
Abstract
CRISPR-associated (Cas) transposases (CAST) are RNA-guided systems capable of programmable integration of large segments of DNA without creating double-strand breaks. Engineered Cascade CAST function in human cells but are challenging to deploy due to the complexity of the targeting components. Unlike Cascade, which require three Cas proteins, type V-K CAST require a single Cas12k effector for targeting. Here, we show that compact type V-K CAST from uncultivated microbes are repurposable for programmable DNA integration into the genome of human cells. Engineering for nuclear localization and function enables integration of a therapeutically relevant transgene at a safe-harbor site in multiple human cell types. Notably, off-targets are rare events reproducibly found in specific genomic regions. These CAST advancements are expected to accelerate applications of genome editing to therapeutic development, biotechnology, and synthetic biology.
Similar content being viewed by others
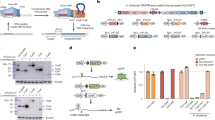


Introduction
Transposons and other mobile genetic elements have been harnessed for use in genetics, biotechnology, and therapeutics to introduce large fragments of DNA into genomes1,2. Many types of transposons randomly integrate into multiple genomic loci by recognizing short, frequent target motifs (e.g., Tc1/mariner, retrotransposons3,4,5), while others are able to integrate into a single, well-conserved site on the host genome (Tn7)6. With any of these systems, the ability to programmably deliver DNA cargo with high specificity has not been possible. Recently, two new families of transposons distantly related to both Tn7 and Tn5053 transposons were found to associate with RNA-guided nuclease-dead Cas proteins for targeted genomic integration of large DNA cargos (CRISPR-associated transposases or CAST)7,8,9. These two types of CAST systems have been shown to differ in their Cas and transposase components, as reviewed by Peters10. The type I-F Cascade complex is derived from Tn7-like elements that have co-opted CRISPR-associated proteins into CAST systems7,8,9, whereas the type V-K CAST evolved from transposon proteins distantly related to Tn5053 transposons7,8. Both systems contain the transposase TnsB, the AAA+ ATPase TnsC, and acquired a Cas recognition component, TniQ, while the Cascade CAST systems contain an additional TnsA that enables cut-and-paste integration7,8,11,12. Additionally, transposon targeting in Cascade CAST requires the proteins Cas6, Cas8, and multiple moieties of Cas7, whereas targeting by type V-K CAST is dependent on a single Cas12k effector7,13,14,15,16. Both systems are promising for the development of biotechnological applications, but translation for use in human cells has been challenging due to their multicomponent nature and need for additional host factors. For example, the small prokaryotic ribosomal subunit S15 is necessary for efficient integration of type V-K CAST13,14,15,16, a discovery that enabled episomal integration in human cells17. Moreover, Lampe and King et al. recently demonstrated that the addition of bacterial chaperone ClpX was necessary for Cascade CAST DNA integration into single-copy targets in human cells, albeit at low efficiencies18.
In this work, we focus on type V-K CAST with the hypothesis that their simpler composition compared to Cascade systems would simplify translation for human genome editing applications. We identify diverse type V-K CAST systems from an extensive metagenomic dataset, confirm their activity both in vitro and in cells, and engineer them for targeted integration in human cells. Furthermore, we address system specificity by developing an unbiased assay and evaluate CAST off-target effects in human cells. Additional optimization results in a system active in multiple cell types, including the integration of a full therapeutically relevant gene (Factor IX) at a safe harbor locus. These advances with the type V-K CAST system illustrate the promise of this editing system over CRISPR-Cas9 and Cascade CAST approaches for achieving site-specific and programmable gene-sized integrations in human cells.
Results
Diverse type V-K CAST systems from metagenomics
To explore the diversity of type V-K CAST, we analyzed thousands of high-quality metagenomic assemblies to identify genes encoding CRISPR type V proteins in the genomic vicinity of transposons. Over 70 phylogenetically diverse Cas12k effectors were encoded in genomic fragments containing complete and partial type V-K CAST systems (Fig. 1A, B, Supplementary Fig. 1, and Supplementary Data 1, 2). Boundaries of the type V-K CAST transposon were determined by analyzing intergenic regions flanking the CRISPR locus and the transposon machinery. These intergenic regions were aligned among several homologs, and regions of conservation were used to predict the transposon boundaries (Supplementary Fig. 2). The 3′ end of type V-K CAST CRISPR repeats (crRNA) contain a conserved motif 5′-GNNGGNNTGAAAG-3′ (Supplementary Data 2) predicted to bind to the tracrRNA anti-repeats to form active guide RNA structures. Upstream from the antirepeat in the tracrRNA is a conserved “CCYCC(n4-n6)GGRGG” stem-loop structure (Supplementary Fig. 3). This feature may be evolutionarily maintained for protein recognition or RNA folding and to position the downstream spacer sequence for target pairing. The secondary structure of tracrRNA and crRNA repeat sequences were determined and trimmed to design single guide RNAs (sgRNA). Notably, the sgRNA designs display conserved structural features despite sharing less than 70% pairwise nucleotide identity (Fig. 1C, supplementary text and Supplementary Fig. 4). It has been previously shown that self-matching spacers within the CAST transposon are frequently encoded adjacent to a pseudo CRISPR repeat in the vicinity of the CRISPR arrays19,20 and are emblematic of active type V-K CAST systems. These self-matching spacers were identified within a subset of our metagenomic-derived systems (Supplementary Fig. 1A), suggesting that these are likely part of functional CAST transposons. Therefore, we selected 13 predicted complete type V-K CAST systems for screening activity in vitro (Supplementary Data 1).

A Type V-K CAST MG64-1 genomic context. The transposon is characterized by terminal inverted repeats (TIR, light salmon bars) that define the transposon left end (LE) and right end (RE), transposon genes TnsB, TnsC, and TniQ, the dead effector Cas12k (orange arrow), a tracrRNA (pink arrow), and CRISPR array. A predicted “TAAA” target site duplication (dark blue bars flanking the transposon’s TIRs), a pseudo repeat (trimmed wine-colored bar), and a self-targeting spacer (teal bar) were also identified. B Unrooted phylogenetic tree of Cas12k effector sequences. Cas12k effectors recovered here are shown as orange (confirmed transposon features) and black branches (only Cas effector), while reference Cas12k effector sequences are shown in gray. Reference sequences ShCas12k and AcCas12k are highlighted with red arrows. C Active single guide RNA design for MG64-1 and MG64-6. D Schematic of integration products assayed in in vitro experiments. Four junction PCR products are expected based on the orientation of integration: forward (Fwd) or reverse (Rev). E Integration junction PCR products for CAST systems MG64-1 (left lanes) and MG64-6 (right lanes) in vitro. Experiments were independently replicated twice. Product labels are derived from the schematic in (D). F SeqLogo representations of determined PAMs for MG64-1 (left) and MG64-6 (right).
Characterization of active type V-K CAST
In vitro experiments were conducted to determine the activity of predicted systems and their protospacer adjacent motif (PAM) preference. Integration reactions were conducted using in vitro expressed CAST proteins and guide RNA, a linear donor fragment, and a target plasmid library containing a PAM library (Fig. 1D). Active CAST systems will selectively integrate the donor DNA downstream of the target when a PAM motif from the library is recognized. The product of the integration reaction is detected by PCR amplification of each donor-target junction for both possible integration orientations (Fig. 1D). The type V-K CAST systems MG64-1 and MG64-6 were capable of integration in a programmable, sgRNA-dependent manner under the well defined in vitro conditions (Fig. 1E). Sanger-based sequencing of the PCR integration fragments confirmed the bioinformatic predictions of the transposon boundaries for both systems (Supplementary Fig. 5A).
To determine the PAM of the type V-K CAST systems, PCR products of successful integration events were sequenced via next-generation sequencing (NGS). PAM preferences were calculated by identifying enriched recognition motifs from the original population of the 8 N PAM library. Visualization of the enriched motifs identified a 5′ GTN PAM for MG64-1 and a 5′ rGTN PAM for MG64-6 (lowercase “r” denotes a weak A/G preference) (Fig. 1F). To determine the precision of integration events at the target site, we quantified NGS reads and observed that 90% of the integration events for MG64-1 and MG64-6 occurred between 57 and 67 base pairs away from the PAM (Supplementary Fig. 5 B, C).
Given that a large number of partial type V-K CAST systems were recovered from metagenomic data, we evaluated whether additional Cas12k targeting components were active with MG64-1 and MG64-6 transposase components. We observed that the Cas12k effector MG64-2 and its sgRNA (56 nt smaller than the endogenous MG64-1 sgRNA) were active with the MG64-1 transposase components, while MG64-57 Cas12k effector and its sgRNA were active with the MG64-6 transposase components (Supplementary text and Supplementary Figs. 6, 7). In addition, we showed that reduction of the endogenous sgRNA by 20% (Supplementary text and Supplementary Fig. 6) and 50% reduction of the terminal inverted repeats (TIR, Supplementary text and Supplementary Fig. 8) could be achieved without impacting in vitro activity.
Efficient and targeted genomic integration in E. coli
To benchmark the integration efficiency of the active CAST systems at diverse sites in the E. coli genome, we selected four endogenous target sites with an rGTN-5’ PAM in open reading frames (ORFs) and intergenic regions. Three separate plasmids containing protein-coding components, the single guide RNA, and donor DNA, were transformed into an engineered E. coli strain and maintained on triple antibiotic selection plates (Fig. 2A). The resulting colonies obtained from the transformation were then pooled and sequenced in order to analyze the population of genomes for on- and off-target integration. Probe-based qPCR and unbiased whole genome sequencing indicated integration efficiencies up to 80% at the engineered and endogenous loci (Fig. 2B and Supplementary Fig. 9). We extended these observations to evaluate multi-locus targeting in E. coli and observed simultaneous integration at both loci with activity as high as 80% at the engineered target and up to 50% at an endogenous intergenic region (Fig. 2C).

A Schematic representation of E. coli integration experiments. Plasmids harboring protein-coding elements (pHelper), integration cargo (pDonor), and sgRNA targeting locus (pGuide) are transformed into an engineered E. coli strain BL21(DE3). Antibiotic resistance is non-selective for integration. B On-target efficiencies at engineered loci targeting intergenic or coding targets (n = 3 biological replicates with one standard deviation from the mean). C On-target integration efficiencies at the engineered locus when multiplexed with integrations targeted to endogenous intergenic sites and protein-coding region targets (n = 3 biological replicates with one standard deviation from the mean). Source data are provided as a Source Data file.
Previously, S. hoffmanni type V-K CAST (ShCAST) activity has been shown to result in a mixture of integration events when the donor is delivered as a circular plasmid7,12,17,21. In addition to the expected integration of the transposon cargo, up to 80% of integrations have been shown to include two copies of the cargo along with the plasmid backbone in what are referred to as co-integration events3,7,12. This occurs due to the absence of TnsA for second-strand donor cleavage7,17. Both MG64-1 and MG64-6 systems lack TnsA and, as expected, result in both single (20-30%) and cointegration (70–80%) events upon delivery of a circular plasmid donor (Supplementary Fig. 9D).
In addition to co-integration events, off-targets of ~40–60% integration have been observed for ShCAST7,12,22. Notably, our multiplexed experiments with different sgRNAs simultaneously indicated fewer than 7% off-targets across all conditions, as demonstrated using unbiased whole genome sequencing (Supplementary Fig. 9C).
Engineering CAST for nuclear localization and function
One of the crucial steps to translate gene editing tools derived from bacteria into human cells is to ensure proper localization of the components into the nucleus, achieved through tagging with a nuclear localization signal (NLS). To determine the optimal orientation of NLS fusions to CAST proteins, we fused NLS tags to the N- or C-terminus of each CAST protein and evaluated expression and integration in vitro. System-specific improvements were observed, including tolerance for NLS tags at specific orientations determined for MG64-1 and flexible NLS fusions observed for MG64-6 (Table 1 and Supplementary Fig. 10). By using an in vitro expression and transposition procedure, we were able to rapidly determine a starting set of NLS constructs for further investigation.
While active in vitro, we sought to confirm that intracellular expression of each NLS-protein fusion would result in nuclear localization using immunofluorescent staining of epitope tags in HEK293T cells. Each NLS-tagged protein for both CAST systems efficiently translocated into the nuclear environment, with the exception of TnsC (Fig. 3A and Supplementary Fig. 11). However, co-expression of TniQ with TnsC decreased the cytoplasmic restriction of TnsC, resulting in nearly full localization of TnsC and TniQ into the nucleus (Supplementary Fig. 11B, C). These results are in line with observations of the cryo-EM structure of ShCAST, suggesting direct interactions between TnsC and TniQ as an RNP complex23.

A Immunofluorescence of MG64-6 components localizing to the nucleus of human cells. Left: components tagged with an HA flag. Right: components tagged with a FLAG tag. Arrows indicate residual localization of TnsC in the cytoplasm, scale bars represent 10 µm. B Schematic representation for in vitro testing of nuclear extracts to determine the functionality of CAST components post-localization. Cells expressing components of interest are differentially lysed to yield cytoplasmic and nuclear extracts. Upon incubation with the addition of sgRNA, donor fragment, and pTarget, PCR junctions are amplified. C Integration junction PCR products of in vitro transposition reactions using nuclear extract inputs from cells expressing Cas12k-NLS with (lanes 3–6) and without (lanes 8–10) sso7d, NLS-TnsB, NLS-TnsC, and either NLS-HMGN1-TniQ or NLS-H1 core-TniQ. In vitro, control reactions contain all components expressed exclusively in vitro and tested in vitro. Expected bands for the forward (Fwd) left end (LE) and right end (RE) or reverse (Rev) LE and RE, with added sgRNA (+), indicate positive integration (arrows). D Genomic search space (bp) for single targets at high-copy loci in the human genome. The search space required to target the LINE-1a 3’ (10,000 copies), SVA (1000 copies), Mariner (50 copies), HERV (4 copies), or a unique human target (1) are compared to the search space required to target a unique E. coli site. E In vitro integration junction PCR products at high-copy targets in purified human genomic DNA. Expected bands for the Fwd LE or Rev LE rows indicate positive integration (arrows). Experiments in A, C, E were independently replicated twice.
In order to confirm the activity of CAST protein components after successful nuclear localization, we selected the MG64-1 system for its higher efficiencies in E. coli for evaluating if nuclear extracts obtained for each CAST component were active for integration activity in vitro (Fig. 3B). Active CAST components from nuclear extracts include TnsB with either N- or C- terminal NLS tags, and TnsC with an N-terminal NLS (Table 1 and Supplementary Fig. 12A). However, NLS-tagged Cas12k and TniQ from both CAST systems were not active despite proper localization (Supplementary Fig. 12B). Complementing with in vitro expressed Cas12k was sufficient to achieve transposition (Supplementary Fig. 12B). This indicated that Cas12k, while localizing to the nuclear compartment, may not function robustly in the nuclear environment. Given that the activity of CRISPR enzymes can be improved with chromodomain or processivity factor fusions24,25, we sought to evaluate whether this approach could be extended to unlock type V-K CAST function in nuclear extracts. We constructed NLS-tagged Cas12k and TniQ fusions with and without sso7d, human H1-core, and human HMGN1 domains and assayed for their activity in vitro after in-cell expression. Results indicate that combining Cas12k-sso7d with either H1 core-TniQ or HMGN1-TniQ fusions activates CAST extracted from the nuclear environment (Fig. 3C). With all type V-K CAST protein components localized to the nucleus and confirmed active after in-cell expression, we evaluated the ability of the system to integrate into human genomic DNA.
Target availability impacts CAST integration directionality
One potential limit to CAST system activity in human cells is the ability of Cas12k to navigate the target landscape of the human genome. We sought to test two components of this search: the ability of the system to integrate when targets are rare, and the ability of the system to navigate the complex sequence and size of the human genome. We conducted an in vitro experiment where the amount of target plasmid was titrated across six orders of magnitude, and found that integration favored a single orientation (“forward”) as the system was challenged to find more rare targets (Supplementary Fig. 13A). Furthermore, we found that adding complex background DNA derived from human cells did not prevent integration (Supplementary Fig. 13B), but as the concentration of background DNA increased we similarly saw only the “forward” orientation junction products. Together, these results demonstrate that CAST have a favored directionality of integration, which is consistent with data showing that the forward orientation is also favored when the ShCAST system is used in E. coli7,17 (Supplementary Fig. 9E). The observed directionality of integration will be advantageous for most applications of CAST technology.
In vitro integration at multicopy loci in the human genome
Given the three orders of magnitude increase in the size of the human genome compared with the native cyanobacterial genome, we simplified the search for target sites in the human genome by selecting multicopy sites that occur at various frequencies. We surveyed a conserved family of Long Interspersed Nuclear Elements (LINE-1a 3’), SINE-VNTR-Alu Elements (SVA), and Human Endogenous Retrovirus (HERV) elements because they are present at approximately 10,000, 1000, and 4 copies per haploid HEK293T genome, respectively (Fig. 3D). Notably, the SVA site is found at a frequency that mimics single-target abundance in the E. coli genome. Integration activity to high-copy elements across the human genome were detected in vitro at three target sites in the LINE-1a 3’ element, in both forward and reverse orientations (Fig. 3E). Furthermore, we were able to detect integration to SVA and HERV elements (Fig. 3E), which represents a range of in vitro activity across three orders of magnitude of target concentrations. With these promising results, we then tested integration directly in immortalized human cells.
Programmable genomic integration in human cells at multicopy loci
For initial experiments, all CAST components and the recently discovered host factor S1515,16 with a C-terminal NLS were cloned into two plasmids for expression in HEK293T cells (see Methods, Fig. 4A, left, and Supplementary Fig. 14). sgRNAs were designed to target multiple LINE-1a 3’ sites and a single SVA target. For the donor DNA, a 2.5 kb sequence was designed and flanked with the transposon TIR (Fig. 4A). Post-transfection, cells were harvested for genomic DNA, and target-donor junctions were successfully detected at all LINE-1a 3′ targets, exclusively in the forward orientation (Fig. 4B). Sanger sequencing traces demonstrated a clean signal up to the integration junction, where the signal expectedly degrades in quality due to the heterogeneity of integration sites across the genomic population (Supplementary Fig. 15A). NGS reads of target-donor junction PCR reactions confidently mapped single molecule readouts confirming integration in the forward orientation for all expected LINE-1a 3′ and SVA targets (Supplementary Fig. 15B, E).

A, B Schematic representation of the dual plasmid system (left) and all-in-one plasmid system (right) expressing CAST and either an untargeted or targeted single guide RNA. The donor plasmid (pDonor) contains LE and RE, flanking either a reporter gene, a therapeutic transcript, or a selection marker. B Integration junction PCR products in the forward (Fwd) LE direction using the dual plasmid system indicate programmable integration to the human genome. Mock: transfection control (no bands expected). Lanes labeled 8, 12, and 15 correspond to LINE-1a 3’ targets 8, 12, and 15. C Junction PCR of Fwd integration events targeting two different AAVS1 sites (T1 and T2) confirm integration of the donor in the human genome. D Sanger sequencing trace of T1 target junction PCR products. Reference sequence (topmost sequence) with annotated Target and LE represents the expected integration junction sequence 63 bp away from the PAM. A 3-bp deletion is observed. E, F On-target integration efficiency determined from NGS for the expected LE integration junction product for MG64-1 with and without ClpX (E, n = 6 biological replicates for the target condition (T1) and n = 3 biological replicates for the non-targeting control (Null), with one standard deviation from the mean), and for MG64-1 and ShCAST (F, n = 4 biological replicates for both target condition (T1) and non-targeting control (Null), with one standard deviation from the mean). G Schematic of the on and off-target integration assay. Integration reads containing the target-to-LE junction will align upstream from the integration site (blue arrows), while reads containing the RE-to-target junction will align downstream from the integration site (pink arrows), leaving a target site duplication (TDS). H Convergence of AAVS1-T1 on-target reads, indicated by LE and RE reads aligning to the human genome with overlap due to the TDS confirms complete integration of the donor to the target site (T1). Experiments were done in duplicate. I Venn diagram of independent and shared off targets from two biological replicates of AAVS1-T1 sgRNA-containing samples. Source data are provided as a Source Data file.
In order to simplify CAST delivery, we tested whether a single transcript expression of all five protein-coding components necessary for in-cell integration could be used to integrate large, multi-kilobase cargo. We designed a single plasmid containing all the protein-coding components of type V-K CAST and S15 separated by IRES and 2A elements for concerted transcription and independent translation (Fig. 4A, right). The all-in-one plasmid also contains a single guide expression cassette. The design of the all-in-one plasmid reduced the design constraint for a second plasmid for cargo delivery. We validated human genomic integration to high-copy elements by targeting LINE-1a 3′ with the all-in-one plasmid construct (Supplementary Fig. 16A).
Recently, the bacterial protein ClpX, an AAA+ ATPase, was identified as a host factor that significantly improves Cascade CAST integration18. It is hypothesized that this improvement comes from the destabilization of the transposition complex, which enables resolution of the integration event due to increased accessibility to DNA repair machinery18. However, it is unclear whether the role of ClpX can be extended to the type V-K CAST. We tested if ClpX could improve efficiency by expressing NLS fusion proteins encoded on a third plasmid. Both N- and C- terminal fusions enhanced integration efficiency at the high-copy locus LINE-1a 3’ (Table 1, Fig. 4E, and Supplementary Fig. 16B). Given the qualitative, albeit subtle, improvement of the C-terminal versus N-terminal fusion, we chose to use the ClpX-NLS fusion to test targeted integration to single-copy loci with type V-K CAST.
CAST efficiency improvements enable integration at a single-copy safe harbor locus in human cells
AAVS1 was selected as a target because it is a single-copy site that has been extensively studied as a safe harbor locus for targeted transgene integration for human therapeutic applications26. When cells were transfected with the all-in-one plasmid, donor plasmid, and ClpX plasmid, we were able to detect successful integration of the 2.8 kb donor for two sgRNAs targeting distinct AAVS1 sites by junction PCR (T1 and T2, Fig. 4C). Sanger sequencing and NGS of the resulting target-donor junction PCR reactions verified the ability of the MG64-1 CAST system to integrate gene-sized DNA cargos to AAVS1 target site T1 (Fig. 4D and Supplementary Fig. 16D, E).
Through quantification by NGS, we determined the fraction of reads that corresponded with an integration event to be over 1% on average across AAVS1-T1 as measured by junction PCR NGS sequencing where the addition of ClpX enhanced editing rates by five-fold (Fig. 4E). Additional NGS analysis aimed at profiling all on-target editing events did not detect InDel formation or any other unexpected modifications (Supplementary Fig. 17). These integration efficiency measurements across the full population of cells are in the range observed for genomic integration in human cells with Cascade CAST18
In order to determine if the optimizations we conducted were system-specific, we applied our findings for MG64-1 translation to the components of the best characterized S. hoffmanni Cas12k system (ShCAST). Cognate components were synthesized with NLS fusions in the same orientation as those for MG64-1, where chromodomains sso7d and H1-core were fused to the C-terminus of Cas12k and N-terminus of TniQ, respectively. We also applied sgRNA optimization for expression in human cells and designed cargoes using the previously described full-length LE and RE elements7. Given that ShCAST recognizes a 5’ NGTN PAM7, we used the same AAVS1-T1 targeting spacer that was validated with MG64-1. When tested under the same experimental conditions with ClpX co-expression, MG64-1 CAST showed 2% targeted integration as measured by NGS, while for ShCAST, no reads could be identified that would indicate target-specific integration (Fig. 4F).
Specificity assay development
Although off-target CAST integration was measured in E. coli using whole genome sequencing, this approach could not be applied to the human genome due to the sequencing depth that would be needed to accurately capture rare off-target events given the system efficiencies observed. In order to overcome this challenge, we developed a method that uses deep sequencing of enriched LE and RE junction sites to identify complete integration events. This is achieved by fragmenting the genome after a transposition experiment and then ligating an adapter containing a unique molecular index (UMI) that enables unbiased PCR-based amplification and detection (Fig. 4G). Using this method, we compared integration events across two replicate conditions containing the AAVS1-T1 single guide and a negative control lacking the entire single guide cassette. We confirmed on-target transposition by observing reads containing both the LE and RE elements aligning at the expected primary sequence of AAVS1, within a 50 bp window (Fig. 4H). This bi-directional convergence of reads suggests complete integration with the expected overlap caused by a target site duplication22. Successful observation of the on-target integration event provided confidence that this method could be used to evaluate off-targets in the human genome.
Off-target integrations are rare and localized in genomic ‘hot spots’
Using the developed assay, we determined that most off-target events correspond to sites within the pHelper plasmid, including the H1-core chromodomain fused to TniQ (H1-0), the sgRNA expression promoter (RNU6-1 promoter), and other regulatory elements in the plasmid backbone (HBB insulator and intron) (Supplementary Fig. 18A). This suggests that most detected off-targets may be due to plasmid to plasmid, versus plasmid to genomic, integration events. Off-targets involving integration into human chromosomal DNA included up to eight target sites observed in conditions containing a single guide RNA targeting AAVS1-T1. Five sites were reproducible in both conditions (Fig. 4I) but were not detected in negative controls. The five off-target sites detected had a similar observed frequency of integration to the on-target site (Supplementary Fig. 18B). Poor alignment between the sgRNA spacer sequence of AAVS1-T1 and adjoining regions of detected transposition events (9 - 14 mismatches or bulges) indicated that these integration events are not driven by spacer homology. Interestingly, reads aligning to off-target sites are broader in their distribution compared with the on-target site, suggesting multiple distinct integration events occurring in a confined genomic region (Supplementary Fig. 18C). Integrations detected in both experiments containing sgRNA were found in regulatory elements (promoter of EEF1A1 and FTL and 3′ UTR of HBB). The reproducibility of these events across the two experiments suggests that off-target integrations are largely constrained to genomic “hot spots,” and are not driven by spacer homology. Off-target integrations observed here can likely be addressed by tuning expression levels of TnsC or with further system optimization, as observed for ShCAST in E. coli17,22.
CAST are active in multiple human cell types, and with donor templates delivered on linear DNA
Initial CAST integration experiments in human cells were conducted using plasmids that include an SV40 origin that allows them to replicate in HEK293T cells. Plasmid replication may improve integration efficiencies, both by increasing the expression of CAST components and by providing additional copies of donor DNA. Given that dependence on plasmid replication would be a barrier to the therapeutic deployment of CAST, we sought to determine whether or not CAST could be translated to cell types where integration was not dependent on having replicating plasmids. Immortalized myelogenous K562 cells and liver carcinoma Hep3b cells do not express the SV40 large T antigen, and thus transfected CAST plasmids would not be able to replicate. We measured targeted integration by NGS as high as 0.3% (Fig. 5A, B) at AAVS1 in both K562 and Hep3b cell lines, confirming that integration is not dependent on plasmid replication.

A On-target integration efficiency of type V-K CAST in K562 cells at AAVS1 target T1 (n = 3). B Integration efficiencies in Hep3b at AAVS1 target T1 at varying doses of an all-in-one helper plasmid (AIO), donor plasmid, and ClpX plasmid concentrations (Supplementary Data 3) (n = 2). C Integration efficiencies determined by NGS when delivered with a linear donor at AAVS1 target T1 (single experiment). D Schematic of Factor IX (FIX) donor plasmids. E On target efficiencies of FIX donor with varying ratios of CAST AIO expression plasmid and FIX donor plasmids. F Relative efficiencies with FIX pDonor plasmid with and without multiple copies of the sgRNA cassette. Fold increases were normalized to the mNeon donor plasmid control with one copy of the sgRNA. G Relative rates of LE and RE junctions normalized to the LE from FIX integration experiments. Quantified integration efficiencies in A–C, E–G are shown as the mean (bar) of biological replicates (dots) with one standard deviation (whisker). Null represents the non-targeting spacer (negative control) and T1 represents the AAVS1 target 1. Source data are provided as a Source Data file.
Based on the promising results with non-replicating plasmids, we also sought to determine whether or not integration could be accomplished with a linear DNA donor. Given that using a linear donor would enable bacteria-free manufacturing, the use of a linear donor would be easier to scale and could potentially be safer to deliver than bacterially-produced plasmids27. We were able to detect integration at the AAVS1 locus in HEK293T cells with a linear donor, albeit at lower levels than with the plasmid-based experiments (Fig. 5C). These results validate the use of type V-K CAST systems for programmable genome editing, including with gene-size cargo.
Improving CAST delivery for integration of therapeutically relevant cargo
Bleeding disorders such as hemophilia have been an important target for gene therapies because it is possible to express corrective proteins in the liver that are secreted into the bloodstream. Both Hemophilia A and B have received considerable interest for this approach; however, gene editing that involves stable integration would have significant therapeutic advantages in terms of treatment durability. As a proof-of-concept, we sought to determine whether a complete copy of the gene for the Factor IX clotting factor (FIX), for which mutations are known to cause Hemophilia B, could be integrated into the AAVS1 safe-harbor locus using type V-K CAST. We used the all-in-one CAST expression plasmid containing the sgRNA targeting AAVS1-T1, a plasmid expressing ClpX, and a third donor plasmid containing the full human FIX gene (1383 bp) expressed under the control of a chimeric phAAT promoter amounting to 3.6 kb of donor cargo (Fig. 5D, top). We observed >1% targeted integration of the FIX cargo at the AAVS1-T1 site, as measured by NGS when tested using multiple ratios of the all-in-one and donor plasmids (Fig. 5E).
To evaluate whether increased expression of the sgRNA could increase transposition rates, we assembled a donor construct with two identical sgRNA cassettes outside of the LE and RE regions of the FIX donor (Fig. 5D, bottom). Titration of the dual sgRNA-containing plasmid with the all-in-one plasmid containing one copy of the sgRNA resulted in a linear increase in integration efficiency of FIX, with a maximum threefold increase (Fig. 5F).
Finally, when integrating a therapeutic payload into the human genome, the rate at which complete integration of the donor DNA is achieved will affect the amount of functional protein produced as a result of the genomic edit. Full integration of a donor would be characterized by the detection of the LE-target and RE-target junctions at a 1:1 ratio. Integration experiments of FIX in cells indicate that the relative integration efficiency from LE and RE target to donor junctions approximate a 1:1 ratio, confirming that a majority of integration events are complete (Fig. 5G). Results demonstrate the versatility of compact type V-K CAST for complete programmable integration of gene-size DNA constructs, including therapeutically relevant cargo, to a safe harbor locus across multiple human cell types, and their potential for development into tools for therapeutic gene editing.
Discussion
CRISPR-associated transposons (CAST) are promising systems for therapeutic and biotechnological applications, enabling target-specific integration of large DNA constructs or transgenes. Systems for targeted integration of full-length genes have the potential to treat genetic diseases caused by allelic heterogeneity in a single therapeutic approach. While the translation of CAST systems has been challenging, we report on a compact, RNA-guided type V-K CAST system capable of integration of gene-sized DNA cargos into the genome of human cells. This was achieved by discovering active systems from uncultivated microorganisms through metagenomic analysis and engineering the components for nuclear localization and targeted integration activity.
Compared to DNA integration strategies requiring dsDNA breaks and DNA repair, where integration events may not be predictable or reproducible, we find that integrations from CAST occur in a predictable orientation when tested in E. coli and human cells. This agrees with other reports on type V-K CAST systems previously described7,28. The strong preference for directionality and narrow integration window may enable applications where an exogenous donor requires an endogenous or native promoter for transcriptional control of the therapeutic cargo or simultaneous knock-in/knock-out editing. Similarly, the high levels of multi-locus targeting capability observed in E. coli and in cells may streamline cell and strain engineering for biotechnology and synthetic biology, for example, by the integration of entire biosynthetic pathways at multiple loci simultaneously.
Using a modular approach to CAST component testing, we determined the requirements for the activity of CAST discovered here and demonstrated that sgRNA and Cas12k effectors of inactive and partial CAST systems are functional with active CAST integration machinery. These observations were unexpected given the multicomponent co-evolution of the CAST systems but open the possibility of swapping components to further optimize integration activity and specificity. Previous work on type V-K ShCAST indicated that high rates of off-target integrations in the E. coli genome were driven by high expression of TnsC, and that by titering TnsC expression down the relative ratio of on-to-off-target integrations increased22. One potential explanation for the relatively low rates of off-target transposition observed here compared to literature reports may be the attenuated strength of TnsC expression driven by our construct design.
Off-target analyses for CAST systems integrating cargo in human cells have not been previously reported. To address this gap, we developed an assay capable of detecting integration events while enriching transposition-based junctions across the human genome. Our results revealed consistent off-target integrations clustered in specific transposition hot spots. Notably, we observed plasmid-to-plasmid integrations, which may occur more frequently than genomic integrations, possibly due to the higher availability of plasmid substrates. Additional experimental work will be needed to determine the dynamics between on-target versus off-target insertions and to identify paths to reduce off-target integration for further CAST development. Overall, we anticipate that further engineering of CAST components and optimization of delivery methods will reduce off-target integrations in human cells.
One unique aspect of type V-K CAST is that the lack of TnsA can result in co-integration events, where additional cargo is integrated beyond the LE and RE sequences when circular donors are used for delivery. This has been mitigated in the ShCAST system through fusions with an endonuclease that mimics the activity of TnsA in the transposition process17. Here, we demonstrated that type V-K CAST can use a linear DNA donor, which is expected to result in a consistent single integration event that avoids the co-integration product. This finding simplifies the delivery and integration process for future applications.
Although previous studies showed that type V-K CAST systems promote integration into target plasmids in human cells17, we achieved genomic integration by fusing Cas12k and TniQ components with chromodomains and processivity factors. These additions likely stabilize the targeting components in the human nuclear compartment or compensate for missing host factors. However, these enhancements were not universally effective for other CAST systems, as they failed to activate ShCAST in human cells. Further research to identify additional host factors remains ongoing. Recently, Lampe and colleagues observed that integration at multiple single-copy target sites in human cells with a multicomponent targeting Cascade CAST improved with the addition of the bacterial protein ClpX18. Our ability to integrate cargo into the LINE-1a 3′ locus with a type V-K CAST indicates that the use of ClpX is not strictly necessary for genomic integration in human cells, suggesting that further optimization may dispense with the need for this additional component.
Through targeted optimization and selection of genomic sites, we demonstrated programmable integration of gene-sized DNA donors into high-copy target sites, as well as successful integration of therapeutically relevant DNA donors at single-copy loci in the human genome. Expanding on this foundation, we achieved improvements in integration efficiency and extended the application of the system to additional immortalized human cell types. Further, results address specificity while showcasing the robustness and versatility of the system. Our findings provide a path for advancing and optimizing the specificity and efficiency of CAST, laying the groundwork for application in therapeutics and biotechnology.
Methods
In silico discovery of CAST
Thirteen sediment samples were collected and stored on ice or in Zymo DNA/RNA Shield after collection. DNA was extracted from samples using either the Qiagen DNeasy PowerSoil Kit or the ZymoBIOMICS DNA Miniprep Kit. DNA sequencing libraries were constructed (Illumina TruSeq) and sequenced on an Illumina HiSeq 4000 or Novaseq at the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, with paired 150 bp reads with a 400–800 bp target insert size. Publicly available metagenomic sequencing data from diverse environments were downloaded from the NCBI SRA. Sequencing reads were trimmed using BBMap29 (sourceforge.net/projects/bbmap/) and assembled with MEGAHIT30. Protein sequences were predicted with Prodigal31. HMM profiles of known Type V CRISPR nucleases and Tn7-like transposons were built and searched against all predicted proteins using HMMER3 (hmmer.org). Predicted proteins were annotated by searching Pfam (http://pfam.xfam.org/) HMMs using HMMER3. CRISPR arrays were predicted on assembled contigs with Minced (https://github.com/ctSkennerton/minced). Taxonomy was assigned to proteins with Kaiju32 and contig taxonomy was determined by finding the consensus of all encoded proteins. Protein alignments were done with MAFFT33 with parameters G-INSI, and phylogenetic trees were inferred using FastTree234. Active tracrRNA sequences were aligned with MAFFT with parameters X-INSI and the secondary structure of the alignment was predicted with RNAalifold from the Vienna Package35. These structural alignments were used to build covariance models with cmbuild and cmcalibrate from the Infernal suite36. Covariance models were used for tracrRNA prediction on all contigs containing a Cas12k protein with the program cmsearch from the Infernal suite.
Plasmid and cloning assembly
CAST expression plasmids were synthesized and cloned into pET21(+) expression vectors under the control of the T7 promoter. Cas12k was codon optimized for E coli expression and synthesized alone on a vector, while TnsB, TnsC, and TniQ were codon optimized for E. coli expression except for the first and last 40 bp of every open reading frame and assembled on a pET 41(+) plasmid. Guides were synthesized on a p15a cloning plasmid under the control of the J23119 constitutive promoter. Donors were cloned on a sc101 origin where the kanamycin cloning cassette was flanked with TIR elements. A bacterial helper plasmid containing Cas12k, TnsB, TnsC, and TniQ was cloned with Gibson assembly onto the pET21(+) backbone.
Cas12k, TnsB, TnsC, and TniQ with N and C terminal NLS peptides were synthesized on Twist Lentivirus transfer vectors for transduction into HEK293T and K562 (ATCC CCL-243) cells. Active versions of TnsB were cloned with TnsC in a polycistronic transcript separated by an IRES element. Cas12k and TniQ variations were cloned in a polycistronic transcript for mammalian cell expression.
Transient transfection plasmids were synthesized in a polycistronic transcript for Cas12k-sso7d-NLS-2A-S15-NLS with NLS-TniQ, and NLS-TnsB with NLS-TnsC on Twist replicating mammalian expression plasmids. pU6 driven 64-1 single guide RNA was cloned into the Cas12k-TniQ plasmid backbone and LE and RE elements were added to the bacterial origin and selection cassette of the TnsB-TnsC expression plasmid.
In vitro targeted integrase activity
Single guide RNAs (sgRNAs) were designed as follows: tracrRNA and crRNA sequences were jointly folded using RNAfold (Andreaceau 2007). The repeat-antirepeat annealing region was trimmed to maintain 8 to 10 full base pairs and a GAAA tetraloop was appended on the 3′ end of the tracrRNA to join the 5′ end of the trimmed crRNA. Integrase activity is assayed with an 8N PAM library substrate adjacent to the spacer sequence. T7 promoter sequences were introduced by PCR amplification of all transposase, single guide, and Cas components, and expressed independently in an in vitro transcription/translation system (PURExpress, NEB). Purified in vitro transcribed single guide RNA (HiScribe, NEB) was refolded in duplex buffer (10 mM Tris pH 7.0, 150 mM NaCl, 1 mM MgCl2) and normalized to 1 µM. Donor fragments were PCR amplified from pDonors of the respective systems, a plasmid bearing a kanamycin or tetracycline resistance marker flanked by the left end (LE) and right end (RE) transposon motifs for integration and normalized to 50 ng/µL.
After expression, 1 µL of Cas12k PURExpress reaction is added to 0.5 pmol of sgRNA and incubated for 20 min at 25 ˚C. Individually expressed Tns proteins were then added volumetrically at 1 µL per expression. About 50 ng target DNA, and 50 ng donor DNA are then added to the transposition reaction in a reaction buffer, with final concentrations of 26 mM HEPES pH 7.5, 4.2 mM TRIS pH 8, 50 ug/mL BSA, 2 mM ATP, 2.1 mM TCEP, 0.05 mM EDTA, 0.2 mM MgCl2, 28 mM NaCl, 21 mM KCl, 1.35% glycerol,(final pH 7.5), and 15 mM MgOAc2. In vitro transposition reactions were performed at 37 ˚C for 2 h, and transposition reactions were diluted tenfold in water, and used subsequently as a template for junction analysis. NLS-tagged CAST components were tested similarly to in vitro experiments explained above except single NLS-tagged components or multiple tagged components, where noted, were swapped for WT versions. For in vitro human gDNA experiments, 1 µg of purified HEK293T genomic DNA was added as the target substrate to the reaction unless otherwise indicated.
To test for S15-NLS tagging, TnT (R) Coupled Wheat Germ Extract (Promega, #L4140) was used as the expression system. Constructs were amplified as above using a T7 promoter, with a T40 reverse primer to add a 40A poly-A tail. All PCR constructs were expressed at 10 ng/µL final concentration and subsequently assembled similar to PURExpress reactions with IVT guide RNA, Donor, pTarget, and Buffer
Junction analysis
Junction PCR was performed with Q5 polymerase (NEB) and amplified with primers (Supplementary Data 2) for: Target (LA179 & LA125), Donor (oJL220 & oJL221), Forward LE (LA125 & LA155), Forward RE (LA179 & LA156), Reverse LE (LA179 & LA155), Reverse LE (LA125 & LA156). PCR fragments were then run on a 2% agarose gel in 1x TAE and analyzed for size discrimination. Appropriately sized bands of each PCR junction were then gel excised, and the PCR fragment recovered and sanger sequenced using both amplification primers. The resulting Sanger sequencing was mapped to a putative forward or reverse integration at 60 bp away from the PAM.
PAM determination and transposition distance
PCR junctions containing the PAM were indexed and sequenced using a V2 300-cycle MiSeq read kit. PAMs transposition positive reads were normalized to an untransposed library and visualized using SeqLogo. Reads were subsequently mapped and quantified using CRISPResso237 using an amplicon sequence of a putative transposition sequence with a 60 bp distance of integration from the PAM (guideseq = 20 bp 3′ end of LE or RE, center of window = 0, window size = 20) Indel histogram was normalized to total indel reads detected, and frequencies were plotted relative to the 60 bp reference sequence
Both PCR reactions of Target-LE-RE transpositions were plotted on the sequence and distance from the PAM for MG64-1. Analysis of the integration window indicates that 95% of the integrations that occur at the spacer PAM site are within a 10 bp window between 58 and 68 nucleotides away from the PAM. Differences in the integration distance between the distal and the proximal frequencies reflect the integration site duplication.
LE/RE and single guide engineering
Repeated elements of the Terminal Inverted Repeats were predicted using RepeatFinder algorithm (Geneious) and minimized LE/RE were designed through sequential deletion from the cargo end of the TIR using HiFi NEBuilder (NEB). Resulting engineered TIR were then cloned into pDonor formats and amplified for in vitro transposition reactions.
pGuide plasmids for MG64-1 and MG64-6 single guides were truncated using HiFi NEBuilder (NEB), and PCR amplified for in vitro transcription, and transcribed as mentioned above. Split guides were PCR amplified from the WT single guide MG64-2 pGuide plasmid and transcribed as mentioned above.
Construction of E. coli with engineered target
For testing of effector-assisted integrase activity in bacterial cells, strain MGB0034 was constructed from BL21(DE3) E. coli cells. A target sequence of 5′-GTCGAGGCTTGCGACGTGGTGGCT-3′ was inserted into the lacZ locus immediately downstream of a 5′-AGTC-3′ PAM sequence. MGB0034 E. coli cells were then transformed with two plasmids: pHelper and pGuide. pHelper is an ampicillin-resistant plasmid that expresses the effector and the Tns proteins suite for either MG64-1 or MG64-6 Tns proteins and Cas12k. In multiloci experiments, the pHelper of MG64-1 also contained a single guide targeted to the engineered target sequence driven by the T7 promoter. pGuide is a chloramphenicol-resistant plasmid that expresses the single guide RNA sequence for the engineered or endogenous target of interest driven by a J23119 promoter.
E. coli transposition experiments
A culture containing pHelper or pHelper and pGuide plasmids was then grown to at 37 ˚C until saturation, diluted at least 1:10 into LB with appropriate antibiotics (OD <0.2), and incubated at 37 °C until OD of ~0.6. Cells from this growth stage were made chemically competent by washing four times in a 1x volume of ice-cold 0.1 M calcium chloride. They were transformed with 100 ng pDonor, a plasmid bearing a kanamycin or tetracycline resistance marker flanked by the left end (LE) and right end (RE) transposon motifs for integration. Heat-shocked cells were then recovered for 2 h on LB medium at 37 °C before being plated on LB-agar-ampicillin-chloramphenicol-kanamycin with or without 0.02 mM IPTG, and incubated 2 days at 37 °C. Plates were scraped into LB medium, and a cell pellet was collected by centrifugation at 14,000 rpm for 30 min. The pellet was resuspended in ~1 mL of LB. Approximately 200 μL of this suspension was set aside for genomic DNA extraction, while the remainder was re-pelleted and stored at −20 °C. Genomic DNA was extracted using the PureLink Genomic DNA Kit (Invitrogen) and quantified by QuBit (dsDNA HS kit, Invitrogen). Transposition experiments were performed and analyzed in triplicate.
qPCR
Genomic DNA isolated from transposition experiments was then used to test for transposition efficiency by qPCR and NGS. qPCR experiments were performed in 96 well plates where each well contained 2.5 μL of 2 ng/μL genomic DNA template, 1 μL each of 10 μM primers and 4 μM probe, 4.5 μL UltraPure DNAse RNAse—Free Distilled Water (Invitrogen), and 10 μL of PrimeTime Gene Expression MasterMix (IDT). Plates were sealed and inserted into an AriaMx Real-Time PCR System (Agilent). The qPCR protocol consisted of an activation step at 95 °C for 3 min, and then 40 cycles of 5 s at 95 °C followed by 10 s at 60 °C. Cq values were determined using Agilent AriaMx software (Agilent). For each genomic DNA template, six replicate reactions were prepared with transposition primers and a probe, and six replicates were prepared with reference primers and a probe. The highest and lowest Cq value of these replicates were thrown out, and the remaining four were averaged to determine the final Cq. The transposition efficiency was calculated as 100*2^(ΔCq) where ΔCq = Cq of the reference reaction minus the Cq of the transposition reaction. Primers and Probes are listed in Supplementary Data 2. All were efficiency tested prior to their use in qPCR experiments.
E. coli NGS transposition efficiency, on/off target determination, co-integration analysis
Whole genome sequencing with Illumina short reads was performed by building DNA sequencing libraries from extracted E. coli genomic DNA (Illumina TruSeq) and sequenced using the Illumina MiSeq platform. Whole genome sequencing with Oxford Nanopore was performed using the rapid barcoding kit (SQK-RBK110.96, Oxford Nanopore) and sequenced on the MinION Mk1B. The results of short-read whole genome sequencing following transposition experiments were analyzed by custom python scripts identifying moving averages of an on-target integration normalized to coverage of the integration site. Briefly, filtered reads were aligned against the MGB0034 genome sequence, pHelper, pTCM, and pDonor plasmids using BWA38. Chimeric reads mapping to both pDonor TIR sequences and genomic sequence were filtered from the alignment and normalized to the number of reads spanning at least 20 bp on either side of the genomic breakpoint. The total efficiency of transposition was calculated by performing a weighted average of local breakpoints across relative transposition efficiency. Breakpoint analysis was performed at the 5’-TGTACA-3’ motif of the LE and RE, and on-target relative transposition efficiency was summed for breakpoints across a 20 bp on-target window between 50–70 bp distance from the PAM. All other breakpoints outside the 20 bp window were counted as off-target.
Co-integration events were detected by aligning the Oxford Nanopore long reads against the MGB0034 genome sequence, pHelper, pTCM, and pDonor plasmids using Minimap239. Chimeric reads were extracted from the alignment and coordinates of the DNA cargo within the pDonor plasmid were projected onto read segments that aligned to the pDonor plasmid using custom python scripts. Chimeric reads in which a single segment of the read aligned to the full length of the DNA cargo, flanked on both sides by read segments that aligned to the E. coli genome, were counted as single integration events. Chimeric reads in which a single segment of the read aligned to the full length of the DNA cargo, flanked on one side by a read segment that aligned to the E. coli genome and on the other side a read segment that aligned to the pDonor backbone, were counted as co-integration events. Due to the error profile of these long reads, aligned segments in chimeric reads shorter than 25 bp were ignored during this analysis.
Lentiviral transduction and intracellular fractionation
To test the functionality of the NLS constructs in a physiologically relevant environment, constructs cloned with active NLS-tagged CAST components were integrated into K562 cells using lentiviral transduction. Briefly, constructs cloned into lentiviral transfer plasmids were transfected into 293T cells with envelope and packaging plasmids, and virus-containing supernatant was harvested from the media after 72 h incubation. Media containing the virus was then incubated with K562 or HEK293T cell lines with 8 µg/mL of polybrene for 72 h, and transfected cells were then selected for integration in bulk using Puromycin at 1 µg/mL for 4 days. Cell lines undergoing selection were harvested at the end of 4 days, and differentially lysed for nuclear and cytoplasmic fractions. Subsequent fractions were then tested for transposition capability with a complementary set of in vitro expressed components.
10 million cells are harvested and washed once with 1xPBS pH 7.4. Supernatant wash is aspirated completely to the cell pellet, and flash-frozen at −80 °C for 16 h. After thawing on ice, cell pellet size is measured by mass, and appropriate extraction volumes of cell fractionation and nuclear extraction reagent (NE-PER) is used to natively extract proteins in cell fractions. Briefly, cytoplasmic extraction reagent is used at 1:10 mass of cells to the volume of extraction reagent. The cell suspension is mixed by vortexing and lysed with non-ionic detergent. Cells are then centrifuged at 16,000×g at 4 ˚C for 5 min. Cytoplasmic extraction supernatant is then decanted and saved for in vitro testing. Nuclear extraction reagent is then added 1:2 original cell mass to nuclear extraction reagent, and incubated on ice for 1 h on ice with intermittent vortexing. Nuclear suspension is then centrifuged at 16,000×g for 10 min at 4 ˚C, and the supernatant nuclear extract is decanted and tested for in vitro transposition activity. Using 4 µL of each cell and nuclear extract for each condition, we performed the in vitro transposition reaction with a complementary set of in vitro expressed proteins, IVT sgRNA, donor DNA, pTarget, and buffer. Evidence of transposition activity was assayed by PCR amplification of donor-target junctions.
Immunofluorescence in HEK293T cells
HEK293T or Lentiviral transduced HEK293T cells were plated on a collagen-coated coverslip at 50,000 cells per 24-well plate. Cell cultures were left to adhere to the coverslip overnight. For each of the active MG64-1 NLS fusion proteins, we in vitro transcribed the template with a poly-A tail, and transfected the mRNA of these constructs in WT HEK293T at 500 ng/well. After 48 h of expression, RNA transfected cells and Lentiviral expression cells were fixed using 4% formaldehyde, cell membranes were permeabilized with Triton X-100, then washed with 2% BSA and probed overnight with anti-HA (BioLegend #901501) or anti-FLAG antibody (Sigma #F3165). Cells were then washed with 2% BSA in PBS and subsequently stained with Alexa Fluor 488 conjugated goat anti-Mouse secondary antibody (Invitrogen # A32723). Post-secondary antibody exposure, cells were washed with PBS, mounted on DAPI mounting epoxy (Invitrogen), and cured overnight. Visualization of cells was performed on an EVOS M5000 (Thermo Fisher Scientific) for fluorescence and nuclear localization was determined by Alexa Fluor 488 co-localization with DAPI staining.
Plasmid transient transfection and screening
MG64-1 CAST proteins were expressed on two high-expression plasmids for transposition experiments in human cells (dual plasmid system). One plasmid expresses the protein targeting complex under the control of a pCAG promoter. Two versions of the protein targeting complex were designed (Fig. 4A, left). One version contains a Cas12k-sso7d functional domain fusion, with a 2A peptide fused to S15-NLS, IRES, and NLS-H1-core-TniQ. A second version contains Cas12k-sso7d-2A-S15-NLS with an NLS-HMGN1-TniQ fusion. The targeting plasmids also contained a pU6 PolIII promoter driving transcription of a humanized single guide. MG64-1 sgRNA contains four sequential uridine bases, expression under control of the RNA polymerase III promoter U6 would normally signal termination for 75% of transcripts40. We engineered the sgRNA to mutate the UUUU motif to UCUU and to contain a “UUUUAUUUUUU” termination signal, known to enhance sgRNA termination of type V guides in human cells41 MG64-1 sgRNA was programmed for targeting one of the LINE-1 targets 8, 12 and 15, or SVA target 3. The second plasmid transfected into cells is the donor plasmid containing NLS-TnsB and NLS-TnsC, separated with an IRES under the expression of the pCAG promoter (Fig. 4A, left). On this plasmid, 2.5 kb of DNA cargo was contained between the LE and RE terminal inverted repeats. The all-in-one pHelper plasmid was constructed off the Cas12k-S15 and TniQ targeting plasmid using IRES elements to separate the targeting constructs with TnsB-IRES-TnsC (Fig. 4A, right). The total length of the all-in-one pHelper plasmid is 16 kb. Donor plasmids were then constructed using a pCMV-BetaGlobin promoter driving mNeon (Fig. 4A, right).
Targeting and cargo plasmids were transfected into 10 cm plates seeded with 2.5 million cells for 24 h at a ratio of 9 µg: 9 µg Targeting:Cargo plasmids using 45 µL LT1 transfection reagent (Mirus Bio). pHelper and pDonor transfections were assembled with 12 µg pHelper: 6 µg pDonor: 54 µL LT1 transfection reagent to high-copy loci, with ClpX-NLS, additional plasmid was added to each transfection reaction without added increases of LT1 transfection reagent. At a single-copy locus, 12 µg pHelper: 6 µg pDonor: 1 µg ClpX-NLS: 54 µL LT1 was used for each transfection reaction.
Transfected cells were incubated at 37 ˚C for 72 h and harvested for genomic DNA using a Midi Blood L Kit (Macherey-Nagel). About 2 µg of gDNA was used as the input for a 100 µL Q5 PCR reaction (NEB) using 500 nmol primers of oJL1059 & oJL1060 for Line1, oJL1057 & oJL1058 for SVA, oJL1055 & oJL1056 for HERV with oJL1023 as the in cargo primer. For AAVS1, oJL1109 and oJL1110 was used for genomic target, oJL1108 was used as a primer for LE cargo, and oJL1125 was used for NGS quantification with oJL1109. PCR reactions were run at 30 s for 98 ˚C, [10 s at 98 ˚C, 30 s at 65 ˚C, 60 s at 72 ˚C] × 35, and 2 min. at 72 ˚C, held at 4 ˚C and visualized on a 2% agarose gel. Primers and Probes are listed in Supplementary Data 2.
To generate a linear donor fragment, the non-replicative donor plasmid containing the AAVS1 target 5 Primer binding sequence (PBS) was cut with the enzyme, SphI-HF (NEB). Cut donor molecules were then concentrated and purified using DNA Clean and Concentrator Kit - 100 (Zymo).
To deliver to K562 cells, three-plasmid system was delivered by nucleofection using a ratio of 1.3 µg pHelper: 3.2 µg pDonor: 0.1 µg pClpX, along with 20 µL of SF Nucleofector Solution with Supplement, in a Nucleocuvette Strip containing either 2e5 or 5e5 K562 cells, using the FF-120 program on the 4D nucleofector (Lonza). Nucleofected cells were incubated for 72 h at 37 ˚C in 24-well plates, followed by genomic DNA extraction using 100 µL of Lucigen QuickExtract, and quantification was performed by NGS using LE primers (oJL1109 and oJL1125). To deliver to Hep3B cells, the three-plasmid system was transfected using Lipofectamine 3000 (L3K) between 1.5 and 3.75 µL for 200k cells or 3.75–8.25 µL for 500k cells. pHelper: pDonor: pClpX dosing varied between 0.3–1.3 µg pHelper, 0.051–0.1 µg pDonor, and 0.8–3.2 µg pClpX in a 12-well for 200k cells or six-well plate 500k cells. Transfected cells were incubated for 72 h at 37 ˚C, and genomic DNA was extracted at post-72 h transfection using the Kingfisher MagMax 2.0 Extraction Kit (Thermo Fisher Scientific), then quantified by NGS using LE primers (oJL1109 and oJL1125). Primers and Probes are listed in Supplementary Data 2.
In the study comparing ShCAST with MG64-1, the ShCAST components were synthesized as a direct replacement with the MG64-1 all-in-one plasmid (Supplementary Data 2). The full ShCAST LE and RE were synthesized on the donor plasmid where binding of oJL1125 was maintained for distance from the LE breakpoint for transposition. sgRNA for ShCAST was synthesized with a “UUUCGUU” replacing the “UUUUGUU” motif in the tracrRNA. The three-plasmid system was delivery by LT1 transfection reagent (Mirus) in a 12 µg pHelper: 6 µg pDonor: 1 µg pClpX on 2,500,000 HEK293T cells in a 10 cm petri dish. Transfected cells were recovered for 72 h at 37 ˚C, genomic DNA (gDNA) was extracted from the cells at 72 h post-transfection using the Macherey-Nagel L Blood Kit, and then quantified by NGS using LE primers (oJL1109 and oJL1125). Primers and Probes are listed in Supplementary Data 2.
CAST NGS quantification
Target-specific donors were made where a genomic fragment of the AAVS1 locus was cloned into the pDonor 152 bp away from the 5′ LE. This genomic fragment allows for the simultaneous amplification of the AAVS1 transposition product and the genomic sequence with the same PCR primer set. Genomic and transposed LE forward sequences were amplified using 100 µL PCR reactions of Q5 polymerase (NEB) for 25 cycles using 500 nM oJL1109 and oJL1125 primers. Primers included a Nextera adapter sequence and a 5 bp diversity stub. Illumina sequencing adapters were then amplified for ten cycles onto the PCR product library after 1x SPRI cleanup (Beckman Coulter) and sequenced with a 2 × 300 cycle V3 kit on MiSeq (Illumina). Resulting NGS reads were analyzed with CRISPResso2, using the 63 bp target 1 transposition sequence as the reference amplicon, the genomic fragment as the HDR amplicon, and the Left End 5′ sequence as the spacer within a 20 bp window. Resulting alignments were then filtered for reads passing “-amas 95 filter”. Unmodified reference sequences, NHEJ sequences, HDR sequences, and modified HDR sequences were then pulled from the editing profile. Transposition frequencies were calculated by summing reference aligned sequences and NHEJ sequences over the total amount of reads.
Human cell off-target detection
About 400 ng of HEK293T gDNA in 26 µL volume was sheared using 2 µL NEBNext Ultra II FS enzyme mix and 7 µL buffer (NEB) for 10 min at 37 ˚C and quenched with a 30 min incubation at 65 ˚C. Ligation adapter (TA_Adaptor_Top/Bottom) with a (10 bp UMI) and oligo were heated to 95 ˚C for 3 min and slow cooled to room temperature over the course of 1 h to make a partial double-stranded substrate for ligation to sheared ends. About 3.6 µM UMI adapter was ligated to sheared ends for 15 min at 20 ˚C using 30 µl FS Ligation Mix and 1 µL Enhancer, and ligation reactions were selected by double-sided selection at 0.2x and 0.3x using Hiprep PCR size selection beads (Magbio). 1 µM P7 adapter specific oligo (Lig_Enrich_P7) was used to amplify the UMI-ligation adapter along with 1 µM P5 specific primers amplifying the LE or RE (P5-LE/P5-RE) junction in separate PCR reaction: 30 s at 98 ˚C initial denaturation, [10 s at 98 ˚C, 30 s at 70/−1 ˚C every cycle, 60 s at 72 ˚C] × 7, [10 s at 98 ˚C, 30 s at 63 ˚C, 60 s at 72 ˚C] × 13, and hold at 4 ˚C. About 1 µL of each PCR of LE and RE samples were pooled together and fully indexed with Nextera based primer adapters (500 nM) for ten cycles to form a pool library of comprehensive off targets, and sequenced at 100 million reads per sample on a 2 × 150 paired end read Nextseq flowcell.
Sequencing reads were then filtered with fastp42 to remove low-quality reads and trim Illumina adapters. Further filtering was performed by read sequence to remove R1 reads that did not contain either the LE or RE ends of the cargo and R2 reads that did not contain the ligation adapter. UMIs were extracted, and reads were tagged with UMI-tools43. Sequencing reads were aligned with BWA44 to a reference of the human genome (hg38) plus all plasmids used in the experiment. The alignments were converted to the BAM file format, indexed, and sorted using SAMtools45. Read pileups were identified and then start-mapping positions for reads with mapping quality >20 were counted, start-mapping positions were merged using a 50-bp sliding window. To ensure that detected sites were from full-length integration products, we imposed two requirements, first, there must be three read starts within a window, and second there must be at least one read aligned in the forward and reverse orientation. Then the UMIs were counted to get the number of unique integration events detected in the site.
ddPCR quantification
Prior to ddPCR, gDNA derived from 10 cm plates was digested with EcoRI-HF (NEB) for 1 h at 37 ˚C. A total of 20 µL of reaction mix containing 125 ng digested gDNA, 2x ddPCR Supermix for Probes (no dUTP), 18 µM forward and reverse primers, and 5 µM probe designed for each predicted integration site from LE, and RE was dropletized with Generation Oil for Probes (Biorad 1864110) and PCR amplified in the following manner: 95 ˚C at 10 min initial denaturing; 40 cycles of [94 ˚C at 30 s; 55.2 for 90 s for LE, 60.8 for 90 s for RE], final denaturing at 98 ˚C for 10 min, and then 4 ˚C droplet hardening for 20 min. Data were then generated and analyzed using the QX200 Direct Quantification program using FAM detection for LE and RE with normalization to the housekeeping gene, RPL13A under HEX channel (Biorad ddPCR Copy Number Assay:RPL13A, Human, Assay ID: dHsaCNS189783948). Primers and Probes are listed in Supplementary Data 2.
Statistics and reproducibility
Integration efficiencies are presented as percentages of the mean of biological replicates ± one standard deviation, with sample sizes indicated in figure legends. No statistical method was used to predetermine sample size, and no data were excluded from the analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated here, including protein and non-coding sequences, and dataset accession numbers, are provided in the Supplementary Information and Source Data files. Raw metagenomic data generated in this study have been deposited in the SRA database under BioProject accession number PRJNA874471. For all other materials, interested parties should contact Metagenomi, Inc. for more information. Correspondence and requests for materials should be addressed to Christopher T. Brown (ctb@metagenomi.co). Source data are provided with this paper.
References
Sandoval-Villegas, N., Nurieva, W., Amberger, M. & Ivics, Z. Contemporary transposon tools: a review and guide through mechanisms and applications of sleeping beauty, piggyBac and Tol2 for genome engineering. Int. J. Mol. Sci. 22, 5084 (2021).
Tipanee, J., VandenDriessche, T. & Chuah, M. K. Transposons: moving forward from preclinical studies to clinical trials. Hum. Gene Ther. 28, 1087–1104 (2017).
Hickman, A. B. & Dyda, F. DNA transposition at work. Chem. Rev. 116, 12758–12784 (2016).
Ochmann, M. T. & Ivics, Z. Jumping ahead with sleeping beauty: mechanistic insights into cut-and-paste transposition. Viruses 13, 76 (2021).
Goodier, J. L. & Kazazian, H. H. Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135, 23–35 (2008).
Peters, J. E. Tn7. Microbiol. Spectr. 2 (2014).
Strecker, J. et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science 365, 48–53 (2019).
Klompe, S. E., Vo, P. L. H., Halpin-Healy, T. S. & Sternberg, S. H. Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature 571, 219–225 (2019).
Peters, J. E., Makarova, K. S., Shmakov, S. & Koonin, E. V. Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc. Natl Acad. Sci. USA 114, E7358–E7366 (2017).
Peters, J. E. Targeted transposition with Tn7 elements: safe sites, mobile plasmids, CRISPR/Cas and beyond. Mol. Microbiol. 112, 1635–1644 (2019).
Hoffmann, F. T. et al. Selective TnsC recruitment enhances the fidelity of RNA-guided transposition. Nature 609, 384–393 (2022).
Vo, P. L. H., Acree, C., Smith, M. L. & Sternberg, S. H. Unbiased profiling of CRISPR RNA-guided transposition products by long-read sequencing. Mob. DNA 12, 13 (2021).
Querques, I., Schmitz, M., Oberli, S., Chanez, C. & Jinek, M. Target site selection and remodelling by type V CRISPR-transposon systems. Nature 599, 497–502 (2021).
Xiao, R. et al. Structural basis of target DNA recognition by CRISPR-Cas12k for RNA-guided DNA transposition. Mol. Cell 81, 4457–4466.e5 (2021).
Schmitz, M., Querques, I., Oberli, S., Chanez, C. & Jinek, M. Structural basis for the assembly of the type V CRISPR-associated transposon complex. Cell 185, 4999–5010.e17 (2022).
Park, J.-U. et al. Structures of the holo CRISPR RNA-guided transposon integration complex. Nature 613, 775–782 (2023).
Tou, C. J., Orr, B. & Kleinstiver, B. P. Precise cut-and-paste DNA insertion using engineered type V-K CRISPR-associated transposases. Nat. Biotechnol. 41, 968–979 (2023).
Lampe, G. D. et al. Targeted DNA integration in human cells without double-strand breaks using CRISPR-associated transposases. Nat. Biotechnol. 42, 87–98 (2023).
Petassi, M. T., Hsieh, S.-C. & Peters, J. E. Guide RNA categorization enables target site choice in Tn7-CRISPR-Cas transposons. Cell 183, 1757–1771.e18 (2020).
Saito, M. et al. Dual modes of CRISPR-associated transposon homing. Cell 184, 2441–2453.e18 (2021).
Chen, W. et al. Targeted genetic screening in bacteria with a Cas12k-guided transposase. Cell Rep. 36, 109635 (2021).
George, J. T. et al. Mechanism of target site selection by type V-K CRISPR-associated transposases. Science 382, eadj8543 (2023).
Park, J.-U. et al. Structural basis for target site selection in RNA-guided DNA transposition systems. Science 373, 768–774 (2021).
Wang, Y. et al. A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro. Nucleic Acids Res. 32, 1197–1207 (2004).
Ding, X. et al. Improving CRISPR-Cas9 genome editing efficiency by fusion with chromatin-modulating. Pept. Crispr J. 2, 51–63 (2019).
Lombardo, A. et al. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat. Methods 8, 861–869 (2011).
Bishop, D. C. et al. CAR T cell generation by piggyBac transposition from linear doggybone DNA vectors requires transposon DNA-flanking regions. Mol. Ther. Methods Clin. Dev. 17, 359–368 (2020).
Vo, P. L. H. et al. CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat. Biotechnol. 39, 480–489 (2021).
Brian, B. BBMap: a fast, accurate, splice-aware aligner. In Conference: 9th Annual Genomics of Energy & Environment Meeting (2014).
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. Bmc Bioinformatics 11, 119–119 (2010).
Menzel, P., Ng, K. L. & Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7, 11257 (2016).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLos ONE 5, e9490 (2010).
Lorenz, R. et al. ViennaRNA package 2.0. Algorithm Mol. Biol. 6, 26 (2011).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv:1303.3997 (2013).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Gao, Z., Herrera-Carrillo, E. & Berkhout, B. RNA polymerase II activity of type 3 Pol III promoters. Mol. Ther. Nucleic Acids 12, 135–145 (2018).
Moon, S. B. et al. Highly efficient genome editing by CRISPR-Cpf1 using CRISPR RNA with a uridinylate-rich 3′-overhang. Nat. Commun. 9, 3651 (2018).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Smith, T., Heger, A. & Sudbery, I. UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Acknowledgements
We thank Benjamin Freeman for the helpful discussion around human target sites. We thank Christopher Hann-Soden, Karen Lundy, and Shana L McDevitt from QB3 at UC Berkeley for their help with library preparation and sequencing. This work used the services provided by the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, which is supported by the NIH S10 OD018174 Instrumentation Grant.
Author information
Authors and Affiliations
Contributions
A.E.D. and C.T.B. processed and curated metagenomics datasets. A.E.D., D.S.A.G., and C.T.B. conducted CRISPR system searches. D.S.A.G. and S.L. conducted data mining. DSAG performed in silico characterization and candidate nomination. J.L. and C.N.B. designed in vitro experiments. J.L. and L.M.A. designed and tested guide RNAs. L.M.A. determined rules for guide RNA design. J.L., L.M.A., and L.G.-O. conducted in vitro activity and PAM determination. J.L., C.A.R., and K.K. performed E. coli integration experiments. J.L., K.K., J.M., and R.F.O. designed and tested in vitro NLS fusion constructs. J.L., M.T.D., and S.C. performed mammalian nuclear localization. J.L., K.K., and J.H.H. performed in vitro integration experiments in human genomic DNA. J.O.-O., O.P.J., and D.T.D. developed methods and calculated integration efficiency analyses. J.L., L.M.A., and L.G.-O. performed TIR and gRNA engineering. L.M.A., C.A.R., K.T., and C.J.C. designed and performed CAST swapping experiments. J.L., J.H.H., D.T.D., K.K., and J.R. designed and performed cell integration experiments. J.L., D.S.A.G., C.N.B., G.J.C., C.T.B., and B.C.T. designed the study. J.L., D.S.A.G., and C.T.B. wrote the paper. All authors contributed to and approved of the manuscript.
Corresponding author
Ethics declarations
Competing interests
J.L., D.S.A.G., L.M.A., K.K., J.H.H., D.T.D., C.A.R., M.M.T.-D., S.C., R.F.O., J.O.-O., O.P.J., K.T., J.M., J.R., L.G.O., S.M.L., A.E.D., C.J.C., C.N.B., G.J.C., C.T.B., and B.C.T. are or were employees of Metagenomi, Inc. Metagenomi Inc has submitted patent applications to the US patent office related to the manuscript (application No. PCT/US2021/047195, PCT/US2021/047196, PCT/US2023/063181, PCT/US2023/063184, PCT/US2024/028988, and US63/679,302).
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, J., Aliaga Goltsman, D.S., Alexander, L.M. et al. Integration of therapeutic cargo into the human genome with programmable type V-K CAST. Nat Commun 16, 2427 (2025). https://doi.org/10.1038/s41467-025-57416-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57416-2
This article is cited by
-
Advances in large-scale DNA engineering with the CRISPR system
Experimental & Molecular Medicine (2025)
-
Powerful CRISPR system inserts whole gene into human DNA
Nature (2025)
-
Structure-guided engineering of type I-F CASTs for targeted gene insertion in human cells
Nature Communications (2025)
