
Abstract
Protein synthesis by ribosomes produces functional proteins but also serves diverse regulatory functions, which depend on the coding amino acid sequences. Certain nascent peptides interact with the ribosome exit tunnel to arrest translation and modulate themselves or the expression of downstream genes. However, a comprehensive understanding of the mechanisms of such ribosome stalling and its regulation remains elusive. In this study, we systematically screen for unidentified ribosome arrest peptides through phenotypic evaluation, proteomics, and mass spectrometry analyses, leading to the discovery of the arrest peptides PepNL and NanCL in E. coli. Our cryo-EM study on PepNL reveals a distinct arrest mechanism, in which the N-terminus of PepNL folds back towards the tunnel entrance to prevent the catalytic GGQ motif of the release factor from accessing the peptidyl transferase center, causing translation arrest at the UGA stop codon. Furthermore, unlike sensory arrest peptides that require an arrest inducer, PepNL uses tryptophan as an arrest inhibitor, where Trp-tRNATrp reads through the stop codon. Our findings illuminate the mechanism and regulatory framework of nascent peptide-induced translation arrest, paving the way for exploring regulatory nascent peptides.
Similar content being viewed by others


Introduction
Translation is an essential process that converts nucleotide sequences into amino acid sequences, facilitating the expression of a diverse array of genes with various functions inside the cell. This complex process is orchestrated by the ribosome, a universal protein factory found in all cellular forms of life on Earth. In bacteria, ribosomes assemble an initiation complex upon encountering the Shine-Dalgarno sequence in mRNA upstream of the initiation codon1. Subsequently, ribosomes catalyze the transfer of the growing peptide chain from an aminoacyl/peptidyl-tRNA at the P-site to an aminoacyl-tRNA at the A-site2. Upon encountering a stop codon, peptide release factors (RF) recognize the stop codon to trigger the hydrolysis of the ester bond between the nascent peptide and tRNA3,4,5,6,7,8. This event leads to the dissociation of the polypeptide chain and terminates translation.
While ribosomes were once considered robust protein factories capable of translating any mRNA sequence, it is now evident that this is not always the case. For example, the nascent polypeptide chain (nascent peptide) within the ribosome tunnel can cause translational difficulties9,10,11. The nascent peptide exit tunnel (NPET) is primarily composed of negatively charged rRNA and contains a constriction site formed by ribosomal proteins12. Certain amino acid sequences in the nascent peptide closely interact with the complex tunnel structures, causing elongation stalling, a phenomenon termed “translation arrest”13,14. Although translation arrest appears to be disadvantageous and might be eliminated during evolution, conserved translation arrests persist among organisms, which may utilize it as part of their survival strategies15,16,17,18,19.
The majority of ribosome arrest peptides (RAPs) discovered so far are encoded within upstream ORFs (uORFs)9,10. The occurrence of translation arrest often depends on environmental changes inside and outside the cell and serves to regulate the expression of downstream genes. For example, Escherichia coli TnaC induces translation arrest only when the intracellular tryptophan concentration is high20. In this arrest, excessive tryptophan enters the ribosome tunnel and alters the structure of the peptidyl transferase center (PTC) through interactions with the nascent peptide and the tunnel, thus inhibiting translation termination21,22. In this way, TnaC regulates the expression of tryptophanase (TnaA) in a manner dependent on intracellular tryptophan as an “arrest inducer”. RAPs that control the expression of downstream genes like TnaC are widely found from prokaryotes to eukaryotes and are considered useful and universal gene expression regulation mechanisms in life9,10,11. The search for such unidentified RAPs has been advanced through sequence and gene structure conservation analyses23,24,25,26, as well as large-scale translatome analyses using ribosome profiling27,28,29. However, the overall landscape of nascent peptide-dependent translation regulation remains enigmatic.
In addition to the diversity of the sequences that cause translation arrest, the molecular mechanisms of arrest also vary considerably. Recent cryo-electron microscopy (cryo-EM) structural analyses have significantly clarified the molecular details of translation arrest, revealing the intricate interactions between RAPs and the ribosome tunnel at resolutions approaching 2.0 Å21,22,30,31,32,33,34,35,36,37,38. Structural studies of arrest peptides, including TnaC, SpeFL, and MsrDL have revealed case examples of their mechanisms for sensing metabolites and antibiotics, as well as their inhibition of release factors. However, structural analyses of ribosomes arrested by RAPs are currently limited to typical examples, and further studies are needed to systematically understand the mechanism of translation arrest.
In this study, we comprehensively examined whether small ORFs recently identified from ribosome profiling exhibit RAP activity. Through phenotype characterization, proteomic analysis, and mass spectrometry (MS) analyses of nascent peptides, E. coli PepNL (14 aa) and NanCL (14 aa) were demonstrated to induce translation arrest at their stop codons. Focusing on PepNL, our cryo-EM analysis revealed an unusual structure in which the nascent PepNL peptide folds back towards the entrance of the ribosome tunnel, contrary to the usual orientation. This distinctive conformation distorts the structure of the subsequent nascent peptide, leading to a steric clash with the GGQ motif of RF2 and thereby inhibiting translation termination. Unlike previously studied RAPs such as TnaC, PepNL arrested the ribosome solely based on its amino acid sequence, without requiring any arrest inducer. This study not only introduces a distinct mode of translation arrest but also provides insights into the regulatory mechanism of the PepNL-dependent arrest, which is achieved by tryptophanyl-tRNA.
Results
Screening of uncharacterized ribosome arrest peptides by overexpression phenotype
The overexpression of TnaC, a tryptophan-dependent ribosome arrest peptide (RAP), reportedly impedes cell growth due to the depletion of tRNAPro captured within the arrested ribosome (Fig. 1a)39. To identify uncharacterized RAPs, we investigated whether the overexpression of RAPs other than TnaC would inhibit cell growth, as observed with TnaC in nutrient-rich media. Notably, the overexpression of established RAPs in E. coli cells growing on LB agar, such as SecM13 and TnaC14, resulted in significant growth inhibition (Fig. 1b). In contrast, SpeFL, which requires a sufficient supply of ornithine for arrest induction (Fig. 1b)34, or the TnaC variant harboring the arrest-attenuating P24A mutation, failed to induce growth inhibition (Supplementary Fig. 1a). MgtL, another type of regulatory nascent peptide that triggers premature translation abortion, had a negligible effect on cell growth (Fig. 1b)40. Based on these results, we inferred that the cytotoxicity induced upon overexpression could serve as one of the indicators of RAP activity.

a Working hypothesis for screening uncharacterized ribosome arrest peptides. Serial dilution spot assay for evaluating the cytotoxicity when established (b) or recently identified (c) sORFs were overexpressed in E. coli cells. A representative of three independent experiments (n = 3) is shown. d Schematic illustration of a comparative quantitative proteomic analysis. Each sORF was overexpressed in E. coli cells, and all samples were subjected to the LC-MS/MS-based proteome analysis. The proteomic changes by the overexpression were quantified as the fold changes relative to the cells harboring an empty vector. e PCA score plot of PC1 and PC2. The numbers in the x- and y-axis labels represent the proportion of the variances of PC1 and PC2, respectively. Vector means the data from fold changes of another vector control against a vector control. Colors (green, blue, and red) correspond to the clustering by a hierarchical clustering analysis (Supplementary Fig. 1i).
In this study, we analyzed a total of 38 candidates, including 26 recently annotated small open reading frames (sORFs)27,28,29,41,42,43 and 12 putative sORFs identified by the GWIPS-viz browser (Supplementary Fig. 1b–d)44. Upon overexpression of these 38 candidates in E. coli cells, 18 sORFs induced growth inhibition (Fig. 1c). The presence or absence of cytotoxicity did not correlate with their regulatory effects on downstream gene expression (Fig. 1c, colored frame, Supplementary Fig. 1e).
Subsequently, we investigated whether the examined sORFs induce stress responses due to excessive translation arrest. We anticipated that TnaC overexpression would inhibit translation elongation by tRNAPro depletion. In such a situation, the expression of cold shock proteins (CSPs) is expected to increase, as observed in E. coli cells exposed to sublethal concentrations of chloramphenicol (Cm) or tetracycline (Tet), which globally inhibit translational elongation45,46,47,48. Based on this hypothesis, we conducted a comparative and quantitative proteomic analysis for E. coli cells overexpressing each of the sORFs, and analyzed their proteomic landscape rearrangement (Fig. 1d). The obtained dataset (Supplementary Data 1 and 2) was subjected to PCA (principal component analysis), and a plot illustrating the similarity of the proteomic rearrangements is depicted in Fig. 1e. Parallel clustering analysis categorized our datasets into three clusters: I. induction of CSPs expression (blue circles), II. induction of HSPs expression (red circles), and III. limited variation (green circles) (Fig. 1e, Supplementary Fig. 1f–i). Overexpression of RAPs such as TnaC and SecM, as well as treatment with translation inhibitors like Cm, induced the expression of CSPs, including CspA, DeaD, and InfC (Supplementary Fig. 1h)49,50,51,52. These datasets were grouped in the same cluster, indicating that the inhibition of translation elongation often induces the expression of CSPs (Fig. 1eblue circles, Supplementary Fig. 1i). A similar “cold shock”-like response was observed for 12 sORF candidates, suggesting the possible occurrence of translation arrest during their expression. Meanwhile, several sORFs such as YqgB induced the expression of heat shock proteins (HSPs) that strongly respond to aggregation, including IbpA and ClpB (Fig. 1ered circles, Supplementary Fig. 1h and i)45,53,54. Overexpression of these sORFs, unlike TnaC, might inhibit cell growth due to accumulation of the short peptides.
LC-MS/MS measurements of the peptidyl-tRNA molecules
We further evaluated the RAP activities of candidate sORFs by an LC-MS/MS analysis of the peptidyl-tRNA intermediates that accumulate due to translation arrest. After the overexpression of sORFs in E. coli cells, total RNA was extracted and purified by silica adsorption. Peptide fragments ester-bonded to tRNA were then subjected to alkaline hydrolysis, peptidase digestion, and LC-MS/MS analysis (Fig. 2a)55,56. Identification of the MS2 spectra of the 19thK / IVDHRP25th fragment of TnaC (Fig. 2b) or 149thK / GSTPVWISQAQGIRAG165th fragment of SecM (Fig. 2c), which coincides with the established translation arrest site, demonstrates the effectiveness of this method. Additionally, the identification of Tris-adducted peptide fragments at the C-terminus, which occurs upon alkaline hydrolysis of peptidyl-tRNA, further supports the RAP activities of TnaC and SecM (Supplementary Fig. 2a and b).

a Schematic illustration of the experimental procedure for the identification of peptidyl-tRNA-derived peptides. Each candidate sORF was overexpressed in E. coli cells, and the total RNA fraction in the cell lysate was concentrated with a silica column. The RNA fraction was hydrolyzed by alkaline treatment, and the obtained peptides were identified by protease digestion and subsequent LC-MS/MS. b–e MS/MS spectra of peptides derived from peptidyl-tRNAs. Amino acid sequences above the graphs represent the positions of the detected peptides (blue areas indicate the detected peptide). P and A below the amino acid sequence represent the plausible positions of the P- and A-sites in the ribosome during translation arrest, respectively. In the graphs, the peaks of the b- and y-fragment ions are shown in red and blue, respectively.
The LC-MS/MS analysis of peptidyl-tRNA was extended to include sORF candidates, resulting in the identification of peptide fragments indicative of stalling at various sites (Supplementary Data 3). Many of the identified peptide fragments were inconsistent with the distribution of ribosomes on the mRNA in the ribosome profiling. In contrast, the MS2 spectra indicating translation arrest at stop codons of the sORF candidates pepNL and nanCL were in line with the profiling results (Fig. 2d and e, Supplementary Fig. 1b). In addition, Tris-adducted peptide fragments at the C-terminus were detected for these two sORFs, as in the cases of TnaC and SecM (Supplementary Fig. 2c and d). Based on these LC-MS/MS results and the growth inhibition results shown in Fig. 1, we concluded that pepNL and nanCL encode RAPs that induce translation arrest at stop codons.
Structure of the PepNL-arrested ribosome
In this study, we focused on PepNL, which induced a particularly robust translation stress response (Fig. 1e). PepNL, the 14 amino acid ORF whose AUG initiation codon was verified by the reporter assay (Supplementary Fig. 3a and b), is encoded upstream of the aminopeptidase pepN gene. The frameshift mutation abolished the cytotoxicity (Fig. 3a) and the accumulation of peptidyl-tRNA (Fig. 3b), indicating that PepNL arrests translation through the context of its amino acid sequence. Moreover, the frameshift mutation increased the expression of PepN, underscoring the regulatory role of pepNL in a translation arrest-dependent manner (Supplementary Fig. 1e and 3c).

a Serial dilution spot assay to assess the cytotoxicity upon overexpression of wild-type PepNL (WT) or its frameshift mutant (FS) in E. coli cells. b The PepNL-tRNA accumulated in E. coli cells expressing the indicated pepNL variants was detected by northern blotting using an anti-tRNAAsp probe. An asterisk indicates the unprocessed rrnC or rrnH transcripts that include the unprocessed tRNAAsp sequence. (#) c The wild-type or frameshifted pepNL mRNA was translated by PUREfrex in the absence of tryptophan, and the PepNL-arrested ribosome was visualized by toeprint analysis. Thiostrepton, which inhibits translation elongation, was pre-included where indicated. The pepNL mRNA was translated in the absence of release factors (RF−) to prepare the ribosomes stalled at the stop codon for the position marker (lane 5). (#) d Overall cross section of the cryo-EM density map at the peptide exit tunnel of the 70S ribosome (gray), showing P-tRNA (green), RF2 (cyan), and PepNL peptide (orange). e, f PepNL peptide and interacting 23S rRNA nucleotides in the ribosome exit tunnel. The N-terminus and C-terminus of PepNL were labeled as “N” and “C”, respectively. Close-up views of the intramolecular interactions within the PepNL peptide (g) and intermolecular interactions between the PepNL peptide and 23S rRNA nucleotides (h). g, panel 1: A hydrophobic interaction between Ile3 and Tyr9. g, panel 2: β-sheet-like interactions involving Lys2 with Ala10 and Leu4 with Ile8. h, panel 1: A hydrophobic interaction between Ile3 and U2609. h, panel 2: A hydrophobic interaction between Leu4 and A2062. h, panel 3: Hydrophobic interactions between Ile8 and A2058-A2059. h, panel 4: A hydrophobic interaction between Tyr9 and U2610. i Structural comparison of the PepNL nascent peptide (orange) with nascent peptides that lack RAP activity {PDB: 8CVJ (nascent peptide sequence: fMSEAC, pink) and 8CVL (fMTHSMRC, purple)}. j Serial dilution spot assay to assess the cytotoxicity upon overexpression of wild-type PepNL (WT) or its variants carrying the indicated amino acid substitution in E. coli cells. (#) A representative of three independent experiments (n = 3) is shown.
To elucidate the molecular mechanism of PepNL-dependent translation arrest, we analyzed the structure of the ribosome arrested by the PepNL nascent peptide. We translated pepNL mRNA using the reconstituted cell-free translation system (PURE system: PUREfrex v1.0)57 deprived of tryptophan, as discussed later. A toeprint analysis confirmed the accumulation of ribosomes stalled at the stop codon of the pepNL mRNA (Fig. 3c, Supplementary Fig. 3d). This result is further supported by the alleviation of PepNL toxicity when RF2 or tRNAAsp, both of which are trapped in the arrested ribosome, were overexpressed (Supplementary Fig. 3e). To avoid dissociation or release of the stalled ribosomes, we directly applied the in vitro translation mixture to the cryo-EM grids without purification and performed the cryo-EM data collection. The contrast of the ribosomes on the grid was sufficient for the cryo-EM analysis even without purification, resulting in the 3D reconstruction of ribosomes. We obtained four states with different components in the A and P-sites (P-tRNA only: 31%, RF2 and P-tRNA: 25%, EF-Tu•tRNA and P-tRNA: 23%, and empty: 21%, Supplementary Fig. 4). We further analyzed the particles in the RF2 and P-tRNA bound state and P-tRNA bound state, and determined their structures at 2.9 Å resolution (Fig. 3d, Supplementary Fig. 5). In the structure determined with RF2, the arrested 70S ribosome was bound with the mRNA, RF2 at the A-site, and tRNAAsp carrying the nascent PepNL peptide at the P-site (Supplementary Fig. 6a).
All 14 amino acid residues of PepNL were successfully traced in the density, indicating that the PepNL nascent peptide adopts a stable conformation in the exit tunnel. The PepNL peptide forms a mini-hairpin conformation with residues Lys2 to Ala10, which directs the N-terminal residues back toward the PTC but not far enough to reach the constriction site of the exit tunnel (Fig. 3e and f). This distinctive structure is stabilized by intramolecular interactions, including a hydrophobic interaction between Ile3 and Tyr9 (Fig. 3g, panel 1), as well as β-sheet-like main-chain interactions involving Lys2 with Ala10 and Leu4 with Ile8 (Fig. 3g, panel 2). Moreover, intermolecular hydrophobic interactions between PepNL (Ile3, Leu4, Ile8, and Tyr9) and 23S rRNA (U2609, A2062, A2058, A2059, and U2610) also contribute to supporting the structure (Fig. 3h). While the N-terminal portion of PepNL exhibits a compact conformation, the C-terminal part is distorted, as illustrated by a large main-chain shift as compared to nascent peptides that lack RAP activity58 (Fig. 3i) {PDB: 8CVJ (nascent peptide sequence: fMSEAC) and 8CVL (fMTHSMRC)}. The distortion of PepNL further interrupts the hydrogen-bond network between rRNA (A2506 and A2062) and the nascent peptides without RAP activity, resulting in the conformational rearrangement of A2506 and A2062 in the 23S rRNA (Supplementary Fig. 6b and c). These structural observations are supported by the fact that the arrest activity of PepNL was abolished by alanine substitutions of Ile3, Leu4, Ile8, and Tyr9, which are involved in the formation of the β-hairpin loop (Fig. 3j, Supplementary Fig. 3f).
RF2 undergoes a conformational rearrangement
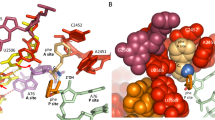
We next focused on the structural rearrangement of RF2. In canonical translation termination, the recognition of the stop codon by domain II of RF2 triggers the positional extension of domain III, inserting the 250thGGQ252nd catalytic motif into the PTC to hydrolyze the ester bond of the peptidyl-tRNA (Fig. 4a)5,6,7,8. Within the PepNL-arrested ribosome, domain II of RF2 properly recognizes the UGA stop codon and the extension of domain III is also observed (Fig. 4a). However, we found a significant difference in the conformation of the apical loop (residues 246-257) in a comparison of our structure with the canonical termination complex59 (PDB: 6C5L) (Fig. 4b). In the canonical structure, the methylated Gln252 in the GGQ motif enters the narrow pocket formed by A2451, C2452, and U2506 of rRNA (Figs. 4b and f). However, in the arrested structure, Gln252 undergoes a 16 Å relocation, as measured between the Nε2 atoms (Fig. 4b). This relocation causes a drastic rearrangement of the apical loop, which adopts an inactive conformation stabilized by hydrogen bonds between the residues in the apical loop (Gly251, Gly252, and Arg256) and rRNA nucleotides (Fig. 4c). In addition to the conformational change of the apical loop, we observed a notable shift of the entire domain III of RF2, by approximately 3 Å toward the L1 stalk, in comparison with its position in the canonical termination complex (Figs. 4a and b). Residues within the shifted region (Arg245, Glu258, Arg262, Gln280, and His281) further stabilize this conformation through hydrogen-bonding interactions with rRNA nucleotides (Figs. 4c–e). Since there are several hydrogen-bonding interactions between domain III and rRNA that are absent in the canonical termination complex, the rearranged and shifted RF2 conformation observed in our structure could be functionally important for general termination regulation.

a Overview of the RF2 structure within the PepNL-arrested ribosome (cyan) compared with the active RF2 (purple, PDB: 6C5L). The P-tRNAAsp (green) and pepNL mRNA (brown) are also shown. b Close-up view of the apical loop conformations of the rearranged and active RF2 states. Gln252s of the GGQ motif are highlighted as sticks with sphere models. The ribosome is shown as a surface model. Hydrogen bond interactions between the apical loop of the rearranged RF2 and 23S rRNA. c: Arg245 and G2508, Gln252 and G2583, Val254 and C2573, Arg256 and U2554, Gly251 and G2553. d: Glu258-Gln280 and U2492, His281 and U2460. e: Arg262 and G2557. f Superimposition of the active RF2 (purple) indicates a steric clash of its Gln252 with Ile13 of PepNL. The distorted Ile13 of the PepNL peptide occupies the pocket formed by A2451, C2452, and U2506 (not shown) of 23S rRNA, blocking the proper accommodation of the RF2 apical loop. The rRNA nucleotides and Ile13 are shown as sphere models. g Schematics representing the mechanism of RF2 inhibition by the PepNL nascent peptide.
The PepNL nascent peptide inhibits RF2 activity through a steric clash with Ile13
Further structural comparisons showed that the side chain of Ile13 in the distorted C-terminus of PepNL nascent peptide is accommodated in the pocket formed by A2451, C2452, and U2506 (Fig. 4f). This pocket is crucial for translation termination, as the 250thGGQ252nd motif of RF2 enters this pocket to cleave the ester bond5,6,7,8,59. Consequently, the distortion of Ile13 shown in Fig. 3i leads to a steric clash with Gln252 of RF2, hampering the accommodation of the GGQ motif in the A2451/C2452/U2506 pocket (Fig. 4f). This steric clash between Ile13 of PepNL and Gln252 of RF2 would induce a drastic rearrangement of the apical loop and the subsequent rearrangement of the entire domain III of RF2. These considerations are supported by the finding that the Ile13 mutation abolished the arrest activity of PepNL (Fig. 3j, Supplementary Fig. 3f).
Taken together, our cryo-EM reconstruction revealed the molecular details underlying the PepNL nascent peptide-mediated inhibition of the translation termination activity of RF2. The translated nascent PepNL peptide forms a compact hairpin structure, with its N-terminus directed back toward the PTC, and stacks inside the exit tunnel (Fig. 4g, left). This stacking distorts the C-terminus of the PepNL nascent peptide, leading to the steric clash between Ile13 of PepNL and Gln252 of RF2 (Fig. 4g, middle). Consequently, this steric clash induces a drastic conformational change in RF2, shifting it into an inactive conformation (Fig. 4g, right).
Read-through of the stop codon serves as an inhibitory mechanism for PepNL-induced translation arrest
Despite our success in determining the structure of the PepNL-arrested ribosome, we initially faced difficulties in preparing the stalled ribosome within the complete PUREfrex system (Fig. 5a, lanes 1 and 2). However, our experiments with the pepNL mutant, harboring a UAG stop codon instead of UGA, revealed robust ribosome stalling both in vitro and in vivo (Fig. 5a, lanes 5 and 6, Supplementary Fig. 7a, and b). Furthermore, the pepNL mutants carrying the altered stop codon exhibited increased cytotoxicity (Supplementary Fig. 7c). These results indicated that the UGA stop codon of pepNL plays a role in attenuating ribosome stalling.

a The wild-type (UGA) or mutated (UAG) pepNL mRNA was translated by PUREfrex in the presence or absence of tryptophan (Trp), and the PepNL-arrested ribosome was visualized by toeprint analysis as shown in Fig. 3c. (#) b Schematics of pepNL mRNAs analyzed. c The pepNL mRNAs indicated in Fig. 5b were translated by PUREfrex in the absence (lanes 3 and 4) or presence of tryptophan (lanes 1, 2, 5, 6, 7, and 8). The 35S-methionine-labeled translation products were separated by neutral pH SDS-PAGE with optional RNase A (RN) pretreatment. The truncated pepNL NS−1 (14 aa) and NS−2 (27 aa) mRNA were also analyzed to serve as size markers (lanes 5 to 8). The PepNL (14 aa) or PepNL (read-through: RT) peptidyl-tRNA and PepNL (14 aa) or PepNL (RT) peptide are schematically indicated. The asterisk denotes the fMet-tRNA. (#) d The pepNL-stop (UGA/UAA/UAG)-lacZ mRNA was expressed in E. coli cells, and the frequency of stop codon read-through was calculated as described in the Methods section. The mean values ± SE estimated from three independent biological replicates (n = 3) are shown. e The pepNL mRNA was translated by PUREfrex without tryptophan and release factors for 30 min at 37°C. Afterward, a final 25 µM of tryptophan was added and further incubated for the indicated duration. The 35S-methionine-labeled translation products were analyzed as shown in Fig. 5c. The asterisk denotes the fMet-tRNA. (#) f Schematic illustration of the Trp-tRNATrp−dependent inhibition of the PepNL-induced translation arrest. RF2 inefficiently terminates the translation of pepNL due to the steric clash shown in Fig. 4. However, in the presence of sufficient tryptophan, the Trp-tRNATrp decodes the UGA of pepNL, leading to the stop codon read-through. Two potential scenarios could explain this event: 1) Trp-tRNATrp initiates read-through before the hairpin folds, which could otherwise inhibit Trp-tRNATrp accommodation (Supplementary Fig. 7f); or 2) Trp-tRNATrp releases the ribosome stalled by the hairpin-shaped PepNL, alleviating the translation arrest at a moderate rate. (#) A representative of three independent experiments (n = 3) is shown.
To investigate this, we conducted experiments where Trp-tRNATrp, which is known to induce read-through at the UGA codon60,61,62, was depleted from the in vitro translation system by excluding tryptophan. Remarkably, even with the wild-type pepNL (UGA), robust ribosome stalling was observed (Fig. 5a, lanes 3 and 4). We further analyzed the polypeptide products and detected an accumulation of the peptidyl-tRNA with a 14-aa-length polypeptide when tryptophan was absent, consistent with the toeprint analysis. (Fig. 5b and c, lanes 3, 4, 5 and 6). Contrary, in the presence of tryptophan, we observed a longer peptidyl-tRNA compared to full-length PepNL (14 aa)-tRNA (Fig. 5c, lane 1). This longer peptidyl-tRNA exhibited almost identical electrophoretic mobility to the peptidyl-tRNA containing 27-aa-length polypeptide up to the downstream stop codon (Fig. 5c, lane 7, Supplementary Fig. 7a). These results indicated that ribosomes could read through the UGA stop codon in the presence of the Trp-tRNATrp and then stall at downstream stop codons. Consistent with this, in vivo reporter assays demonstrated that the UGA codon, but not UAA or UAG, exclusively induced the read-through of the pepNL stop codon (Fig. 5d). Moreover, when we mutated the UGA15 stop codon of pepNL to those of 20 canonical amino acids, we observed varying degrees of inhibition in the peptidyl transfer of the 14-amino-acid-long PepNL nascent peptide. Mutations to Asp and Glu resulted in pronounced inhibition, while Ile, Lys, Leu, Asn, Pro, Arg, Val, and Trp showed moderate inhibitory effects. The remaining amino acid mutations exhibited little or no inhibition (Supplementary Fig. 7d). A similar observation, where peptidyl transfer inhibition depends on the identity of the A-site aminoacyl-tRNA, has been reported previously63,64. Nonetheless, these data indicate that PepNL nascent peptide blocks not only RF2-mediated termination but also interferes with transpeptidation involving specific aminoacyl-tRNAs at the A-site.
Finally, the addition of tryptophan triggered stop codon read-through even after the ribosome was arrested by PepNL (Fig. 5e). However, the migration rate under these conditions is relatively slow (approximately one aa/min). Moreover, toeprint analysis revealed that the read-through becomes less efficient when tryptophan is reintroduced after depletion, with ribosome progression halted at multiple sites beyond the UGA15 stop codon (Supplementary Fig. 7e). Structural comparison further revealed that Ile13 in the hairpin-shaped PepNL potentially clashes with not only RF2 but also Trp-tRNATrp (Supplementary Fig. 7f). Collectively, these findings suggest that Trp-tRNATrp reads through the UGA15 stop codon more efficiently “before” the formation of the PepNL’s mini hairpin (Fig. 5f, discussed in the following section).
Discussion
Our study demonstrated that the small ORFs pepNL and nanCL in E. coli encode ribosome arrest peptides (RAP) (Figs. 1 and 2). Furthermore, we successfully determined the cryo-EM structure of the ribosome arrested by PepNL (Figs. 3 and 4). Our structure revealed that the nascent PepNL peptide adopts a distinctive “mini hairpin” conformation near the entrance of the exit tunnel, causing the distortion of its C-terminal segment, including the Ile13 residue. This distorted structure prevents the proper accommodation of the GGQ motif of RF2, inhibiting peptide release. Importantly, PepNL-dependent stalling does not require an “arrest inducer”; instead, the stalling is inhibited via stop codon read-through induced by Trp-tRNATrp (Fig. 5).
The PepNL nascent peptide within the tunnel adopts a mini-hairpin fold to enable a distinct arresting mechanism, by employing only 14 amino acid residues. Generally, the N-terminus of the nascent peptide elongates toward the exit of the tunnel9,12,58. In contrast, in our structure, the N-terminus of PepNL is oriented in the opposite direction, toward the tunnel entrance. In the previous studies, the nascent peptides of VemP, XBP1u, and MsrDL were reported to adopt S-shaped or hook-like conformations that partially folded back within the exit tunnel32,33,36. However, a U-shaped conformation with the overall reversal of the nascent peptide, as in the present PepNL structure, has not yet been reported. Moreover, the translation inhibition mechanism by this “mini hairpin” structure might raise a concept at the early stage of protein synthesis. The N-terminal amino acid sequences immediately adjacent to the initiation codon can affect translation efficiency65,66. Therefore, the stochastic formation of mini hairpin-like structures could potentially inhibit the expression of proteins, and thus not be limited to PepNL.
Previous cryo-EM studies elucidated the details of arrest peptide prevention of translation termination21,22,33,36. In the case of TnaC, U2585 and A2602 of the 23S rRNA and Arg23 (Phe23) of the TnaC nascent peptide cooperatively inhibit the proper accommodation of RF222. In SpeFL, the distortion of U2585 by Asn32 of the SpeFL nascent peptide causes a steric clash with RF1, thereby inhibiting peptide release33. In MsrDL, U2585 and U2584 of the 23S rRNA and Phe5 of the MsrDL nascent peptide prevent the accommodation of RF1 and RF236. In the present structure, the steric clash between Ile13 of the PepNL nascent peptide and the GGQ motif of RF2 inhibits peptide release. These four RAPs commonly prevent the proper accommodation of the GGQ motif by forming distinct compact conformations (Supplementary Fig. 8a–c). Moreover, our study once again emphasizes the significance of the penultimate amino acid residue (Ile13 in PepNL) for peptide release inhibition22,67. The structural comparison of PepNL with other RAPs that inhibit translation termination revealed that, although the overall shape of the nascent peptides is distinct from one another, the distortion of the penultimate amino acid shows a striking similarity (Supplementary Fig. 8d–f). Earlier studies noted that the efficiency of RF-mediated termination is affected by the amino acid sequence of the nascent peptide68,69,70. Notably, Isaksson and colleagues pointed out the importance of the penultimate C-terminal residue in canonical translation termination67. The inhibition mechanisms revealed by recent structural analyses may contribute to a deeper understanding of the universal translation cycle. Our successful examination of the entire structure of distorted RF2 could offer several insights. We observed that Ile13 of PepNL excluded the GGQ motif, causing a drastic rearrangement of the apical loop within domain III of RF2. In addition, this noncanonical structure of RF2 is stabilized by extra hydrogen bonds (Fig. 4c–e). Therefore, the GGQ motif can be regarded as a sensor to fine-tune the translation termination efficiency in response to the structural state within the PTC. When a mismatch exists between the GGQ motif and the PTC structure, it could result in the stabilization of the non-canonical RF2 conformation via additional interactions, potentially delaying translation termination.
In contrast to sensory arrest peptides such as TnaC, the folding of the PepNL nascent peptide within the tunnel is independent of an arrest inducer. Instead, PepNL utilizes the stop codon read-through as an “arrest inhibitor”. This framework makes PepNL a metabolite-sensing arrest peptide that functions in a distinct way from the previously studied RAPs. However, the PepNL nascent peptide, which forms a mini hairpin, could cause steric clashes not only with RF2 but also with Trp-tRNATrp (Supplementary Fig. 7f). Furthermore, after stop codon read-through, the ribosome exhibited a slower migration rate (approximately one aa/min, Fig. 5e) compared to the reported elongation velocity in the PURE system (4.8 ~ 26.4 aa/min71,72,73). These suggest that the hairpin structure may inhibit not only the termination process but also stop codon read-through and subsequent translation elongation (Fig. 5e, Supplementary Fig. 7e). These together indicate that the read-through hardly releases the PepNL-arrested ribosome. What accounts for this discrepancy? Although we have not obtained direct evidence in this study, we predict that the PepNL nascent peptide does not form the hairpin structure immediately after synthesis (the pre-arrest state in Fig. 5f). At this stage, Trp-tRNATrp could efficiently read through the UGA15 stop codon, thereby preventing translation arrest (Fig. 5f, top). However, once the hairpin structure forms, it inhibits termination by RF2 and read-through by Trp-tRNATrp (Fig. 5f, bottom). Therefore, since PepNL relies on its unique hairpin structure for translation arrest, PepNL may utilize the “time” required for folding as an arrest inducer. This concept is reminiscent of how SpeFL relies on non-optimal rare codons for effective regulation, potentially promoting the folding of its N-terminal sensor domain by slowing translation34. Further analyses are necessary to clarify this hypothesis in greater detail.
The translation of pepNL regulates the expression of the downstream gene, pepN (Supplementary Fig. 1e, 3c). The ViennaRNA prediction program74,75 indicated the presence of stem-loop structures surrounding the stop codon of pepNL, as well as near the Shine-Dalgarno (SD) sequence and the initiation codon of pepN (Supplementary Fig. 9a). However, the SD-masking stem-loop does not seem to be affected by translation arrest at the pepNL stop codon (Supplementary Fig. 9b). Further studies are needed to clarify the detailed regulatory mechanism involved. PepN, an endopeptidase, degrades not only oligopeptides but also full-length polypeptides with broad substrate specificity76. In nutrient-limited conditions, PepN plays a crucial role in adaptation through the reproduction of free amino acids77. These characteristics suggest that the correlation between the PepN activity and the intracellular amino acid concentration holds significance for environmental adaptation.
Our approaches identified two uncharacterized RAPs, PepNL and NanCL, with functions in living E. coli cells. However, it should be noted that our evaluations of the RAP activity for each sORF are limited to the standard laboratory conditions (LB medium, 37 °C, aerobic conditions). In fact, SpeFL, which responds to ornithine, did not exhibit a significant result under our testing conditions, indicating the limitations of our approaches. Meanwhile, our analysis did not exclude the possibility that sORFs other than pepNL and nanCL encode sensory arrest peptides. In our study, the translation of 19 out of 39 sORFs significantly influenced the expression of downstream genes, implying that they harbor translation-coupled functions (Supplementary Fig. 1e). Further analyses of these sORFs would expand our understanding of translation regulation.
To explore the unidentified “arrest inducer” for each RAP candidate, monitoring the expression of Cold Shock Proteins (CSPs) could serve as a valuable indicator. While the stalling of translation elongation commonly triggers the expression of CSPs, the precise mechanism has not yet been elucidated45. This enigmatic phenomenon should be studied in the future, and if similar responses occur across species78,79, then it could be a convenient method for evaluating RAP activity. In addition, this study also demonstrated the effectiveness of LC-MS/MS measurements of nascent peptidyl-tRNA species to identify RAPs. Our approaches to identifying PepNL and NanCL, as well as the distinct molecular mechanism of translation stalling and regulation, provide valuable insights into deciphering the hidden genetic codes within polypeptide sequences.
Methods
E. coli strains, plasmids, and primers
E. coli strain BW25113 {∆(araD-araB)567, ∆lacZ4787(::rrnB-3), λ-, rph-1, ∆(rhaD-rhaB)568, hsdR514} was used as the experimental standard strain. Plasmids used in this study are listed in Supplementary Data 4. Plasmids were constructed using standard cloning procedures, including Gibson assembly and site-directed mutagenesis. The sequence files of the plasmids constructed in this study are available in the Mendeley repository [https://data.mendeley.com/datasets/2bkz2xnnn5/1].
Spot assay
E. coli cells harboring pCA24N with each sORF were grown overnight at 37 °C in LB medium supplemented with 20 µg/ml chloramphenicol. On the next day, cultures were serially diluted with fresh LB medium (10−1, 10−2, 10−3, 10−4, 10−5, and 10−6) and spotted onto LB agar plates containing 20 µg/ml chloramphenicol, with or without 1 mM IPTG (isopropyl β-D-thiogalactopyranoside, Nacalai Tesque). The plates were incubated overnight at 37 °C, and the growth of colonies was recorded.
In the complementation assay shown in Supplementary Fig. 3e, E. coli cells harboring pCA24N-pepNL and pBAD30 or its derivatives harboring the indicated genes under the control of the PBAD promoter were grown overnight at 37 °C in LB medium supplemented with 20 µg/ml chloramphenicol and 100 µg/ml ampicillin. On the next day, cultures were serially diluted and spotted onto LB agar plates containing 20 µg/ml chloramphenicol, 100 µg/ml ampicillin, and 2×10−3 % arabinose, with or without 500 µM IPTG. Plates were incubated overnight at 37 °C, and the growth of colonies was recorded.
β-galactosidase assay
E. coli cells harboring the lacZ reporter plasmid were grown overnight at 37 °C in LB medium supplemented with 100 µg/ml ampicillin. On the next day, the cultures were inoculated into fresh LB medium containing 2 ×10−3% arabinose and 100 µg/ml ampicillin, and grown at 37 °C until the A660 reached 0.5. Afterward, 20 µl portions were subjected to a β-galactosidase assay to calculate the Miller units (m.u.), as described80. The average of three independent experiments is presented with a standard error (SE) value.
sORF-dependent regulation score
The sORF-dependent regulation score was calculated using the following formula.
Plasmid-A: A plasmid carrying the PBAD promoter, the 5’ region of the downstream main ORF (mORF) relative to the small ORF, spanning from the transcription start site to the initiation codon of the mORF, and the lacZ reporter translationally fused with the mORF. Plasmid-B: A derivative of plasmid-A with a mutation (ATG to ACG) to disrupt the translation initiation codon (AUG) or the insertion of a stop codon to prematurely terminate the translation of the small ORF. Detailed information on these mutations can be found in Supplementary Data 4, and sequence files are available in the Mendeley repository [https://data.mendeley.com/datasets/2bkz2xnnn5/1].
Frequency of stop codon read-through
E. coli cells expressing the pepNL-stop-lacZ or its derivative lacking the stop codon of the pepNL reporter were grown and subjected to a β-galactosidase assay. The frequency of stop codon read-through was calculated with the following formula.
In vitro translation and product analysis
The coupled transcription-translation reaction was performed using PUREfrex v1.0 (GeneFrontier) in the presence of 35S-methionine {EasyTag L-[35S]-Methionine (PerkinElmer)} at 37 °C, as described previously40. DNA templates were prepared by PCR, as summarized in Supplementary Data 5. Tryptophan or release factors (RF: RF1, RF2, RF3, and RRF) were excluded from the in vitro translation mixture where indicated. The reaction was stopped by the addition of ice-cooled 5% TCA, washed with ice-cooled acetone, and dissolved in SDS sample buffer (125 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 50 mM DTT) that had been treated with RNAsecure (Ambion). Finally, the sample was divided into two portions, one of which was incubated with 50 µg/ml of RNase A (Promega) at 37 °C for 60 min, and separated by a WIDE RANGE Gel SDS-PAGE system (Nacalai Tesque). Radioactive bands were detected by using an imaging plate and an Amersham™ Typhoon™ scanner RGB system (GE Healthcare).
Toeprint analysis
The toeprint analysis was performed as described previously40,81. The in vitro translation reaction sample was mixed with an equal volume of reverse transcription mixture [50 mM HEPES-KOH, pH 7.6, 100 mM potassium glutamate, 2 mM spermidine, 13 mM magnesium acetate, 1 mM DTT, 2 µM fluorescently labeled oligonucleotide (pe-lacZ-N-rv with Alexa 647 at 5’-terminus), 40 µM of each dNTP, 10 unit/µl ReverTra Ace (Toyobo)] and incubated at 37 °C for 10 min. The reverse transcription products were purified by a NucleoSpin Gel and PCR clean-up kit equilibrated with NTC buffer (Macherey-Nagel). Dideoxy DNA sequencing samples were prepared using Thermo Sequenase DNA polymerase (Cytiva) and the same templates and primer (pe-lacZ-N-rv) as used for the toeprint analysis. Samples were subjected to 8% polyacrylamide-7 M urea-TBE gel electrophoresis. Fluorescent images were visualized and analyzed by an Amersham™ Typhoon™ scanner RGB system (GE Healthcare), using a 635 nm excitation laser and LPR emission filter. Thiostrepton was added at a final concentration of 100 µg/ml beforehand, as indicated. Tryptophan or release factors (RFs: RF1, RF2, RF3, and RRF) were excluded from the in vitro translation mixture as indicated.
Northern blotting
E. coli cells were grown in LB medium until the A660 reached ~0.5. The expression of pepNL was then induced by 1 mM IPTG for 10 min. After centrifuging and collecting the bacterial cells, total RNA was extracted using TriPure Isolation Reagent (Roche), according to the supplier’s instructions. RNA samples were dissolved in SDS sample buffer, separated by 11% WIDE RANGE Gel SDS-PAGE, transferred onto a BrightStar™-Plus Positively Charged Nylon Membrane (InvitrogenTM), and hybridized with a biotinylated oligonucleotide complementary to tRNAAsp; CGGAACGGACGGGACTCGAACCCGCGACC. Hybridization experiments were performed using ULTRAhyb™ Ultrasensitive Hybridization Buffer (InvitrogenTM) and the Chemiluminescent Nucleic Acid Detection Module (Thermo Scientific), according to the manufacturers’ instructions. Images were visualized and analyzed by a LAS4000 LuminoImager (GE Healthcare).
Preparation of the lysate for proteomic analysis
Samples for proteomic analysis were prepared as described previously82. E. coli cells harboring pCA24N with each sORF were grown in LB medium supplemented with 20 µg/ml of chloramphenicol until the A660 reached ~0.2. Subsequently, 1 mM of IPTG was added to induce the expression of small ORFs, and cells were further incubated for 1 h with shaking at 37 °C. The cells were then harvested and resuspended in PBS buffer (137 mM NaCl, 8.1 mM Na2HPO4, 2.68 mM KCl, 1.47 mM KH2PO4, pH 7.4). The suspension was mixed with an equal volume of 10% TCA. After standing on ice for at least 10 min, the samples were centrifuged, and the supernatant was removed by aspiration. Precipitates were washed twice with acetone, by vigorous mixing. Proteins were dissolved in PTS buffer (12 mM sodium deoxycholate, 12 mM sodium lauryl sulfate, 100 mM Tris-HCl, pH 9.0), and 50 µg of total protein at a concentration of 1 µg/µl was processed by reduction with dithiothreitol (DTT), alkylation with iodoacetamide, and limited digestion by Trypsin/Lys-C Mix (Promega). The samples were then extracted with ethyl acetate, evaporated, dissolved in MS buffer-A (0.1 % TFA and 2% acetonitrile), desalted by a StageTip composed of an SDB-XC Empore disk (3 M, U.S.A.), eluted with MS buffer-B (0.1% TFA and 80% acetonitrile), evaporated again, and dissolved in MS buffer-A. Finally, the samples were subjected to the LC-MS/MS measurement.
LC-MS/MS analysis for comparative quantitative proteomic analysis
In a series of quantitative proteomic analyses, we used the Eksigent NanoLC 415 nanoflow HPLC system and the TripleTOF 4600 tandem-mass spectrometer (AB Sciex, U. S. A.) in the DIA/SWATH acquisition mode83. The detailed settings for the LC-MS/MS measurements are summarized in Supplementary Table 1. The measurement was conducted three times for each sample prepared from one biological replicate.
Data analysis was performed by the DIA-NN software (version 1.8.1, https://github.com/vdemichev/diann, downloaded on 1 November 2023)84. The spectral library for DIA/SWATH analysis was obtained from the SWATH atlas (http://www.swathatlas.org/, accessed on 30 April 2021); the original data were acquired by Midha et al.85. All downstream statistical analyses were performed using in-house R scripts (R.app for Mac, version 4.3.1). Only the proteins with intensities obtained in all three measurements in both samples were used to calculate fold changes. P-values for the volcano plots were calculated using Welch’s t-test with the Benjamini-Hochberg correction (using the “p.adjust” function in R.app). PCA and hierarchical clustering analyses were performed using the data from 1,205 proteins for which foldchange values were obtained under all 36 conditions.
Preparation of the peptidyl-tRNA for LC-MS/MS analyses
E. coli cells harboring pCA24N with each sORF were grown in LB medium supplemented with 20 µg/ml of chloramphenicol until the A660 reached approximately 0.5. Subsequently, 1 mM of IPTG was added to induce the expression of small ORFs, and cells were further incubated for 30 min with shaking at 37 °C. Afterward, the cell culture was mixed with an equal volume of ice-cooled 10% TCA to precipitate the cellular components. The precipitate was washed twice with ice-cooled acetone, and dissolved in PTS buffer.
The peptidyl-tRNAs in the PTS buffer were isolated by using a High Pure miRNA Isolation Kit (Roche), as described previously56. After adding binding buffer (provided by the manufacturer), the lysate was vortexed and incubated at 37 °C for 30 min with shaking. Then binding enhancer (provided by the manufacturer) was added and the lysate was centrifuged at 12,000 × g for 3 min at 4 °C to remove the precipitates. The supernatant was loaded into silica columns, and the following steps were performed according to the manufacturer’s instructions. The silica-bound RNA was eluted with PTS buffer. This purification was repeated twice.
The purified RNA sample was mixed with 0.1x volume of 2 M Tris base (pH= ~11) and incubated at 80 °C for 20 min to hydrolyze the ester bond of the peptidyl-tRNA. After the addition of 0.9x volume of deionized water, the sample was reduced by DTT, alkylated with iodoacetamide, and digested with Lys-C (Fujifilm-Wako) or Lys-C / Trypsin mix (Promega). The samples were then extracted with ethyl acetate, dried, dissolved in MS buffer-A (0.1% TFA and 2% acetonitrile), desalted by a StageTip composed of an SDB-XC Empore disk, eluted with MS buffer-B (0.1% TFA and 80% acetonitrile), dried again, and resolved in MS buffer-A.
LC-MS/MS analysis for peptides derived from peptidyl-tRNAs
For the detection of the peptides derived from peptidyl-tRNAs, the Easy-nLC 1000 nanoflow HPLC system and the Q-Exactive tandem-mass spectrometer (Thermo Fisher Scientific, U. S. A.) were used in the data-dependent acquisition (DDA) mode. The detailed settings for the LC-MS/MS measurements are summarized in Supplementary Table 2. The data analysis of the DDA measurements was performed with the Proteome Discoverer 2.4 software bundled with the SEQUEST HT search engine (Thermo Fisher Scientific, U. S. A.). To detect stalled peptides in translation, the Trypsin (semi) or LysC (semi) setting was used for enzymatic digestion.
Statistical analyses
Statistical analyses were conducted by using R and Python (https://www.python.org).
GWIPS-viz browser
We configured the settings of the GWIPS-viz browser44 as follows: Organism: Escherichia coli K12; Elongating Ribosome (A-site): Global Aggregates; mRNA-seq Reads: Global Aggregates.
Grid preparation and data acquisition
For the cryo-EM analysis, 3 µL of the PURE system reaction solution containing stalled ribosomes was applied onto a glow-discharged holey-carbon grid coated with a continuous 2 nm-thick carbon film (Quantifoil Au 300 mesh, R2/1 + 2 nm C) and incubated for 30 s in a controlled environment of 100% humidity at 4°C. The grids were blotted for 4 s, and then plunge frozen in liquid ethane, using a Vitrobot Mark IV (FEI).
The datasets were collected using a Titan Krios G4 microscope (Thermo Fisher Scientific), running at 300 kV and equipped with a Gatan Quantum-LS Energy Filter (GIF). A Gatan K3 Summit direct electron detector was used at a pixel size of 0.83 Å (magnification of × 105,000) with a dose of approximately 30 electrons per Å 2 with 30 frames. The data were automatically acquired using the EPU software (Thermo Fisher Scientific), with a defocus range of −0.8 to −1.6 µm, and 15,342 movies were obtained. Detailed parameters are listed in Supplementary Table 3.
Image processing
The data were processed by the cryoSPARC v4.4.0 software platform86. The dose-fractionated movies were subjected to beam-induced motion correction and dose weighting using patch motion correction, and the contrast transfer function (CTF) parameters were estimated using patch-based CTF estimation. From the 15,342 preprocessed micrographs, 2,521,444 particles were automatically picked by the template picker, using the references created by the blob picker and several rounds of 2D classification. The particles were subjected to several rounds of reference-free 2D classifications to create particle sets. The curated particles were aligned by non-uniform refinement using the E. coli ribosome map (EMD-22586) as the reference, followed by unsupervised 3D classification and a successive 2D classification to eliminate the ratcheted ribosomes and junk particles. The remaining particles were further classified by no-align focused 3D classification using the mask covering the P-site, A-site, and the binding sites of EF-Tu:tRNA and RF2, resulting in four classes with RF2-bound, A-tRNA-bound, EF-Tu:tRNA-bound, and empty ribosomes, respectively. The particles of the RF2-bound ribosome (71,980 particles) were subjected to CTF refinement (beam-tilt, trefoil, fourth-order aberrations, and per-particle defocus) and reference-based motion correction, followed by the final non-uniform refinement. The global resolution of the final map is 2.9 Å based on the gold-standard, applying the 0.143 criterion on the Fourier shell correlation (FSC) between the reconstructed half maps87.
Model building and refinement
The reported high-resolution model of the E. coli ribosome88 (PDB 7K00), including bound ligands and metal ions, was used as the starting model, while that of RF2 was from 6OUO. The models were docked into our map by rigid body fitting, followed by jiggle fitting89 and manual revision in Coot90. The PepNL peptide was built manually based on the density. Metal ions were revised and added based on the coordination pattern and the density, which is stronger in the map without sharpening due to their positive charge. Geometrical restraints of modified residues and ligands were calculated by Grade Web Server (http://grade.globalphasing.org). The final model was subjected to the refinement of energy minimization and atomic displacement parameters (ADP) estimation by Phenix.real_space_refine v1.2091 with Ramachandran and rotamer restraints, against the map with B-factor sharpening and local-resolution filtering by cryoSPARC. Metal-coordination restraints were generated by ReadySet implemented in PHENIX, and non-canonical covalent bond restraints for modified residues were manually prepared. The refined model was validated with MolProbity92 and EMRinger93 in the PHENIX package. The models used for the structure comparison are docked into our map by rigid body fitting. Model refinement statistics are listed in Supplementary Table 3. UCSF ChimeraX 1.694 was used to make the figures.
RNA secondary structure prediction in silico
The secondary structure of pepNL-pepN mRNA was predicted using the Vienna RNAfold WebServer74,75 with the following settings: Folding Constraints: no constraint; Fold algorithms and basic options: minimum free energy and partition function; Dangling end options: dangling energies on both sides of a helix in any case; Energy Parameters: RNA parameters (Turner model, 2004); Temperature: 37 °C; Salt concentration: 1.021 M NaCl; Modified Bases: no modification; SHAPE reactivity data: no designation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry data have been deposited in the jPOST repository with the dataset identifier PXD052430 [https://repository.jpostdb.org/entry/JPST003118.1]95. The atomic coordinates have been deposited in the Protein Data Bank under the accession codes 8ZTU and 8ZTV. The cryo-EM density maps have been deposited in the Electron Microscopy Data Bank under the accession codes of EMD-60473 and EMD-60474. Source Data are provided with this paper. Raw data files are available in the Mendeley Data repository [https://data.mendeley.com/datasets/2bkz2xnnn5/1]. Source data are provided with this paper.
References
Shine, J. & Dalgarno, L. Determinant of cistron specificity in bacterial ribosomes. Nature 254, 34–38 (1975).
Rodnina, M. V. Translation in Prokaryotes. CSH Perspect. Biol. 10, a032664 (2018).
Capecchi, M. R. Polypeptide chain termination in vitro: isolation of a release factor. Proc. Natl. Acad. Sci. Usa. 58, 1144–1151 (1967).
Scolnick, E., Tompkins, R., Caskey, T. & Nirenberg, M. Release factors differing in specificity for terminator codons. Proc. Natl. Acad. Sci. Usa. 61, 768–774 (1968).
Weixlbaumer, A. et al. Insights into translational termination from the structure of RF2 bound to the ribosome. Science 322, 953–956 (2008).
Laurberg, M. et al. Structural basis for translation termination on the 70S ribosome. Nature 454, 852–857 (2008).
Korostelev, A. et al. Crystal structure of a translation termination complex formed with release factor RF2. Proc. Natl. Acad. Sci. Usa. 105, 19684–19689 (2008).
Korostelev, A., Zhu, J., Asahara, H. & Noller, H. F. Recognition of the amber UAG stop codon by release factor RF1. EMBO J. 29, 2577–2585 (2010).
Ito, K. & Chiba, S. Arrest Peptides: Cis-Acting Modulators of Translation. Annu. Rev. Biochem. 82, 171–202 (2013).
Dever, T. E., Ivanov, I. P. & Sachs, M. S. Conserved Upstream Open Reading Frame Nascent Peptides that Control Translation. Annu. Rev. Genet. 54, 1–28 (2020).
Chiba, S., Fujiwara, K., Chadani, Y. & Taguchi, H. Nascent chain-mediated translation regulation in bacteria: translation arrest and intrinsic ribosome destabilization. J. Biochem. 173, 227–236 (2023).
Nissen, P., Hansen, J., Ban, N., Moore, P. B. & Steitz, T. A. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Science 289, 920–930 (2000).
Nakatogawa, H. & Ito, K. The Ribosomal Exit Tunnel Functions as a Discriminating Gate. Cell 108, 629–636 (2002).
Gong, F. & Yanofsky, C. Instruction of Translating Ribosome by Nascent Peptide. Science 297, 1864–1867 (2002).
Gaba, A., Wang, Z., Krishnamoorthy, T., Hinnebusch, A. G. & Sachs, M. S. Physical evidence for distinct mechanisms of translational control by upstream open reading frames. EMBO J. 20, 6453–6463 (2001).
Onouchi, H. et al. Nascent peptide-mediated translation elongation arrest coupled with mRNA degradation in the CGS1 gene of Arabidopsis. Genes Dev. 19, 1799–1810 (2005).
Chiba, S., Lamsa, A. & Pogliano, K. A ribosome-nascent chain sensor of membrane protein biogenesis in Bacillus subtilis. EMBO J. 28, 3461–3475 (2009).
Yanagitani, K., Kimata, Y., Kadokura, H. & Kohno, K. Translational Pausing Ensures Membrane Targeting and Cytoplasmic Splicing of XBP1u mRNA. Science 331, 586–589 (2011).
Ishii, E. et al. Nascent chain-monitored remodeling of the Sec machinery for salinity adaptation of marine bacteria. Proc. Natl. Acad. Sci. Usa. 112, E5513–E5522 (2015).
Cruz-Vera, L. R., Rajagopal, S., Squires, C. & Yanofsky, C. Features of Ribosome-Peptidyl-tRNA Interactions Essential for Tryptophan Induction of tna Operon Expression. Mol. Cell 19, 333–343 (2005).
Su, T. et al. Structural basis of l-tryptophan-dependent inhibition of release factor 2 by the TnaC arrest peptide. Nucleic Acids Res. 49, 9539–9547 (2021).
van der Stel, A.-X. et al. Structural basis for the tryptophan sensitivity of TnaC-mediated ribosome stalling. Nat. Commun. 12, 5340 (2021).
Takahashi, H. et al. Comprehensive genome-wide identification of angiosperm upstream ORFs with peptide sequences conserved in various taxonomic ranges using a novel pipeline. Esuca. BMC Genom. 21, 260 (2020).
Sakiyama, K., Shimokawa-Chiba, N., Fujiwara, K. & Chiba, S. Search for translation arrest peptides encoded upstream of genes for components of protein localization pathways. Nucleic Acids Res. 49, 1550–1566 (2021).
Fujiwara, K., Tsuji, N., Yoshida, M., Takada, H. & Chiba, S. Patchy and widespread distribution of bacterial translation arrest peptides associated with the protein localization machinery. Nat. Commun. 15, 2711 (2024).
Imamichi, T. et al. Phylogeny-linked occurrence of ribosome stalling on the mRNAs of Arabidopsis unfolded protein response factor bZIP60 orthologs in divergent plant species. Nucleic Acids Res. 52, 4276–4294 (2024).
Meydan, S. et al. Retapamulin-Assisted Ribosome Profiling Reveals the Alternative Bacterial Proteome. Mol. Cell 74, 481–493.e6 (2019).
Weaver, J., Mohammad, F., Buskirk, A. R. & Storz, G. Identifying Small Proteins by Ribosome Profiling with Stalled Initiation Complexes. mBio 10, e02819–18 (2019).
Stringer, A., Smith, C., Mangano, K. & Wade, J. T. Identification of Novel Translated Small Open Reading Frames in Escherichia coli Using Complementary Ribosome Profiling Approaches. J. Bacteriol. 204, e00352–21 (2021).
Arenz, S. et al. Molecular basis for erythromycin-dependent ribosome stalling during translation of the ErmBL leader peptide. Nat. Commun. 5, 3501 (2014).
Sohmen, D. et al. Structure of the Bacillus subtilis 70S ribosome reveals the basis for species-specific stalling. Nat. Commun. 6, 6941 (2015).
Su, T. et al. The force-sensing peptide VemP employs extreme compaction and secondary structure formation to induce ribosomal stalling. Elife 6, e25642 (2017).
Shanmuganathan, V. et al. Structural and mutational analysis of the ribosome-arresting human XBP1u. Elife 8, e46267 (2019).
Herrero Del Valle, A. et al. Ornithine capture by a translating ribosome controls bacterial polyamine synthesis. Nat. Microbiol. 5, 554–561 (2020).
Beckert, B. et al. Structural and mechanistic basis for translation inhibition by macrolide and ketolide antibiotics. Nat. Commun. 12, 4466 (2021).
Fostier, C. R. et al. Regulation of the macrolide resistance ABC-F translation factor MsrD. Nat. Commun. 14, 3891 (2023).
Gersteuer, F. et al. The SecM arrest peptide traps a pre-peptide bond formation state of the ribosome. Nat. Commun. 15, 2431 (2024).
Morici, M. et al. RAPP-containing arrest peptides induce translational stalling by short circuiting the ribosomal peptidyltransferase activity. Nat. Commun. 15, 2432 (2024).
Gong, M., Gong, F. & Yanofsky, C. Overexpression of tnaC of Escherichia coli inhibits growth by depleting tRNA2Pro availability. J. Bacteriol. 188, 1892–1898 (2006).
Chadani, Y. et al. Intrinsic Ribosome Destabilization Underlies Translation and Provides an Organism with a Strategy of Environmental Sensing. Mol. Cell 68, 528–539.e5 (2017).
Hemm, M. R., Paul, B. J., Schneider, T. D., Storz, G. & Rudd, K. E. Small membrane proteins found by comparative genomics and ribosome binding site models. Mol. Microbiol. 70, 1487–1501 (2008).
VanOrsdel, C. E. et al. Identifying New Small Proteins in Escherichia coli. Proteomics 18, 1700064 (2018).
Adams, P. P. et al. Regulatory roles of Escherichia coli 5’ UTR and ORF-internal RNAs detected by 3’ end mapping. eLife 10, e62438 (2021).
Michel, A. M. et al. GWIPS-viz: development of a ribo-seq genome browser. Nucleic Acids Res. 42, D859–D864 (2013).
VanBogelen, R. A. & Neidhardt, F. C. Ribosomes as sensors of heat and cold shock in Escherichia coli. Proc. Natl. Acad. Sci. Usa. 87, 5589–5593 (1990).
Jiang, W., Jones, P. & Inouye, M. Chloramphenicol induces the transcription of the major cold shock gene of Escherichia coli, cspA. J. Bacteriol. 175, 5824–5828 (1993).
Wilson, D. N. The A-Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 44, 393–433 (2009).
Lin, J., Zhou, D., Steitz, T. A., Polikanov, Y. S. & Gagnon, M. G. Ribosome-Targeting Antibiotics: Modes of Action, Mechanisms of Resistance, and Implications for Drug Design. Annu. Rev. Biochem. 87, 1–28 (2018).
Lim, B., & Gross, C. A. Cellular response to heat shock and cold shock. In G. Storzand & R. Hengge (Eds.), Bacterial stress responses (2nd Ed., pp. 93-114). Washington, DC: ASM Press. (2011).
Goldstein, J., Pollitt, N. S. & Inouye, M. Major cold shock protein of Escherichia coli. Proc. Natl. Acad. Sci. Usa. 87, 283–287 (1990).
Jones, P. G., Mitta, M., Kim, Y., Jiang, W. & Inouye, M. Cold shock induces a major ribosomal-associated protein that unwinds double-stranded RNA in Escherichia coli. Proc. Natl. Acad. Sci. Usa. 93, 76–80 (1996).
Giuliodori, A. M., Brandi, A., Giangrossi, M., Gualerzi, C. O. & Pon, C. L. Cold-stress-induced de novo expression of infC and role of IF3 in cold-shock translational bias. RNA 13, 1355–1365 (2007).
Kitagawa, M., Wada, C., Yoshioka, S. & Yura, T. Expression of ClpB, an analog of the ATP-dependent protease regulatory subunit in Escherichia coli, is controlled by a heat shock sigma factor (sigma 32). J. Bacteriol. 173, 4247–4253 (1991).
Miwa, T., Chadani, Y. & Taguchi, H. Escherichia coli small heat shock protein IbpA is an aggregation‐sensor that self‐regulates its own expression at posttranscriptional levels. Mol. Microbiol. 115, 142–156 (2021).
Ito, Y. et al. Nascent peptide-induced translation discontinuation in eukaryotes impacts biased amino acid usage in proteomes. Nat. Commun. 13, 7451 (2022).
Yamakawa, A., Niwa, T., Chadani, Y., Kobo, A. & Taguchi, H. A method to enrich polypeptidyl-tRNAs to capture snapshots of translation in the cell. Nucleic Acids Res. 51, e30–e30 (2023).
Shimizu, Y. et al. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 19, 751–755 (2001).
Syroegin, E. A., Aleksandrova, E. V. & Polikanov, Y. S. Insights into the ribosome function from the structures of non-arrested ribosome–nascent chain complexes. Nat. Chem. 15, 143–153 (2023).
Zeng, F. & Jin, H. Conformation of methylated GGQ in the Peptidyl Transferase Center during Translation Termination. Sci. Rep. 8, 2349 (2018).
Weiner, A. M. & Weber, K. Natural Read-through at the UGA Termination Signal of Qβ Coat Protein Cistron. Nat. N. Biol. 234, 206–209 (1971).
Engelberg-Kulka, H. UGA suppression by normal tRNATrP in Escherichia coli: codon context effects. Nucleic Acids Res. 9, 983–991 (1981).
Lovett, P. S., Ambulos, N. P., Mulbry, W., Noguchi, N. & Rogers, E. J. UGA can be decoded as tryptophan at low efficiency in Bacillus subtilis. J. Bacteriol. 173, 1810–1812 (1991).
Ramu, H. et al. Nascent Peptide in the Ribosome Exit Tunnel Affects Functional Properties of the A-Site of the Peptidyl Transferase Center. Mol. Cell 41, 321–330 (2011).
Chevance, F. F. V., Guyon, S. L. & Hughes, K. T. The Effects of Codon Context on In Vivo Translation Speed. PLoS Genetics 10, e1004392 (2014).
Verma, M. et al. A short translational ramp determines the efficiency of protein synthesis. Nat. Commun. 10, 5774 (2019).
Osterman, I. A. et al. Translation at first sight: the influence of leading codons. Nucleic Acids Res. 48, 6931–6942 (2020).
Mottagui-Tabar, S., Björsson, A. & Isaksson, L. A. The second to last amino acid in the nascent peptide as a codon context determinant. EMBO J. 13, 249–257 (1994).
Björnsson, A., Mottagui-Tabar, S. & Isaksson, L. A. Structure of the C-terminal end of the nascent peptide influences translation termination. EMBO J. 15, 1696–1704 (1996).
Sunohara, T., Abo, T., Inada, T. & Aiba, H. The C-terminal amino acid sequence of nascent peptide is a major determinant of SsrA tagging at all three stop codons. RNA 8, 1416–1427 (2002).
Hayes, C. S., Bose, B. & Sauer, R. T. Proline Residues at the C Terminus of Nascent Chains Induce SsrA Tagging during Translation Termination* 210. J. Biol. Chem. 277, 33825–33832 (2002).
van Nies, P. et al. Unbiased Tracking of the Progression of mRNA and Protein Synthesis in Bulk and in Liposome‐Confined Reactions. Chem. Bio. Chem. 14, 1963–1966 (2013).
Kazuta, Y., Matsuura, T., Ichihashi, N. & Yomo, T. Synthesis of milligram quantities of proteins using a reconstituted in vitro protein synthesis system. J. Biosci. Bioeng. 118, 554–557 (2014).
Matsuura, T., Tanimura, N., Hosoda, K., Yomo, T. & Shimizu, Y. Reaction dynamics analysis of a reconstituted Escherichia coli protein translation system by computational modeling. Proc. Natl. Acad. Sci. USA. 114, E1336–E1344 (2017).
Gruber, A. R., Lorenz, R., Bernhart, S. H., Neuböck, R. & Hofacker, I. L. The Vienna RNA Websuite. Nucleic Acids Res. 36, W70–W74 (2008).
Lorenz, R. et al. ViennaRNA Package 2.0. Algorithms Mol. Biol. 6, 26 (2011).
Chandu, D., Kumar, A. & Nandi, D. PepN, the Major Suc-LLVY-AMC-hydrolyzing Enzyme in Escherichia coli, Displays Functional Similarity with Downstream Processing Enzymes in Archaea and Eukarya IMPLICATIONS IN CYTOSOLIC PROTEIN DEGRADATION*. J. Biol. Chem. 278, 5548–5556 (2003).
Bhosale, M. et al. Catalytic activity of Peptidase N is required for adaptation of Escherichia coli to nutritional downshift and high temperature stress. Microbiol. Res. 168, 56–64 (2013).
Evers, S. et al. Mechanism‐related changes in the gene transcription and protein synthesis patterns of Haemophilus influenzae after treatment with transcriptional and translational inhibitors. Proteomics 1, 522–544 (2001).
Bandow, J. E., Brötz, H., Leichert, L. I. O., Labischinski, H. & Hecker, M. Proteomic Approach to Understanding Antibiotic Action. Antimicrob. Agents Chemother. 47, 948–955 (2003).
Miller, J. H. Experiments in molecular genetics. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory (1972).
Chiba, S. & Ito, K. Multisite Ribosomal Stalling: A Unique Mode of Regulatory Nascent Chain Action Revealed for MifM. Mol. Cell 47, 863–872 (2012).
Onodera, H., Niwa, T., Taguchi, H. & Chadani, Y. Prophage excision switches the primary ribosome rescue pathway and rescue‐associated gene regulations in Escherichia coli. Mol. Microbiol. 119, 44–58 (2023).
Gillet, L. C. et al. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteom. 11, O111.016717 (2012).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Midha, M. K. et al. A comprehensive spectral assay library to quantify the Escherichia coli proteome by DIA/SWATH-MS. Sci. Data 7, 389 (2020).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. CryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Watson, Z. L. et al. Structure of the bacterial ribosome at 2 Å resolution. eLife 9, e60482 (2020).
Brown, A. et al. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr. Sect. D: Biol. Crystallogr. 71, 136–153 (2015).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 74, 531–544 (2018).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010).
Barad, B. A. et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Okuda, S. et al. jPOSTrepo: an international standard data repository for proteomes. Nucleic Acids Res. 45, D1107–D1111 (2017).
Acknowledgements
We thank Takashi Kanamori for providing customized PUREfrex v1.0, and the anonymous reviewers for their valuable comments. Additionally, we extend our thanks to the Integrative Bioscience Facility at the Institute of Science Tokyo for DNA sequencing, and the Cell Biology Center Research Core Facility at the Institute of Science Tokyo for the Q-Exactive mass spectrometry measurements. This work was supported by MEXT Grants-in-Aid for Scientific Research (Grant Number JP20H05925 to H.T.), Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP22ama121012 (support number 4857 to H.T.) and JP23ama121002 (support number 3273 to O.N.), Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant Numbers 22H02553 to Y.I., 23H02410 to Y.C.), and Core Research for Evolutional Science and Technology (CREST) of the Japan Science and Technology Agency (JST) (Grant Number JPMJCR20E2 to O.N.), Pioneering Research Initiated by the Next Generation (SPRING) of Japan Science and Technology Agency (JST) (Grant Number JPMJSP2106 to A.K.), and by grants from the Ohsumi Frontier Science Foundation, the Japan Foundation for Applied Enzymology, the Takeda Science Foundation, the Yamada Science Foundation, and the Senri Life Science Foundation to Y.C.
Author information
Authors and Affiliations
Contributions
A.K., Y.C., S.K., and Y.K. performed genetic and biochemical experiments. T.N. and A.Y. performed mass-spectrometry measurements and data analyses. Y.A. collected the cryo-EM data and built the model. Y.A. and Y.I. analyzed the structure. Y.C., Y.A., A.K., T.N., Y.I., O.N., and H.T. wrote the manuscript. Y.C., Y.I., H.T., and O.N. supervised the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ando, Y., Kobo, A., Niwa, T. et al. A mini-hairpin shaped nascent peptide blocks translation termination by a distinct mechanism. Nat Commun 16, 2323 (2025). https://doi.org/10.1038/s41467-025-57659-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57659-z
