344 were defined as hvKp-p (n = 127)14. All other strains were classified as cKp-p (n = 1052).
DNA extraction and whole genome sequencing
Briefly, the whole genomic DNA for all isolates was extracted and sequenced using the Illumina sequencing method on the NovaSeq platform by constructing a paired-end (PE) library (NEB dsDNA Fragmentase) with average insertion lengths of 150 bp9. We obtained the clean data by using fastp and assembly using SPAdes v3.1320.
Detection of virulence-associated genes, serotypes, sequence types, and AMR genes
The determination of virulence-associated genes (VF), serotypes, O-antigen types, and sequence types (STs) was conducted using genome sequences analyzed as described previously21,22,23. A complete minimum spanning tree was generated based on the allelic profiles of the STs by the goeBURST algorithm using PHYLOViZ V2.0 software (http://www.phyloviz.net/). To further understand the distribution of AMR genes, we evaluated the AMR classes, resistance score, and genes using Kleborate software v2.3.223. The definition of the resistance score was shown in the Supplementary Methods.
Whole genome phylogenetic analysis
The complete genome sequence of K. pneumoniae HS11286 (accession no.: NC_016845) was used as the reference to perform phylogenetic analysis1,9,21. The sequencing read of all Kp isolates was mapped to the reference by using bowtie 2 v2.2.8 with the --sensitive mode (https://github.com/BenLangmead/bowtie2). We identified single nucleotide polymorphisms (SNPs) by using Samtools v1.9 (https://github.com/samtools/samtools) and the polymorphic sites among these genomes were distilled according to the reference using the iSNV-calling pipeline (https://github.com/generality/iSNVcalling)9, then detected and retained the high-quality SNPs defined as those supported by more than 5 reads of mapping quality > 20 in the query genome. Subsequently, recombination sites were further identified and filtered by Gubbins24. The concatenated sequences of remained polymorphic sites conserved in all genomes (core genome SNPs) were used to perform phylogenetic analysis using the maximum likelihood method with a generalized time-reversible substitution model by FastTree25 and visualized using iTOL (https://itol.embl.de/).
Statistical analysis
Continuous variables were represented as means ± standard deviation (SD) or median ± interquartile range (IQR), and comparisons between the hvKp-p and cKp-p cohorts employed either the independent t test or Wilcoxon rank sum test as appropriate. For multiple group comparisons, the Kruskal-Wallis test was used, followed by Dunn’s test for pairwise comparisons. For categorical variables, cohort comparisons were analyzed by Pearson’s chi-square test, Yates’ Correction for Continuity, or Fisher exact test. Risk factors were assessed using univariate analysis. Variables with P < 0.1 were included in multivariable logistic regression to determine independent risk factors.
To minimize the potential bias, we performed a 1:1 propensity score matching study between the hvKp-p cohort (all 5 biomarkers) and the cKp-p cohort (0-4 biomarkers) (n = 126). The cKp-p cohort was further subcategorized as cKp-p sub-cohort 1 (1-4 biomarkers) (n = 115) or as cKp-p sub-cohort 2 (0 biomarkers) (n = 118). The propensity scores were calculated using multiple logistic regression models including all prespecified covariates. The balance of baseline covariates between groups was assessed using standardized mean differences (SMDs). The cohorts were then subjected to further matching comparison of clinical outcomes. The Kaplan-Meier (KM) survival curves were constructed to compare the outcomes between matched cohorts.
All tests were two-tailed, and the P < 0.05 were considered to be statistically significant. All analysis were performed by using the SPSS software v29.0 and R v4.4.1 (packages: ggplot2, MatchIt, survival, survminer, dplyr).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing data generated in this study have been deposited in the National Center for Biotechnology Information (NCBI) database under the accession number PRJNA1219236 (http://www.ncbi.nlm.nih.gov/bioproject/1219236) and in the National Microbiology Data Center (NMDC) under the accession number NMDC10019410. Source data are provided in this paper.
Code availability
The source code for SNP and iSNV calling is available on GitHub (http://github.com/generality/iSNV-calling/). The source code for data analysis and visualization in this study is available on GitHub (https://github.com/Wendy-Tang01/data_analysis).
References
Liu, C. et al. An increased prevalence of carbapenem-resistant hypervirulent Klebsiella pneumoniae associated with the COVID-19 pandemic. Drug Resist. Updat. 77, 101124 (2024).
Wang, Q. et al. Phenotypic and genotypic characterization of carbapenem-resistant enterobacteriaceae: data from a longitudinal large-scale CRE study in China (2012-2016). Clin. Infect. Dis. 67, S196–S205 (2018).
Russo, T. A. & Marr, C. M. Hypervirulent Klebsiella pneumoniae. Clin. Microbiol. Rev. 32, https://doi.org/10.1128/cmr.00001-19 (2019).
Liu, C., Guo, J., Lu, M., Shen, N. & Du, P. Dissemination of the mobilised RND efflux pump gene cluster tmexCD-toprJ among Klebsiella pneumoniae. The Lancet. Microbe 4, e135 (2023).
Liu, C. et al. Emergence and Inter- and Intrahost Evolution of Pandrug-Resistant Klebsiella pneumoniae Coharboring tmexCD1-toprJ1, bla(NDM-1), and bla(KPC-2). Microbiol. Spectr. 11, https://doi.org/10.1128/spectrum.02786-22 (2023).
Liu, C., Shi, J. & Guo, J. High prevalence of hypervirulent Klebsiella pneumoniae infection in the genetic background of elderly patients in two teaching hospitals in China. Infect. Drug Resist. 11, 1031–1041 (2018).
WHO. Antimicrobial Resistance Hypervirulent Klebsiella Pneumoniae-Global Situation. https://www.who.int/emergencies/disease-outbreak-news/item/2024-DON527 (2024).
Wang, R. et al. Increase in antioxidant capacity associated with the successful subclone of hypervirulent carbapenem-resistant Klebsiella pneumoniae ST11-KL64. Nat. Commun. 15, 67 (2024).
Liu, C. et al. Hypervirulent Klebsiella pneumoniae is emerging as an increasingly prevalent K. pneumoniae pathotype responsible for nosocomial and healthcare-associated infections in Beijing, China. Virulence 11, 1215–1224 (2020).
Emergence of hypervirulent Klebsiella pneumoniae ST23 carrying carbapenemase genes in EU/EEA countries. European Centre for Disease Prevention and Control. ECDC: Stockholm (2024).
Liu, C. & Guo, J. Characteristics of ventilator-associated pneumonia due to hypervirulent Klebsiella pneumoniae genotype in genetic background for the elderly in two tertiary hospitals in China. Antimicrob. Resist. Infect. Control 7, 95 (2018).
Li, W. et al. Increasing occurrence of antimicrobial-resistant hypervirulent (hypermucoviscous) Klebsiella pneumoniae isolates in China. Clin. Infect. Dis. 58, 225–232 (2014).
Russo, T. A. & MacDonald, U. The Galleria mellonella Infection Model Does Not Accurately Differentiate between Hypervirulent and Classical Klebsiella pneumoniae. mSphere 5, https://doi.org/10.1128/msphere.00850-19 (2020).
Russo, T. A. et al. Differentiation of hypervirulent and classical Klebsiella pneumoniae with acquired drug resistance. mBio 15, e0286723 (2024).
Russo, T. A. et al. Identification of biomarkers for differentiation of hypervirulent Klebsiella pneumoniae from classical K. pneumoniae. J. Clin. Microbiol. 56, https://doi.org/10.1128/jcm.00776-18 (2018).
Liu, C. & Guo, J. Hypervirulent Klebsiella pneumoniae (hypermucoviscous and aerobactin positive) infection over 6 years in the elderly in China: antimicrobial resistance patterns, molecular epidemiology and risk factor. Ann. Clin. Microbiol. Antimicrob. 18, 4 (2019).
Liu, C. et al. Phenotypic and genomic characterization of virulence heterogeneity in multidrug-resistant ST11 Klebsiella pneumoniae during inter-host transmission and evolution. Infect. Drug Resist. 13, 1713–1721 (2020).
Liu, C. et al. Emergence of extensively drug-resistant and hypervirulent KL2-ST65 Klebsiella pneumoniae Harboring bla(KPC-3) in Beijing, China. Microbiol. Spectr. 10, e0304422 (2022).
Grosjean, V. et al. Community-acquired Klebsiella pneumoniae pneumonia in ICU: a multicenter retrospective study. Ann. Intensive Care 14, 69 (2024).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Liu, C. et al. Dynamic within-host cefiderocol heteroresistance caused by bla(SHV-12) amplification in pandrug-resistant and hypervirulent Klebsiella pneumoniae sequence type 11. Drug Resist. Updat. 73, 101038 (2023).
Chen, L. et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328 (2005).
Lam, M. M. C. et al. A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat. Commun. 12, 4188 (2021).
Croucher, N. J. et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15 (2015).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650 (2009).
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 82200012 and 82470010) (Dr. Liu), NIH 1R21AI141826-01A1 (Dr. Russo), the Department of Veterans Affairs VA Merit Review (I01 BX004677-01) (Dr. Russo), Key clinical project of Peking University Third Hospital (BYSY2022066) (Dr. Liu), Peking University Clinical Scientist Training Program (BMU2024PYJH011) (Dr. Liu) and the Fundamental Research Funds for the Central Universities (Dr. Liu). The authors acknowledge the assistance provided by the Clinical Microbiology Laboratory (Researcher: Jiajia Zheng) of Peking University Third Hospital. The authors also thank the members of Ning Shen’s Lab for cohort construction.
Author information
Authors and Affiliations
Contributions
Chao Liu, Yunfei Tang, and Pengcheng Du analyzed the data, wrote the manuscript draft, and performed critical reviews. Yunfei Tang collected clinical data. Yunfei Tang, Chunjing Du, and Ping Yang constructed the cohort. Ping Yang helped with the experiments and checked the cases in revision. Ning Shen reviewed the manuscript, provided suggestions, and verified the infection cases. Thomas A. Russo, Chao Liu, and Pengcheng Du conceptualized and designed the study. All authors reviewed and approved the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, Y., Du, P., Du, C. et al. Genomically defined hypervirulent Klebsiella pneumoniae contributed to early-onset increased mortality. Nat Commun 16, 2096 (2025). https://doi.org/10.1038/s41467-025-57379-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57379-4
