
Abstract
Antimicrobial resistance is a global health concern, and methicillin-resistant Staphylococcus aureus (MRSA) is one of the highest-priority organisms exhibiting this phenotype. Here, we performed a national surveillance integrating patient clinical data of S. aureus isolated from bloodstream infections. We performed genome sequencing, standardized antimicrobial susceptibility testing, and collected clinical metadata of 580 S. aureus isolates collected during 2019–2020. We focused on three predominant clonal complexes (CC1, CC5, and CC8) and assesses their microbiological and clinical significance, as well as their distribution across eastern and western Japan. Furthermore, we conducted a genomic comparison of the isolates of 2019–2000 with those of 1994–2000 and investigated the evolutionary trajectory of emerging clones from the three dominant clonal complexes. We revealed that the emerging MRSA ST764-SCCmecII showed the highest mortality rate within 30 days of hospitalization. This high-risk clone diverged from the New York/Japan clone (ST5-SCCmecII), which was inferred to have undergone repeated infections with phages carrying superantigen toxin genes and acquired antimicrobial resistance genes via mobile genetic elements, leading to its emergence around 1994. Overall, we provide a blueprint for a national genomic surveillance study that integrates clinical data and enables the identification and evolutionary characterization of a high-risk clone.
Similar content being viewed by others

Introduction
Antimicrobial resistance (AMR) is one of the greatest threats to human health, necessitating effective surveillance. Staphylococcus aureus is an important antimicrobial-resistant bacterium. It's spread in healthcare and community settings, and in the livestock industry, is a global challenge because S. aureus causes diverse pathologies, from skin and soft tissue infections to systemic invasive diseases, such as pneumonia, septicemia, infective endocarditis, osteomyelitis, toxic shock syndrome, and food poisoning through its enterotoxin1. Bloodstream infection (BSI) caused by S. aureus is a severe condition with an increased mortality risk2,3. An estimated 119,247 S. aureus BSIs with 19,832 associated deaths occurred in the United States4 in 2017. The number of BSI deaths attributed to S. aureus in Japan was 17,412 in 2011 and 17,157 in 2017, wherein 5924 (34%) deaths were attributed to methicillin-resistant S. aureus (MRSA) in 2011, which significantly decreased to 4224 (24.6%) in 20175.
In recent years, various MRSA clones isolated from BSIs have emerged in Japan. However, studies on the molecular epidemiology and genomic characterization of MRSA isolates from BSIs are limited to certain healthcare facilities or regions. A pioneering national genomic surveillance study in the Philippines included S. aureus, although it was limited by inconsistencies in the antimicrobial panels that were tested at sentinel sites, which highlighted the importance of standardized phenotypic antimicrobial susceptibility testing6.
The ST5-Staphylococcal Cassette Chromosome mec (SCCmec) II is present in the New York/Japan (N/J) clone, the most common clone responsible for healthcare-associated MRSA (HA-MRSA) BSIs in Japan, although infections by this clone have recently been declining7,8,9. ST764-SCCmecII, belonging to clonal complex (CC) 5, was first isolated from the blood cultures of patients with invasive infections in Japan in 2005 and was reported as a hybrid variant of ST5-SCCmecII and community-acquired MRSA (CA-MRSA) with acquired virulence genes10. Recently, ST764-SCCmecII has been isolated from invasive infections in Asia, including Thailand and China11,12. In contrast, the proportion of CA-MRSA-SCCmecIV has increased in isolates from invasive infections9,13 and other SCCmecIV MRSA clones (ST1-SCCmecIV or ST8-SCCmecIV)9,14. However, CA-MRSA SCCmecIVa USA300 lineages (including USA300-LV, a Latin American variant), which are predominant in nosocomial settings in North and South America and cause severe infections with high mortality rates, are still not common in Japan9. The ST8-SCCmecIVl with the CA-MRSA genotype, represented by the MRSA/J clone, was first isolated in Japan from the bullous impetigo of a child in 200315, and one case of death caused by a strongly invasive pathotype was reported in 201216. During the same period, we reported a case of systemically disseminated MRSA/J infection17,18. ST2725-SCCmecIV, belonging to CC1, which has never been reported outside Japan, was recently isolated from the blood cultures of patients in Japanese regional hospitals19,20.
Here, we conducted a large-scale national genomic surveillance of S. aureus isolated from patients with BSI during 2019–2020, performed antimicrobial susceptibility testing using the same panel in the same reference laboratory, collected clinical metadata of these S. aureus isolates, and investigated the clinical risk of the clones. We also conducted genomic comparisons with isolates collected during 1994–2000 to delineate the evolutionary trajectories of BSI-derived S. aureus clones. By combining the national genome sequencing data with the standardized antimicrobial susceptibility testing results and clinical metadata, this report highlighted dynamic changes in MRSA in 26 years (1994–2020) in Japan and the emergence of ST764-SCCmecII with high mortality risk associated with BSI.
Results
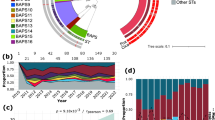
Phylogenetic relationship and geographic distribution of 580 S. aureus isolated from blood during 2019–2020
Population structure analysis of S. aureus isolated from BSIs during 2019–2020 using Bayesian hierarchical clustering of the core genome alignment showed 13 distinct sequence clusters (SCs) ranging in size from 7 to 155 genomes (Fig. 1a and Supplementary Data 1). Phylogenetic analyses revealed three dominant SCs, namely SC1, SC3, and SC8, comprising 155 (26.7% of the total population), 127 (21.9% of the total population), and 73 genomes (12.6% of the total population), respectively (Fig. 1a, pie chart 1). We performed in silico multilocus sequence typing to identify the sequence types (STs) that comprised the SC. The CCs of these STs were examined using the eBURST analysis. SC1 comprised CC1, which included ST1 (104 genomes, 67.1% of SC1), ST2725 (28 genomes, 18.1% of SC1), and ST81 (10 genomes, 6.5% of SC1) (Supplementary Data 1). SC3 comprised CC8 and, almost exclusively, ST8 (127 genomes, 84.3% of SC3). SC8 comprised CC5, which included ST5 (73 genomes, 57.5% of SC5) and ST764 (25 genomes, 34.2% of SC5). The most dominant STs of the 580 S. aureus were ST8 (18.4%), ST1 (17.9%), and ST5 (7.2%) (Fig. 1a, pie chart 2). BSI-derived S. aureus from 2019 to 2020 contained 343 isolates (59.1%) from western and 237 strains (40.9%) from eastern Japan (Fig. 1a, pie chart 3). The most dominant STs in western Japan were ST8 (23.0%), ST1 (11.9%), and ST5 (7.0%) (Fig. 1b, pie chart 1). However, the most dominant ST in eastern Japan were ST1 (26.6%), ST8 (11.8%), and ST188 (7.6%). Additionally, the major STs of CC8 were ST8 (approximately 80%) and ST630 (less than 10%) in both western and eastern Japan (Fig. 1c, middle panel). Similarly, the major STs of CC5 were ST5 (approximately 57%) and ST764 (approximately 34%) in both western and eastern Japan (Fig. 1c, right panel). A geographic comparison of the major STs in CC1 revealed significantly higher proportions of ST1 (75.9%) and ST81 (10.8%) in eastern Japan compared to in western Japan (p < 0.001 for ST1 and ST81, respectively, χ2 test), whereas ST2725 (33.3%) was more common in western Japan (p = 0.0263, χ2) (Fig. 1c, left panel). The proportion of MRSA of BSI-derived S. aureus during 2019–2020 was 46.4%, mostly SCCmecIV (76.5% of total SCCmec) (Fig. 1a, pie chart 4). The proportions of MRSA in western and eastern Japan were 47.5 and 44.7%, respectively (Fig. 1b, pie chart 2). SCCmecIV was the most common MRSA of BSI-derived S. aureus in both regions, followed by SCCmecII. The major STs of MRSA (n = 269) were ST1 (34.6%) and ST8 (26%) (Supplementary Fig. 1, left panel). In eastern Japan, the major STs of MRSA were ST1 (55.7%) and ST8 (14.2%). In addition, ST81, which belongs to CC1, was detected in eastern Japan, although in small numbers, and not in western Japan. In contrast, in western Japan, the major STs of MRSA were ST8 (33.5%) and ST1 (21.3%). The number of STs of methicillin-susceptible S. aureus (MSSA) was more diverse than those of MRSA (Supplementary Fig. 1, right panel). These results indicate that the representative CCs of BSI-derived S. aureus in Japan were CC1, CC8, and CC5 (Fig. 1a), with ST1 and ST2725 having different distributions in the eastern and western regions.

a Midpoint-rooted maximum-likelihood (ML) tree showing the phylogenetic structure of 580 S. aureus isolates during 2019–2020. The scale bar represents the number of nucleotide substitutions per site. Matrix shows the metadata; 1, Bayesian analysis of population structure (BAPS) sequence cluster (SC) number identified by FastBAPS; 2, Sequence type (ST) number of each S. aureus isolate; 3, indicates the eastern or western Japan where S. aureus was isolated; 4, indicates the SCCmec type of MRSA. The pie graph shows the proportion of the metadata 1–4. ST with less than 3 isolates included in other. b Genotypic distribution in eastern or western Japan. ST under top 10 included in other. c Comparison of geographic proportions of STs is shown in SC1, SC3, and SC8. The color tone is shown in the same pattern in Fig. 1. *p < 0.05; western Japan vs. eastern Japan. P-value (p) was calculated using the χ2 test. Metadata information of 580 S. aureus isolated in 2019–2020 is summarized in Supplementary Data 1.
ST764 infection is associated with high mortality
Subsequently, we investigated the association between these lineages and 30-day mortality rates. Notably, the 30-day mortality rate of S. aureus BSIs caused by the ST764-SCCmecII clone was 48% (Table 1), which was significantly higher than the 22% of the other clones (p = 0.0088, log-rank test) (Table 2). The 30-day mortality rate associated with the ST8-SCCmecI clone was also high (50%) compared to that of the other clones, although the difference was not statistically significant owing to the small sample size (n = 10). Further analysis of the ST764-SCCmecII clone revealed that age was a confounding factor significantly associated with the ST764-SCCmecII clone (p = 0.026, Wilcoxon’s rank-sum test) and 30-day mortality rate (p < 0.0001, univariate Cox regression) with p-values < 0.05. The association between the ST764-SCCmecII clone and 30-day mortality rate remained significant even after controlling for the confounding effect of age (Table 2) (p = 0.04, hazard ratio 1.86, 95% CI: 1.03–3.38). There were no confounding factors in the following clinical variables: sex, diabetes, hemodialysis, surgery within 30 days before blood culture, artificial respirator use, and atopic dermatitis.
Phylogenetic relationship of BSI-derived 183 S. aureus during 1994–2000
We performed a comparative phylogenetic analysis of S. aureus isolated from BSIs during 2019–2020 and 1994–2000. We collected the BSI-derived S. aureus isolates from post-marketing surveillance conducted nationwide during 1994–2000 after levofloxacin (LVFX) was released in Japan21. Population structure analysis of 183 BSI-derived S. aureus isolated during 1994–2000 using Bayesian hierarchical clustering of the core genome alignment showed 10 distinct SCs, ranging in size from 2 to 109 genomes (Supplementary Fig. 2 and Supplementary Data 2). Phylogenetic analysis revealed that SC-A (109 genomes, 59.6% of the total population) was dominant (Supplementary Fig. 2, pie chart 1) and comprised CC5, consisting almost exclusively of ST5 (97 genomes, 90% of SC-A, 53% of the total population). The Shannon diversity index of the STs significantly increased from 3.08399 in 1994–2000 to 4.767595 in 2019–2020. The proportion of MRSA in BSI-derived S. aureus in 1994–2000 was over 56%, mostly SCCmecII (50.8% of the total population) (Supplementary Fig. 2, pie chart 4). The proportion of STs of MRSA was almost exclusively ST5 (84.6%) (Supplementary Fig. 3, left upper panel). The most common ST of MSSA was ST5 (12.7%); however, the ST types of MSSA were more diverse than those of MRSA (Supplementary Fig. 3, right upper panel). The predominant CC of 183 S. aureus during 1994–2000 was the CC5 lineage, including the N/J clone (ST5-SCCmecII) (Supplementary Fig. 2). These results confirm the dynamic changes in the population structure of MRSA within 20 years when comparing the phylogenetic analysis data of the isolates of 1994–2000 to those of 2019–2020.
Comparative analysis of antimicrobial-resistant (AMR) pattern of BSI-derived S. aureus
We examined the differences in the number of AMR genes (ARGs) between the isolates of 2019–2020 and 1994–2000 (Supplementary Fig. 4a). The median number of ARGs was 3 in 2019–2020 and 6 in 1994–2000 (Supplementary Fig. 4a, left). For MRSA, the number of ARGs differed significantly between the two periods (median of 8 in 1994–2000 and 4 in 2019–2020; p < 2.2e-16, Wilcoxon’s rank-sum test). In contrast, no significant difference was observed for MSSA, with a median of 1 in both periods (p > 0.56, Wilcoxon’s rank-sum test) (Supplementary Fig. 4a, right panel). The results of the proportion of each ARG revealed that the prevalence of the aminoglycoside resistance gene aadD, tetracycline resistance gene tet(M), bleomycin resistance gene bleO, fosfomycin (FOM) resistance gene fosB, chlorhexidine resistance gene qacA decreased significantly between the two periods in the MRSA (p < 0.05) (Supplementary Fig. 4b). Conversely, the prevalence of the aminoglycoside resistance genes ant(9)-Ia, macrolide-resistant genes erm(A), erm(C), and the quinolone resistance-determining region (QRDR) mutations (GrlA/S80F and GyrA/S84L) increased in isolates of 2019–2020 in MSSA (n = 311, p < 0.05) (Supplementary Fig. 4b, Supplementary Table 1). Antimicrobial susceptibility testing showed that resistance to cefazolin (CEZ), cefmetazole (CMZ), gentamicin (GM), clindamycin (CLDM), and minocycline (MINO) significantly decreased in isolates of 2019–2020 in the total samples or MRSA (p < 0.02) (Supplementary Fig. 4c, upper and middle panels). In MSSA, the level of resistance to each antibiotic was very low; however, resistance to erythromycin (EM) and LVFX increased slightly from 1994–2000 to 2019–2020 (Supplementary Fig. 4c, lower panel). We subsequently examined the distribution of ARGs in CC1, CC5, and CC8, the dominant clones of BSI-derived MRSA of 2019–2020 (Fig. 2). In CC1, the ST1-SCCmecIV and ST2725-SCCmecIV lineages contained the same number of ARGs (median: 4) (Fig. 2a, CC1) and exhibited very similar AMR patterns (Fig. 2b, C1; Supplementary Table 1; Fig. 2c, upper panel). Consistent with the presence of ARGs and QRDR mutations in ST1-SCCmecIV and ST2725-SCCmecIV, the resistance rates of these strains to oxacillin (MPIPC), EM, and LVFX were high (Fig. 2c, upper panel). In CC5, the median number of ARGs in ST5-SCCmecII was 8 in 1994–2000 and 7 in 2019–2020 (p < 0.01) (Fig. 2a, CC5). In contrast, the median number of ARGs in ST764-SCCmecII in 2019–2020 was 5, significantly lower than that in ST5-SCCmecII (median: 7; p < 0.01). In ST5-SCCmecII, blaZ, aac(6’)-aph(2”), tet(M), cat, fosB, and qacA showed decreasing trends in 2019–2020 compared to in 1994–2000 (p < 0.05) (Fig. 2b, CC5; Supplementary Table 1). In ST764-SCCmecII, aac(6’)-aph(2”), tet(M), fosD, and qacB were detected at higher prevalence compared to ST5-SCCmecII (p < 0.02). Resistance rates to MPIPC, CEZ, CMZ, EM, CLDM, and LVFX remained consistently high in ST5-SCCmecII during both periods, whereas resistance rates to GM and MINO were significantly lower in 2019–2020 compared to in 1994–2000 (p < 0.01) (Fig. 2c, middle panel). In contrast, ST764-SCCmecII showed very high rates of resistance to these antibiotics, except for ABK. In CC8, the median number of ARGs in ST8-SCCmecI was 6, significantly higher than those in ST8-SCCmecIVj and ST8-SCCmecIVa, both with a median of 5 (p < 0.05) (Fig. 2a, CC8). In ST8-SCCmecIVj, blaZ, ant(9)-Ia, erm(A), tet(M), and QRDR mutations (GrlA/S80F and GyrA/S84L) were prevalent, whereas aac(6’)-aph(2”) was less common (Fig. 2b, CC8 and Supplementary Table 1). ST8-SCCmecIVl showed lower proportions of ant(9)-Ia, erm(A), tet(M), and QRDR mutations (GrlA/S80F and GyrA/S84L), but higher proportions of aac(6’)-aph(2”), aadD, bleO, and qacB. Unique to ST8-SCCmecIVa, mph(C) and msr(A) were present in approximately 38% of the isolates. ST8-MRSA showed varying patterns of resistance to each antibiotic depending on the SCCmec type; however, ST8-SCCmecI was more resistant than the other clones (Fig. 2c, lower). The small number of BSI-derived USA300 isolates in Japan makes the comparative assessment difficult.

a The number of antimicrobial-resistant genes (ARGs) per strain is shown, but duplicate ARG genes are not counted. The triangle in the box indicates mean, the box indicates the 25th and 75th percentiles, and the whiskers indicate the highest and the lowest values of the results. The line in the box indicates median. P-value (p) was calculated using the Student’s t-test. b Presence heatmap of major ARGs and quinolone resistance-determinant region (QRDR) mutations identified. The numbers in the heatmap columns are shown as percentages. *p < 0.05; ST5-II_1994-2000 vs. ST5-II_2019-2020. #p < 0.02; ST5-II_2019-2020 vs. ST764-II_2019-2020. P-value (p) was calculated using the χ2 test. c Antimicrobial susceptibility profiles of BSI-derived MRSA. The radar chart shows the resistance rates of isolates to MPIPC, CEZ, CMZ, GM, ABK, EM, CLDM, MINO, and LVFX in 1994–2000 and 2019–2020.
We calculated Kendall’s concordance coefficient (W), as described in our previous study22, and we observed a certain degree of concordance between genotype and phenotype in CC1, CC5, and CC8 (Supplementary Table 2). High concordance (Kendall’s W > 0.8) was observed for EM, GM, CLDM, MINO, and LVFX, along with their corresponding resistance determinants.
Comparative analysis of virulence factor genes (VFGs) of BSI-derived S. aureus
We investigated the presence of VFGs in S. aureus isolates from BSIs. The proportion of toxic shock syndrome toxin-1 gene (tst-1) decreased significantly in 2019–2020 compared to in 1994–2000 (p < 0.001) (Supplementary Fig. 4d). Additionally, the proportion of the staphylococcal enterotoxin gene cluster EGC (seg, sei, sem, sen, and seo) decreased drastically in total samples and MRSA isolates in 2019–2020 compared to in 1994–2000 (p < 0.001) but remained at approximately 30% in MSSA isolates. In contrast, the proportions of sea, seh, sek, and seq in the total samples or MRSA of 2019–2020 increased compared to 1994–2000 (p < 0.05). A few Panton–Valentine leukocidin (PVL) genes (lukF/S-PV), exfoliative toxin (ET) genes (eta, etb, and etd), and epidermal cell differentiation inhibitor (EDIN) genes (ednA, ednB, and ednC) were detected during both periods (Supplementary Fig. 4d). For a human-specific immune evasion cluster, the proportion of the staphylococcal complement inhibitor gene (scn) and staphylokinase gene (sak) in the two periods remained at the same level; however, that of the chemotaxis inhibitory protein Chp gene (chp) was markedly decreased in the total and MRSA.
Subsequently, we examined the distribution of VGFs in CC1, CC5, and CC8, the dominant BSI-derived MRSA in 2019–2020 (Fig. 3). In CC1, most ST1-SCCmecIV and ST2725-SCCmecIV had a high prevalence of sea, seh, sek, seq, scn, and sak, with the same distribution pattern (Fig. 3, CC1). In CC5, ST5-SCCmecII showed high proportions of sec, tst-1, EGC, and sel during both periods (Fig. 3, CC5). The proportion of seb in ST5-SCCmecII in 1994–2000 was 12% but was not detected in 2019–2020. ST764-SCCmecII exhibited high proportions of seb, seg, sei, sem, sen, and seo but lacked sec, tst-1, and sel. In CC8, sep was prevalent in ST8-SCCmecI, IVj, and IVl (Fig. 3, CC8). ST8-SCCmecIVl showed high proportions of sec, tst-1, and sel, along with additional se1 and ednA. ST8-SCCmecIVa carried sek, seq, and PVL genes (lukF/S-PV), which are characteristic of the USA300 lineage.

The proportion of major virulence factor genes (VFGs) of each MRSA type is shown in a heatmap. The numbers in the heatmap columns are shown as percentages. *p < 0.02; ST5-II_1994-2000 vs. ST5-II_2019-2020. P-value (p) was calculated using the χ2 test.
Evolutionary origins and population dynamics of three CC lineages of blood-derived S. aureus in Japan
To provide a historical perspective on the emergence of the three dominant CC lineages of S. aureus causing bacteremia in Japan, we constructed time-calibrated phylogenies of CC1 (ST1 and ST2725), CC5 (ST5 and ST764), and CC8 (MRSA/J and USA300) using Bayesian coalescent analysis implemented in the BEAST software. Each phylogenetic tree was calibrated using the isolation dates of the strains, which ranged from 1997 to 2020 (CC1) and 1982 to 2020 (CC5 and CC8), and public collection isolates with identifiable isolation dates (Supplementary Fig. 5 and Supplementary Data 3). Root-to-tip regression analyses revealed a positive correlation between genetic distance and sampling date for each lineage (Supplementary Fig. 6). Given the presence of a temporal structure in each dataset, we performed dated coalescent phylogenetic analysis. Consequently, we estimated the time to the most recent common ancestor (tMRCA) of the ST1-SCCmecIV and ST2725-SCCmecIV lineages to be approximately 1998 (95% highest posterior density [HPD] intervals: 1995–2002) (Fig. 4a). The results of this analysis indicated that the ancestor of ST1-MRSA-t1784 acquired SCCmecIV, Tn554 harboring ant(9)-Ia, and erm(A)23 after 1965 (95% HPD intervals: 1958–1974). ST1-MRSA-t1784 emerged around 1998 (95% HPD intervals: 1995–2002) and started to circulate in Japan after 2000. Six years later, around 2004 (95% HPD intervals: 2001–2006), the ST2725-SCCmecIV-t1784 lineage diverged from ST1 and spread primarily to western Japan (Fig. 4a square light blue and Fig. 1c bar graph in the left panel). However, the ST1-t127 lineage diverged in 1953 and acquired the genomic island νSa4, carrying the seb and fusidic acid resistance gene fusC-harboring SCCfus24 by phage infections somewhere up to 1970. Through a timescale phylogenetic analysis, we first estimated the age of SCCfus acquisition in the ST1-t127 lineage.

The three major MRSA clones are shown in (a), CC1; (b), CC8; and (c), CC5. Upper panel indicates the Bayesian maximum clade credibility time-calibrated phylogenies based on non-recombining regions of the core genome of each CC. Arrow nodes indicate the divergence date (median estimate with 95% highest posterior). Blue horizontal bars at each node represent 95% confidence intervals. Lower panel, Bayesian skyline plots showing changes in effective population size over time. Median is represented by a dark blue line, and 95% confidence intervals are in light blue. Index No. 1–5 represents the STs, SCCmec type, ACME type, spa typing, and VFGs/AGRs, respectively. In index No. 5 of each CC, ant(9)-Ia and erm(A) harbored in Tn554 are indicated in red. In CC8 (b), MRSA/J acquired SCCmecIVl, genomic island vSa4, and se1-harboring plasmid somewhere between 1957 and 1986. sec, tst-1, and sel on vSa4 and se1-harboring plasmid are indicated in blue (see also Supplementary Fig. 7a and b). Some strains of ST8-SCCmecIVj and ST8-SCCmecI acquired tet(M)-harboring Tn916-like transposable unit somewhere between the late 1980s and 1990s. tet(M) is indicated in green (see also Supplementary Fig. 7c). In CC5 (c), in the process leading to the emergence of ST5-SCCmecII (N/J clone) and ST764-SCCmecII, it is estimated that SCCmecII, genomic islands, and transposons were acquired stepwise. The related VFGs are indicated in blue, and tet(M) is indicated in green (see also Supplementary Fig. 8).
In CC8, USA300 isolated from BSIs in Japan belonged to the USA300-NAE (North American Epidemic) lineage25 and appeared around 1989 (95% HPD intervals: 1986–1992) (Fig. 4b). This is consistent with the previously reported data25. The trajectory of the emergence of ST8-SCCmecIVl (MRSA/J) (Fig. 4b, square light blue) was somewhere between 1957 and 1986, when it acquired SCCmecIVl and νSa4 (carrying sec, tst-1, and sel) by phage infections (Fig. 4b, light blue of index No. 2 and 5, and Supplementary Fig. 7a) and plasmids carrying se1 (Fig. 4b, light blue of index No. 5, and Supplementary Fig. 7b), emerging around 1986 (95% HPD intervals: 1983–1989) contemporaneously and in parallel with the USA300-NAE lineage (SCCmec IVa-arginine catabolic mobile element [ACME] type I). In addition, the ST8-SCCmecI and ST8-SCCmecIVj lineages emerged around 1979 (95% HPD interval: 1976–1982) and 1988 (95% HPD interval: 1985–1991), respectively. Some strains of the ST8-SCCmecI and ST8-SCCmecIVj lineages were estimated to have acquired tet(M)-harboring Tn916-like transposable units in the late 1980s and 1990s (Fig. 4b and Supplementary Fig. 7c). These multiple ST8 lineages emerged one after another during the 1980s–1990s, and the effective population size of CC8 increased in the late 1990s–2000s (Fig. 4b, lower).
In CC5, we estimated the tMRCA of the ST5-SCCmecII (N/J clone) and ST764-SCCmecII lineages to be around 1959 (95% HPD interval: 1956–1962) (Fig. 4c). Our genome sequence comparison inferred the trajectory of the emergence of the ST764-SCCmecII clone from the N/J clone based on recombination events (index No. 5 in Fig. 4c and Supplementary Fig. 8). At the first step, somewhere between 1928 and 1959, SCCmecII, genomic island νSa3 (carrying sec, tst-1, and sel) (Fig. 4c and Supplementary Fig. 8a) and tet(M)-harboring Tn916-like transposable unit (Supplementary Fig. 8b) were acquired by phage infection or transposition, respectively, resulting in the emergence of ST5-SCCmecII (N/J clone). In the second step, after 1974 (95% HPD interval: 1971–1976), N/J clones acquired the genomic island νSa4 carrying seb by phage infection and emerged as a seb-positive-N/J lineage, which evolved into the ST764-SCCmecII lineage (Fig. 4c and Supplementary Fig. 8c). At the third step, genomic island νSa3 (carrying sec, tst-1, and sel) was replaced by a completely different phage, followed by a recombination event at the same location at the νSa3 of N/J clone, loosing blaZ (Fig. 4c and Supplementary Fig. 8a). Finally, approximately 20 years after the emergence of the N/J clone, around 1979 (95% HPD intervals: 1977–1982), the ST764-SCCmecII lineage diverged from ST5 and spread throughout Japan after 1994. One sub-lineage of ST764-SCCmecII acquired the genomic island νSa1 carrying sek and seq by phage infection somewhere between 1998–2001 (Fig. 4c and Supplementary Fig. 8d). Another ST764-SCCmecII sub-lineage acquired ACME type II somewhere between 1996 and 1999 (Fig. 4c and Supplementary Fig. 8e). However, the ST5-SCCmecI ancestor diverged from the N/J lineage around 1928, and somewhere between 1932–1967, the ST5-SCCmecI lineage emerged by acquiring SCCmecI and νSa4 (carrying sec, sek, and seq) by phage infections but did not spread as far in Japan (Supplementary Data 1 and 2). ST5-SCCmecIV followed a different phylogenetic trajectory from the clones described above, emerging somewhere between 1966 and 1994 with the acquisition of SCCmecI, which was also not widespread in Japan (Fig. 4c). The effective population size of CC5 increased in a staircase-like manner during the emergence of ST5-SCCmecII and ST764-SCCmecII (Fig. 4c, lower panel).
Discussion
More than 80 years since the antimicrobial-resistant S. aureus (including MRSA) was first recognized, MRSA has rapidly evolved following the widespread use of antimicrobials and has been reported to have a remarkable potential to spread globally26. Here, we used 580 S. aureus genomes from the late 2010s and 183 genomes from the late 1990s, derived from BSIs, to determine the evolutionary history and geographical distribution of Japanese BSI-derived S. aureus, focusing on three CC lineages. We previously reported that the prevalence of MRSA in inpatients decreased from 40.3 to 35.1% between 2011 and 2019, according to data from the national phenotypic antimicrobial surveillance (Japan Nosocomial Infections Surveillance, JANIS), and the analysis of resistance profiles indicated that isolates resistant to three antimicrobials (MPIPC, EM, and LVFX) increased, whereas those resistant to six antimicrobials (MPIPC, GM, EM, CLDM, MINO, and LVFX) decreased26. A comparison of the resistance profiles and genome data of BSI-derived isolates in a single university hospital between 2011 and 2019 revealed that increased resistance to the three drugs corresponded to CC8-SCCmecIV27. Here, we compared BSI-derived MRSA isolates of 1994–2000 with those of 2019–2020, wherein the proportion of isolates resistant to the six drugs decreased from 33.7 to 10.8%, whereas the proportion of isolates resistant to three drugs sharply increased from 1 to 41.6%, confirming the change in the multidrug-resistant phenotype of MRSA (Supplementary Table 3 and Supplementary Fig. 9). Additionally, our genome analysis indicated that ST5-SCCmecII (N/J clone) showed a significant reduction in the proportion of isolates resistant to the six drugs, whereas ST764-SCCmecII appeared as a new clone resistant to the six drugs during 2019–2020. Conversely, 96.4% (109/112 strains) of the increased proportion of isolates resistant to the three drugs belonged to the CC1-SCCmecIV lineage, which was different from the results of the previous study in a single university hospital. The reason for this difference is that the single university hospital was located in western Japan, where CC8-SCCmecIV is more prevalent (Supplementary Fig. 1); however, in the nationwide surveillance of BSI-derived S. aureus used in this study, the predominant CC1-SCCmecIV had a significant impact on the resistance to the three antimicrobials. ST1-SCCmecIV and ST2725-SCCmecIV had extremely high rates of erm(A) and QRDR mutations, and these genes contributed significantly to resistance to the three antimicrobials (Fig. 2b, CC1). Another notable feature is the significant decrease in rates of resistance to CEZ and CMZ among BSI-derived MRSA isolates in 2019–2020. Similar findings have been reported by Yamaguchi et al.28. This reduction in MICs for CEZ and CMZ in BSI-derived MRSA isolates is likely attributable to the decline in multidrug-resistant MRSA-SCCmecII, which was predominant in the past in Japan, and the corresponding increase in MRSA-SCCmecIV. However, the detailed mechanism remains unknown. This drastic change in MRSA clones reflected not only the resistance gene repertoire but also the virulence gene repertoire of MRSA (Fig. 3, Supplementary Fig. 1). A significant decrease in the proportion of tst-1 and EGC (seg, sei, sem, sen, and seo) and an increase in the proportion of sea, seh, sek, and seq could be attributed to an increase in ST1-SCCmecIV and ST8-SCCmecIV and a decrease in ST5-SCCmecII.
Our timescale phylogenetic and population dynamics analysis showed that ST5-SCCmecII emerged around 1960 and reigned as a representative of HA-MRSA in Japan until 2000. However, after 2000, it was replaced by some sublineages of CC1-SCCmecIV and CC8-SCCmecIV, which are representative clones of CA-MRSA. This was further supported by several previous epidemiological studies8,12,29.
Regional differences in the distribution of CC1 during 2019–2020 were observed: ST1 and ST81 tended to be slightly more common in eastern Japan, whereas ST2725-SCCmecIV circulated slightly more frequently in western Japan (Fig. 1c). These STs-SCCmecIV possessed almost identical patterns of ARGs, VFGs, and AMR patterns (Figs. 2b, c, and 3). Notably, the interval between the emergence of ST1-SCCmecIV and ST2725-SCCmecIV was very short, only approximately 5 years, as revealed for the first time in our study (Fig. 4a). Most ST1-SCCmecIV-t1784 isolates in Japan belong to a different lineage from the PVL-negative ST1-SCCmecIV-t127, which is widespread in Europe30. Moreover, some subclades of European ST1-SCCmecIV-t127 have SCCmecfus carrying the fusidic acid resistance gene fusC but not ST1-SCCmecIV-t1784. The Japanese fusC-positive ST1-MSSA-t127 strain is rare and mecA negative (Supplementary Data 2). ST1-SCCmecIV-t1284 is a more modern lineage than European ST1-t127, according to timescale phylogenetic analysis.
Japanese ST2725-SCCmecIV-t1784 has been detected in both inpatients and outpatients, suggesting that it has spread to hospitals and the community19. To the best of our knowledge, the ST2725-SCCmecIV-t1784 clone has not been reported overseas and is a relatively young CA-MRSA unique to Japan. In contrast to CC1, CC5 was distributed nationally without an eastern-western regional bias. Although CC5 in Japan has been replaced, its proportion is still high, ranking third (approximately 12%) after CC1 and CC8. The dominant CC5, ST5-SCCmecII (the N/J clone), has spread31 and circulated in medical centers throughout Japan since its emergence in 1959. Over a period of approximately 35 years, our time-calibrated phylogenic analysis inferred that the ST5-SCCmecII lineage underwent repeated acquisition or shedding via superantigen toxin-harboring phage infection and ARG-carrying transposons, leading to the emergence of the ST764-SCCmecII lineage around 1994. Furthermore, herein, we found that several strains of ST764-SCCmecII were independently acquired by phage infection carrying sek/seq or ACME-II and the ACME-related cassette JR1 (cJR1) around 2000 (Figs. 4c, 3 blue bar). ACME-II was first identified in S. epidermidis32. Urushinbara et al. reported several variants of the ACME-II-SCCmec composite island (ACME-II-SCCmec-CI) in ST764, isolated from Hokkaido, Japan33. Therefore, we presume that the ST764-SCCmecII lineage isolated in Japan is becoming increasingly diverse. ST764-ACME-SCCmec-CI may be distributed throughout Northern-Eastern Japan, particularly in Hokkaido.
We demonstrated that ST764-SCCmecII was the highest-risk clone, with statistical significance, according to the risk assessment of the 30-day mortality rate (Table 1). The spread of seb-positive ST764-SCCmecII clones in long-term care facilities (LTCFs) in Japan was recently reported by Kawamura34. In our study, 72.4% (21/29) of the registered ST764-SCCmecII cases were ST764-SCCmecII-t002 (Fig. 4c, 4 ocher bar). In China, ST764-SCCmecII-t1084, a lineage similar to the Japanese ST764-SCCmecII-t002, has recently been reported to increase as a hypervirulent clone11. Our study indicates that age is a confounding factor significantly associated with ST764 and 30-day mortality. This is in agreement with a previous observational study demonstrating the spread of ST764 in Japanese LTCF residents with multiple comorbidities and increased susceptibility to infections34. The association between ST764 and the 30-day mortality rate remained significant, even after controlling for the confounding effect of age. A previous regional surveillance study demonstrated that all MRSA-SCCmecII isolates from patients with pneumonia belonged to ST76435. Our recent epidemiological risk assessment study of isolates from patients admitted to the intensive care unit36 and isolates from BSIs demonstrated that the primary focus of ST764 was pneumonia. Furthermore, ST764 was the only lineage that emerged and increased in number within 20 years and was resistant to six antimicrobials: MPIPC, GM, EM, CLDM, MINO, and LVFX. Overall, these results strongly suggest that ST764 is an emerging highest-risk MRSA clone with multiple AMRs that cause BSI.
Intriguingly, a comparison of the complete genome sequences of ST764 with its ancestral lineage ST5 revealed frequent detection of a large chromosomal inversion (24% in 25 complete genomes of ST764 compared to the reference genome N315 in this study vs. 3.4% in 147 of ST5 in the NCBI public database) (Supplementary Fig. 10 and Supplementary Data 4). In prokaryotes, large chromosomal inversions are associated with phenotypic changes in bacterial virulence through phase variation37,38,39, thereby contributing to bacterial survival strategies and defining evolutionary trajectories. ST764 may have been in the middle of an evolutionary process. CC8 tends to be slightly more common in western Japan; however, it has spread almost nationwide, and when considering the MRSA surveillance results of previous research8,13, it is reconfirmed that CC8 circulates in community8 and hospital settings. Most CC8 isolates from BSIs in 2019–2020 were ST8-SCCmecIV. We revealed through timescale phylogenic analysis that ST8-SCCmecI and multiple ST8-SCCmecIV clones emerged around the same period in the 1980s. In recent years, the USA300 lineage, which is increasingly found in dermatology clinics in Japan, has been isolated from BSIs in healthcare settings at a low rate8. All the detected USA300 strains belonged to the USA300-NA lineage. We first revealed that the emergence of MRSA/J clones associated with invasive infections occurred in Japan almost during the same period as USA300-NA.
This study has some limitations. First, we performed in-depth genomic analysis of only three major clonal lineages—CC1, CC5, and CC8—while excluding minor lineages. Second, geographic and clinical data for isolates from the p1994–2000 period were not available. Therefore, we were unable to compare the geographical distribution of isolates between the two datasets (2019–2020 and 1994–2000) or assess the clinical significance of ST764-SCCmecII relative to ST5-SCCmecII (the N/J clone), which was dominant in 1994–2000 and is the ancestor clone of ST764-SCCmecII. Third, we were not able to collect isolates from the Tohoku-region in eastern Japan and the Shikoku-region in western Japan, leaving a possibility of bias.
Despite these limitations, this is the first study revealing that MRSA populations causing BSIs in Japan are primarily composed of three clonally expanded CC lineages, each emerging and spreading at distinct times and places. This study further identified ST764-SCCmecII-t1084 as a clone associated with a high mortality rate and characterized its evolutionary trajectory. In summary, our study provides a blueprint for national genomic surveillance that integrates clinical data and enables the identification and evolutionary characterization of a high-risk clone.
Methods
Study design
This study integrates BSI-derived S. aureus isolates with their associated clinical data collected through our genomic surveillance (the Japan Antimicrobial Resistant Bacterial Surveillance for S. aureus, JARBS-SA). Therefore, the present study complies with ethical guidelines for research involving human subjects. This study was approved by the Medical Research Ethics Committee of the National Institute of Infectious Diseases (NIID) (approval no. 1251). Although approval was granted for the overall study, each participating hospital obtained approval from the respective ethical approval committee. Regarding informed consent, an opt-out approach (a method where research information is disclosed on websites, providing research subjects the opportunity to decline participation) was implemented at each participating medical institution. All S. aureus isolates were anonymized and individually numbered when isolated from blood cultures. All data and isolates were fully anonymized before being sent to the Antimicrobial Resistance Research Center (AMR-RC)-NIID.
We recruited 63 Japanese medical institutions through the National Hospital Organization. S. aureus isolates were detected in two or more blood samples obtained at the same time, collected at each hospital between April 2019 and July 2020. In total, 798 isolates were collected from 55 hospitals by AMR-RC-NIID. Of the 798 strains collected, eight S. argenteus and one S. epidermidis isolates were re-identified by MALDI biotyper (Bruker Daltonics, Billerica, MA) at the AMR-RC-NIID and excluded from subsequent analyses. Patient information used in this study included age, sex and/or gender, race, date of admission, underlying medical condition, presence or absence of diabetes, presence or absence of dialysis, medical history, surgical history (30 days to the time of blood culture), presence or absence of injection drug use, presence or absence of HIV infection, history of influenza (within 2 weeks), infection site, date of discharge or death, and 30-day mortality rate (from the time of blood culture collection). Although two sets of blood culture tests are usually performed, for samples with positive strains in both vials, the S. aureus isolate from the first vial was used for whole-genome sequencing and integrated analysis with clinical data, and the isolate from the second vial was excluded. Only first-time isolates from the same patient were used, and S. aureus re-isolated multiple times were excluded. Ultimately, of the 789 S. aureus strains, 209 strains were excluded, and 580 isolates were analyzed. The following data regarding S. aureus isolates were collected from participating medical institutions: day of isolation from blood culture, day of admission, infection focus, underlying disease, and day of discharge or death (Supplementary Data 1). For comparative analysis of BSI-derived S. aureus isolates from decades ago, we used a nationwide collection (also known as the YK-Collection) of BSI-derived S. aureus isolates from 1994 to 2000 described in a previous study21. This collection was stored in the Japan Antimicrobial Resistant Bacterial Bank (JARBB) (https://jarbb.jp/en/about/) at NIID-AMR-RC. We selected 183 BSI-derived S. aureus isolates from 1994 to 2000 (Supplementary Data 2). All isolates were stored at −80 °C in a preservation medium supplemented with 30% (vol/vol) glycerol and cultured at 37 °C in tryptone soy broth.
DNA extraction and whole-genome sequencing
All isolates were subcultured from glycerol stocks onto tryptic soy agar at 37 °C overnight. A single colony was picked up and cultured at 37 °C in TSB for 12 h. Genomic DNA was extracted from the liquid culture using lysostaphin (FUJIFILM Wako Pure Chemical Corp., Osaka, Japan) and Agencourt AMPure XP (Beckman Coulter Inc., Brea, CA, USA), according to the manufacturer’s instructions. Short-read DNA libraries were prepared for Illumina sequencing using the Enzymatic 5X WGS Fragmentation Mix, 5X WGS Ligase Mix (BioStream Corp, Tokyo, Japan), and the automated NGS preparation system Biomek i7 Workstation (Beckman Coulter Inc., Brea, CA, USA). Short-read Illumina sequencing was performed on the Illumina HiSeq X FIVE platform to generate 150-bp paired-end reads at Macrogen Japan Corp., Tokyo, Japan, and on Illumina MiSeq to generate 300-bp paired-end reads at the NIID-AMR-RC according to the manufacturer’s instructions. For long-read sequencing, genomic DNA from each strain was purified using the Monarch HMW DNA Extraction Kit for Tissue (#T3060; New England BioLabs, Ipswich, MA, USA) following the manufacturer’s instructions. A long-read DNA library was prepared using the SQK-RBK004 Rapid Barcoding Kit [Oxford Nanopore Technologies (ONT), Oxford, UK], and barcoding was performed on GridION (ONT, Oxford, UK) using MinKNOW v21.05.25 and an FLO-MIN106 flow cell (ONT, Oxford, UK).
De novo assembly and annotation
Quality control of the raw sequenced reads was performed using FastQC v0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Illumina reads were assembled into contigs using Shovill v1.0.9 pipeline (available at https://github.com/tseemann/shovill) with the option –trim to produce high-quality draft genomes. The Shovill program performs subsampling of read depth down to 150×, trimming adapters, correcting sequencing errors, and assembling using SPAdes v3.15.540. ONT reads were trimmed using FiltLong (https://github.com/rrwick/Filtlong), assembled with trimmed long reads using Flye v2.9.141, and polished with trimmed Illumina reads using Pilon v1.2442. The quality of the assembled genome sequences was assessed using QUAST v4.043 and CheckM v1.1.044. We performed the taxonomy check of each genome using the dfast_qc v0.4.2 (https://github.com/nigyta/dfast_qc) and confirmed S. aureus. Genome annotation of all the isolates was performed using DFAST-core v1.2.1645.
In silico sequence typing and detection of virulence factor and AMR genes
The ST of each isolate was defined using mlst v2.22.1 (https://github.com/tseemann/mlst), which extracts seven housekeeping genes (arcC, aroE, glpF, gmk, pta, tpi, and yqiL) from the sequence contigs and matches them against characterized STs in the S. aureus PubMLST database (https://pubmlst.org/organisms/staphylococcus-aureus/). PHYLOViZ v2.046 with the geoBURST Full MST algorithm was used to determine CC in the MLST database (as of July 2022). The presence of virulence factors and AMR genes was detected by ABRicate v1.0.1 (https://github.com/tseemann/abricate) with ResFinder47 database 2022-06-09 and VFDB48 database 2022-06-09 and custom virulence factor database for S. aureus49 with 90% identity and 90% query coverage cutoffs. PointFinder v4.1.1150 was used to detect QRDR mutations in the DNA topoisomerase IV subunit A (GrlA) and DNA gyrase A (GyrA) on the chromosome, also with 90% identity and 90% query coverage cutoffs. SCCmec and spa typing was performed using the web-based SCCmecFinder51 and spaTyper v0.2.1 (https://github.com/HCGB-IGTP/spaTyper) with default settings. GenomeMatcher v3.0.252 was used for chromosome sequence comparison and visualization.
Phylogenetic and clustering analysis
A phylogenetic tree of the core genome alignment was constructed using the kSNP3.053 algorithm without reference genomes. Genetic population structure analysis was performed by partitioning the isolates into SCs of genetically similar individuals, using the Bayesian hierarchical clustering program FastBAPS package v1.0.854 in R v4.2.2 (R Core Team. 2019. R: Language and environment for statistical computing; available at https://www.R-project.org/). A maximum-likelihood (ML) phylogenetic tree was constructed using RAxML-NG v1.0.155 with the best model inferred by ModelTest-NG v0.1.756 and 100 bootstrap replicates. The ML trees were rooted at the midpoint and visualized using Figtree v1.4.4 (available at http://tree.bio.ed.ac.uk/software/figtree).
Time-calibrated phylogeny and population demographic analysis
To perform temporal analysis of the CC1, CC5, and CC8 lineages individually using our collection and NCBI public database (Supplementary Data 3), snippy v4.6.0 (available at https://github.com/tseemann/snippy) was used to perform reference-based mapping and identify SNPs for CC1, CC5, and CC8 with snippy-multi script and default parameters; MSSA476 (Assembly accession no. BX571857), N315 (accession no. BA000018), and JH4899 (accession no. AP014921) were used as references for CC1, CC5, and CC8, respectively. The Core SNP aligned sequence files of each CC were generated using snippy-core and snippy-clean_full_aln scripts. Recombination-free aligned sequences were generated using Gubbins v3.2.057 for each SC lineage. ML trees were checked using a Figtree (Supplementary Fig. 5). To reduce the computational intensity, we constructed dated phylogenies from randomly sampled datasets, each of which includes 150–190 isolates. We then investigated the temporal signals in the ML trees for each SC lineage using TempEst58 to assess the linear relationship between the root-to-tip distance and year of isolation. A time-calibrated phylogenetic tree and effective population size over time (including the age of the most recent common ancestor [MRCA]) were estimated using BEAST2 v2.6.759 and BEAST X v10.5.060. Different models were tested for HKY nucleotide substitution, molecular clock (strict and relaxed), and demographic growth (Bayesian skyline and SkyGrid). Each model was run for 500 million generations with sampling every 5000 steps on the Markov Chain Monte Carlo (MCMC). The models with a relaxed clock failed to achieve convergence in the MCMC, as more than half of the model parameters had an ESS <200. We selected the skyline tree prior with a strict clock model for CC1, CC5, and CC8 as the best-fitting model because a comparison of the models, based on the Bayes factor, strongly supported the skyline model over the SkyGrid model (Bayes factor >20). The effective sample size for all parameters was >200, and sufficient chain mixing was confirmed using Tracer v1.7.161. The log-combiner program in the BEAST package was subsequently used to combine the results of these runs. The final tree was output and annotated using FigTree v1.4.4.
Antimicrobial susceptibility testing
The minimum inhibitory concentrations of ampicillin (ABPC), MPIPC, CEZ, CMZ, imipenem (IPM), ampicillin/sulbactam (ABPC/SBT), GM, ABK, EM, CLDM, MINO, LVFX, vancomycin (VCM), teicoplanin (TEIC), daptomycin (DAP), sulfamethoxazole/trimethoprim, FOM, rifampicin (RFP), linezolid (LZD), mupirocin (MUP), cefoxitin (CFX), and I-CLDM were measured via broth microdilution testing using a MicroScan Pos series panel for MicroScan WalkAway96 System (Beckman Coulter Inc., Brea, CA, USA) according to the manufacturer’s instructions. AMR to MPIPC, CEZ, CMZ, GM, EM, CLDM, MINO, and LVFX was determined according to the breakpoints defined in the CLSI guidelines (Thirty-First Edition: M100). Since the CLSI guidelines do not provide interpretive criteria for ABK MICs, GM was used as a reference.
Statistical analysis
Statistical analyses were performed using Pearson’s χ2 test in R program v4.0.3 (R Core Team, 2019; R: Language and environment for statistical computing, https://www.R-project.org/) and JMP version 13.2.1 (SAS Institute, Cary, NC, USA). The diversity parameters of ST types were determined with the Shannon diversity index using R. Statistical analyses of the number of ARGs were conducted using the ggplot2 package ver. 3.3.662 in R. Survival and regression analyses were conducted using JMP.
Kendall’s W was calculated in the R program.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All genome sequence raw data generated in this study have been deposited in GenBank/EMBL/DDBJ under the BioProject accession number PRJDB15501. The metadata used in this study are provided in the Supplementary Information/Supplementary Data files. All other relevant data are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Turner, N. A. et al. Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat. Rev. Microbiol. 17, 203–218 (2019).
Anderson, D. J. et al. Bloodstream infections in community hospitals in the 21st century: a multicenter cohort study. PLoS ONE 9, e91713 (2014).
Almeida Junior, E. R. D., Braga, I. A., Filho, P. P. G. & Ribas, R. M. Multicentre surveillance of epidemiologically important pathogens causing nosocomial bloodstream infections and pneumonia trials in Brazilian adult intensive care units. J. Med. Microbiol. https://doi.org/10.1099/jmm.0.001654 (2023).
Kourtis, A. P. et al. Vital signs: epidemiology and recent trends in methicillin-resistant and in methicillin-susceptible Staphylococcus aureus bloodstream infections – United States. MMWR Morb. Mortal. Wkly Rep. 68, 214–219 (2019).
Tsuzuki, S. et al. National trend of blood-stream infection attributable deaths caused by Staphylococcus aureus and Escherichia coli in Japan. J. Infect. Chemother. 26, 367–371 (2020).
Argimón, S. et al. Interating whole-genome sequencing within the National Antimicrobial Resistance Surveillance Program in the Philippins. Nat. Commun. 11, 2719 (2020).
Yamada, K. et al. Clinical features of bacteremia caused by methicillin-resistant Staphylococcus aureus in a tertiary hospital. Tohoku J. Exp. Med. 224, 61–67 (2011).
Zuo, H., Uehara, Y., Lu, Y., Sasaki, T. & Hiramatsu, K. Genetic and phenotypic diversity of methicillin-resistant Staphylococcus aureus among Japanese inpatients in the early 1980s. Sci. Rep. 11, 5447 (2021).
Kaku, N., Sasaki, D., Ota, K., Miyazaki, T. & Yanagihara, K. Changing molecular epidemiology and characteristics of MRSA isolated from bloodstream infections: nationwide surveillance in Japan in 2019. J. Antimicrob. Chemother. 77, 2130–2141 (2022).
Takano, T. et al. A new local variant (ST764) of the globally disseminated ST5 lineage of hospital-associated methicillin-resistant Staphylococcus aureus (MRSA) carrying the virulence determinants of community-associated MRSA. Antimicrob. Agents Chemother. 57, 1589–1595 (2013).
Kondo, S. et al. Molecular characterization of methicillin-resistant Staphylococcus aureus genotype ST764-SCCmec type II in Thailand. Sci. Rep. 12, 2085 (2022).
Xiao, Y. et al. Phylogenetic analysis and virulence characteristics of methicillin-resistant Staphylococcus aureus ST764-SCCmec II: an emerging hypervirulent clone ST764-t1084 in China. Emerg. Microbes Infect. 12, 2165969 (2023).
Hamada, M. et al. Increased incidence and plasma-biofilm formation ability of SCCmec type IV methicillin-resistant Staphylococcus aureus (MRSA) isolated from patients with bacteremia. Front. Cell. Infect. Microbiol. 11, 602833 (2021).
Kishita, M. et al. Increase in the frequency of community-acquired methicillin-resistant Staphylococcus aureus clones among inpatients of acute care hospitals in the Kyoto and Shiga regions, Japan. J. Infect. Chemother. 29, 458–463 (2023).
Iwao, Y. et al. The emerging ST8 methicillin-resistant Staphylococcus aureus clone in the community in Japan: associated infections, genetic diversity, and comparative genomics. J. Infect. Chemother. 18, 228–240 (2012).
Ishitobi, N. et al. Fatal case of ST8/SCCmecIVl community-associated methicillin-resistant Staphylococcus aureus infection in Japan. New Microbes New Infect. 26, 30–36 (2018).
Hagiya, H. et al. Comprehensive analysis of systemically disseminated ST8/non-USA300 type community-acquired methicillin-resistant Staphylococcus aureus infection. Intern. Med. 53, 907–912 (2014).
Hisatsune, J., Hagiya, H., Shiota, S. & Sugai, M. Complete genome sequence of systemically disseminated sequence type 8 staphylococcal cassette chromosome mec type IVl Community-acquired methicillin-resistant Staphylococcus aureus. Genome Announc. 5, e00852–17 (2017).
Tsujiwaki, A. et al. Epidemiology of methicillin-resistant Staphylococcus aureus in a Japanese neonatal intensive care unit. Pediatr. Int. 62, 911–919 (2020).
Mitsuboshi, S. et al. Regional outbreak of methicillin-resistant Staphylococcus aureus ST2725-t1784 in rural Japan. Infect. Control Hosp. Epidemiol. 42, 1294–1296 (2021).
Tateda, K. et al. Investigation of the susceptibility trends in Japan to fluoroquinolones and other antimicrobial agents in a nationwide collection of clinical isolates: a longitudinal analysis from 1994 to 2016. J. Infect. Chemother. 25, 594–604 (2019).
Kayama, S. et al. National genomic surveillance integrating standardized quantitative susceptibility testing clarifies antimicrobial resistance in Enterobacterales. Nat. Commun. 14, 8046 (2023).
Murphy, E., Huwyler, L., de Freire Bastos. & Mdo, C. Transposon Tn554: complete nucleotide sequence and isolation of transposition-defective and antibiotic-sensitive mutants. EMBO J. 4, 3357–3365 (1985).
Baines, S. L. et al. Rapid emergence and evolution of Staphylococcus aureus clones harbouring fusC-containing staphylococcal cassette chromosome elements. Antimicrob. Agents Chemother. 60, 2359–2365 (2016).
Strauß, L. et al. Origin, evolution, and global transmission of community-acquired Staphylococcus aureus ST8. Proc. Natl Acad. Sci. USA 114, E10596–E10604 (2017).
Chambers, H. F. & Deleo, F. R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 7, 629–641 (2009).
Hosaka, Y. et al. Surveillance of multidrug resistance phenotypes in Staphylococcus aureus in Japan and correlation with whole-genome sequence findings. J. Hosp. Infect. 123, 34–42 (2022).
Yamaguchi, T. et al. Changes in the genotypic characteristics of community-acquired methicillin-resistant Staphylococcus aureus collected in 244 medical facilities in Japan between 2010 and 2018: a nationwide surveillance. Microbiol. Spectr. 10, e0227221 (2022).
Miura, Y. et al. Epidemiological trends observed from molecular characterization of methicillin-resistant Staphylococcus aureus isolates from blood cultures at a Japanese university hospital, 2012-2015. Microb. Drug Resist. 24, 70–75 (2018).
Earls, M. R. et al. A novel multidrug-resistant PVL-negative CC1-MRSA-IV clone emerging in Ireland and Germany likely originated in south-eastern Europe. Infect. Genet. Evol. 69, 117–126 (2019).
Piao, C., Karasawa, T., Totsuka, K., Uchiyama, T. & Kikuchi, K. Prospective surveillance of community-onset and healthcare-associated methicillin-resistant Staphylococcus aureus isolated from a university-affiliated hospital in Japan. Microbiol. Immunol. 49, 959–970 (2005).
Miragaia, M. et al. Genetic diversity of arginine catabolic mobile element in Staphylococcus epidermidis. PLoS ONE 4, e7722 (2009).
Urushibara, N. et al. Novel structures and temporal changes of arginine catabolic mobile elements in methicillin-resistant Staphylococcus aureus genotypes ST5-MRSA-II and ST764-MRSA-II in Japan. Antimicrob. Agents Chemother. 60, 3119–3122 (2016).
Kawamura, K. et al. Spread of seb-positive methicillin-resistant Staphylococcus aureus SCCmec type II-ST764 among elderly Japanese in nonacute care settings. Microb. Drug Resist. 25, 915–924 (2019).
Mitsumoto-Kaseida, F. et al. Clinical and pathogenic features of SCCmec type II and IV methicillin-resistant Staphylococcus aureus in Japan. J. Infect. Chemother. 23, 90–95 (2017).
Inagawa, T. et al. Genome characterization of Staphylococcus aureus isolated from patients admitted to intensive care units of a tertiary care hospitals: epidemiological risk of nasal carriage of virulent clone during admission. Microbiol. Spectr. 12, e0295023 (2024).
Kojic, M. et al. Large chromosome flip-flop reversible inversion mediates phenotypic sqitching of expression of antibiotic resistance in Lactococci. Microbiol. Res. 241, 126583 (2020).
Le, V. V. H., León-Quezada, R. I., Biggs, P. J. & Rakonjac, J. A large chromosomal inversion affects antimicrobial sensitivity of Escherichia coli to sodium deoxycholate. Microbiology. https://doi.org/10.1099/mic.0.001232 (2022).
Li, H. et al. Polymyxin resistance caused by large-scale genomic inversion due to IS26 intramolecular translocation in Klebsiella pneumoniae. Acta Pharm. Sin. B 13, 3678–3693 (2023).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Tanizawa, Y., Fujisawa, T. & Nakamura, Y. DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 34, 1037–1039 (2018).
Ribeiro-Gonçalves, B., Francisco, A. P., Vaz, C., Ramirez, M. & Carriço, J. A. PHYLOViZ Online: web-based tool for visualization, phylogenetic inference, analysis and sharing of minimum spanning trees. Nucleic Acids Res. 44, W246–W251 (2016).
Florensa, A. F., Kaas, R. S., Clausen, P. T. L. C., Aytan-Aktug, D. & Aarestrup, F. M. ResFinder – an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb. Genom. 8, 000748 (2022).
Chen, L., Zheng, D., Liu, B., Yang, J. & Jin, Q. VFDB 2016: hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 44, D694–D697 (2016).
Obata, S. et al. Comprehensive genomic characterization of Staphylococcus aureus isolates from atopic dermatitis patients in Japan: correlations with disease severity, eruption type, and anatomical site. Microbiol. Spectr. 11, e0523922 (2023).
Zankari, E. et al. PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 72, 2764–2768 (2017).
Kaya, H. et al. SCCmecFinder, a web-based tool for typing of staphylococcal cassette chromosome mec in Staphylococcus aureus using whole-genome sequence data. mSphere 3, e00612–e00617 (2018).
Ohtsubo, Y., Ikeda-Ohtsubo, W., Nagata, Y. & Tsuda, M. GenomeMatcher: a graphical user interface for DNA sequence comparison. BMC Bioinform. 9, 376 (2008).
Gardner, S. N., Slezak, T. & Hall, B. G. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31, 2877–2878 (2015).
Tonkin-Hill, G., Lees, J. A., Bentley, S. D., Frost, S. D. W. & Corander, J. Fast hierarchical Bayesian analysis of population structure. Nucleic Acids Res. 47, 5539–5549 (2019).
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019).
Darriba, D. et al. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294 (2020).
Croucher, N. J. et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15 (2015).
Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2, vew007 (2016).
Bouckaert, R. et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15, e1006650 (2019).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, New York, 2016).
Acknowledgements
We are grateful to all the hospitals participating in JARBS-SA. We are grateful to Yumiko Hosaka for the discussions and to the following staff for technical contributions to this project: Manami Tsunoi, Eiko Anzai, Takahisa Ishizuka, Mayumi Sasada, Koichi Shimakawa, Sayoko Kawakami, Yoshie Taki, Satoyo Wakai, Sadao Aoki, Mikako Nakazawa, Emi Fujimura, Noriko Sakamoto, Elahi Shaheem, and Chika Arai. This work was supported by the Research Program on Emerging and Reemerging Infectious Diseases of the Japan Agency for Medical Research and Development (AMED) under grant number 21fk0108604.
Author information
Authors and Affiliations
Consortia
Contributions
M.S. (Sugai) conceptualized this study. M.S. (Sugai), M.S., J.H. (Hisatsune), T.K. (Kajihara), H.K. (Kitagawa), H.K. (Ohge), T.M. (Mizukami), T.T. (Takahashi), and F.K. (Kawano) designed this study. T.M., T.T., F.K., and JARBS-SA Consortium contributed to collecting the isolates and clinical data. S.K. (Kutsuno), Y.I. (Iwao), and K.I. (Ishida-Kuroki) were involved in species identification. J.H., Y.S. (Sugawara) contributed to genome sequencing. J.H., N.K. (Kitamura), T.K., and K.Y. (Yahara) analyzed the data. J.H. contributed to the information processing and database construction. J.H. was the major contributor to the writing of the manuscript, and M.S., K.Y., and S.K. (Kayama) significantly contributed to editing the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Taj Azarian, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hisatsune, J., Kutsuno, S., Iwao, Y. et al. Staphylococcus aureus ST764-SCCmecII high-risk clone in bloodstream infections revealed through national genomic surveillance integrating clinical data. Nat Commun 16, 2698 (2025). https://doi.org/10.1038/s41467-025-57575-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57575-2
