
Abstract
Laboratory mice (Mus musculus domesticus) harbor gut bacterial strains that are distinct from those of wild mice but whose evolutionary histories are unclear. Here, we show that laboratory mice have retained gut bacterial lineages that diversified in parallel (co-diversified) with rodent species for > 25 million years, but that laboratory-mouse gut microbiota (LGM) strains of these ancestral symbionts have experienced accelerated accumulation of genetic load during the past ~ 120 years of captivity. Compared to closely related wild-mouse gut microbiota (WGM) strains, co-diversified LGM strains displayed significantly faster genome-wide rates of nonsynonymous substitutions, indicating elevated genetic drift—a difference that was absent in non-co-diversified symbiont clades. Competition experiments in germ-free mice further indicated that LGM strains within co-diversified clades displayed significantly reduced fitness in vivo compared to WGM relatives to an extent not observed within non-co-diversified clades. Thus, stochastic processes (e.g., bottlenecks), not natural selection in the laboratory, have been the predominant evolutionary forces underlying divergence of co-diversified symbiont strains between laboratory and wild house mice. Our results show that gut bacterial lineages conserved in diverse rodent species have acquired novel mutational burdens in laboratory mice, providing an evolutionary rationale for restoring laboratory mice with wild gut bacterial strain diversity.
Similar content being viewed by others


Introduction
Mammalian lineages share gut bacterial taxa, but bacterial strains within these taxa differ systematically among host populations and species1,2,3,4,5,6,7. For example, laboratory lines of house mice (Mus musculus domesticus), the most common of which were derived from wild mice ~ 120 years ago (e.g., C57BL/6 J)8, harbor gut-microbiota strains distinct from those of the same taxa found within wild house mice9. Understanding the divergence of laboratory-mouse gut microbiota (LGM) from wild-mouse gut microbiota (WGM) is critical, because LGM and WGM have been previously shown to differentially affect mouse immune-cell proliferation10,11, infection resistance12, cancer progression10, and ability to model drug outcomes for humans13. However, the evolutionary histories of LGM strains remain poorly understood. LGM strains may have been acquired in captivity from external sources, such as other laboratory animals, humans, or the indoor environment. Alternatively, LGM strains may be descendants of ancestral WGM symbionts that have been reshaped by laboratory-specific evolutionary forces, such as natural selection or genetic drift.
In principle, LGM strains descended from symbioses ancestral to laboratory and wild mice could be identified through co-phylogenetic analyses, which test for parallel diversification (co-diversification) between symbionts and hosts. Discovering ancestral, co-diversifying symbionts in the gut microbiota (GM) would also provide a phylogenetic framework for interrogation of the histories of symbiont adaptation and genetic drift within host lineages, as has been possible in simpler host-microbe symbioses, such as those between insects and bacterial endosymbionts14,15. However, co-phylogenetic tests have not been feasible for rodent gut microbiota due to the limited phylogenetic information provided by 16S rDNA sequencing and assembly-free shotgun metagenomics16. Analyses of high-quality bacterial genomes assembled directly from metagenomes17,18 provide increased power to test for co-diversification between gut microbiota and hosts. These approaches have recently revealed that multiple GM strains have co-diversified with humans and other primate species6,7 but have yet to be leveraged to assess the evolutionary histories of mouse GM strains, owing to a lack of genome-resolved datasets from closely related rodent species.
Results and discussion
A genome-resolved bacterial phylogeny from the rodent gut microbiota
Here, we employed comparative, genome-resolved metagenomics approaches to test for co-diversification between GM strains and rodents, to assess the extent to which ancestral GM symbionts have persisted within laboratory mice, and to interrogate how natural selection and genetic drift have driven divergence of LGM strains from their wild relatives. To enable tests for co-diversification between gut microbiota and rodents in the super family Muroidea, we used a combination of long-read Oxford Nanopore and short-read Illumina sequencing to generate 504 metagenome-assembled genomes (MAGs) from individuals of six species/subspecies of deer mice (genus Peromyscus) descended from wild populations in the United States and reared in a common facility on a common diet (Methods). Information on read depth per sample and other relevant metadata are presented in Supplementary Data 1. We combined these MAGs with previously published MAGs from the gut microbiota of 307 host individuals from 14 rodent species9,10,13,19,20,21,22,23,24,25,26,27,28 (Fig. 1a; Supplementary Data 2, Supplementary Data 3) and generated a curated host tree from timetree.org29. We used IQ-TREE 230 to construct a maximum likelihood phylogenetic tree of all MAGs based on single-copy core genes (Methods), which define bacterial lineages’ taxonomic classifications31. The final tree included 5567 MAGs (> 50% complete, < 5% contamination) from 20 rodent host species, allowing assessment of GM-strain diversification coincident with rodent speciation.

a Phylogeny shows evolutionary relationships among rodent host species for which gut bacterial genomes were included in this study. b Phylogeny shows relationships among gut bacterial genomes assembled from the rodent gut microbiota. Color of outer ring denotes bacterial phyla. Inner bar surrounding phylogeny marks clades displaying significant evidence of co-diversification (nonparametric permutation test, r > 0.75, p-value < 0.01). Raw p-values and p-values corrected for multiple comparisons are presented in Supplementary Data 4. c–e Tanglegrams show concordance between gut bacterial (left) and rodent (right) phylogenetic trees. Dashed lines connect bacterial genomes with the host species from which they were recovered. Genomes derived from mice (Mus musculus) sampled in the lab or the wild are labeled. ‘W’ and ‘eW’ indicate ‘wildling’ and ‘ex-wild’ mice, respectively. Numbers left and right of tree are bootstrap support values and genome IDs, respectively. f Phylogeny shows relationships among all bacterial genomes belonging to co-diversifying clades shown in Fig. 1 that contain genomes from murids and non-murids, i.e., co-diversifying clades that could be inferred to be ancestral to murids. Circles indicate clades (colored by phylum, left) or MAGs derived from laboratory mice (grey, right). Sizes of circles indicate the number of MAGs. Identification numbers are listed to the right of each clade. Source data are provided as a Source Data file. Created in BioRender. Moeller, A. (2025) https://BioRender.com/s37r208.
Widespread co-diversification between symbionts and rodents
To test for co-diversification between GM strains and rodent species, we employed an approach previously used to identify GM strains that co-diversified with primate species7. We tested every node in the distal 1/4th of the bacterial phylogeny (1245 nodes) for co-diversification with host species using the method developed by Hommola et al.32 (Methods). The choice to test nodes in the distal 1/4th of the bacterial phylogeny was motived by molecular clock analyses indicating that more basal diversification events likely occurred before the most-recent common ancestors of rodents7. We identified 158 gut bacterial clades showing evidence of co-diversification with host species following previously employed significance thresholds of Mantel’s r > 0.75 and p-value < 0.01 (Supplementary Data 4, Fig. 1b)7. Co-diversifying gut bacterial lineages comprised 22.6% of the total branch length on the MAG phylogeny and spanned 8 phyla. MAGs belonging to co-diversifying clades were enriched within the phyla Bacteroidota, Spirochaetota, and Deferribacterota relative to the entire MAG phylogeny (Pearson’s Chi-squared test, p-value < 0.05 for each phylum). Perfect or near perfect concordance between host and symbiont phylogenies was observed, indicative of ancestral relationships between host lineages and host species–specific symbionts spanning > 25 million years (Fig. 1c–e, Supplementary Fig. 1). These symbionts included strains of Helicobacter ganmani (a common commensal of laboratory mice33), an unclassified species of Lactobacillus (paralleling results from studies of this taxon in other hosts34), several unclassified genera, and others (Supplementary Data 4).
To further assess the evidence for co-diversification in these clades, we conducted tests after subsampling a single MAG from each monophyletic clade derived from a single host species. These analyses assessed only the association between the backbone of each symbiont clade and the host phylogeny, eliminating the possibility of pseudoreplication caused by repeated sampling of individual bacterial clades from the same host species35. We employed multiple tests for co-diversification, including PACo36, ParaFit37, and Hommola’s test32. We found that 324 clades displayed significant evidence of co-diversification in at least one test (Supplementary Data 4, Supplementary Data 5, Supplementary Fig. 2), that the results of these different tests were significantly associated with one another (Supplementary Fig. 2a–c), and that 156 clades displayed significant evidence for co-diversification based on at least two of the tests (Supplementary Fig. 2d). We observed between eight-fold and twelve-fold more significantly co-diversifying clades (based on a Mantel test p-value of 0.01) than expected under the null hypothesis (i.e., 1% of tests) (Supplementary Fig. 3a), depending on the specific test employed. In addition, we assessed the false discovery rate of the initial scan for co-diversification based on the complete dataset using a previously developed permutation test7,38, finding that the scan based on the real data always detected more co-diversifying clades than did the permuted scans, which detected 53.74 clades on average (Supplementary Fig. 3b). Sensitivity analyses further indicated that the detection of 73–100% of co-diversifying clades was robust to the removal of MAGs from individual host species (depending on the host species removed) (Supplementary Fig. 4), and molecular clock analyses corroborated contemporaneous diversification of symbiont and rodent lineages (Supplementary Fig. 5, Supplementary Data 6). These results demonstrate widespread co-diversification between gut microbiota and rodent species.
Retention and extinction of co-diversifying mouse symbionts
The discovery of co-diversifying GM strains enabled identification of ancestral GM lineages that have either been retained in or lost from laboratory house mice. Of the 158 co-diversifying clades identified, 40 phylogenetically independent (i.e., non-nested) clades were inferred to be ancestral to house mice and other murids (i.e., present in Mus musculus domesticus, at least one non-M. m. domesticus murid, and at least one outgroup to murids) (Supplementary Information). Of these 40 ancestral clades, 24 contained MAGs from laboratory house mice (Fig. 1f, Supplementary Fig. 6). Previous work showed that wild-derived inbred mouse lines could retain subsets of the microbiota from their wild population of origin for > 10 host generations39. The observation of 24 ancestral, co-diversifying clades in laboratory mice (Fig. 1f) shows that these symbioses have persisted since their hosts’ derivation from wild stock ~ 120 years ago8.
These analyses also indicated that laboratory house mice have experienced elevated rates of loss of ancestral symbionts relative to wild house mice. Only 7 ancestral clades lacked MAGs from wild house mice, whereas 33 clades contained MAGs from wild house mice (compared to 16 and 24 clades, respectively, for laboratory house mice). These results support significantly accelerated loss of co-diversified GM symbionts from laboratory house mice compared to wild mice (chi-squared test p-value = 0.0262), in agreement with previous results showing reduced GM diversity in lab mice compared to wild mice9. This difference could not be explained by sampling effort, which favored laboratory house mice (217 laboratory versus 90 wild house-mouse samples). The loss of co-diversifying lineages from lab mice has coincided with increases in the relative abundances of non-co-diversifying lineages: 1418 out of 5567 MAGs (25.5%) belonged to co-diversifying clades, whereas only 103 out of 1162 MAGs (8.9%) that showed significantly higher relative abundances in lab mice (based on previous analyses1) belonged to co-diversifying clades (chi-squared test p-value < 0.0001) (Supplementary Data 7). MAGs from ‘wildling’13 (born to a wild-caught mother via embryo transfer) or ‘rewilded’11,12 (released outdoors) mice were contained within a subset of clades lacking MAGs from laboratory mice (Supplementary Fig. 6), indicating that ancestral clades absent from laboratory mice can be regained through ‘wildling’ or ‘rewilding’ approaches.
Altered patterns of positive selection in LGM strains
Within co-diversifying clades ancestral to murids (Fig. 1f), MAGs derived from laboratory mice formed monophyletic clades to the exclusion of MAGs derived from wild (e.g., Fig. 1c, e), rewilded, wildling, and ‘ex-wild’ (wild-caught but housed in a laboratory) mice (Supplementary Data 4) (5/6 of ancestral co-diversifying clades containing two or more LGM strains and at least one WGM strain showed this pattern). These phylogenetic relationships indicate genomic distinctiveness of co-diversified LGM strains from related WGM strains. We next interrogated the evolutionary forces that have driven this divergence. We reasoned that LGM strains may be experiencing altered forces of natural selection compared to their wild relatives due to inbred host genetic backgrounds, laboratory-mouse diets, and myriad other factors. To test this idea, we conducted genome-wide scans for positive selection on each protein-coding gene in each co-diversifying clade ancestral to murids that contained MAGs from laboratory and wild house mice. For each gene, we calculated the ratio of the rates of nonsynonymous and synonymous substitutions per site, i.e., dN/dS, which is expected to be 1 under neutral evolution, > 1 under positive selection to change the protein product, and < 1 under purifying selection against non-synonymous mutations. Results showed that most co-diversifying GM strains’ genes have evolved under purifying selection in both wild and laboratory mice (Fig. 2, Supplementary Data 8, Supplementary Data 9, Supplementary Data 10), as expected. However, distinct sets of genes exhibited evidence of positive selection in LGM and WGM strains (Fig. 2). Significantly more genes displayed dN/dS > 1 in laboratory mice (76 genes) but dN/dS < 1 in wild mice than dN/dS > 1 in wild mice but dN/dS < 1 in laboratory mice (50 genes) (binomial test p-value = 0.0255), consistent with novel selective forces driving accelerated evolution of a minority of protein sequences encoded by the genomes of LGM strains. Genes showing evidence of positive selection in the laboratory but purifying selection (or near neutrality) in the wild included hpt (Borkfalkiaceae) involved in the purine salvage pathway, tsaD (Muribaculaceae) involved in tRNA metabolism, oppC (Schaedlerella) involved in oligopeptide transport, kbl (Odoribacter) involved in amino acid degradation, carB_2 (Duncaniella freteri) involved in pyrimidine metabolism, pgi (unclassified genus 1XD8-76) involved in carbohydrate degradation, and pflB (Lachnospira) involved in pyruvate fermentation (Fig. 2; Supplementary Data 8). Laboratory-specific positive selection on these metabolic genes may result from the compositionally distinct, ad libitum diets of laboratory mice, although our analyses were not able to identify specific selective agents. Genes experiencing positive selection in the wild but purifying selection in the laboratory included dnaE (catalytic α subunit of DNA polymerase III) (Lachnospira), the transcriptional repressor nrdR (unclassified genus Zag111), and genes of unknown function (Supplementary Data 8). We also tested for differences in gene functional annotations between laboratory and wild GM strains (and between co-diversifying and non-co-diversifying GM strains) while controlling for bacterial phylogenetic history (Supplementary Information). The left-skewed distributions of p-values obtained from tests comparing laboratory and wild GM strains indicated that these analyses lacked power (Supplementary Fig. 7, Supplementary Data 11) (whereas tests comparing co-diversifying and non-co-diversifying GM strains revealed more significant differences than expected based on the false discovery rate; Supplementary Fig. 7, Supplementary Data 12). In total, these results identify genes in ancestral, co-diversifying symbionts of house mice showing evidence of laboratory-specific adaptation.

Scatter plot shows the log of the dN/dS ratio for bacterial genes in co-diversifying clades in laboratory house mice (y-axis) and wild house mice (x-axis). Each point represents a bacterial gene. Positive values indicate evidence of positive selection, values near zero indicate neutral evolution, and negative values indicate purifying/negative selection. Points in the upper left quadrant show evidence of positive selection in laboratory house mice but purifying/negative selection in other rodents. Source data are provided as a Source Data file.
Significantly elevated genetic drift in LGM strains
In addition to testing for divergent natural selection between LGM and WGM strains, we also tested for divergence in the strength of genetic drift. Laboratory breeding and animal care procedures may exert bottlenecks on LGM strains, which would elevate the strength of genetic drift. For example, C57BL/6 J, which is the most widely used laboratory mouse line (ancestry of 92% Mus musculus domesticus, 6% M. m. musculus, and 1% M. m. castaneus)40, was derived from a single mating pair (https://www.jax.org/strain/000664). Previous work has also shown that moving wild mice into the laboratory and establishing inbred lines is associated with a precipitous loss of GM diversity39, a hallmark of drift. Moreover, once in the laboratory, GM strains are vertically inherited from mother to offspring within inbred mouse lines39 through stochastic sampling processes that can exert bottlenecks on GM diversity41.
Theory predicts that stronger genetic drift will reduce the efficacy of purifying selection, increasing the rate of fixation of weakly deleterious mutations. In bacterial lineages with low effective population sizes (Ne), stochastic sampling leads to elevated genome-wide rates of fixation of nonsynonymous substitutions (dN) that would otherwise be efficiently purged by purifying selection in lineages with large Ne, elevating dN/dS genome-wide42,43. Whereas elevated dN/dS values in individual genes can be driven by the action of positive selection, elevated genome-wide dN/dS is indicative of reduced Ne and increased genetic drift44,45.
To test whether co-diversifying GM strains in laboratory mice have experienced elevated genetic drift, we compared genome-wide dN/dS values in LGM and WGM strains within each co-diversifying clade ancestral to murids. We observed significant genome-wide elevation of dN/dS in LGM strains in tests based on all genes (paired-test p-value = 0.000356) and in tests based only on genes under purifying selection (bottom-left quadrant of Fig. 2) (paired t-test p-value = 0.00511) (Fig. 3a). The genome-wide signatures of genetic drift became more evident when individual co-diversifying clades were tested separately (Fig. 3b–e). Significantly elevated dN/dS in LGM strains was observed in clades of Schaedlerella (class: Clostridia) (paired t-test FDR-corrected p-values = 2.05e-07, all genes; 5.87e-06, genes under purifying selection) (Fig. 3b), unclassified Lachnospiraceae genus CAG-95 (paired t-test FDR-corrected p-values < 5.5e-15) (Fig. 3c), Anaerotingum (class: Clostridia) (paired t-test FDR-corrected p-values = 0.00390 and 0.0228) (Fig. 3d), unclassified Borkfalkiaceae UMGS1004 (paired t-test FDR-corrected p-values = 0.00569 and 0.00685), and others (Supplementary Data 8). Only one co-diversifying clade, unclassified Oscillospiraceae UMGS1872, displayed significantly higher dN/dS in the wild than in the laboratory (paired t-test FDR-corrected p-value = 7.93e-05 and 4.51e-05). Unclassified Oscillospiraceae were previously shown to be two of seven bacterial taxa found at significantly higher frequency in laboratory mice than in wild mice (compared to 68 taxa found at significantly higher frequency in wild mice)9. Larger populations sizes of this taxon may buffer against stronger genetic drift in the laboratory. Elevated genetic drift in LGM strains was not observed for non-co-diversifying symbiont clades (Mantel’s r < 0) (Supplementary Information; Supplementary Data 13), indicating that elevated genetic drift in the laboratory has primarily affected co-diversifying bacterial symbionts. Moreover, genome-wide dN/dS was significantly higher for co-diversified clades (r > 0.75) than for non-co-diversified clades (r < 0) in both laboratory and wild house mice (Supplementary Information, Supplementary Fig. 8), indicating stronger genetic drift in co-diversifying compared to non-co-diversifying GM symbiont lineages. In addition, within co-diversifying clades containing > 2 LGM strains and > 2 WGM strains, average nucleotide diversity (pi) within clades was significantly lower among LGM strains than among WGM strains (paired t-test p-value = 0.026) (LGM pi was lower than WGM pi in every case; Supplementary Data 5). Cumulatively, these results show that LGM strains within co-diversifying clades display stronger genomic signatures of genetic drift compared to their WGM relatives.

a Density plot shows significant genome-wide elevation of dN/dS (a hallmark of genetic drift) in co-diversifying GM strains in laboratory house mice relative to wild house mice. dN/dS values for all genes from all co-diversifying clades ancestral to murids and containing MAGs from both laboratory and wild mice are shown. b–e Density plots show the distribution of per-gene dN/dS values for four co-diversifying clades displaying the most significant genome-wide elevation of dN/dS in laboratory house mice relative to wild house mice. In a–e, significance of paired, two-sided t-tests for differences in mean is denoted by asterisks; **p-value < 0.01; ***p-value < 0.001; **** p-value < 0.0001 (Benjamini-Hochberg corrected p-values are provided in Supplementary Data 10). Top asterisks denote significance of tests considering all genes, and bottom asterisks denote significance of tests considering only genes displaying dN/dS < 1 (log dN/dS < 0) in both laboratory and wild mice. Source data are provided as a Source Data file.
Co-diversified WGM strains out-compete LGM strains in vivo
The observations that co-diversified LGM strains show genomic evidence of both laboratory-specific adaptation (Fig. 2) and elevated genetic drift (Fig. 3) raised two conflicting hypotheses regarding the relative fitness of LGM and WGM strains. Laboratory-specific adaptation is expected to increase the fitness of LGM strains compared to WGM relatives in the laboratory-mouse environment. In contrast, elevated genetic drift (Fig. 3) is expected to reduce fitness via the accelerated fixation of weakly deleterious mutations (i.e., genetic load). To assess the net consequences of these evolutionary forces in driving divergence between LGM and WGM strains, we analyzed data from previously reported competition experiments in which the relative fitness of strains was assessed directly in germ-free laboratory mice13.
In these experiments, individual wildling (C57BL/6 J harboring wild-derived microbiota), laboratory (C57BL/6 J from either The Jackson Laboratory or Taconic Biosciences), and germ-free (GF) (C57BL/6 J) mice were co-housed in trios and then sampled over the course of 17 days (3 replicate cages in 4 independent experiments; n = 6 co-housed trios containing Jackson Laboratory mice and n = 6 co-housed trios containing Taconic mice)13. We assessed the relative fitness in germ-free mice of wildling- and laboratory mouse–derived gut bacteria from co-diversified GM taxa, enabling comparisons to previous analyses that assessed relative fitness of wildling- and laboratory mouse–derived gut bacteria across all GM taxa13. First, we identified all genera and species containing MAGs that showed evidence of co-diversification with hosts (Fig. 1b; Supplementary Data 4) and that were detected in both wildling and laboratory mice before co-housing (i.e., on day 0). We then identified all Amplicon Sequence Variants (ASVs) within these shared genera and species that were detected in either wildling or laboratory mice, but not both, at day 0 (i.e., ASVs within shared co-diversifying taxa that could be unambiguously defined as derived from either wildling or laboratory mice). In total, we identified 11 genera present in Jackson or Taconic mice at day 0 and containing ASVs that could be unambiguously identified as wildling- or laboratory mouse–derived.
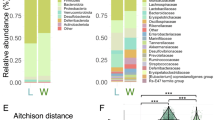
Based on these uniquely wildling- or laboratory mouse–derived ASVs within co-diversifying taxa (349 and 383 ASVs, respectively), beta-diversity dissimilarities between wildling and laboratory mice sampled at day-0 (before co-housing) and co-housed GF mice sampled from day 1 to day 17 (after co-housing) indicated that wildling ASVs displayed significant competitive advantages over closely related LGM ASVs (Fig. 4; Supplementary Fig. 9, Supplementary Data 14–19). Results from the experiments using Jackson mice are shown in Fig. 4, and results from the experiments using Taconic mice are shown in Supplementary Fig. 9. Moreover, comparing differences between beta-diversities (which can assess degrees of fitness advantages in microbiota-competition experiments46) indicated stronger fitness advantages for wildling-derived ASVs within co-diversified taxa than within non-co-diversified taxa (Fig. 4b, Supplementary Fig. 9, Supplementary Data 14; Wilcoxon Test * < 0.05, ** < 0.01). These results corroborate that co-diversified GM lineages have experienced elevated genetic drift in laboratory mice (Fig. 3) to a greater extent than that experienced by non-co-diversified lineages (Supplementary Data 13). Longitudinal relative abundances of wildling- and laboratory mouse-derived ASVs within the co-diversified genera and species tested also indicated significant wildling advantage. The individual co-diversified taxa in which wildling-derived ASVs showed fitness advantages included the genera Muribaculum, Bacteroides, CAG − 485, Parasutterella, Duncaniella, UMGS1004 (whose elevated genome-wide dN/dS is shown in Fig. 3), AM07 − 15, Acetatifactor, UMGS1872 (Fig. 4d), and Parabacteroides (Supplementary Fig. 9d) and the species CAG − 485 sp002362485 (Supplementary Fig. 10) (Wilcoxon Test on day 17, p-values < 0.0001). All taxa that were tested are shown in Fig. 4 and Supplementary Figs. 9 and 10. The fitness advantages of the wildling-derived ASVs could not be explained by differences in relative abundance in the wildling or laboratory cage mates at day zero, because in several cases laboratory mouse–derived ASVs displayed higher relative abundances than wildling-derived ASVs in their respective hosts at day 0 (before co-housing) (Fig. 4c, Supplementary Fig. 9c). These results show that co-diversified GM taxa displayed disproportionate (relative to non-co-diversified GM taxa) advantages for wildling ASVs, supporting accelerated accumulation of genetic load in co-diversified LGM strains.

a Principal coordinates plot shows beta-diversity (Jaccard) dissimilarities among microbiota profiles based on laboratory mouse– and wildling-derived ASVs within co-diversified taxa. Plot shows data from the Wildling/Jackson co-housing experiments. Shapes and colors denote host of origin as indicated in the key. Samples from ex-germ-free mice (n = 6) are colored according to the day of sample collection. Arrows connect longitudinal samples from the same germ-free mouse. b Line plots show that ex-germ-free mice (n = 6) sampled on days 2–17 harbored a greater fraction of wildling-derived ASVs from co-diversified taxa than laboratory mouse–derived ASVs from co-diversified taxa. The y-axis displays the microbiota similarity (1 minus Jaccard dissimilarity) of ex-germ-free mouse samples to their wildling cage mates minus the similarity of the ex-germ-free mouse samples to their laboratory-mouse cage mates. Colors of lines denote whether they are based on ASV profiles within co-diversifying taxa or other taxa, as indicated by the key. Datapoints are jittered along the x-axis. Asterisks indicate significant differences between the analyses conducted using ASVs from co-diversified taxa and the analyses conducted using all other ASVs; Wilcoxon Test, p-value = ns not significant, * < 0.05, ** < 0.01. c Box plots show the normalized read counts (log10[read count + 1]) of all laboratory mouse–specific or wildling-specific ASVs from co-diversified genera in the laboratory and wildling mice sampled at day 0 (before co-housing) (n = 6). For boxplots, the middle line represents the median value, the lower and upper hinges correspond to the first and third quartiles, respectively, and the upper and lower whiskers extend to the largest and smallest values, respectively, within 1.5 times the inter-quartile range. Significant differences in mean abundance are shown (Wilcoxon Test, p-value = ns not significant, * < 0.05, ** < 0.01). d Line plots show higher relative abundance in ex-germ-free mice of wildling-derived ASVs from bacterial genera showing evidence of co-diversification (yellow) compared to laboratory mouse–derived ASVs from the same genera (blue). Each line indicates the normalized relative abundance (log10[read count + 1]) within individual ex-germ-free mice (n = 6) sampled between days 1 and 17 of wildling-derived (yellow) or laboratory mouse–derived (blue) ASVs from a co-diversified genus. Significant differences in mean were assessed with modified Wilcoxon rank sum tests for zero-inflated data, *p-value < 0.05, ****p-value < 0.0001. Results from experiments with Taconic mice are presented in Supplementary Fig. 9. Raw data and metadata used for these analyses are presented in Supplementary Data 14 and Supplementary Data 16. Source data are provided as a Source Data file. P-values are provided in Supplementary Data 15.
In summary, we found that laboratory mice have retained > 25-million-year-old symbiont lineages that co-diversified with rodent species, and that these ancestral laboratory-mouse symbionts have experienced elevated levels of genetic drift during ~ 120 years of captivity. Co-diversifying symbionts in laboratory mice were close relatives of symbionts from wild house mice (as expected based on the predominantly M. m. domesticus genetic ancestry of laboratory mice) but represented only a subset of the strain diversity found in the wild and displayed elevated genome-wide rates of nonsynonymous nucleotide substitutions (Fig. 3). The observation that LGM strains from ancestral, co-diversifying taxa displayed increased genetic load provides an evolutionary basis for their reduced fitness when competed in germ-free mice against relatives from wild mice (Fig. 4; Supplementary Figs. 9 and 10). These findings suggest that genetic drift—rather than positive selection—has been the predominant evolutionary force driving the divergence of LGM strains from their ancestors in wild mice.
Methods
Ethical statement
All procedures conformed to guidelines established by the U.S. National Institutes of Health and have been approved by the Cornell University Institutional Animal Care and Use committee (IACUC: Protocol #2015-0060).
Sampling of Peromyscus gut microbiota
Samples from Permoyscus species sequenced in this study were collected from the University of South Carolina Peromyscus Stock Center. Host species sampled included P. maniculatus bardii (n = 1), P. leucopus (n = 1), P. polionotus (n = 2), P. errimicus (n = 1), P. californicus (n = 1), and P. maniculatus sonoriensis (n = 1). Each host lineage was reared in a common laboratory environment on a standard laboratory chow diet ad libitum (~18% protein, ~58% carbohydrates, ~6.2% fat, ~5% fiber). No host lineage was rederived under sterile conditions (e.g., through embryo transfer to a laboratory mouse lineage). These husbandry practices have been previously shown to enable the retention of wild-derived gut microbiota in the laboratory animal-facility environment for > 10 host generations39. For sampling, individual rodents were transferred to clean cages and monitored for 1–2 h, after which fecal samples deposited during that time were collected with sterile tweezers. Fecal samples were immediately placed in empty sterile tubes on dry ice and shipped to Cornell University, where they were stored at -80C until DNA extraction.
DNA extraction and library preparation
For Peromyscus samples sequenced in this study, DNAs were extracted for Nanopore sequencing using a three-step extraction protocol. Steps included 1) osmotic lysis, 2) enzymatic lysis, and 3) bead beating following previously described methods7,47. For each extraction, ~100 mg of starting fecal material was used to ensure sufficient yield for Nanopore sequencing. Libraries were prepared using the Nanopore Ligation Sequencing kit (SQK-LSK110) following the manufacturer-supplied protocols. Separate extractions from the same fecal samples were made for Illumina short-read metagenome sequencing using Qiagen PowerSoil microbiome extraction kits.
Illumina metagenome sequencing
Libraries for Illumina short-read metagenome sequencing of Peromyscus samples were prepared at the Cornell Biotechnology Resource Center (BRC) using their Illumina TruSeq-equivalent ligation library prep protocol (https://www.biotech.cornell.edu/). Libraries were sequenced on an Illumina NovaSeq sequencer at the UC Davis DNA Technologies Core.
Nanopore sequencing and base calling
Each library was sequenced on the MinION platform using an entire flow cell. Base calling was conducted either in real time or post-sequencing with the Guppy base caller48 v3.1.5 using two nVIDIA RTX 3090 Graphical Processing Units (GPUs). The following settings were employed in guppy_basecaller --device “cuda:all” --chunk_size 3000 --chunks_per_runner 768 --gpu_runners_per_device 4 --qscore_filtering --min_qscore 7 --config dna_r9.4.1_450bps_hac.cfg --calib_detect --compress_fastq --recursive.
Assembly of Peromyscus MAGs
Contiguous sequences from Peromyscus MAGs were assembled from nanopore sequence data and polished with Illumina short-read sequencing data using the snakemake49 reticulatus workflow available at https://github.com/SamStudio8/reticulatus. We used the MetaFlye v2.8 ‘spell’ within reticulatus50, followed by a polishing pipeline employing four rounds of polishing by Racon51, one round of Medaka v1.0.1 [https://github.com/nanoporetech/medaka], and two rounds of Pilon v1.23 polishing with Illumina short-read metagenome data52. Finally, contigs likely to be derived from hosts were removed from the polished assembly using the ‘dehumanizer’ step of the reticulatus pipeline against an assembly from the corresponding host species (P. leucopus: GCA_004664715.1; P. polionotus: GCA_003704135.2; P. maniculatus: GCA_003704035.1; P. californicus: GCA_007827085.2). Assembled and polished contigs were binned in Anvi’o v6.2 using CONCOCT53 and refined manually using anvi-summarize and anvi-refine54.
Phylogenomic analyses
Phylogeny was constructed from the combined set of MAGs from all host species for which > 20 MAGs were available as well as MAGs from Castor canadensis, for which only 16 MAGs were available but which represents a basally branching rodent lineage that was not otherwise represented in the data. Core genes from the bac120 collection were identified for each MAG with the Genome Taxonomy Database Toolkit (GTDB-Tk) v1.4.1 using the ‘identify’ function31. Concatenated core genes were then aligned in GTDB-Tk using the ‘align’ function with default settings31. The alignment was then used to infer a phylogenetic tree of the combined set of rodent MAGs in IQTree2 version 2.1.3 using the settings -mset LG,WAG, --seed 0, and -B 1000.
Scans for co-diversification
To identify co-diversified clades in the rodent MAG phylogeny, we employed an extension of the method developed by Hommola et al.32, which uses permutation tests to estimate non-parametric p-values based on the null hypothesis of no association between the symbiont and host evolutionary distances. This workflow yielded a Mantel’s r correlation coefficient for each clade of gut bacteria tested as well as a non-parametric p-value indicating the probability of observing by chance a Mantel’s r greater than or equal to that observed in the real data.
Here, we applied these tests to nodes that contained ≥ 3 hosts and ≥ 7 symbionts and spanned less than 1/4th of the total bacterial phylogeny, as we reasoned that more deeply diverging clades represent bacterial diversification events that predate the most recent common ancestor of rodents. For each node, the test employed 999 permutations. Only clades with a resulting p-value of < 0.01 and an r coefficient > 0.75 were considered “co-diversifying” for downstream analyses. All code used to conduct these analyses is available in Python at https://github.com/CUMoellerLab/codiv-tools and in R at https://github.com/DanielSprockett/codiv.
In addition, we conducted scans for co-diversification based on dereplicated clades containing only a single MAG per monophyletic clade of MAGs derived from the same host species. For these tests, we randomly selected a MAG from each monophyletic clade and performed PACo36, ParaFit37, and Hommola’s test32 using default settings. All code used for these analyses is available at https://github.com/DanielSprockett/codiv.
Permutation tests for whether extent of co-diversification exceeds null expectation
In some cases, the MAG clades tested in the co-diversification scan were non-independent due to the underlying tree structure, complicating the adjustment of p-values based on false discovery rate correction. Moreover, in some cases MAGs belonging to the same co-diversifying clade were sampled from multiple individuals per host species, thereby introducing pseudo-replication into tests for co-diversification between host-species lineages. To address these issues and to assess whether there was greater evidence for co-diversification of MAG clades with rodent species in the MAG phylogeny than expected by chance, we conducted additional permutation tests in which the host-species labels were permuted on the host-species phylogeny 100 times and the co-diversification scans were reperformed for each permutation. These analyses yielded a null distribution of the proportion of co-diversifying clades expected to reach significance thresholds (r > 0.75, p-value < 0.01) by chance given the underlying structure of and pseudoreplication in the MAG phylogeny. This null distribution was used to calculate a non-parametric p-value indicating the probability of observing, by chance under the null hypothesis of no association between bacterial and host evolutionary distances, a number of significantly co-diversifying clades (r > 0.75, p-value < 0.01) that was equal to or greater than the number observed in the analyses based on the host phylogeny containing the correct host-species tip labels. All code used to conduct these analyses is available in Python at https://github.com/CUMoellerLab/codiv-tools and in R at https://github.com/DanielSprockett/codiv.
Molecular clock analyses
We regressed symbiont divergence estimates based on protein-sequence divergence in clades that displayed the strongest evidence of co-diversification (that is, Mantel’s r > 0.95) and known divergence times of host species based on timetree.org29. These regression analyses and calculations of 95% confidence intervals were conducted in base R55 (version 4.2.3).
Phylogenetic ANOVA
Phylogenetic ANOVA was performed using the rodent gut bacterial phylogeny and the KEGG annotations for each MAG to identify gene annotations enriched in co-diversifying clades relative to non-co-diversifying clades or in laboratory-mouse symbionts relative to wild-mouse symbionts. For each test, the abundance of each KEGG module in every MAG from M. m. domesticus was used as the response variable, and the group variable was either co-diversifying vs. non-co-diversifying or laboratory vs. wild. These analyses were conducted using the phylANOVA function in the R package phytools56 v2.1. Benjamin-Hochberg correction was performed to account for multiple testing across classes of annotations. Furthermore, we also developed a permutation-based analysis to generate a non-parametric p-value that does not assume a normal distribution of KEGG annotations. For each KEGG module, the phylogenetic ANOVA test was repeated 99 times using randomly permuted group labels. The distribution of F-statistics generated from tests conducted on randomly permuted group labels was used to calculate a non-parametric p-value for the F-statistic generated from the test conducted on the observed data. Analysis details are available in the Supplementary Code and at https://zenodo.org/records/14181931.
dN/dS analyses
For each co-diversifying clade containing MAGs from laboratory and wild house mice, we used Roary57 v3.12.0 to identify and codon-align each gene family containing orthologs from at least one laboratory-derived house-mouse MAG and at least one wild-derived house-mouse MAG. Codon alignments were then used to construct a phylogenetic tree for each gene family using RAxM58 v8.2.12. Codon alignments and gene trees were then used in CODEML within PAML59 v4.10.6 to estimate the proportion of nonsynonymous substitutions per nonsynonymous site (dN) to synonymous substitutions per synonymous site (dS) (i.e., dN/dS) for every branch leading to a laboratory-derived house-mouse gene copy and every branch leading to a wild-derived house-mouse gene copy. The averages for laboratory-derived house-mouse gene copies and wild-derived house-mouse gene copies were calculated for each gene family. Differences in genome-wide dN/dS across the whole dataset and in individual clades were conducted with paired t-tests in base R. All genes with nonzero dN and dS are shown in Figs. 2 and 3.
Reanalysis of co-housing experiments
We downloaded raw sequencing reads from accessions from PRJNA54089313, which contained results of an experiment in which C57BL/6 J mice from The Jackson Laboratory or Taconic Biosciences (sources from which genomes of laboratory-mouse GM strains displaying evidence of genetic drift were assembled, e.g., Fig. 3) containing either a laboratory-derived or wild-derived (wildling) microbiota were co-housed with germ-free mice for 17 days. Forward and reverse reads were denoised with dada260 in R (v4.4.0)55 using the following settings: trimLeft = c(19, 20), truncLen = c(170, 165), maxEE = 2, truncQ = 2, maxN = 0, rm.phix = TRUE. Chimeric sequences were removed using the “consensus” method, and ASVs that were not 250–255 bp in length were removed. GTDB ribosomal sequences and taxonomy (bac120_ssu_reps_r202.tar.gz and bac120_ssu_reps_r202.tar.gz) were imported into qiime2 (v2023.9.1) using ‘qiime tools import’, and a classifier was trained using ‘qiime feature-classifier fit-classifier-naive-bayes’61,62. The resulting set of 1450 ASVs were then classified using ‘qiime feature-classifier classify-sklearn’61. Samples from experiments involving Jackson laboratory mice were rarefied to a common depth of 40,000 reads, while samples from experiments involving Taconic laboratory mice were rarified to 30,000 reads in order to retain the full set of laboratory and wildling samples. A total of 349 ASVs were identified as derived from wildling mice (i.e., found in at least one wildling mouse but no laboratory mice on day 0), while 383 ASVs were identified as derived from laboratory mice (i.e., found in either Jackson or Taconic laboratory mice but no wildling mice at day 0). Beta diversity (Jaccard) analyses were performed using the phyloseq63 (v1.48.0) and vegan64 (v 2.6.8) R packages, and results were plotted using ggplot265 (v 3.5.1). Further details of this analysis and the R code is available in the Supplementary Code and at https://zenodo.org/records/1418193166.
Statistics & reproducibility
No statistical method was used to predetermine sample size. MAGs from host species for which > 20 MAGs were available were include in the analysis. No samples were excluded from the analysis, and investigators were not blinded to the experimental outcome during re-analysis. No manipulative experiments were conducted.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All sequence data generated in this study have been deposited to the National Center for Biotechnology Information Sequence Read Archive under accessions BioProject ID PRJNA1089132. Additional metadata about the genome assemblies generated by previous studies are available at BioProject PRJNA725899 and DOI: 10.1038/s43705-021-00053-9. All MAGs generated by this study are available at BioProject ID PRJNA1221925 and at Zenodo DOI: 10.5281/zenodo.14181931. All display items presented in the main manuscript and supplementary information can be reproduced from raw data and code provided. Source data are provided with this paper.
Code availability
Code used for analyses and for generating figures is available at https://github.com/DanielSprockett/codiv and at Zenodo https://doi.org/10.5281/zenodo.14859771.
References
Ley, R. E. et al. Evolution of Mammals and Their Gut Microbes. Science 320, 1647–1651 (2008).
Moeller, A. H. et al. Cospeciation of gut microbiota with hominids. Science 353, 380–382 (2016).
Moeller, A. H. et al. Dispersal limitation promotes the diversification of the mammalian gut microbiota. Proc. Natl. Acad. Sci. 114, 13768–13773 (2017).
Groussin, M. et al. Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat. Commun. 8, 14319 (2017).
Levin, D. et al. Diversity and functional landscapes in the microbiota of animals in the wild. Science 372, eabb5352 (2021).
Suzuki, T. A. et al. Codiversification of gut microbiota with humans. Science 377, 1328–1332 (2022).
Sanders, J. G. et al. Widespread extinctions of co-diversified primate gut bacterial symbionts from humans. Nat. Microbiol. 8, 1039–1050 (2023).
Beck, J. A. et al. Genealogies of mouse inbred strains. Nat. Genet. 24, 23–25 (2000).
Bowerman, K. L. et al. Effects of laboratory domestication on the rodent gut microbiome. ISME Commun. 1, 49 (2021).
Rosshart, S. P. et al. Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell 171, 1015–1028.e13 (2017).
Chen, Y.-H. et al. Rewilding of laboratory mice enhances granulopoiesis and immunity through intestinal fungal colonization. Sci. Immunol. 8, eadd6910 (2023).
Yeung, F. et al. Altered Immunity of Laboratory Mice in the Natural Environment Is Associated with Fungal Colonization. Cell Host Microbe 27, 809–822.e6 (2020).
Rosshart, S. P. et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, eaaw4361 (2019).
McCutcheon, J. P. & Moran, N. A. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10, 13–26 (2012).
Perreau, J. & Moran, N. A. Genetic innovations in animal–microbe symbioses. Nat. Rev. Genet. 23, 23–39 (2022).
Groussin, M., Mazel, F. & Alm, E. J. Co-evolution and Co-speciation of Host-Gut Bacteria Systems. Cell Host Microbe 28, 12–22 (2020).
Parks, D. H. et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542 (2017).
Bickhart, D. M. et al. Generating lineage-resolved, complete metagenome-assembled genomes from complex microbial communities. Nat. Biotechnol. 40, 711–719 (2022).
Hildebrand, F. et al. A comparative analysis of the intestinal metagenomes present in guinea pigs (Cavia porcellus) and humans (Homo sapiens). BMC Genom. 13, 514 (2012).
Kohl, K. D., Weiss, R. B., Cox, J., Dale, C. & Dearing, M. D. Gut microbes of mammalian herbivores facilitate intake of plant toxins. Ecol. Lett. 17, 1238–1246 (2014).
Wang, J. et al. Dietary history contributes to enterotype-like clustering and functional metagenomic content in the intestinal microbiome of wild mice. Proc. Natl. Acad. Sci. 111, E2703–E2710 (2014).
Xiao, L. et al. A catalog of the mouse gut metagenome. Nat. Biotechnol. 33, 1103–1108 (2015).
Finlayson-Trick, E. C. L. et al. Taxonomic differences of gut microbiomes drive cellulolytic enzymatic potential within hind-gut fermenting mammals. PLoS ONE 12, e0189404 (2017).
Kohl, K. D. et al. Metagenomic sequencing provides insights into microbial detoxification in the guts of small mammalian herbivores (Neotoma spp.). FEMS Microbiol. Ecol. 94, fiy184 (2018).
Pan, H. et al. A gene catalogue of the Sprague-Dawley rat gut metagenome. GigaScience 7, giy055 (2018).
Zhao, L. et al. Saturated long-chain fatty acid-producing bacteria contribute to enhanced colonic motility in rats. Microbiome 6, 107 (2018).
Zhang, S. et al. Shen-Ling-Bai-Zhu-San alleviates functional dyspepsia in rats and modulates the composition of the gut microbiota. Nutr. Res. 71, 89–99 (2019).
Donovan, M. et al. Metagenome-Assembled Genome Sequences of Five Strains from the Microtus ochrogaster (Prairie Vole) Fecal Microbiome. Microbiol. Resour. Announc. 9, e01310–e01319 (2020).
Kumar, S. et al. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol. Biol. Evol. 39, msac174 (2022).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. G. T. D. B.- Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2020).
Hommola, K., Smith, J. E., Qiu, Y. & Gilks, W. R. A permutation test of host–parasite cospeciation. Mol. Biol. Evol. 26, 1457–1468 (2009).
Robertson, B. R., O’Rourke, J. L., Vandamme, P., On, S. L. & Lee, A. Helicobacter ganmani sp. nov., a urease-negative anaerobe isolated from the intestines of laboratory mice. Int. J. Syst. Evol. Microbiol. 51, 1881–1889 (2001).
Li, F. et al. A phylogenomic analysis of Limosilactobacillus reuteri reveals ancient and stable evolutionary relationships with rodents and birds and zoonotic transmission to humans. BMC Biol 21, 53 (2023).
Nishida, A. H. & Ochman, H. Captivity and the co-diversification of great ape microbiomes. Nat. Commun. 12, 5632 (2021).
Hutchinson, M. C., Cagua, E. F., Balbuena, J. A., Stouffer, D. B. & Poisot, T. paco: implementing Procrustean Approach to Cophylogeny in R. Methods Ecol. Evol. 8, 932–940 (2017).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2018).
Moeller, A. H., Sanders, J. G., Sprockett, D. D. & Landers, A. Assessing co‐diversification in host‐associated microbiomes. J. Evol. Biol. 36, 1659–1668 (2023).
Moeller, A. H., Suzuki, T. A., Phifer-Rixey, M. & Nachman, M. W. Transmission modes of the mammalian gut microbiota. Science 362, 453–457 (2018).
Yang, H. et al. Subspecific origin and haplotype diversity in the laboratory mouse. Nat. Genet. 43, 648–655 (2011).
Sonnenburg, E. D. et al. Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215 (2016).
Kuo, C.-H., Moran, N. A. & Ochman, H. The consequences of genetic drift for bacterial genome complexity. Genome Res 19, 1450–1454 (2009).
Ohta, T. The nearly neutral theory of molecular evolution. Annu. Rev. Ecol. Syst. 23, 263–286 (1992).
Daubin, V. & Moran, N. A. Comment on “The origins of genome complexity. Science 306, 978a–978a (2004).
Moran, N. A. Accelerated evolution and Muller’s rachet in endosymbiotic bacteria. Proc. Natl. Acad. Sci. 93, 2873–2878 (1996).
Sprockett, D. D. et al. Home-site advantage for host species–specific gut microbiota. Sci. Adv. 9, eadf5499 (2023).
Quick, J., Nicholls, S. & Loman, N. The “Three Peaks” faecal DNA extraction method for long-read sequencing v2. https://doi.org/10.17504/protocols.io.7rshm6e (2019).
Wick, R. R., Judd, L. M. & Holt, K. E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20, 129 (2019).
Mölder, F. et al. Sustainable data analysis with Snakemake. F1000Research 10, 33 (2021).
Kolmogorov, M. et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 17, 1103–1110 (2020).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Eren, A. M. et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6 (2021).
R Core Team (2024). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/.
Revell, L. J. phytools 2.0: an updated R ecosystem for phylogenetic comparative methods (and other things). PeerJ 12, e16505 (2024).
Page, A. J. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693 (2015).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90 (2018).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
McMurdie, P. J. & Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 8, e61217 (2013).
Oksanen, J. et al. vegan: Community Ecology Package. R package version 2.7-0, https://github.com/vegandevs/vegan (2025).
Wickham, H. ggplot2, Elegant Graphics for Data Analysis. (2016).
Sprockett, D. D. et al. Recent genetic drift in the co-diversifying gut bacterial symbionts of laboratory mice. Zenodo. https://doi.org/10.5281/zenodo.14859771 (2025).
Acknowledgements
We thank Dr. Weiwei Yan for assistance with DNA extractions from rodent fecal samples. We thank the staff at Peromyscus Stock Center for providing fecal samples. Funding was provided by the National Institutes of Health grants R35 GM138284 and R01 DK139214 (AHM) and NIAID T32 AI145821 (DDS).
Author information
Authors and Affiliations
Contributions
A.H.M. supervised the research, performed analyses, and wrote and edited the paper. D.D.S. performed analyses and wrote and edited the paper. B.A.D., A.A.L., and J.G.S. performed analyses and edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Liang Xiao, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sprockett, D.D., Dillard, B.A., Landers, A.A. et al. Recent genetic drift in the co-diversified gut bacterial symbionts of laboratory mice. Nat Commun 16, 2218 (2025). https://doi.org/10.1038/s41467-025-57435-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57435-z
