
Abstract
Among different age groups, middle-aged individuals are particularly susceptible to obesity, with a 22% higher risk of all-cause mortality. However, the underlying mechanisms remain unclear. In this study, we identify adipose progenitor cells (APCs) in the white adipose tissue (WAT) of middle-aged subjects as potential causes of midlife obesity. Specifically, the extracellular vesicles (EVs) derived from APCs display an impaired ability to mitigate the inflammaging of adipose tissue macrophages (ATMs) in middle-aged individuals. Mechanistically, these EVs, lacking miR-145-5p, fail to suppress the expression of L-selectin in ATMs, thereby facilitating their M1 program via the NF-κB signaling pathway. In contrast, EVs from young APCs effectively inhibit M1 macrophage polarization. Accordingly, targeted liposomes are designed to deliver miR-145-5p mimics to ATMs, which effectively prevent the obesity in middle-aged mice. Collectively, our findings highlight the role of APC-derived EVs in midlife obesity and propose miR-145-5pas a promising therapeutic target for clinical applications.
Similar content being viewed by others

Introduction
Obesity in the midlife (aged 30–49 years) stage is becoming increasingly prevalent worldwide1. Notably, approximately 43% of middle-aged adults in the United States are classified as individuals with obestiy2, with a similar trend observed in other developed and developing countries1. Moreover, emerging epidemiological evidence indicates that middle-aged individuals with obesity face a significantly greater mortality risk than their counterparts in other age groups with similar health conditions3. Despite these clear clinical observations, the molecular mechanisms underlying these phenomena remain largely unexplored.
As the largest energy store and endocrine organ, adipose tissue plays a crucial role in maintaining the systemic homeostasis of glycolipid metabolism4,5,6. Single-cell RNA sequencing data have revealed that white adipose tissue (WAT) may serve as an outpost of systemic aging7. Specifically, during the course of aging, WAT exhibits increased expression of senescence-related genes, which are associated with imbalanced immune homeostasis and metabolic disorders7. Notably, these changes occur much earlier in WAT than in other organs, and adipose progenitor cells (APCs) are likely of great importance in driving these alterations8,9. Indeed, with aging, APCs gradually lose their ability to replicate and differentiate, leading to a declined capability for browning8,10. Furthermore, senescent APCs tend to secrete more proinflammatory factors, ultimately contributing to insulin resistance in adipose tissue11,12. While these findings are exciting, the exact role of APCs in midlife obesity remains poorly explored.
Another hallmark of adipose tissue aging is chronic and low-grade inflammation, known as “inflammaging13. Inflammaging is characterized by an increase in the levels of proinflammatory cytokines and other inflammatory markers, even in the absence of infection or injury. As the largest immune cell subpopulation within WAT, adipose tissue macrophages (ATMs) play a pivotal role in driving inflammaging14,15,16. During the initiation and progression of aging, senescent cells accumulate in WAT and secrete proinflammatory factors to activate ATMs, which amplify inflammation and insulin resistance by secreting additional proinflammatory cytokines17. Aging also alters the composition of ATMs, with an increase in the number of proinflammatory macrophages and a decrease in the number of anti-inflammatory macrophages15,18. This shift toward a proinflammatory phenotype exacerbates chronic inflammation and insulin resistance18,19. Although recent studies have suggested a role for ATMs in inflammaging during the aging process, their involvement in midlife obesity and the underlying regulatory mechanisms are yet to be elucidated.
Extracellular vesicles (EVs) are membrane-structured particles secreted by various cell types to transfer biomolecules such as proteins, lipids or microRNAs (miRNAs), facilitating intercellular communication20. As EVs have been recognized to be involved in the pathogenesis of various metabolic diseases21,22,23, it is plausible to assume the feasibility of their involvement in midlife obesity. To address this possibility, we conducted bulk RNA-seq and single-nucleus RNA-seq analyses and found that APC-derived EVs in WAT could be potential culprits triggering midlife obesity. Specifically, EVs secreted by young APCs alleviated ATM inflammaging in middle-aged mice upon challenge with a high-fat diet (HFD), but this effect decreased with age. Moreover, a greater decrease in miR-145a-5p (miR-145) within the APC-derived EVs was noted during the middle stage of life, which accounted for the reduced efficacy of EVs against obesity in middle-aged individuals. Therefore, cationic liposomes were designed to specifically deliver miR-145 mimics to ATMs in WAT, effectively preventing middle-aged mice from becoming obese. Collectively, our studies suggest the role of APC-derived EVs in midlife obesity and highlight miR-145 as a promising therapeutic target for the prevention and treatment of midlife obesity.
Results
Susceptibility of middle-aged mice to obesity is attributed to altered WAT function
We first aimed to demonstrate that middle-aged mice are more susceptible to obesity. To this end, 3-month-old (young, equivalent to humans aged 20) and 12-month-old (midlife, equivalent to humans aged 43) mice (n = 14) were fed either a standard chow diet or a HFD for 4 weeks. A significantly greater increase in body weight was observed in middle-aged mice than in their young counterparts following HFD challenge (Fig. 1a). Consistently, young mice exhibited increased oxygen consumption (Supplementary Fig. 1a) and energy expenditure (Supplementary Fig. 1b) coupled with a lower respiratory exchange ratio (RER; Supplementary Fig. 1c)and comparable food intake (Supplementary Fig. 1d). Moreover, HFD-challenged middle-aged mice presented impaired glucose tolerance (Fig. 1b) and significantly reduced insulin sensitivity (Fig. 1c and Supplementary Fig. 1e–g). Flow cytometry analysis further revealed a marked reduction in M2 (CD11b+/F4/80+/CD206+/CD11c-) and a significant increase in M1 (CD11b+/F4/80+/CD206-/CD11c+) macrophages in the epididymal WAT of HFD-challenged middle-aged mice (Fig. 1d), along with increased production of proinflammatory cytokines (e.g., Tnf-α, Il-1β, and Il-6; Fig. 1e). Adipocyte hypertrophy was also observed in the inguinal WAT (iWAT) and epididymal WAT (eWAT) from middle-aged mice (Supplementary Fig. 1h). In sharp contrast, only a moderate difference was observed between young and middle-aged mice fed a normal chow diet (Supplementary Fig. 1i-k). These findings suggest that middle-aged subjects, even those with a normal body weight under normal conditions, are more susceptible to obesity upon challenge with metabolic stress.

a Body weight gain of young (3-month-old) and middle-aged (12-month-old) mice fed with a 4-week high-fat diet (HFD) (n = 14 per group). b, c Glucose tolerance test (GTT) (b) and insulin tolerance test (ITT) (c) results of young and middle-aged mice challenged with a 4-week HFD (n = 14 per group). d Flow cytometry analysis of the proportions of M1 (CD11b+/F4/80+/CD11c+/CD206-) and M2 (CD11b+/F4/80+/CD11c-/CD206+) macrophages in the epididymal WAT of young and middle-aged mice (n = 8 biological replicates per group). e RT‒qPCR analysis of the mRNA expression of proinflammatory genes (Tnf-α, Il-1β, and Il-6) in the epididymal WAT of young and middle-aged mice challenged with a 4-week HFD (n = 8 biological replicates per group). f Number of differentially expressed genes (DEGs) in the liver, muscle and WAT of C57BL/6 mice of different ages (9, 12, and 15 months). g–i Western blot analysis of p-AKTSer473/AKT, p-ACCSer79/ACC, p-HSLSer660/HSL and PGC1α in WAT (g), p-AKTSer473/AKT, PGC1α, SDHB and NDUFS1 in skeletal muscle (h), p-AKTSer473/AKT, PEPCK, G6Pase and p-GSK3βSer9/GSK3β in liver (i) from young and middle-aged mice (n = 3 biological replicates per group). j-l Western blot analysis of p-ACCSer79/ACC, p-HSLSer660/HSL and PGC1α in subcutaneous WAT (j), PGC1α, SDHB and NDUFS1 in skeletal muscle (k), PEPCK, G6Pase and p-GSK3βSer9/GSK3β in liver (l) from young and middle-aged humans (n = 10 biological replicates per group). The data were presented as mean ± SEM. Statistical significance was assessed by two-way ANOVA and Bonferroni’s multiple comparisons test (a, left panels in b, c) or a two-sided Student’s t test (right panels in b–e). See also Supplementary Fig. 1. Source data are provided as a Source Data file.
To determine why middle-aged individuals present a greater risk for obesity, we collected data and samples from mice and humans at different ages. Given that factors, such as diet and being overweight, could interfere with the effect of aging, all samples and data were collected from mice or humans with normal diets (dietary quality index score >85 for human subjects) and normal body weights (body mass index between 18.5 and 23.9 for human subjects). To identify the metabolic organ(s) responsible for the increased susceptibility to obesity in midlife subjects, we first analyzed public RNA-seq data (Gene Expression Omnibus, accession code GSE132040) of major metabolic organs (liver, skeletal muscle and WAT) derived from C57BL/6 mice of different ages (3, 9, 12, and 15 months; equivalent to humans aged 20, 36, 43, and 50 years, respectively)7. Pairwise differential gene expression levels in metabolic organs were calculated to determine the metabolic organ that exhibited the most significant functional changes in middle-aged individuals. The number of differentially expressed genes (DEGs) increased markedly in middle-aged mice compared with young mice, and most notably, significant DEGs were detected only in WAT from middle-aged mice (12 and 15 months old) (Fig. 1f), suggesting that WAT could be the primary metabolic organ conferring an increased risk for midlife obesity. In support of this notion, compared with young (3-month-old) mice, middle-aged (12-month-old) mice presented significantly increased lipogenesis along with a marked decrease in insulin sensitivity and lipolytic capacity in WAT (Fig. 1g and Supplementary Fig. 1l). In sharp contrast, no significant change was noted in terms of insulin sensitivity or metabolic capacity in skeletal muscle (Fig. 1h and Supplementary Fig. 1m) or the liver (Fig. 1i and Supplementary Fig. 1n). We then checked the above metabolic markers in human subcutaneous WAT, liver, and skeletal muscle (Fig. 1j‒l and Supplementary Fig. 1o‒q, n = 10, see Supplementary Table 2‒5 for detailed information), and consistent results were obtained, reinforcing the notion that altered WAT function could be the key contributor to increased risk for midlife obesity.
Midlife APC-derived EVs exhibit a reduced capacity to maintain adipose immune homeostasis
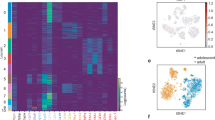
To characterize the cell subpopulation(s) responsible for WAT dysfunction, we collected WAT samples from both young and middle-aged subjects (Supplementary Table 1) for single-nucleus RNA sequencing (snRNA-seq). After standard data processing and quality control, transcriptomic profiles were generated from 26,294 cells (Supplementary Fig. 2a). Twenty-five subclusters were identified (Supplementary Fig. 2a) using a graph-based clustering method. We curated 16 cell type signatures to establish a detailed cell identity annotation (Fig. 2a, b). By comparing the transcriptomic data of different cell subpopulations between young and middle-aged individuals, we obtained DEGs for each cell subgroup (Fig. 2c). In accordance with the above definition criteria (Fig. 1f), APCs were identified as the cell subgroup exhibiting the most significant functional alterations in WAT from middle-aged subjects, followed by macrophages and adipocytes (Fig. 2c).

a A t-SNE plot displaying single-nucleus RNA sequencing data of cells from WAT samples from young and middle-aged humans. b Cell identity annotation in human WAT samples. c Number of DEGs for each identified cell subgroup. d, e Western blot results of p19ARF and p53 in mouse APCs (d), P16Ink4a and P21 in human APCs (e) from the young (mice, 3-month-old; humans, aged 18-29), middle-aged (mice, 12-month-old; humans, aged 30-49) and aged (mice, 24-month-old; humans, aged 50-69) groups (n = 3 independent experiments). f, g Transcriptional levels of senescence-associated genes in the APCs of mice (p16Ink4a, p21, Il-6, and Igfbp3) (f) and humans (P16Ink4a, P21, IL-6, and IGFBP3) (g) from the indicated groups (n = 6 per group). h Number of DEGs annotated through gene ontology (GO) enrichment-based clustering based on bulk RNA-seq analysis of young and middle-aged APCs from mice or humans. i The engulfment of PKH26-labeled EVs (red) by APCs, adipocytes and macrophages was detected by confocal microscopy. The experiments were repeated independently three times. Scale bar, 20 µm. j The engulfment of yEVs and mEVs by macrophages was detected by flow cytometry. k Western blot analysis of iNOS expression from BMDMs of blank, yEVs and mEVs groups in the presence of PBS or LPS (n = 3 per group). l RT‒qPCR results of inflammation-associated genes (Tnf-α, Il-1β, and Il-6) in BMDMs of blank, yEVs and mEVs groups in the presence of PBS or LPS (n = 3 biological replicates per group). The data were presented as mean ± SEM. Statistical significance was assessed by one-way ANOVA (f, g, bottom row in k, l). See also Supplementary Fig. 2 and Supplementary Fig. 3. Source data are provided as a Source Data file.
To validate the above findings, we first assessed the proportion of APCs in WAT from young and middle-aged individuals using flow cytometry. A significant decrease in the proportion of APCs (CD34+/CD31-/CD45-) was detected in the subcutaneous WAT of middle-aged subjects (Supplementary Fig. 2b), which was similar to that observed in the inguinal WATs of middle-aged mice (SCA-1+/CD31-/CD45-/Ter119-) (Supplementary Fig. 2c). The APCs were subsequently isolated using magnetic beads for functional studies (Supplementary Fig. 2d, e). Unlike young APCs (from 3-month-old mice or 18- to 29-year-old subjects), middle-aged APCs (from 12-month-old mice or 30- to 49-year-old subjects) manifested an exacerbated senescent phenotype, resembling those of older APCs (from 24-month-old mice or 50- to 69-year-old subjects), as evidenced by Western blot and RT‒qPCR analysis of senescence markers (Fig. 2d, g, Supplementary Fig. 2f, g). β-Gal staining indeed confirmed the aggravated senescence-associated secretory phenotype (SASP) in both mice and humans (Supplementary Fig. 2h, i), suggesting that the middle stage of life serves as a pivotal checkpoint for the pronounced aging and functional decline of APCs. To elucidate the functional changes in APCs during midlife, we performed comparative bulk RNA-seq analysis of APCs from young and middle-aged humans and mice. The identified DEGs were then functionally annotated by gene ontology (GO) enrichment-based clustering (Fig. 2h). Data from both humans and mice revealed consistent alterations in extracellular vesicles (EVs) from APCs (Fig. 2h).
Next, APCs were isolated from both mice and humans and expanded for 6 generations, after which no perceptible change in terms of the senescent phenotype was observed (Supplementary Fig. 2j). The culture supernatants were then collected for EV extraction. Transmission electron microscopy revealed that these particles exhibited a typical cup-shaped structure (Supplementary Fig. 2k). Dynamic light scattering demonstrated that the particles derived from both young and midlife APCs had a hydrodynamic diameter of 85 nm, with a polydispersity index of 0.22 (Supplementary Fig. 2l). Western blotting also confirmed the presence of EV-specific markers, such as tumor susceptibility gene 101 (TSG101) and the EV-related protein CD63, while the endoplasmic reticulum marker calnexin was absent (Supplementary Fig. 2m, n), indicating that the isolated particles were indeed EVs. BCA assays failed to detect a discernable difference in terms of EV secretion between young and middle-aged APCs (Supplementary Fig. 2o).
To identify the recipient cells of EVs, we examined the uptake of EVs by three cell types (APCs, macrophages and adipocytes) that exhibited the most prominent functional changes in WAT (Fig. 2c). To our surprise, macrophages, but not APCs or adipocytes, exhibited the greatest ability to engulf PKH-26-labeled APC-derived EVs, as determined either by immunostaining (Fig. 2i) or by flow cytometry (Fig. 2j), regardless of the EV type (i.e., young APC-derived EVs [yEVs] or midlife APC derived EVs [mEVs]). We then analyzed the biodistribution of EVs in mice. The mice were injected with DiI-labeled EVs. After 24 h, the mice were sacrificed, and major metabolic organs, such as WAT, liver, and skeletal muscle, were collected. Substantial accumulation of EVs (red) was observed in macrophages (F4/80+, green) within WAT (both iWAT and eWAT), whereas EV+ cells were almost undetectable in the liver (albumin+, green) and skeletal muscle (laminin receptor+, green) (Supplementary Fig. 3a). Consistently, flow cytometry analysis of the stromal vascular fraction (SVF) revealed that macrophages, rather than T cells, dendritic cells, NK cells, or APCs, sufficiently engulfed EVs (Supplementary Fig. 3b–e). Moreover, the depletion of macrophages in middle-aged mice effectively alleviated weight gain and insulin resistance (Supplementary Fig. 3f–h), highlighting the essential role of macrophages in midlife obesity. These findings prompted us to hypothesize that ATMs might serve as the primary recipient cells of APC-derived EVs.
The above results prompted us to assess the impact of yEVs and mEVs on macrophages. We first treated bone marrow-derived macrophages (BMDMs) with equal amounts of yEVs or mEVs, followed by LPS stimulation to induce M1 polarization. Western blotting (Fig. 2k) and RT‒qPCR analysis (Fig. 2l) demonstrated that EVs alone did not impact the M1 program. However, yEVs exhibited a markedly greater ability to suppress the BMDM M1 program as compared to that of mEVs (Fig. 2k, l), and similar results were observed in isolated ATMs (Supplementary Fig. 3i). Finally, BMDMs were cultured with conditioned medium from yAPCs in the presence of LPS, and an attenuated M1 program was noted, while the suppressive effect was abolished once EVs were removed from the conditioned medium (Supplementary Fig. 3j, k), indicating that APCs regulate the macrophage program through secreted EVs. Collectively, our data suggest that mEVs have a reduced ability to suppress the M1 macrophage program, which may be a key factor contributing to WAT inflammaging.
APC-derived EVs suppress WAT inflammaging to prevent obesity
We then sought to address whether APC-derived EVs regulate WAT inflammaging, which renders middle-aged mice more susceptible to obesity upon challenge with metabolic stress. We first semiquantified the distribution of yEVs and mEVs across various tissues in mice following intravenous injection. No significant difference in biodistribution between yEVs and mEVs was noted (Supplementary Fig. 4a). Consistent with the results shown in Supplementary Fig. 3a, both yEVs and mEVs preferentially accumulated in iWAT and eWAT (Supplementary Fig. 4a). Next, middle-aged mice (12 months old) were challenged with a HFD for 4 weeks, during which yEVs or mEVs (30 μg each) were intravenously administered weekly as previously described (Fig. 3a, n = 10). Although both yEVs and mEVs were comparably enriched in WAT, yEVs displayed superior efficacy against HFD-induced obesity compared with mEVs (Fig. 3b). Consistently, yEV-treated mice presented increased energy expenditure (Fig. 3c) and oxygen consumption (Supplementary Fig. 4b) along with a lower RER (Supplementary Fig. 4d), whereas food intake remained comparable between the groups (Supplementary Fig. 4c). Moreover, the mice that received yEVs presented significantly improved glucose tolerance (Fig. 3d) and insulin sensitivity (Fig. 3e, f, Supplementary Fig. 4e), a marked reduction in iWAT and eWAT mass (Supplementary Fig. 4f, g), and attenuated adipocyte hypertrophy (Supplementary Fig. 4h). Increased lipolysis and attenuated lipogenesis were also observed in yEV-treated mice (Supplementary Fig. 4i, j), which was coupled with an increase in free fatty acids (FFAs) in eWAT (Supplementary Fig. 4k).

a A schematic diagram of the mouse treatments. b Body weight gain of 12-month-old male mice fed a NCD or HFD and treated with PBS, yEVs or mEVs (n = 10 per group). c Energy expenditure of 12-month-old male mice treated with mEVs or yEVs (n = 4 per group). GTT (d) and ITT (e) of 12-month-old male mice fed a NCD or HFD and treated with PBS, yEVs or mEVs (n = 10 per group). f Western blot of p-AKTSer473/AKT in the eWAT, liver and skeletal muscle of 12-month-old male mice fed a NCD or HFD and treated with PBS, yEVs or mEVs. g Confocal images and quantitative analysis of crown-like structures (CLSs, arrows) in eWAT from 12-month-old male mice fed a NCD or HFD and treated with PBS, yEVs or mEVs (n = 10 biological replicates per group). Scale bar, 25 µm. h Flow cytometry analysis of M1 (CD11b+/F4/80+/CD11c+/CD206-) and M2 (CD11b+/F4/80+/CD11c-/CD206+) macrophage subsets in the eWAT of 12-month-old male mice fed a NCD or HFD and treated with PBS, yEVs or mEVs (n = 10 per group). i RT‒qPCR analysis of Tnf-α, Il-1β, and Il-6 in eWAT from 12-month-old male mice fed a NCD or HFD and treated with PBS, yEVs or mEVs (n = 10 biological replicates per group). j Western blot analysis of p19ARF, p53 in eWAT from 12-month-old male mice fed a NCD or HFD and treated with PBS, yEVs or mEVs (n = 4 biological replicates per group). k RT‒qPCR analysis of p16Ink4a, p19Arf, and p21 in eWAT from 12-month-old male mice fed a NCD or HFD and treated with PBS, yEVs or mEVs (n = 10 biological replicates per group). l Representative SA-β-Gal staining of the iWAT and eWAT of 12-month-old male mice fed a NCD or HFD and treated with PBS, yEVs or mEVs. The data were presented as mean ± SEM. Statistical significance was assessed by two-way ANOVA and Bonferroni’s multiple comparisons test (b, left panels in d and e), two-sided ANCOVA with body weight as co-variate (left panel in c), two-sided Student’s t-test (right panel in c), or one-way ANOVA (i, k and right panels in d, e, g, h and j). See also Supplementary Fig. 4 and Supplementary Fig. 5. Source data are provided as a Source Data file.
The next key goal was to address whether the reduced ability of mEVs to prevent HFD-induced obesity was related to inflammaging. Therefore, we first checked the crown-like structures (CLSs) in eWAT. An increased number of CLSs was observed in eWAT from mice that received mEVs compared with mice that received yEVs(Fig. 3g, arrows), indicating that mEVs likely have a reduced capacity to attenuate macrophage infiltration. Indeed, flow cytometry analysis confirmed a marked increase in M1 macrophages and a reduction in M2 population in the eWAT of mEV-treated mice (Fig. 3h). In line with these observations, mice that received mEVs presented increased expression of proinflammatory cytokines in eWAT (Fig. 3i), which was further confirmed by ELISA of eWAT and serum samples (Supplementary Fig. 4l, m). In addition to attenuating the inflammatory microenvironment in WAT, yEVs also suppressed the senescent phenotype, as evidenced by the decreased expression of senescence markers (Fig. 3j, k) and attenuated SASP (Fig. 3l) in WAT. To further confirm these observations, we conducted the same studies in female middle-aged mice, and consistent results were obtained (Supplementary Fig. 5a–f). Next, we investigated the dose effect by administering a higher dose of mEVs (100 μg each/week) to middle-aged mice. Notably, a high dosage of mEVs was more effective against HFD-induced obesity than a low dosage (30 μg each/week; Supplementary Fig. 5g, l). Taken together, these findings suggest that the reduced capacity of mEVs to suppress WAT inflammaging could be attributed to the reduced levels of beneficial cargo in mEVs.
EV-derived miR-145 serves as a critical factor that suppresses the ATM M1 program
Given that miRNAs act as age-specific disease biomarkers and are recognized as regulators of aging-associated phenotypes24,25, we prioritized our focus on miRNAs to identify the component responsible for impaired mEV function. The expression profiles of miRNAs in both yEVs and mEVs were clarified by sequencing, and differentially abundant miRNAs between yEVs and mEVs were identified (Fig. 4a). On the basis of the expression levels and fold changes, six candidate miRNAs were box-selected (Fig. 4a), and their levels in yEVs and mEVs were validated by RT‒qPCR (Fig. 4b). Among these 6 candidates, miR-145a-5p (miR-145) showed the highest abundance and the most significant fold change in yEVs. Notably, the amount of miR-145 in EVs derived from human APCs dramatically decreased with age (Fig. 4c). miR-541 and miR-351 in human EVs also tended to decrease with age, but the magnitude of decrease was much less pronounced than that of miR-145 (Supplementary Fig. 6a, b). Consistently, as the donor cells that secrete those EVs, APCs exhibited a dramatic reduction in miR-145 levels as the aging process progressed (Fig. 4d). Critically, a similar trend was observed in ATMs, the recipients of APC-derived EVs (Supplementary Fig. 6c), further suggesting that miR-145 is likely a key EV component responsible for modulating the ATM program.

a Heatmap of miRNA expression profiles in yEVs and mEVs (n = 3 per group). b RT‒qPCR analysis of the candidate miRNAs in yEVs and mEVs (n = 3 biological replicates per group). c Correlation analysis between age and relative miR-145 expression in EVs derived from APCs humans of different ages. d Relative miR-145 expression in APCs from mice of different aged mice (3, 6, 9, and 12 months old) (n = 3 biological replicates per group). Flow cytometry analysis of CD11c (e), Western blotting results for iNOS (f) and RT‒qPCR results for the proinflammatory genes Tnf-α, Il-1β, and Il-6 (g) in BMDMs treated with PBS, PBS + miR-145 mimics, LPS, LPS + scramble, LPS + miR-145 mimics, LPS + miR-145 mimics + miR-145 inhibitor (n = 3 biological replicates per group). Flow cytometry results of CD11c (h), Western blotting results for iNOS (i) and relative mRNA expression of Tnf-α, Il-1β, and Il-6 (j) in BMDMs treated with PBS, LPS, LPS + yEVs, LPS + miR-145 inhibitor + yEVs (n = 3 biological replicates per group). The data were presented as mean ± SEM. Statistical significance was assessed by two-sided Student’s t-test (b), two-sided Spearman’s correlation (c), or one-way ANOVA (d–j). See also Supplementary Fig. 6. Source data are provided as a Source Data file.
To confirm this notion, we first assessed whether miR-145 regulates the macrophage program. Flow cytometry (Fig. 4e) and Western blotting (Fig. 4f) revealed that miR-145 alone did not affect macrophage polarization, but significantly suppressed the LPS-induced M1 program, which was consistent with the effect of APC-derived EVs (Fig. 2k). Moreover, miR-145 mimics (miR-145 mic) markedly suppressed the production of inflammatory cytokines in M1 macrophages, which was abolished in the presence of miR-145 inhibitors (miR-145 in; Fig. 4g). To further address whether the effects of yEVs on macrophages depend on miR-145, rescue experiments were conducted. As expected, the miR-145 inhibitor markedly reversed the phenotype of yEV-treated macrophages, as evidenced by the enhanced M1 program coupled with the upregulated M1 markers (Fig. 4h, i) and increased inflammatory cytokines (Fig. 4j). These findings suggest that yEVs attenuate the macrophage M1 program, at least in part, through miR-145.
miR-145 attenuates the macrophage M1 program by inhibiting SELL expression
Bulk RNA-seq was subsequently conducted, followed by comparative analysis between miR-145 mimics-treated and PBS-treated M1 macrophages. Fragments per kilobase of exon per million fragments mapped (FPKM) values were calculated to quantify mRNA expression, with average FPKM values of 36 and 35 for the PBS-treated group and the miR-145 mimics-treated group, respectively (Fig. 5a). The DEGs (fold change >2, p value ≤ 0.05) between the two groups were visualized using a volcano plot, through which 428 downregulated genes were identified in the miR-145 mimics-treated group (Fig. 5b). By integrating these genes with the predicted miR-145 target genes from TargetScan, we selected 20 potential target genes (Fig. 5c), and L-selectin (Sell) showed the highest fold change (Fig. 5b, d). Critically, RT‒qPCR and Western blotting confirmed that miR-145 mimics effectively suppressed Sell expression and M1 markers in LPS-stimulated macrophages, whereas miR-145 inhibitors rescued this phenotype (Supplementary Fig. 7a, b). Furthermore, age-dependent upregulation of Sell was noted in human WAT ATMs (Fig. 5e), which was negatively correlated with the miR-145 level in human APC-derived EVs (Fig. 4c). Next, a Sell 3’-UTR plasmid containing the miR-145 binding site (3’UTR-WT) and a mutant plasmid in which the binding site was mutated (3’UTR-Mut) were constructed for luciferase reporter assays (Supplementary Fig. 7c). Each plasmid was cotransfected with miR-145 mimics into HEK293T cells. Luciferase activity was significantly lower in 3’UTR-WT-transfected cells than in 3’UTR-NC- or 3’UTR-Mut-transfected cells (Supplementary Fig. 7d), indicating that miR-145 suppresses Sell expression by binding to its 3’UTR mRNA region.

a Average FPKM values of the PBS-treated group and the miR-145 mimics-treated group (n = 3 per group). Box plots present the means (lines inside the boxes), the 1st and 3rd quartiles (bottom and top bounds of the boxes). b Volcano plot of DEGs (fold change >2, p value ≤ 0.05) between the PBS-treated group and the miR-145 mimics-treated group. c Potential target genes of miR-145 predicted by TargetScan. d Heatmap of potential differentially expressed target genes in the PBS- and miR-145 mimics-treated groups. e Correlation analysis between age and the relative mRNA expression of Sell in the WAT ATMs of humans of different ages. f Protein expression levels of iNOS in BMDMs treated with either scrambled or Sell siRNA (n = 3 independent experiments). g Protein expression levels of iNOS in BMDMs subjected to specific treatments (n = 3 independent experiments). h KEGG analysis of all genes in M1 macrophages treated with scrambled or Sell siRNA, with a focus on the top 20 categories that exhibited the most significant changes. i, j Western blot results for iNOS, p-NF-κB p65, NF-κB p65, p-IκBα, and IκBα in BMDMs subjected to certain treatments (n = 3 independent experiments). The data were presented as mean ± SEM. Statistical significance was assessed by two-sided Student’s t-test followed by Benjamini-Hochberg false discovery rate (FDR) test (b), two-sided Spearman’s correlation (e), two-sided Hypergeometric test with Fisher’s exact test (h). Scr (scrambled siRNA), Si-Sell (Sell siRNA), Sell-Ctrl (rAd-ZsGreen), Sell-OE (rAd-ZsGreen-mSell-3Flag), CU-T12-9 (an NF-κB agonist). See also Supplementary Fig. 7. Source data are provided as a Source Data file.
To determine whether that miR-145 suppresses the M1 program by suppressing Sell expression, we first confirmed that the knockdown of Sell expression by short interfering RNAs (siRNAs) effectively attenuated the LPS-induced M1 program in macrophages (Fig. 5f) and reduced the secretion of inflammatory cytokines (Supplementary Fig. 7e). In contrast, ectopic expression of Sell by adenoviral transduction (Supplementary Fig. 7f) significantly promoted the M1 program as evidenced by the increased expression of M1 markers (Fig. 5g) and inflammatory cytokines (Supplementary Fig. 7g), which abolished the impact of the miR-145 mimics. Similarly, the inhibition of Sell expression in macrophages mitigated the effect of the miR-145 inhibitor on the M1 program (Supplementary Fig. 7h, i). Collectively, these data provide evidence that miR-145 inhibits the macrophage M1 program predominantly by suppressing SELL expression.
To further elucidate how SELL regulates macrophage M1 polarization, bulk RNA-seq was used to analyze the transcriptome of M1 macrophages treated with Sell siRNA. The DEGs (fold change > 2, p-value ≤ 0.05) between scrambled siRNA-treated macrophages and Sell siRNA-treated macrophages were visualized using a volcano plot (Supplementary Fig. 7j). GO analysis revealed that the DEGs were involved primarily in macrophage immune responses (Supplementary Fig. 7k), which is consistent with the results of the functional studies (Fig. 5f, g). KEGG analysis revealed that the DEGs were enriched in the TNF/NF-kappa B (NF-κB) signaling pathway (Fig. 5h). Gain- and loss-of-function studies confirmed a positive correlation between SELL expression levels and NF-κB signaling (Fig. 5i). Rescue experiments further demonstrated that NF-κB signaling could reverse the inhibitory effect of Sell siRNA on the macrophage M1 program (Fig. 5j). These results collectively indicate that SELL promotes the macrophage M1 program by activating NF-κB signaling.
Construction of cationic liposomes for targeted delivery of miR-145
To translate the above findings into a clinical setting, we designed cationic liposomes capable of efficiently delivering miR-145 mimics to ATMs. A peptide (CKGGRAKDC)26 was conjugated to the surface of liposomes (miR-145@lip) through a thiol–Michael addition click reaction (Fig. 6a), enabling targeted delivery to WAT (miR-145@tar-lip). The final obtained liposomes were well dispersed in water, with a hydrodynamic diameter of approximately 96 nm and a polydispersity index (PDI) of 0.12, as measured by dynamic light scattering (DLS; Fig. 6b, d). Owing to the electronegative nature of miRNA, the zeta potential of the miRNA-loaded liposomes was reduced to 3.2 mV (25 °C), which is approximately neutral (Fig. 6b). The liposomes possessed a high miRNA entrapment efficiency of over 90% (Fig. 6b). Transmission electron microscopy (Fig. 6c) confirmed that the liposomes had a spherical shape with a uniform distribution (Fig. 6c, d) and maintained stability for at least 24 h (Fig. 6e).

a Schematic illustration of miR-145@tar-lip via the binding of a peptide (CKGGRAKDC) to the surface of liposomes (miR-145@lip). b Hydrodynamic diameter, polydispersity index (PDI), zeta potential, and entrapment efficiency of the prepared liposomes. c Representative transmission electron microscopy (TEM) image of the liposomes. The experiments were repeated independently three times. Scale bar, 500 nm. d Hydrodynamic diameter distribution of the liposomes. e Colloidal stability of the liposomes within 24 h (n = 3 per group). f Biodistribution of liposomes at 0 h, 0.5 h, 2 h, 8 h, and 24 h tracked by an in vivo imaging system (IVIS) after intravenous injection of miR-145@lip or miR-145@tar-lip. g Accumulation of miR-145@lip and miR-145@tar-lip in iWATs and eWATs. h Flow cytometry analysis of miR-145@lip and miR-145@tar-lip uptake by SVF cells and ATMs. The data were presented as mean ± SEM (e). See also Supplementary Fig. 8. Source data are provided as a Source Data file.
Next, we labeled those liposomes with a fluorescent dye and tracked their pharmacokinetics and biodistribution in mice. Pharmacokinetic analysis indicated that miR-145@tar-lip exhibited a prolonged time course in the circulation (Supplementary Fig. 8a). Using an in vivo imaging system (IVIS, negative control shown in Supplementary Fig. 8b), we observed that miR-145@tar-lip accumulated predominantly in the abdominal WAT (Fig. 6f, upper row), whereas the nontargeted miR-145@lip was located primarily in the liver (Fig. 6f, bottom row). The ex vivo results further confirmed the substantial accumulation of miR-145@tar-lip in WAT, including both iWAT and eWAT (Fig. 6g, negative control shown in Supplementary Fig. 8c). To determine their biodistribution among cellular subpopulations within WATs, we digested the adipose tissues and isolated the stromal vascular fraction (SVF) for flow cytometry analysis. Notably, the number of miR-145@tar-lip positive cells was twice as high as that of nontargeted (miR-145@lip) liposomes (Fig. 6h, left panel), and more critically, the number of ATMs positive for miR-145@tar-lip was 2-fold greater than that of ATMs positive for miR-145@lip (Fig. 6h, right panel). Finally, immunostaining of adipose sections (F4/80+ macrophages, green) confirmed that liposomes (DiI labeled, red) predominantly accumulated in ATMs, with miR-145@tar-lip exhibiting markedly greater targeting efficiency (Supplementary Fig. 8d). Taken together, these results support the effective enrichment of miR-145@tar-lip in WAT, which allows the targeted delivery of miR145 mimics into ATMs.
miR-145@tar-lip efficiently attenuates HFD-induced obesity in middle-aged mice
Finally, 12-month-old mice were challenged with a HFD for 4 weeks, with random intravenous administration of PBS, NC miR@tar-lip (targeting liposomes containing negative control miRNA mimics), miR-145@lip, or miR-145@tar-lip on day 0, and then every three days (Fig. 7a, n = 7). As expected, compared with the other three treatments, treatment with miR-145@tar-lip had the most significant inhibitory effect on weight gain in middle-aged mice (Fig. 7b). Consistently, mice treated with miR-145@tar-lip presented increased oxygen consumption (Supplementary Fig. 9a) and energy expenditure (Fig. 7c) coupled with a decreased RER (Supplementary Fig. 9c), while their food intake remained comparable across all groups (Supplementary Fig. 9b). In line with the attenuated obese phenotype, the administration of miR-145@tar-lip resulted in notable improvements in glucose tolerance (Fig. 7d) and insulin sensitivity (Fig. 7e, f, Supplementary Fig. 9d) following the HFD challenge. Furthermore, miR-145@tar-lip led to a decrease in eWAT and iWAT mass (Supplementary Fig. 9e, f) as well as a reduction in adipocyte hypertrophy (Supplementary Fig. 9g). The miR-145@tar-lip treated mice also exhibited increased lipolysis, along with reduced lipogenesis, and lower SELL expression (Supplementary Fig. 9h), leading to an increased amount of FFAs in eWAT (Supplementary Fig. 9j).

a Schematic diagram of the mouse treatments. b Body weight gain of middle-aged mice treated with PBS, NC miR@tar-lip, miR-145@lip or miR-145@tar-lip (n = 7 per group). c Energy expenditure of mice treated with miR-145@lip or miR-145@tar-lip (n = 4 per group). GTT (d) and ITT (e) of results for middle-aged mice treated with PBS, NC miR@tar-lip, miR-145@lip or miR-145@tar-lip (n = 7 per group). f Western blot results of p-AKTSer473/AKT in eWAT, liver and skeletal muscle from the indicated groups (n = 3 independent experiments). g Macrophage infiltration (green color) and crown-like structures (CLSs, arrows) in eWAT of mice treated with PBS, NC miR@tar-lip, miR-145@lip or miR-145@tar-lip and the statistics for the CLSs (n = 7 biological replicates per group). Scale bar, 25 µm. h Flow cytometry analysis of M1 (CD11b+/F4/80+/CD11c+/CD206-) and M2 (CD11b+/F4/80+/CD11c-/CD206+) macrophage subsets in eWAT of mice treated with PBS, NC miR@tar-lip, miR-145@lip or miR-145@tar-lip (n = 7 biological replicates per group). i RT‒qPCR results for the proinflammatory genes Tnf-α, Il-1β, and Il-6 in eWAT of mice treated with PBS, NC miR@tar-lip, miR-145@lip or miR-145@tar-lip (n = 7 biological replicates per group). j, k Protein expression of p19ARF and p53 (n = 4 biological replicates per group) (j) and the mRNA levels of p16Ink4a, p19Arf, and p21 (k) in eWAT of mice treated with PBS, NC miR@tar-lip, miR-145@lip or miR-145@tar-lip (n = 7 biological replicates per group). l Representative β-gal staining of iWAT and eWAT from the indicated groups. The data were presented as mean ± SEM. Statistical significance was assessed by two-way ANOVA and Bonferroni’s multiple comparisons test (b, left panels in d and e), two-sided ANCOVA with body weight as co-variate (left panel in c), two-sided Student’s t test (right panel in c), or one-way ANOVA (i, k, right panels in d, e, g, h, j), and p values indicate statistical significance. See also Supplementary Fig. 9 and Supplementary Fig. 10. Source data are provided as a Source Data file.
Moreover, in contrast to that from mice receiving miR-145@tar-lip, eWAT from mice receiving PBS or NC miR@tar-lip presented elevated CLS formation (Fig. 7g, arrows), whereas eWAT from mice receiving miR-145@tar-lip exhibited significantly diminished CLSs (Fig. 7g), indicating a decreased macrophage infiltration. A marked reduction in SELL expression was also noted in eWAT ATMs originating from miR-145@tar-lip treated mice (Supplementary Fig. 9i), which was coupled with a marked decrease in M1 macrophages but a significant increase in M2 macrophages (Fig. 7h). Similarly, miR-145@tar-lip reduced the expression of proinflammatory cytokines in eWAT (Fig. 7i) and decreased the levels of inflammatory mediators in eWAT and the circulation (Supplementary Fig. 9k, l). Similar to yEVs, miR-145@tar-lip mitigated the senescent phenotype of WAT, as evidenced by the decreased expression of senescence markers (Fig. 7j, k) and attenuated SASP (Fig. 7l). We replicated these experiments in middle-aged female mice and observed consistent results, in which miR-145@tar-lip also exhibited superior efficacy in attenuating HFD-induced obesity in female recipients (Supplementary Fig. 10a–f). Collectively, these results suggest that miR-145@tar-lip could have the potential to suppress the ATM M1 program and attenuate adipose inflammaging to reduce the risk of midlife obesity in clinical settings.
Discussion
Herein, we revealed that APC-derived EVs in WAT could potentially contribute to the risk of midlife obesity. A significant reduction in miR-145 was observed in EVs secreted from midlife APCs, which resulted in an increase in the ATM M1 program coupled with WAT inflammaging and insulin resistance. Mechanistically, miR-145 inhibited SELL expression to attenuate NF-κB signaling, through which it suppressed ATM M1 polarization. Therefore, the targeted delivery of miR-145 mimics by liposomes dramatically inhibited the ATM M1 program and alleviated WAT aging and midlife obesity in the context of metabolic stress. These data support a crucial role for APC-derived EVs in modulating the risk of midlife obesity and suggest that miR-145 could be a promising therapeutic target against obesity in the middle stage of life.
Obesity susceptibility refers to the tendency of certain individuals to gain weight more easily in response to external environmental stimuli, such as high-fat diets or stress27. These individuals are often able to maintain a normal body weight under normal conditions. However, upon challenge with external triggers, they are more prone to obesity owing to functional alterations in specific tissues or organs. In the present study, we sought to address whether age as a variable component contributes to obesity susceptibility. To isolate the effects of age as a single factor, we adopted mice or humans at different ages with normal diets (dietary quality index score >85 for human subjects) and normal body weights (body mass index between 18.5 and 23.9 for human subjects), as the study subjects. The most significant metabolic decline was noted in the WAT of middle-aged individuals, which prompted us to select adipose tissue as the target organ. Subsequent studies revealed that APCs in WAT presented the most significant functional changes (Fig. 2c). Indeed, recent studies revealed that APCs originating from 12-month-old mice (midlife) displayed an outrageous decline (~300-fold) in terms of browning capacity compared with their young counterparts (3months old), followed by a greater reduction during subsequent aging8,10. These results suggest that APCs might serve as an outpost for systemic senescence, with the midlife stage serving as a critical turning point for their functional decline.
In general, extracellular vesicles tend to accelerate disease progression once they are released from pathological cells22. Unlike this recognized dogma, we noted that EVs derived from midlife APCs slightly alleviated, rather than exacerbated, insulin resistance in middle-aged mice following HFD challenge (Fig. 3). This discrepancy is likely due to the fact that aging is a normal physiological process instead of a pathological process. Subsequent studies further revealed that the functional reduction in midlife APCs, was caused at least in part, by the reduced secretion of EV-derived miR-145. Moreover, a marked decrease in the number of APCs in WAT was also noted in middle-aged individuals (Supplementary Fig. 2b, c), which synergistically resulted in an overall reduction in EV secretion, ultimately leading to defective suppression of the ATM M1 program. In support of this assumption, the obese phenotype was alleviated once middle-aged mice were supplemented with a high dose of mEVs (100 μg for each mouse) during the course of HFD challenge (Supplementary Fig. 5g–l). Interestingly, although APC-derived EVs tend to accumulate in WAT due to their homing effect28 (Supplementary Fig. 3a, 4a), yEV-treated mice also showed a significant alleviation of insulin resistance in the liver and skeletal muscle (Fig. 3F). Given that no significant accumulation of APC-derived EVs in the liver or skeletal muscle was detected (Supplementary Fig. 3a, 4a), the alleviation of insulin resistance in these organs could be a secondary effect resulting from improved adipose metabolism (Fig. 3g-l). Collectively, these data support the notion that WAT senescence in middle-aged individuals could be a primary driver of midlife obesity.
EVs contain various cargo components, including miRNAs, lipids, and proteins29,30,31. Adipose tissue is a primary source of circulating miRNAs, as Dicer deficiency in adipose tissue results in a two-thirds reduction in circulating miRNAs32. Recent studies have further demonstrated that the amount of WAT-derived miRNAs decreases with age33. In this study, we identified three key miRNAs, miR-145-5p, miR-541-5p, and miR-351-5p, and ultimately selected miR-145-5p, the most significant one, as the target. Notably, the other two miRNAs also exhibited age-related downregulation in APC-derived EVs (Fig. 4b and Supplementary Fig. 6a, b), suggesting that they may also play important roles in aging-related metabolic diseases. Interestingly, a recent report aligns with our study, showing that miR-541-5p is downregulated with age in various other tissues25. This finding that miR-541-5p may be a critical factor involved in multi-organ interactions during the aging process, which warrants further investigation. The role of miR-145a-5p in adipose tissues remains poorly understood. Our subsequent studies identified SELL as the target gene of miR-145a-5p (Fig. 5a–d). SELL is a cell adhesion molecule from the selectin family that is widely expressed on the surface of leukocytes and is involved in leukocyte adhesion34,35. Indeed, SELL potently promoted the macrophage M1 program (Fig. 5g), which is coupled with activated NF-κB signaling (Fig. 5h, i), a well-recognized pathway for the transcription of proinflammatory cytokines essential for the macrophage M1 program36. Rescue experiments further demonstrated that SELL is upstream of NF-κB signaling (Fig. 5j), but the exact mechanisms by which SELL activates NF-κB signaling have yet to be elucidated and will be a major focus of our follow-up studies.
ATMs play a critical role in regulating metabolic processes. Under normal conditions, ATMs clear apoptotic adipocytes and cellular debris while taking up and processing lipids via lysosomal metabolism, lipolysis, and lipid storage37. Recent studies further highlight the role of ATMs in maintaining adipose tissue vascular integrity38. However, under conditions of obesity, these functions are impaired. Additionally, an increased number of M1-polarized ATMs secrete inflammatory cytokines, inhibit adipocyte lipolysis, and exacerbate lipid accumulation39,40. Although animal studies suggest that inhibiting M1 ATMs can alleviate obesity, the efficacy of inflammation-targeted interventions in clinical studies remains controversial. Salsalate, a pro-drug of salicylate that inhibits the NF-κB pathway, could reduce glucose and HbA1c levels in patients with type 2 diabetes41,42,43. Similarly, a 12-week treatment with amlexanox, an inhibitor of NF-κB kinase ε (IKK-ε) and TANK-binding kinase 1 (TBK1), improved insulin sensitivity, reduced HbA1c levels, and alleviated hepatic steatosis in patients with obesity and diabetes44. Conversely, systemic TNFα blockade exhibited no improvement in insulin resistance in humans45; IL-1β inhibition with canakinumab could not reduce the incidence of diabetes in prediabetic patients with insulin resistance46. These discrepancies may stem from the diverse effects of different inflammatory signaling pathways. Interestingly, the aforementioned studies suggest that targeting the NF-κB pathway has shown promising efficacy in the clinical treatment of T2DM. Our results demonstrated that miR-145 can inhibit SELL to block the NF-κB pathway (Fig. 5), suggesting its potential efficacy in clinical applications.
Given that the collection of sufficient yEVs for clinical application is a formidable challenge, we explored the feasibility of delivering miR-145 mimics using cationic liposomes47. Unlike extracellular vesicles, the generation of sufficient liposomes is much easier. Moreover, liposomal properties, such as size, surface charge, and content, can be modified to meet various drug delivery needs48. The cationic nature of liposomes allows for efficient encapsulation of negatively charged nucleic acid drugs, such as miRNAs and siRNAs47,49. Additionally, our previous studies demonstrated that liposomes can be readily taken up by macrophages50,51. These advantages prompted us to adopt liposomes as the delivery vehicle. Similar to EVs, liposomes can also accumulate in ATMs (Fig. 6h and Supplementary Fig. 8d). Although nontargeted liposomes are taken up by macrophages to a lesser extent than miR-145@tar-lip, they still have the potential to attenuate SELL expression in ATMs (Supplementary Fig. 9h, i) and inhibit the M1 program (Fig. 7g–i), thereby moderately alleviating HFD-induced midlife obesity (Fig. 7). However, a limitation was noted for our EV studies. Specifically, ultracentrifugation, a method that yields EVs with relatively low purity compared to density gradient centrifugation, was employed for our EV extraction. The complexity of density gradient centrifugation renders it extremely difficult to harvest sufficient quantities of EVs for the designed experiments. Therefore, we ultimately adopted the ultracentrifugation approach. Although EVs extracted by ultracentrifugation produced consistent results, future studies with EVs obtained by density gradient centrifugation would be necessary to strengthen the findings.
In summary, APC-derived EVs from middle-aged subjects presented a marked reduction in functionality, which was caused at least in part by overall decreased miR-145 secretion. We then demonstrated experimental evidence that EV-miR-145 plays a pivotal role in modulating the risk of midlife obesity by suppressing the SELL-NF-κB axis in ATMs (Fig. 8). These findings not only demonstrate that APC-derived EVs modulate the risk of midlife obesity, but also suggest that miR-145 could be a viable target to combat midlife obesity in clinical settings.

Senescent adipose progenitor cells (APCs) in middle-aged individuals are the cause of midlife obesity. miR-145 in APC-derived extracellular vesicles improves inflammaging and insulin resistance in middle-aged subjects. miR-145 inhibits the M1 polarization of adipose tissue macrophages by suppressing the SELL‒NF‒κB axis. miR-145 is a proof-of-concept effective target for the intervention of midlife obesity (Created in BioRender. Wang, W. (2025) https://BioRender.com/k41c453).
Methods
Statement of ethical regulations
All animal experiments were approved by Tongji Hospital Animal Care and Use Committee (Approval Code: TJH-201903031). Human investigations were approved by the Institutional Review Board (IRB) of Tongji Medical College, Huazhong University of Science and Technology (TJ-IRB20160601, TJ-IRB20160602, TJ-IRB20230926), as well as the Chengdu Third Hospital Ethics Committee [2022] S-62. All participants provided informed consent and agreed to the study protocol.
Mice
The animal experiments were approved by Tongji Hospital Animal Care and Use Committee (Approval Code: TJH-201903031). Male or female C57BL/6 wild-type (WT) mice, aged 6 weeks, were obtained from HFK Bioscience in Beijing, China. Breeding was conducted at the Animal Center of Tongji Hospital under specific pathogen-free (SPF) conditions. Mice were kept at ambient temperature (20–24 °C) in humidity-controlled (45–65%) room under a 12/12 h light-dark cycle. Mice were housed individually in isolated cages with continuous access to sterile acidified water and irradiated food.
The young mice were raised until they reached 3 months of age. The middle-aged cohort was maintained until 12 months of age and subsequently subjected to a 4-week dietary regimen consisting of either a normal chow diet (9.4% kcal fat; HFK Bioscience, Beijing, China, 1032) or a high-fat diet (60% kcal fat; Research Diet in Gardners, USA, D12492). For the functional study of extracellular vesicles (EVs), 12-month-old C57BL/6 male or female mice were randomly divided into four experimental groups: normal chow diet (NCD), high-fat diet (HFD), HFD + mEVs, HFD + yEVs. In the HFD groups, the NCD was replaced with a 60% HFD. During the 4-week HFD exposure, the mice received intravenous administration of EVs derived from either young or middle-aged adipose progenitor cells (APCs) (30 μg each per week). For the miR-145 studies, 12-month-old C57BL/6 male or female mice were switched to a 60% HFD for 4 weeks, during which the mice were intravenously injected with PBS, NC miR@tar-lip, miR-145@lip or miR-145@tar-liposome (1 mg miRNA/kg body weight, every three days). In experiments with middle-aged mice receiving different doses of mEVs (Supplementary Fig. 5g–l), 12-month-old C57BL/6 male mice were switched to a 60% HFD for 4 weeks, during which the mice were administered with different dose of mEVs (30 or 100 μg each/week). To assess the phosphorylation of AKT in metabolic organs, mice were injected with 0.75 U/kg insulin 15 min prior to sacrifice. For depletion of adipose tissue macrophages (ATMs) studies, NCD-fed 12-month-old C57BL/6 mice were switched to a 60% HFD for 4 weeks. During this period, the mice were intraperitoneally injected with clodronate liposomes (110 mg/kg) or control empty liposomes twice weekly. At the end of each experiment, mice were euthanized via CO2-dependent asphyxiation, blood and tissues were harvested for further analysis.
Human samples
Human investigations were approved by the Institutional Review Board (IRB) of Tongji Medical College, Huazhong University of Science and Technology (TJ-IRB20160601, TJ-IRB20160602, TJ-IRB20230926), as well as the Chengdu Third Hospital Ethics Committee [2022] S-62. All participants provided informed consent and agreed to publish information. Adipose tissues, livers and skeletal muscles were obtained from a cohort of patients who underwent surgical procedures for benign diseases. Sex was not considered in study design due to limitation on the number of samples. Grouping criteria were as follows: young group: 18–29 years old; midlife group: 30-49 years old; aged group: 50–90 years old. The BMI of all the subjects was between 18.5 and 23.9. In this study, we employed the Dietary Quality Index (DQI) to evaluate the nutritional status of selected human samples. To be included, samples were required to have a total DQI score exceeding 85, indicating high dietary quality. Exclusion criteria included: metabolic disorders (such as diabetes, fatty liver, thyroid dysfunction); cardiovascular diseases (including heart failure, coronary heart disease, hypertension and atherosclerosis); acute infectious diseases; moderate to severe anemia; significant renal or hepatic impairment; respiratory failure; gastrointestinal ulcers; inflammatory bowel disease; malignant tumors; pregnancy; hormone replacement therapy. For physical measurement and biochemical assessments, comprehensive clinical and serological data were collected from the participants in this study. Height and weight were measured using a calibrated scale. Body mass index (BMI) was calculated by dividing weight in kilograms by the square of height in meters. Waist circumference (WC) was measured at the midpoint of a horizontal line connecting the axillary line at the level of the rib cage to the iliac crest, with the participants in an upright position. Total fat mass (FM) and body fat percentage (fat%) were accurately assessed using a body composition analyzer (InBody770) based on bioelectrical impedance analysis. Blood samples were collected from participants following a 10 h overnight fast. Subsequently, fasting blood glucose (FPG), glycated hemoglobin A1c (HbA1c), total cholesterol (TC), triglycerides (TG), high-density lipoprotein cholesterol (HDL-c), and low-density lipoprotein cholesterol (LDL-c) levels were measured. The clinical characteristics of individuals are provided in Supplementary Table 1‒5.
Single-nucleus RNA sequencing
Single-nucleus RNA sequencing and analysis were conducted by Majorbio. Cell nuclei were extracted from frozen human adipose tissues and subjected to 10x single-nucleus RNA sequencing (snRNA-seq). All procedures were performed on ice until the isolation of cell nuclei, with maintained in dry ice storage. Frozen adipose tissue samples were placed in centrifuge tubes, and 2 mL nuclear isolation buffer was added, containing freshly prepared TST buffer (0.03% Tween 20, 0.01% BSA, 146 mM NaCl, 21 mM MgCl2, 1 mM CaCl2, and 10 mM Tris-HCl pH 7.5). Subsequently, the tissue underwent disintegration into individual nuclei utilizing the gentle MACS Dissociator (Miltenyi Biotec). The nuclear mixture was then passed through a 40 µm filter and washed with 3 mL freshly prepared ST buffer (146 mM NaCl, 21 mM MgCl2, 1 mM CaCl2, 10 mM Tris-HCl pH 7.5, and 0.2 U/µL RNase Inhibitor) to remove residual tissue fragments and other impurities. After washing, the contents were collected into the same tube. Centrifugation at 4 °C for 5 min at 500 g with low brake setting was performed, followed by removal of the supernatant. The nuclear pellet was resuspended in PBS containing 0.02% BSA at a volume of 50–200 µL. Cell nuclei from each sample were stained with NucBlue (ThermoFisher Scientific), and stained nuclei were observed and quantified using a hemocytometer.
10x library preparation and sequencing
Beads with unique molecular identifier (UMI) and cell barcodes were loaded close to saturation, so that each cell was paired with a bead in a Gel Beads-in-emulsion (GEM). After exposure to cell lysis buffer, polyadenylated RNA molecules hybridized to the beads. Beads were retrieved into a single tube for reverse transcription. On cDNA synthesis, each cDNA molecule was tagged on the 3’end (that is, the 5’end of a messenger RNA transcript) with UMI and cell label indicating its cell of origin. Briefly, 10x beads that were then subject to second-strand cDNA synthesis, adaptor ligation, and universal amplification. Sequencing libraries were prepared using randomly interrupted whole-transcriptome amplification products to enrich the 3’ end of the transcripts linked with the cell barcode and UMI. All the remaining procedures including the library construction were performed according to the standard manufacturer’s protocol (Chromium Single Cell 3ʹ v3.1). Sequencing libraries were quantified using a High Sensitivity DNA Chip (Agilent) on a Bioanalyzer 2100 and the Qubit High Sensitivity DNA Assay (Thermo Fisher Scientific). The libraries were sequenced on Xplus (Illumina) using 2×150 chemistry. The sequencing and bioinformatic analysis were performed on platform of Majorbio Co., Ltd (Shanghai, China).
Single-nucleus RNA sequencing data analysis
Reads were processed using the Cell Ranger V7.1.0 pipeline with default and recommended parameters. FASTQs generated from Illumina sequencing output were aligned to the human genome, version GRCh38 (Ensemble 98), using the STAR algorithm52. Next, Gene-Barcode matrices were generated for each individual sample by counting UMIs and filtering non-cell associated barcodes. Finally, we generate a gene-barcode matrix containing the barcoded cells and gene expression counts. This output was then imported into the Seurat (v3.2.0) R toolkit for quality control and downstream analysis of our single cell RNA-seq data53. All functions were run with default parameters, unless specified otherwise. We first filtered the matrices to exclude low-quality cells using a standard panel of three quality criteria: (1) number of detected transcripts (number of unique molecular identifiers); (2) detected genes; and (3) percent of reads mapping to mitochondrial genes (Quartile threshold screening criteria). The expression of mitochondria genes was calculated using Percentage Feature Set function of the Seurat package53. The normalized data (Normalize Data function in Seurat package) was performed for extracting a subset of variable genes. Variable genes were identified while controlling for the strong relationship between variability and average expression. Next, we integrated data from different samples after identifying ‘anchors’ between datasets using Find Integration Anchors and Integrate Data in the Seurat package53,54. Then we performed principal component analysis (PCA) and reduced the data to the top 30 PCA components after scaled the data. We visualized the clusters on a 2D map produced with t-distributed stochastic neighbor embedding (t-SNE)55. To identify cell types and subtypes, cells were clustered using graph-based clustering of the PCA reduced data with the Louvain Method56 after computing a shared nearest neighbor graph53. For sub-clustering, we applied the same procedure of scaled, dimensionality reduction, and clustering to the specific set of data (usually restricted to one type of cell). For each cluster, we used the Wilcoxon Rank-Sum Test to find significant deferentially expressed genes comparing the remaining clusters. SCINA57 and known marker genes were used to identify cell type.
To identify DEGs (differential expression genes) between two different samples or clusters, was performed using the function Find Markers in Seurat, using a Mann-Whitney-Wilcoxon test (Wilcon). Essentially, DEGs with |log2FC | >0.25 and Q value ≤ 0.05 were considered to be significantly different expressed genes. In addition, functional-enrichment analysis GO were performed to identify which DEGs were significantly enriched in GO terms and metabolic pathways at Bonferroni-corrected P-value ≤ 0.05 compared with the whole-transcriptome background. GO functional enrichment analysis were carried out by Goatools (https://github.com/tanghaibao/Goatools).
Cell culture and treatment
Bone marrow-derived macrophages (BMDMs) were isolated using established methodologies with slight refinements. Briefly, bone marrow cells were obtained from femurs of 6-week C57BL/6 mice and resuspended in RPMI-1640 medium (10% FBS + 1% penicillin/streptomycin) supplemented with 30 ng/mL M-CSF (eBioscience, San Diego, CA, USA). The cells were incubated at 37 °C in a humidified atmosphere with 5% CO2. The complete RPMI-1640 medium containing 30 ng/mL M-CSF was replaced on the second day, followed by a replacement with medium containing 20 ng/mL M-CSF on the fifth day. Mature BMDMs were harvested on the seventh day. In the experimental design, mature BMDMs treated with LPS (100 ng/mL) were identified as M1 macrophages, while those treated with PBS were identified as M0 macrophages. The groups without any stimulation in both cases were designated as blank groups. For the experiment involving macrophage stimulation with EVs, 50 ng/mL young EVs (yEVs) or midlife EVs (mEVs) were added to mature BMDMs for 24 h. Following this, the macrophages were exposed to LPS (100 ng/mL) for an additional 24 h to induce M1 macrophage polarization. To activate the NF-κB pathway, BMDMs was treated with CU-T12-9 (10 μM, S2979, Selleck), an NF-κB agonist, for 12 h58. HEK293T cells were obtained from the American Type Culture Collection (ATCC, USA, CRL-1573) and were cultured in high-glucose DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin. The cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2.
The stromal vascular fraction (SVF) isolation
The digestion medium was prepared by combining Type I collagenase (2 mg/mL), 0.5% BSA, and CaCl2 (1.11 mg/mL). The collected adipose tissue was placed into a clean, dry cell culture dish, and digestion medium (10 mL collagenase per 10 g tissue) was added. The tissue was minced into small pieces of approximately 1-2 mm, and the processed tissues along with the digestion medium were transferred into a 50 mL sterile centrifuge tube. The mixture was then digested at 200 rpm for 20–30 min in a 37 °C air shaker until the cells became completely homogenous. The reaction was terminated with 20 mL PBS solution containing 10% BSA and the mixture was filtered through a 70 μm cell filter. Following centrifuging at 400 x g for 5 min at 4 °C, discard the upper layer of floating mature adipocytes and collect the precipitate at the bottom of the tube. For SVF, cell precipitate was treated with 2 mL red blood cell lysate for 3 min, and the reaction was terminated with by adding 6 mL PBS. After centrifugation at 400 x g for 5 min, the cell pellet at the bottom of the centrifuge tube was considered to be the stromal vascular fraction. The SVF extraction method was identical for both human and mouse white adipose tissues, as described above.
Isolation of Human and Mouse Adipose Progenitor Cells (APCs)
Human APCs Isolation
For the isolation of human APCs, every 107 stromal vascular fraction cells were resuspended in 80 μL flow buffer (PBS pH 7.2 + 0.5% BSA + 2 mM EDTA). Subsequently, 10 μL CD45 MicroBeads (Miltenyi Biotec, #130-045-801) and 10 μL CD31 MicroBeads (Miltenyi Biotec, #130-091-935) were added. Mix well and incubate on ice for 30 min in darkness. The reaction was terminated with 2 mL flow buffer, and the negative cells sorted by magnetic beads were centrifuged at 300 x g for 10 min at 4 °C. The supernatant was discarded, and the cells were then resuspended with 90 μL flow buffer. Next, 10 μL CD34 MicroBeads (Miltenyi Biotec, #130-046-702) were added to the human SVF cells and incubated another 30 min on ice in darkness. The reaction was terminated with 2 mL flow buffer, and the positive cells sorted by magnetic beads were centrifuged at 300 x g for 10 min at 4 °C. The supernatant was discarded, and APCs were precipitated. The cells were then cultured and expanded in high-glucose DMEM medium containing 10% FBS (without extracellular vesicles) and 1% penicillin/streptomycin.
Mouse APCs Isolation
For the isolation of mouse APCs, every 107 stromal vascular fraction cells were resuspended in 90 μL flow buffer and 10 μL Non-adipose Tissue Progenitor MicroBeads (Miltenyi Biotec, #130-106-639). The mixture was mixed thoroughly and incubated on ice in the dark for 30 min. The reaction was terminated by adding 2 mL flow buffer, and the negative cells sorted using magnetic beads were centrifuged at 300 x g, 4 °C for 10 min. The supernatant was discarded, and the cells were resuspended in 90 μL flow buffer. The mouse SVF were then treated with 10 μL Adipose Tissue Progenitor MicroBeads (Miltenyi Biotec, #130-106-639) and incubated for another 30 min on ice in the dark. The reaction was terminated with 2 mL flow buffer, and the positive cells sorted by magnetic beads were centrifuged at 300 x g, 4 °C for 10 min. The supernatant was discarded, and the APCs were precipitated. Cells were cultured and expanded in high-glucose DMEM medium containing 10% FBS (without extracellular vesicles) and 1% penicillin/streptomycin.
Adipose tissue-derived macrophages (ATMs) isolation
For the isolation of adipose tissue macrophages (ATMs), the SVF cells of mice were resuspended in 90 μL flow buffer (PBS pH 7.2, 0.5% BSA, and 2 mM EDTA) and mixed thoroughly. Subsequently, 10 μL CD11b MicroBeads (Miltenyi Biotec, #130-049-601) were added, and the mixture was incubated on ice in the dark for 30 min. The reaction was then terminated by adding 2 mL flow buffer, and positively labeled cells were isolated using magnetic beads. The cells were centrifuged at 300 x g, 4 °C for 10 min, after which the supernatant was discarded, and ATMs were precipitated. The isolated cells were cultured in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin.
Co-incubation of APC supernatant with macrophages
In the co-incubation experiment, young APCs cell culture supernatant, with or without EVs, was incubated with macrophages for 24 h. Following this incubation, the supernatant was discarded, and the macrophages were subsequently treated with LPS at a concentration of 100 ng/mL for an additional 24 h to induce M1 macrophage polarization.
miR-145 overexpression or inhibition
The mechanism by which miRNA inhibitors function is to bind competitively to mature miRNAs, thereby impeding the binding of target genes to the corresponding miRNAs. Hence, higher concentrations of miRNA inhibitors are required for effectively suppressing miRNA activity. Therefore, in vitro studies, miR-145 mimics (50 nM) or miR-145 inhibitor (100 nM) were transfected into macrophages (5 × 106 cells per well) using Lipofectamine 3000 reagent (Thermo Fisher, #L3000015). After 6 h, the supernatant was discarded and replaced with RPMI-1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin. Following a 24 h post-transfection period, the macrophages were utilized for subsequent experiments. The miR-145 scramble, mimics and inhibitor were obtained from Ribobio (Guangzhou, China), and specific details regarding the miR mimics and inhibitors used are provided in Supplementary Table 7.
Transfection of siRNA
In vitro, 100 nM siRNAs were transfected into macrophages (5 × 105 cells per well) using Lipofectamine 3000 reagent (Thermo Fisher, #L3000015). After 6 h, the supernatant was discarded and replaced with RPMI-1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin. Macrophages were harvested for further experiments 48 h post-transfection. The siRNAs used in this study were obtained from Ribobio (Guangzhou, China), with specific details provided in Supplementary Table 7.
Adenoviral transduction
No-load adenovirus (rAd-ZsGreen), Sell adenoviruses (rAd-ZsGreen-mSell-3Flag) were commercially produced by DesignGene Biotechnology (Shanghai, China). The macrophages were isolated from mice and cultured in a 12-well plate at a density of 106 cells per well in 1 mL RPMI-1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin. Control cells were transduced with no-load adenovirus, while experimental cells were treated with 107 Sell adenoviral particles per well. After 12 h incubation, the virus-containing medium was replaced with complete RPMI-1640 medium. After the successful transduction of the virus, macrophages were utilized for further experiment.
miRNA-seq experimental method
For miRNA sequencing, yEVs and mEVs of mice were collected. Each group had 3 replicates. Total RNA was isolated and purified following the manufacturer’s procedure. The RNA amount and purity of each sample was evaluated using NanoDrop ND-1000 (NanoDrop, Wilmington, DE, USA). The RNA fragment integrity was evaluated through Bioanalyzer 2100 (Agilent, CA, USA). The obtained total RNA were denaturated at 70 °C for 2 min with DNA 3’ Adapters, and then mixed with T4 RNA Ligase 2, truncated K227Q (NEB, M0351L, USA) at room temperature. The 3’ adapter ligation reaction was performed at 16 °C more than 8 h. The rest of 3’ Adapters were removed by RTP at 37 °C for 30 min. The 5’ adapters and the products from last step were ligation with T4 RNA Ligase1 (NEB, M0204L, USA) at 37 °C for 60 min following with reverse-transcription reaction carried by SuperScript III Reverse Transcriptase (Thermo, 18064014, USA) at 50 °C for 60 min, and at 80 °C for 10 min. The cDNA products were amplified using Phusion® High-Fidelity DNA Polymerase (NEB, M0530L, USA). Specifically, the denaturation temperature was 98 °C for 30 s with an annealing temperature of 60 °C for 30 s, and the extension was carried out for 15 s at 72 °C for 10–16 cycles. The final 5 min extension was then done at 72 °C. The PCR products were purified and enriched through PAGE electrophoresis. The sequencing strategy was paired-end 150 bp for Illumina Hiseq 2500 following the vendor’s recommended protocol.
miRNA bioinformatics analysis methods
Raw reads were subjected to an in-house program, ACGT101-miR (v4.2) to remove adapter dimers, junk, low complexity, common RNA families (rRNA, tRNA, snRNA, snoRNA) and repeats. Subsequently, unique sequences with length in 18 ~ 26 nucleotide were mapped to specific species precursors in miRBase 22.1 by BLAST search to identify known miRNAs and novel 3p- and 5p- derived miRNAs. Length variation at both 3’ and 5’ ends and one mismatch inside of the sequence were allowed in the alignment. The unique sequences mapping to specific species mature miRNAs in hairpin arms were identified as known miRNAs. The unique sequences mapping to the other arm of known specific species precursor hairpin opposite to the annotated mature miRNA-containing arm were considered to be novel 5p- or 3p- derived miRNA candidates. The remaining sequences were mapped to other selected species precursors (with the exclusion of specific species) in miRBase 22.1 by BLAST search, and the mapped pre-miRNAs were further BLASTed against the specific species genomes to determine their genomic locations. The above two we defined as known miRNAs.
RNA-seq experimental method
For the PBS-treated and miR-145 mimics-treated groups, mature bone marrow-derived macrophages (BMDMs) were pre-treated with PBS or miR-145 mimics (50 nM) for 24 h, followed by exposure to LPS (100 ng/mL) for an additional 24 h. Each group consisted of three replicates. For the Scramble and Si-Sell groups, mature BMDMs were pre-treated with PBS or Sell siRNA (100 nM) for 24 h before being exposed to LPS (100 ng/mL) for another 24 h, with each group also having three replicates. Total RNA was extracted using Trizol Reagent (Takara, Japan, #9109). The quality of the RNA was determined as follows: RNA integrity number (RIN) was analyzed with the Agilent 2100/4200 system. Three micrograms of RNA were used as the input for subsequent sample preparation. Sequencing libraries were constructed following these steps: initially, mRNA was isolated from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was performed with divalent cations at elevated temperatures in fragmentation buffer. The first strand of cDNA was synthesized using random hexamers, followed by the synthesis of the second strand with the addition of buffer, dNTPs, RNase H, and DNA polymerase I. The double-stranded cDNA was then repaired, and an adenine was added to the 3’ end. Purification and fragment selection were conducted using Hieff NGS® DNA Selection Beads. Subsequently, the products underwent amplification and enrichment via PCR, with quantification performed using Qubit. The target region library was denatured, cycled, and digested to produce single-stranded circular DNA, which was amplified through rolling circle amplification (RCA), resulting in DNA nano balls (DNB). Quantification was again carried out with Qubit following library construction. Following library preparation, the sequencing library was analyzed using the DNBSEQ-T7 platform with the PE150 model.
RNA-seq bioinformatics analysis methods
Samples were sequenced on the platform to obtain image files, which were processed by the sequencing platform’s software to generate original data in FASTQ format (Raw Data). The sequencing data contained several adapters and low-quality reads; therefore, fastp (v0.21.0) software was utilized to filter the data to get high-quality sequences (Clean Data) for subsequent analysis. The reference genome and gene annotation files were obtained from the genome database. Filtered reads were aligned to the reference genome using HISAT2 v2.1.0. StringTie (v2.1.5) was employed to quantify the read counts for each gene, serving as the initial expression measure, followed by normalization using FPKM. Differential gene expression was analyzed using DESeq2 (v1.30.1) under the following criteria: |log2FoldChange | > 1 and adjusted p-value (padj) ≤ 0.05. Additionally, bi-directional clustering analysis of all differentially expressed genes across samples was conducted using R language Pheatmap (1.0.8) software package. A heatmap was generated based on the expression levels of the same gene across different samples and the expression patterns of various genes within the same sample. The Euclidean method was used to calculate distances, and the Complete Linkage method was employed for clustering. Furthermore, all genes were mapped to terms in the Gene Ontology database, and the number of differentially enriched genes in each term was calculated. GO enrichment analysis of the differentially expressed genes was conducted using Cluster Profiler, employing the hypergeometric distribution method to calculate p-values (with significant enrichment defined as padj ≤ 0.05). This analysis aimed to identify GO terms associated with significantly enriched differential genes, thereby elucidating the primary biological functions of these genes. Additionally, KEGG pathway enrichment analysis for the differential genes was performed using ClusterProfiler (v3.18.1), focusing on pathways that exhibited significant enrichment (padj ≤ 0.05).
Flow cytometry analysis
Flow cytometry was conducted as previously reported in ref. 59. To determine the proportion of different cell types in the WATs, SVF cells were resuspended in flow buffer (PBS pH7.2 + 0.5% BSA + 2 mM EDTA) and counted, adjusting the cell density to107 cells /mL. A 100 μL aliquot of the cell suspension (106 cells) was incubated with labeled monoclonal antibodies. An appropriate volume of each sample was mixed and allocated into blank and standard tubes. The following antibodies were obtained from Biolegend (San Diego, CA, USA): PE anti-mouse F4/80 (123110, 1:400), BV421 anti-mouse F4/80 (123131, 1:400), FITC anti-mouse CD11b (101206, 1:400), APC anti-mouse CD11c (117310, 1:400), PE/Cy7 anti-mouse CD206 (141720, 1:400), APCs anti-mouse CD31 (102410, 1:400), APCs anti-mouse CD45 (103112, 1:400), FITC anti-mouse Ly-6A/E(Sca-1) (108106, 1:400), APCs anti-mouse TER-119 (116212, 1:400), PE anti-mouse CD3 (100206, 1:400), APC anti-mouse NK1.1 (156506, 1:400), BV421 anti-mouse I-A/I-E (107631, 1:400), APCs anti-human CD31 (303116, 1:400), PE anti-human CD34 (343506, 1:400), APCs anti-human CD45 (368512, 1:400). Cells were incubated in flow buffer with specific antibodies at 4 °C for 30 min in the dark. After washing, the samples were analyzed using a Miltenyi Biotec flow cytometry. SCA-1+/CD31-/CD45-/Ter119- cells were identified as mouse APCs. CD34+/CD31-/CD45- cells were identified as human APCs. In mice, F4/80+/CD11b+ cells were identified as macrophages. In this subpopulation of cells, with M1 (CD11c+/CD206-) and M2 (CD11c-/CD206+) macrophages distinguished based on CD11c and CD206 expression. The complete gating strategy for dendritic cells (DCs), T cells, NK cells, macrophages, and APCs is provided in the supplementary information. MHCII+/CD11c+ cells were identified as mouse dendritic cells (DCs), CD3+ cells as mouse T cells, and CD3-/NK1.1+ cells as mouse natural killer (NK) cells. For flow analysis in BMDMs, cells were digested with 0.25% trypsin, counted, and 5 × 105 cells were transferred to the flow tubes and washed twice with PBS. Cells were then incubated with APC anti-mouse CD11c antibody (117310). All the data were analyzed with FlowJo software.
Extraction and characterization of EVs
Human or mouse APCs were cultured in medium containing FBS without EVs. The cells were passaged every three days, and the culture medium was collected for subsequent EV extraction, up to the sixth passage. For adipose tissue, EVs isolation was performed as previously described60. Briefly, adipose tissues from mice were cut into small pieces and then cultured in serum-free DMEM for 24 h. EVs were purified from the cultured medium. After centrifugation at 500 x g, 4 °C for 10 min, the supernatant was retained. This supernatant underwent sequential centrifugation in a pre-cooled centrifuge at 4 °C, first at 2000 x g for 20 min and then at 10,000 x g for 30 min with the supernatant being collected after each step. Subsequently, the medium was centrifuged at 120,000 x g for 70 min, and the supernatant was discarded, allowing the EVs to precipitate at the bottom of the tube. After resuspended with PBS, the precipitate was centrifuged at 120000 x g, 4 °C for 70 min, and the precipitate at the bottom of the tube was collected, which was the EVs. The particle size of the EVs were analyzed using a laser particle size analyzer, and their morphology was examined and recorded via transmission electron microscope. Total protein was extracted from the EVs, and the presence of EV markers TSG101, CD63, as well as the negative control marker Calnexin, was assessed by Western blot analysis.
Labeling EVs with fluorescent dye
Fluorescent dye (DiI, Beyotime, C1036; DiO, Beyotime, C1038; DiR, MedChemExpress, 100068-60-8) was applied to label the EVs. Two microliters fluorescent dye (100 μg/ml in ethanol) were added into EVs (100 μL, 1 μg/μL). After incubating for 1 h, ethanol and unincorporated fluorescent dye were removed by ultrafiltration centrifugation (3000 rpm, 10 min). Mice were intravenously injected with 100 μL fluorescent dye-labled EVs (30 μg for each mouse). Different organs were collected and rinsed by PBS. After weighing, the samples were homogenized in 200 μL PBS and the fluorescent dye was extracted by chloroform: methanol (3:1). The samples were then centrifuged (4000 rpm, 10 min) and fluorescence intensity of the supernatant (100 μL) was measured by spectrophotometric analysis. All data were normalized to the percentage of injected dose per gram of the tissue (% ID/g). Additionally, the uptake of EVs by various metabolic organs (white adipose tissue, brown adipose tissue, liver and skeletal muscle) and different cell types (macrophages, T cells, dendritic cells, NK cells, and APCs) was assessed by immunofluorescence or flow cytometry.
Co-incubation of PKH26-labeled EVs and cells
To investigate the phagocytosis of APCs-derived EVs in various cell types, APCs-derived yEVs or mEVs were labeled with PKH26 staining solution (MedChemExpress, 154214-55-8). After incubation at room temperature for 1 h, unincorporated PKH26 was removed by ultrafiltration centrifugation (3000 rpm, 10 min). The cells were plated in a 24-well plate at a density of 5 × 104 cells per well and cultured for 24 h. The protein concentration of labeled EVs was measured, and the final protein concentration of 50 ng/mL per well was added into the cells. PKH26-labeled EVs were incubated with cells for an additional 24 h, after which the uptake of EVs was assessed using immunofluorescence or flow cytometry.
Histological and morphological analysis
Hematoxylin and eosin (H&E) staining of epididymal adipose tissue was performed according to the previously described methods6. Then, tissue samples or cell plates were observed and photographed using a light microscope (OLYMPUS Upright microscope BX53, Olympus Corporation). For the analysis of adipocyte hypertrophy, Image J software was used to quantify the adipocyte area. The specific procedure involved utilizing the polygon tool to outline individual adipocytes within the visual field and calculate their areas. This process was repeated for each adipocyte in a consistent manner. Each adipocyte, delineated by H&E staining, was circled, and its area was quantified. After calculating the areas individually, the average area of the circled cells on each section was determined.
Immunofluorescence
For immunofluorescence staining of tissues, paraffin sections of tissues were baked in an incubator at 55 °C for 30 min. Dewaxing was performed by sequentially immersing the sections in 80% ethanol for 15 min to remove paraffin and hydrate the tissue. Antigen retrieval was achieved by completely submerging the sections in antigen retrieval solution and placing them in a pressure cooker. Once the pressure limiting valve activated, the samples were heated for 2 min. After cooling the retrieval solution to room temperature, the sections were washed three times with PBS. Next, the tissue sections were permeabilized in 2.5% Triton X-100 (Sigma-Aldrich, #9036-19-5) for 5 min and subsequently blocked with 5% donkey serum (Sigma-Aldrich, #D9663) for 1 h. The sections were then incubated overnight with the corresponding primary antibodies, which included Perplin2 (Abclonal, #A6276, 1:100), F4/80 (Santa Cruz Biotechnology, #SC-377009, 1:100), Albumin (Abclonal, #A1363, 1:100), Laminin Receptor (Abclonal, #A5968, 1:100). After rewarming at room temperature for 20 min, the slices were washed three times with PBS. Secondary antibodies were then applied and incubated for 1 h at room temperature. Secondary antibodies used are listed as follows: Anti-mouse Alexa Fluor 488 (Jackson ImmunoResearch, 115-545-003, 1:200),
Anti-rabbit Alexa Fluor 594 (Jackson ImmunoResearch, 111-585-003, 1:200), Anti-rabbit Alexa Fluor 488 (Jackson ImmunoResearch, 111-545-003, 1:200).
The cell nuclei were stained with DAPI (Servicebio, G1012) for 10 min, followed by three washes with PBS. After adding the anti-fade reagent, the images were observed under a confocal microscope.
β-Gal staining
Follow the steps described in the Cell senescence β-Galactosidase Staining Kit (Beyotime, #C0602), cells in 12-well plate or extracted adipose tissues were washed once with PBS, then fixed with 500 μL β-Galactosidase staining fixing solution at room temperature for 15 min. After removing the cell fixative, cells or adipose tissues were washed three times with PBS. After discarding the PBS, 1 ml staining solution was added into each well. The 12-well plate was sealed with plastic wrap and incubated at 37 °C in the absence of carbon dioxide overnight. After incubation, the staining solution was removed, and 1 mL PBS was added. The cells were then observed under a microscope. For adipose tissues, β-Gal staining was photographed. For the β-Gal staining of APCs from young, middle-aged and aged mice and humans, since APCs isolated from mice and humans were expanded for 6 generations to collect EVs, the 6th-generation APCs were used for the β-Gal staining. The mice used in Supplementary Fig. 2j were 3 months of age.
RT‒qPCR
RNA was extracted from tissues or cells using Trizol reagent (Takara, Japan, #9109). A total of 1 µg RNA was then subjected to cDNA synthesis using a cDNA Synthesis Kit (Vazyme, China, #R223-01) for mRNA. The primers for qPCR were sourced from Tsingke Biotechnology Co.,Ltd. (Beijing, China). Amplification reactions were performed using the Real-Time PCR System (ABI 7500, Applied Biosystems, CA, USA) using SYBR qPCR Mix (Yeasen, China, #11203). The PCR cycling conditions included an initial denaturation at 95 °C for 1 minute, followed by 40 cycles of denaturation at 95 °C for 15 s and annealing/extension at 60 °C for 1 min. Relative expression levels of each target gene were evaluated using the 2−ΔΔCt approach. The primer sequences were listed in Supplementary Table 6.
For miRNA analysis, 1 µg RNA was subjected to cDNA synthesis using the Bulge-Loop miRNA qRT-PCR Starter Kit (Ribobio, China, C10211-1). The quantification of miRNA levels was then performed, with U6 as the internal control for calculating relative fold-change. Specific primers of miRNA and U6 were purchased from RiboBio (RiboBio, China). The PCR cycling conditions included an initial denaturation at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 2 s, annealing at 60 °C for 2 s and extension at 70 °C for 10 s. Relative expression levels of each target gene were evaluated using the 2−ΔΔCt approach.
Western blotting analysis
Proteins were extracted from cell samples or tissues using RIPA lysis buffer (Beyotime, #P0013B) supplemented with a protease inhibitor cocktail (Sigma-Aldrich, #P8340) and phosphatase inhibitors (Servicebio, #G2007). Protein concentrations were then measured using BCA protein assay kits (Servicebio, #G2026). The proteins were then separated through SDS-PAGE and transferred to a PVDF membrane (Bio-Rad). After blocking with 5% BSA, the PVDF membrane was incubated with primary antibodies for 16 h at 4 °C, followed by incubation with the appropriate secondary antibodies for 1 h at room temperature. The antibodies used are listed as follows: phospho-AKTSer473 (CST, 4060 s, 1:1000), AKT (CST, 4685 s, 1:1000), phospho-HSLSer660 (Abclonal, AP0853, 1:1000), HSL (Abclonal, A15686, 1:1000), phospho-ACCser79 (CST, 3661 s, 1:1000), ACC (CST, 3676 s, 1:1000), PGC1α (Abclonal, A12348, 1:1000), Lamin B1 (Proteintech,12987-1-AP, 1:1000), β-Actin (Abclonal, AC026, 1:1000), iNOS (CST, 13120S, 1:1000), CD62L/L-Selectin Rabbit pAb (Abclonal, A1622, 1:1000), Flag (Proteintech, 20543-1-AP, 1:1000), TSG101 (Abcam, ab125011, 1:1000), CD63 (Abcam, ab134045, 1:1000), CD63 (Abcam, ab217345, 1:1000), Calnexin (Abcam, ab22595, 1:1000), p16 (CST, 18769 s, 1:1000), p21(Abcam, ab109520, 1:1000), p19ARF(Abcam, ab80, 1:1000), P53 (Proteintech, 60283-2-Ig, 1:1000), Ndufs1 (Abcam, ab169540, 1:1000), Sdhb (Abcam,ab14714, 1:1000), PEPCK (Santa Cruz, SC-32879, 1:1000), G6Pase (Abclonal, A21168, 1:1000), phospho-GSK3βSer9 (CST, 9323 T, 1:1000), GSK3β (CST, 12456 s, 1:1000), Phospho-NF-kB p65 (Ser536) (CST, 3033 s, 1:1000), NF-kB p65 (CST, 8242 s, 1:1000), Phospho-IκBα (CST, 2859, 1:1000), IKBα (CST, 9242, 1:1000), HRP-conjugated Goat anti-Rabbit lgG (H + L) (Abclonal, AS014, 1:10000), HRP-conjugated Goat anti-Mouse lgG (H + L) (Abclonal, ASO03, 1:10000). Uncropped and unprocessed scans of the most important blots were provided in the Source Data file.
Metabolic cage analyses
Mice were individually acclimated to metabolic cages attached to a Comprehensive Laboratory Animal Monitoring System (Columbus Instruments, Columbus, OH). Following a 48-hour acclimatization period, measurements of oxygen consumption (VO2), food intake and heat production were conducted using built-in detection system. The metabolic cage experiments were carried out at a controlled temperature of 22 ± 1 °C and 50 ± 5% humidity under a 12-hour light-dark cycle. Adequate food and water were provided at the outset of experiments, after which the chambers were sealed and no further interventions were made throughout the duration of the study.
IGTT, ITT
Following a 16-hour overnight fasting period, mice received intraperitoneal injections of glucose (1.5 g/kg). Blood glucose levels were subsequently measured at specified intervals. After a 6-hour fasting period, insulin (0.75 U/kg) was intraperitoneally administrated, and insulin resistance was assessed through continuous monitoring of blood glucose levels at specified intervals.
ELISA Test
The serum samples were diluted at a ratio of 1:2, and the diluted serum was used for ELISA detection. The epididymal white adipose tissue was dissected, and approximately 50 mg tissue was homogenized in 500 μL phosphate-buffered saline (PBS). The homogenate was then subjected to centrifugation at 4 °C and 12000 g for 20 min. The levels of pro-inflammatory factors, including TNF-α (Biolegend, #430904), IL-6 (Biolegend, #431304), and IL-1β (Biolegend, #432604), in the serum or adipose tissues were quantified using ELISA.
Cationic liposomes construction
MiR-145 mimics were encapsulated in cationic liposomes composed of lipids such as C12-200 by emulsification. Polypeptides targeting white adipose tissue (CKGGRAKDC) were synthesized according to established protocols26. Lipids, cholesterol, DSPC, mPEG-DMG and mal PEG-DMG were dissolved in ethanol in a molar ratio of 50:38.5:10:1:0.5, while miR-145 mimics were dissolved in a 10 mM citric acid buffer (pH=3). The lipid components and dissolved miR-145 mimics were rapidly mixed by vortexing. Uncaptured miR-145 mimics were removed by ultracentrifugation. The polypeptide was subsequently coupled to the liposome surface through a maleimide-thiol reaction. Finally, miR-145 mimics-liposomes are diluted with PBS. The particle size, zeta-potential, polydispersity index, and stability of the liposomes were evaluated using a laser particle size analyzer. The morphology of liposomes was observed and recorded by transmission electron microscope. The drug loading of miR-145 mimics in liposomes was determined by RiboGreen assay. For liposome labeling, two microliters DiI fluorescent dye (100 μg/mL in ethanol) were added into 1 mg diluted liposomes. After 1 h of incubation, ethanol and unincorporated fluorescent dye was removed by ultrafiltration centrifugation (3000 rpm, 10 min).
In vivo biodistribution of the liposomes
DiR was added to the lipid mixture. DiR-loaded liposomes were prepared using the aforementioned method. Following intravenous injection of the liposomes (1 mg/kg body weight), mice (n = 3) were imaged at various time points (0 h, 0.5 h, 2 h, 8 h, 24 h) by an in vivo imaging system (IVIS Lumina XR, Caliper, USA) with excitation at 748 nm and emission at 780 nm. After 24 h, the mice were sacrificed, and the WATs were harvested for ex vivo fluorescence imaging. For pharmacokinetic study, mice (n = 4) were intravenously injected with 100 μL liposomes. Serial blood samples ( ~ 30 μL) were obtained at different time points post injection (0.5 h, 1.5 h, 3 h, 6 h, 24 h) and serum were collected. DiR was extracted from the serum using a chloroform (3:1) solution, followed by centrifugation at 4000 rpm for 10 min. The fluorescence intensity of the supernatant (100 μL) was measured by spectrophotometric analysis. The blood clearance data were expressed as %ID in blood.
Luciferase reporter assays
HEK293T cells were plated into 24-well plates, and the corresponding plasmids (0.1 μg 3’UTR Luciferase plasmid, 0.4 μg miRNA plasmid, 0.02 μg Renilla Luc plasmid) were transfected when cells reached approximately 70% confluency, and the blank control group (untransfected cells) was set up. After co-transfection for 48 h, cells were collected. Cell lysate was placed into the microplate, then firefly luciferase reaction solution and sea kidney luciferase reaction solution were added in sequence, and the fluorescence intensity was detected by microplate reader. The expression multiples were analyzed and calculated.
Statistics and reproducibility
No statistical method was used to predetermine sample size. The sample size was determined based on previous studies5,6. All animals used in this study were randomly allocated to experimental groups without pre-selection. Statistical analyses were performed by the GraphPad Prism 8.0. The data were presented as mean ± SEM. Two-sided Student’s t-test, one-way ANOVA, two-sided ANCOVA, two-way ANOVA followed with Bonferroni’s multiple comparisons test and two-sided Spearman’s correlation were used for statistical analysis. In volcano plot analysis, two-tailed Student’s t-test was used to screen differentially expressed genes, and the false discovery rate (FDR) was corrected using the Benjamini-Hochberg method. In KEGG and GO analyses, hypergeometric test combined with Fisher’s exact test was applied. In all cases, p values < 0.05 were considered as statistical significance. The specific p values and n values are given in the corresponding graphs or legends. All representative experiments are repeated at least three times independently with similar results. The images were created by using PowerPoint 2016, Adobe Illustrator software and Biorender.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the article and/or its supplementary materials. The raw numbers for charts and graphs are available in the Source Data file whenever possible. The RNA-seq data used in Fig. 1f are public datasets available under accession number GSE132040. The snRNA-seq data of human adipose samples PRJNA1135709 (number 5, 6, 7, 8) (https://ncbi.nlm.nih.gov/bioproject/1135709), miRNA-seq data PRJNA1206780, RNA-seq data of young and middle-aged APCs from mice PRJNA1214726, RNA-seq data of young and middle-aged APCs from humans PRJNA1215282, RNA-seq data for PBS-treated and miR-145 mimics-treated M1 macrophages PRJNA1207006, RNA-seq data for Scramble and Si-Sell-treated M1 macrophages PRJNA1209438 have been deposited in the NCBI public repository Sequence Read Archive. Source data are provided with this paper.
References
Wang, L. et al. Body-mass index and obesity in urban and rural China: findings from consecutive nationally representative surveys during 2004-18. Lancet (Lond., Engl.) 398, 53–63 (2021).
Hales, C. M., Fryar, C. D., Carroll, M. D., Freedman, D. S. & Ogden, C. L. Trends in Obesity and Severe Obesity Prevalence in US Youth and Adults by Sex and Age, 2007-2008 to 2015-2016. Jama 319, 1723–1725 (2018).
Chen, C., Ye, Y., Zhang, Y., Pan, X. F. & Pan, A. Weight change across adulthood in relation to all cause and cause specific mortality: prospective cohort study. BMJ (Clin. Res. ed.) 367, l5584 (2019).
Sakers, A., De Siqueira, M. K., Seale, P. & Villanueva, C. J. Adipose-tissue plasticity in health and disease. Cell 185, 419–446 (2022).
Huang, T. et al. Adipocyte-derived kynurenine promotes obesity and insulin resistance by activating the AhR/STAT3/IL-6 signaling. Nat. Commun. 13, 3489 (2022).
Xie, H. et al. SUMOylation of ERp44 enhances Ero1α ER retention contributing to the pathogenesis of obesity and insulin resistance. Metab.: Clin. Exp. 139, 155351 (2023).
Schaum, N. et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 583, 596–602 (2020).
Berry, D. C. et al. Cellular Aging Contributes to Failure of Cold-Induced Beige Adipocyte Formation in Old Mice and Humans. Cell Metab. 25, 166–181 (2017).
Kirstein, A. S. et al. PTEN regulates adipose progenitor cell growth, differentiation, and replicative aging. J. Biol. Chem. 297, 100968 (2021).
Rogers, N. H., Landa, A., Park, S. & Smith, R. G. Aging leads to a programmed loss of brown adipocytes in murine subcutaneous white adipose tissue. Aging Cell 11, 1074–1083 (2012).
Lee, M. J. et al. Proteomic analysis of tumor necrosis factor-alpha-induced secretome of human adipose tissue-derived mesenchymal stem cells. J. Proteome Res. 9, 1754–1762 (2010).
Kumar, R., Sharma, A., Padwad, Y. & Sharma, R. Preadipocyte secretory factors differentially modulate murine macrophage functions during aging which are reversed by the application of phytochemical EGCG. Biogerontology 21, 325–343 (2020).
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C. & Santoro, A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14, 576–590 (2018).
Thevaranjan, N. et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe 21, 455–466.e454 (2017).
Garg, S. K., Delaney, C., Shi, H. & Yung, R. Changes in adipose tissue macrophages and T cells during aging. Crit. Rev. Immunol. 34, 1–14 (2014).
Sawaki, D. et al. Osteopontin promotes age-related adipose tissue remodeling through senescence-associated macrophage dysfunction. JCI Insight. 8, e145811 (2023).
Zhang, Y. X. et al. Adipose tissue aging is regulated by an altered immune system. Front. Immunol. 14, 1125395 (2023).
Lumeng, C. N. et al. Aging is associated with an increase in T cells and inflammatory macrophages in visceral adipose tissue. J. Immunol. 187, 6208–6216 (2011).
Palmer, A. K. et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18, e12950 (2019).
Valadi, H. et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 9, 654–659 (2007).
Su, T. et al. Bone Marrow Mesenchymal Stem Cells-Derived Exosomal MiR-29b-3p Regulates Aging-Associated Insulin Resistance. ACS Nano 13, 2450–2462 (2019).
Ying, W. et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 171, 372–384.e312 (2017).
Zhao, H. et al. Exosomes From Adipose-Derived Stem Cells Attenuate Adipose Inflammation and Obesity Through Polarizing M2 Macrophages and Beiging in White Adipose Tissue. Diabetes 67, 235–247 (2018).
Zhang, Y. et al. Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature 548, 52–57 (2017).
Wagner, V. et al. Characterizing expression changes in noncoding RNAs during aging and heterochronic parabiosis across mouse tissues. Nat. Biotechnol. 42, 109–118 (2024).
Kolonin, M. G., Saha, P. K., Chan, L., Pasqualini, R. & Arap, W. Reversal of obesity by targeted ablation of adipose tissue. Nat. Med. 10, 625–632 (2004).
Kim, M. S. et al. Association of genetic risk, lifestyle, and their interaction with obesity and obesity-related morbidities. Cell Metab. 36, 1494–1503.e1493 (2024).
Yong, T. et al. Tumor exosome-based nanoparticles are efficient drug carriers for chemotherapy. Nat. Commun. 10, 3838 (2019).
Yong, T., Wei, Z., Gan, L. & Yang, X. Extracellular-Vesicle-Based Drug Delivery Systems for Enhanced Antitumor Therapies through Modulating the Cancer-Immunity Cycle. Adv. Mater. 34, e2201054 (2022).
van Niel, G., D’Angelo, G. & Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19, 213–228 (2018).
Mathieu, M., Martin-Jaular, L., Lavieu, G. & Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 21, 9–17 (2019).
Thomou, T. et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542, 450–455 (2017).
Mori, M. A. et al. Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab. 16, 336–347 (2012).
Kerdiles, Y. M. et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat. Immunol. 10, 176–184 (2009).
Ivetic, A., Hoskins Green, H. L. & Hart, S. J. L-selectin: A Major Regulator of Leukocyte Adhesion, Migration and Signaling. Front. Immunol. 10, 1068 (2019).
Liu, T., Zhang, L., Joo, D. & Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2, 17023 (2017).
Lindhorst, A. et al. Adipocyte death triggers a pro-inflammatory response and induces metabolic activation of resident macrophages. Cell Death Dis. 12, 579 (2021).
Chen, Q. et al. Resident macrophages restrain pathological adipose tissue remodeling and protect vascular integrity in obese mice. EMBO Rep. 22, e52835 (2021).
Jaitin, D. A. et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 178, 686–698.e614 (2019).
Lumeng, C. N., Bodzin, J. L. & Saltiel, A. R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 117, 175–184 (2007).
Goldfine, A. B. et al. A randomised trial of salsalate for insulin resistance and cardiovascular risk factors in persons with abnormal glucose tolerance. Diabetologia 56, 714–723 (2013).
Goldfine, A. B. et al. Salicylate (salsalate) in patients with type 2 diabetes: a randomized trial. Ann. Intern. Med. 159, 1–12 (2013).
Goldfine, A. B. & Shoelson, S. E. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J. Clin. Investig. 127, 83–93 (2017).
Oral, E. A. et al. Inhibition of IKKɛ and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab. 26, 157–170.e157 (2017).
Ferraz-Amaro, I. et al. Systemic blockade of TNF-α does not improve insulin resistance in humans. Horm. Metab. Res. = Horm.- und Stoffwechselforschung = Hormones et. Metab. 43, 801–808 (2011).
Dhorepatil, A., Ball, S., Ghosh, R. K., Kondapaneni, M. & Lavie, C. J. Canakinumab: Promises and Future in Cardiometabolic Diseases and Malignancy. Am. J. Med. 132, 312–324 (2019).
Huang, Z. et al. Nucleus-specific RNAi nanoplatform for targeted regulation of nuclear lncRNA function and effective cancer therapy. Exploration (Beijing, China) 2, 20220013 (2022).
Hald Albertsen, C. et al. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 188, 114416 (2022).
Mashel, T. V. et al. Overcoming the delivery problem for therapeutic genome editing: Current status and perspective of non-viral methods. Biomaterials 258, 120282 (2020).
Pan, T. et al. Suppressing Sart1 to modulate macrophage polarization by siRNA-loaded liposomes: a promising therapeutic strategy for pulmonary fibrosis. Theranostics 11, 1192–1206 (2021).
Mou, Y. et al. Macrophage-targeted delivery of siRNA to silence Mecp2 gene expression attenuates pulmonary fibrosis. Bioeng. Transl. Med. 7, e10280 (2022).
Le, D. T. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Stuart, T. et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821 (2019).
Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Blondel, V. D., Guillaume, J. L., Lambiotte R. & Lefebvre, E. Fast unfolding of communities in large networks. J. of Stat. Mech.: Theor.Exp. 10, P10008 (2008).
Zhang, Z. et al. SCINA: A Semi-Supervised Subtyping Algorithm of Single Cells and Bulk Samples. Genes (Basel) 10, 531 (2019).
Guo, Y. et al. NHWD-1062 ameliorates inflammation and proliferation by the RIPK1/NF-κB/TLR1 axis in Psoriatic Keratinocytes. Biomed. Pharmacother. = Biomedecine Pharmacotherapie 162, 114638 (2023).
Chen, L. et al. Kdm2a deficiency in macrophages enhances thermogenesis to protect mice against HFD-induced obesity by enhancing H3K36me2 at the Pparg locus. Cell Death Differ. 28, 1880–1899 (2021).
Wang, J. et al. Extracellular vesicles mediate the communication of adipose tissue with brain and promote cognitive impairment associated with insulin resistance. Cell Metab. 34, 1264–1279.e1268 (2022).
Acknowledgements
We are grateful to those patients for donating their adipose tissue, skeletal muscle and liver for the studies. We thank Jing Liu, Yang Li, Xi Luo, Wenye Mo and Siqian Liu from the Center of Biomedical Research, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology for their help in animal studies. Also, we thank Shanghai Majorbio Bio-pharm Technology for providing single-cell sequencing. Dr. Dong Yachen, Liu weiting, and He Xiaolong from Majorbio Bio-pharm Technology analyzed single-cell sequencing data and visualized all sequencing data. This work was supported by the National Key R&D Program of China (2022YFA0806101 to C-Y.W.), the National Natural Science Foundation of China (81920108009 to C-Y.W., 82130023 to C-Y.W., 82300948 to Q.Z., 82301753 to T.H., 82200923 to F.S., 82270885 to Q-L.Y., 82200926 to T-T.Y., 82270893 to Q.G., 82100892 to J.Z., 82401031 to X-M.F., and 82100823 to F-X.W.), the Postdoctoral Science Foundation of China (54000-0106540081 to F.S. and 54000-0106540080 to F-X.W.), the Fundamental Research Funds for the Central Universities (2022JYCXJJ050 to J.G., YCJJ20242122 to Z-C.G.), China International Medical Foundation (2023-N-03-02 to T.H.), the Research and Innovative Team Project for Scientific Breakthroughs at Shanxi Bethune Hospital (2024AOXIANG03 to C-Y.W.), the Continuous Funding Program for High-Level Research Achievements at Shanxi Bethune Hospital (2024GSPYJ10 to C-Y.W.).
Author information
Authors and Affiliations
Contributions
Q.Z., J.G., G-R.W., and C-W.W. contributed equally to this work. Q.Z., J.G., G-R.W., and C-W.W. designed the experiments and analyzed the data. Q.Z. and J.G. wrote the manuscript. Q.Z. and J.G. performed the majority of the experiments. Y.Y., W.K., W-G.Z., Y-J.L., T-T.Z. and F.X. provided human samples and corresponding clinical information. Q.Z., J.G., G-R.W., T.H., Z-C.G. and H.X. were involved in RT-qPCR and western blot. J.G., T.H. and H.X. contributed toward the fractionation of adipose tissue. J.G., T.H., T-T.Y., Y.W., S.Z., Q-L.Y., K.X., C.C., X-M.F. and G-R.W. assisted with the animal experiments. C-W.W., Z-C.G., T-Y.Y. contributed to the study of RNA-seq analysis. Q.Z., J.G. and C.C. contributed toward the collection of clinical samples. S-W.L., Y-J.L. and C-Y.W. contributed to the study design and manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ana Salazar-Puerta and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, Q., Gao, J., Wu, G. et al. Adipose progenitor cell-derived extracellular vesicles suppress macrophage M1 program to alleviate midlife obesity. Nat Commun 16, 2743 (2025). https://doi.org/10.1038/s41467-025-57444-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57444-y
