
Abstract
The ubiquitin ligase Nedd4 (Nedd4-1), comprised of C2-WW(n)-HECT domains, regulates protein trafficking. We recently described a primate-specific Nedd4-1 splice isoform with an extended N-terminus replacing the C2 domain, called Nedd4-1(NE). Here, we show that while canonical Nedd4-1 is primarily localized to the cytosol, Nedd4-1(NE) localizes to late endosomes. This localization is mediated by the NE region, is dependent on amino acid availability, is independent of mTORC1, and is inhibited by the autophagy inducer IKKβ. We further demonstrate that VPS16B, which regulates late endosome to lysosome maturation, is a unique Nedd4-1(NE) substrate that co-localizes with Nedd4-1(NE) in the presence of nutrients. Importantly, a potentially pathogenic homozygous variant identified in the NE region (E70Q) of a patient with lymphangiectasia and protein-losing enteropathy leads to reduced VPS16B ubiquitination by Nedd4-1(NE). Finally, we report that Nedd4-1(NE) inhibits autophagy, likely by disrupting late endosome to autophagosome maturation. This work identified an mTORC1-independent, IKK-driven mechanism to regulate Nedd4-1(NE) localization to late endosomes in primates in response to nutrient availability, and uncovered suppression of autophagy by this ubiquitin ligase.
Similar content being viewed by others


Introduction
Cellular proteostasis, a phenomenon crucial to ensuring cellular health and survival, is maintained by regulated protein synthesis and degradation. The latter is tightly controlled by ubiquitination, a post-translational modification that commonly results in proteolysis. This process is mediated by ubiquitin ligases, enzymes that are responsible for recognizing and ubiquitinating specific target proteins1. In addition to binding their substrates and catalyzing their ubiquitination, ubiquitin ligases must also be capable of localizing to specific subcellular locations in a regulated manner to bind and degrade substrates only where and when appropriate.
The ubiquitin ligase Nedd4-1 (also known as Nedd4) regulates multiple cellular functions, including endocytosis of growth factor receptors, cell growth and proliferation2. Nedd4-1 is composed of an N-terminal C2 domain that regulates calcium-dependent plasma membrane localization3 and catalytic activity through an intramolecular interaction with the HECT domain4,5,6, 4 WW domains that usually mediate substrate recognition and binding7,8, and a C-terminal HECT domain that catalyzes ubiquitin transfer9. While several splice isoforms of the Nedd4-1 relative Nedd4-2 (Nedd4-like/Nedd4L) have been described, including multiple isoforms lacking a C2 domain10,11, little is known about alternative splicing of Nedd4-1.
We recently reported that alternative splicing of Nedd4-1 can produce functionally distinct isoforms, including the primate-specific Nedd4-1(NE), an N-terminally extended (NE) isoform that contains a 516-amino acid NE region, which replaces most of the C2 domain12. This isoform is ubiquitously expressed in all human tissues and cells tested (including the intestine), albeit at significantly lower levels than the canonical isoform12. Nonetheless, proteomic analyses of human tissues have identified peptide fragments unique to the NE region, confirming the expression of Nedd4-1(NE) at the protein level13,14. We found that Nedd4-1 and Nedd4-1(NE) differ in their catalytic activity, substrates, and mechanisms of regulation; however, differences in their subcellular localization and associated biological consequences have not been characterized. Here, we identify Nedd4-1(NE) as a late endosome-localized ubiquitin ligase. This localization is unique to this isoform, and contrasts with canonical Nedd4-1, which localizes to the cytosol and plasma membrane. Importantly, we show that the localization of Nedd4-1(NE) depends on amino acid availability, but is independent of mTORC1 signaling, instead relying on IKKβ signaling; in the absence of amino acids, Nedd4-1(NE) re-localizes to the cytosol. Through proteomic (BioID/miniTurbo) and biochemical approaches, we identify unique interactors of Nedd4-1(NE) at late endosomes, including VPS16B, which we show is a substrate of Nedd4-1(NE). Moreover, we found that a pathogenic homozygous variant in the NE region (E70Q) of a patient with lymphangiectasia and protein-losing enteropathy leads to impaired VPS16B ubiquitination by Nedd4-1(NE). Finally, we identify Nedd4-1(NE) as a negative regulator of autophagy.
Results
Nedd4-1(NE) localizes to late endosomes
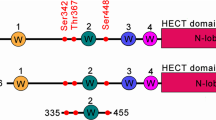
Alternative splicing generates multiple isoforms of Nedd4-1 in humans, including canonical Nedd4-1 and Nedd4-1(NE) (Fig. 1A). To further explore the role of the NE region in Nedd4-1(NE) function, we compared the subcellular localization of Nedd4-1(NE) to that of the canonical isoform. To do so, we transfected HeLa cells with N-terminally Flag-tagged Nedd4-1 or Nedd4-1(NE) and assessed protein localization by immunofluorescence. While canonical Nedd4-1 exhibited diffuse cytosolic and plasma membrane staining (Fig. 1B), Nedd4-1(NE) primarily localized to punctate structures (Fig. 1C). The prominent punctate localization of Nedd4-1(NE) was not restricted to one cell type, as it was also seen in HEK293T cells (Figure S1A) and megakaryocytic Dami cells (Figure S1B). This localization was also independent of its fusion tag, as V5-tagged Nedd4-1(NE) displayed a similar punctate pattern (Figure S1C). Moreover, we found punctate localization of endogenous Nedd4-1(NE), identified with NE-specific antibodies that we raised (Figure S2). This punctate staining was absent or significantly reduced in cells probed with pre-immune serum or secondary antibody alone (Figure S2C–G), as well as in Nedd4-1(NE) knockout (KO) cells (Figure S3), suggesting that this signal is NE-specific. Notably, deletion of the NE region in Nedd4-1(NE) resulted in cytosolic localization (Fig. 1D), while the NE region alone displayed punctate localization (Fig. 1E), suggesting that the NE region is both necessary and sufficient to induce punctate localization of Nedd4-1(NE) in cells.

A Schematic representation of the canonical Nedd4-1 and the Nedd4-1(NE) isoform. Due to alternative splicing, Nedd4-1(NE) contains a 516-amino acid NE region and only a small part of the C2 domain. Images are drawn to scale. B–E HeLa cells were transfected with Flag-tagged constructs, as indicated. Cells were fixed, permeabilized, and immunostained with anti-Flag antibodies (green). Nuclei were stained with DAPI (blue). F HeLa cells were treated as in (B), and fixed samples were additionally immunostained using antibodies against endogenous Rab7 (red). G Quantification of the percentage of overlap of Nedd4-1(NE) with various endogenous protein markers. Data are means ± SD (N = 3 independent experiments, n = 27-36 cells/experiment; precise values are indicated in the Source Data file). All scale bars are 10 μm.
To uncover the identity of these punctate structures, we transfected HeLa cells with Flag-tagged Nedd4-1(NE) and observed the colocalization of tagged protein with endogenous markers of known vesicles and organelles by immunofluorescence microscopy. Nedd4-1(NE) showed a high degree of overlap with a subset of Rab7-positive vesicles (Fig. 1F; see also Figure S4D), indicative of localization at late endosomes. Moreover, we observed Nedd4-1(NE) localization at spherical, ring-shaped structures, consistent with localization at endosomal membranes (Fig. 1C, F). Nedd4-1(NE) showed greater overlap with Rab7-positive structures compared to clathrin-coated vesicles, and much greater overlap compared to caveolae (caveolin-1), early endosomes (EEA1), recycling endosomes (Rab11), lysosomes (LAMP1), or Golgi (RCAS) (Fig. 1G, Figure S4A–G). Despite the high degree of overlap, we observed no detectable co-immunoprecipitation between Nedd4-1(NE) and Rab7 (Figure S4H), suggesting the absence of a complex between the two proteins. Collectively, these data demonstrate that while canonical Nedd4-1 mainly localizes to the cytosol, the Nedd4-1(NE) isoform primarily localizes to late endosomes.
Nedd4-1(NE) vesicular localization is regulated by amino acid abundance
To decipher what triggers the localization of Nedd4-1(NE) to late endosomes, we performed immunofluorescence experiments analyzing its subcellular localization in response to different conditions and stresses. When cells were incubated in complete growth medium (DMEM, supplemented with serum), most Nedd4-1(NE)-expressing cells displayed a punctate staining pattern (Fig. 2A). This distribution was not altered when cells were subjected to various stresses or stimuli, including TNFα, LPS, heat shock, or hyperosmolarity (Figure S5A–D). However, when cells were incubated in Hank’s Balanced Salt Solution (HBSS), a solution devoid of serum and amino acids, most Nedd4-1(NE) was diffusely localized in the cytosol and at the plasma membrane, resembling the localization of canonical Nedd4-1 (Fig. 2B, D).

HeLa cells were transfected with 3xFlag-Nedd4-1(NE) and incubated for 1 hr in (A) DMEM (supplemented with 10% serum), (B) HBSS, or (C) HBSS supplemented with an essential amino acid (EAA) cocktail plus glutamine (Gln; 4 mM). Cells were fixed, permeabilized, and immunostained with anti-Flag antibodies (green). Nuclei were stained with DAPI (blue). D–J Nedd4-1(NE) localization was scored blindly as either “diffuse - cytoplasm/membrane”, “punctate”, or “both”, as indicated by the examples in (H–J). Scale bars are (A–C) 10 μm, or (H–J) 15 μm. Data are means ± SEM of 3 independent experiments. Statistical significance was determined using a chi-squared test (N.S.: not significant; **: p < 0.01; ****: p < 0.0001). In (E), statistical tests are comparing each condition to HBSS. Cell counts for each condition are indicated in Table S2.
We next sought to identify the specific factor that regulates Nedd4-1(NE) localization by comparing the individual components of DMEM and HBSS. These solutions differ mainly in their glucose concentrations, amino acid compositions, and supplementation with serum. Notably, supplementing HBSS with essential amino acids significantly rescued Nedd4-1(NE) punctate localization, a phenomenon that was further enhanced by the addition of glutamine (Fig. 2C, E, F). On the other hand, supplementing HBSS with glucose did not alter the mainly diffuse distribution of Nedd4-1(NE) (Fig. 2E, Figure S5F, G). Moreover, elimination of serum in DMEM did not alter the vesicular localization of Nedd4-1(NE) (Figure S5E). These data demonstrate that amino acids are required for the localization of Nedd4-1(NE) to vesicles.
Given that amino acids are known to activate mTORC1 signaling, we tested whether mTORC1 signaling is involved in mediating Nedd4-1(NE) localization. Unexpectedly, we found that mTORC1 inhibition by rapamycin treatment did not mimic the effect of amino acid deprivation on Nedd4-1(NE) localization (Fig. 2G, Figure S6A–C), despite inducing a similar inhibition of mTORC1 activity, as assessed by immunoblotting for phospho-p70-S6K (Figure S6D). Moreover, co-transfection with RagAQ66L and RagCS75L, which render mTORC1 signaling less sensitive to amino acid deprivation15, had no effect on Nedd4-1(NE) localization (Figure S6E, F). Taken together, these data suggest that amino acids regulate Nedd4-1(NE) localization to late endosomes via a mTORC1-independent mechanism.
The autophagy inducer IKKβ inhibits Nedd4-1(NE) localization to late endosomes
Since the regulation of Nedd4-1(NE) localization to late endosomes is dependent on amino acid availability, but not on mTORC1 activity, we searched for an alternative pathway that may be involved. In search for cellular binding partners unique to the Nedd4-1(NE) isoform, we performed a miniTurbo (modified BioID) screen16,17 to identify proximity/binding partners of Nedd4-1(NE) that do not bind canonical Nedd4-1. The miniTurbo screen identified several proteins related to autophagy, including the autophagy inducer IKKβ (Fig. 3A, Table S3). The inhibitor of nuclear factor-κB (IκB) kinase (IKK) complex, which includes kinases IKKα and IKKβ, was demonstrated earlier to promote autophagy in response to nutrient deprivation, independent of NF-κB activation18.

A 293 T-REx cells expressing 3xFlag-miniTurbo (FmT)-tagged constructs of Nedd4-1 or Nedd4-1(NE) were treated with biotin for 30 min and then collected. Biotinylated proteins in the lysates were identified by mass spectrometry. All raw mass spectrometry data files have been deposited at the MassIVE archive (massive.ucsd.edu), accession ID MSV000093801. Proteins involved in autophagy and protein trafficking were identified using GO-TermFinder and dot plots were generated using ProHits-viz web tool54. Prey abundance across baits and prey confidence are displayed as indicated in the legend (below). B HEK293T cells were transfected with the indicated constructs. Cell lysates were subjected to Flag immunoprecipitation (IP), and the indicated proteins were detected by immunoblotting. Constructs labeled “CS” correspond to full-length, catalytically inactive (Cys → Ser) mutants. C HEK293T cells were transfected with Flag-NE and incubated in the absence or presence of BI605906 (10 μM, 2 hrs) and Calyculin A (50 nM, 10 min). Cell lysates were incubated in 1% SDS and boiled to dissociate non-covalent protein complexes prior to Flag IP. NE phosphorylation was detected by immunoblotting. The markers beside each blot indicate molecular weight (measured in kDa). D–G HeLa cells were transfected with (D) Flag-IKKβ alone, (E), V5-Nedd4-1(NE) alone, (F) both Flag-IKKβ and V5-Nedd4-1(NE), or (G) 3xFlag-Nedd4-1(NE) alone. Cells were incubated in (D–F) DMEM, or (G) HBSS, in the absence or presence of 10 μM BI605906, for 2 hrs. Cells were fixed, permeabilized, and immunostained with (D–F) anti-Flag (IKKβ, red) and/or anti-V5 (Nedd4-1(NE), green) antibodies, or (G) anti-Flag (Nedd4-1(NE), green). Nuclei were stained with DAPI (blue). Scale bars are 10 μm. H–I Nedd4-1(NE) localization was scored as described in Fig. 2. Data are means ± (B, C) SD or (H, I) SEM of 3 independent experiments. Statistical significance was determined using (B) Student’s t-test, (C) one-way ANOVA, or (H, I) a chi-squared test (***: p < 0.001; ****: p < 0.0001). In (I), statistical tests are comparing each condition to the DMSO-treated control. Cell counts for each condition are indicated in Table S2.
Consistent with our miniTurbo data, we show through co-immunoprecipitation (co-IP) experiments that IKKβ binds to Nedd4-1(NE), but not to Nedd4-1 (Fig. 3B, Figure S7A). Moreover, we show that the NE region of Nedd4-1(NE) is phosphorylated in response to amino acid deprivation in an IKKβ-dependent manner (Fig. 3C). While we observed an overall increase in NE phosphorylation under starvation conditions, our phosphoproteomic analysis did not generate sufficient spectral counts of phosphopeptides to compare the phosphorylation status of specific residues between DMEM and HBSS conditions (Figure S7B). However, mutation of the most abundant phosphosite, Ser458Ala, did not affect the subcellular localization of Nedd4-1(NE) (Figure S9A), suggesting that phosphorylation (at least of this site), may not control the mobilization of Nedd4-1(NE) to late endosomes.
Given that Nedd4-1(NE) is phosphorylated by IKKβ under starvation conditions, we next sought to investigate whether IKKβ can regulate Nedd4-1(NE) localization. Our results show that while IKKβ is diffusely localized throughout the cytoplasm, in the absence or presence of Nedd4-1(NE) co-expression (Fig. 3D, F), ectopic expression of IKKβ inhibited Nedd4-1(NE) localization to late endosomes (Fig. 3E, F, H), mimicking the effect of amino acid starvation. Notably, we observed an inverse correlation between the level of expression of IKKβ and the punctate localization of Nedd4-1(NE) (Fig. 3F). In accord, we show that treatment with the IKKβ inhibitor BI605906 partially rescued the punctate localization of Nedd4-1(NE) under conditions of amino acid starvation (Fig. 3G, I, Figure S7C). Collectively, our results suggest that IKKβ regulates the localization of Nedd4-1(NE) in response to amino acid deprivation.
Nedd4-1(NE) interacts with endo-lysosomal proteins including VPS33B and VPS16B
Given the unique subcellular localization of Nedd4-1(NE) compared to Nedd4-1, we hypothesized that Nedd4-1(NE) may have unique substrates in or around late endosomes. Using the above mentioned miniTurbo screen, we identified not only autophagy-related significant hits, but also multiple proteins involved in trafficking (Fig. 3A, Table S3), including many reported to localize to subcellar compartments such as endosomes, lysosomes, and/or Golgi.
Notably, several vacuolar protein sorting (VPS) family proteins were identified as unique interactors of Nedd4-1(NE), including VPS33B, a protein involved in the regulation of intracellular protein trafficking19. Consistent with our proteomics data, we confirmed through co-IP experiments that VPS33B interacts with Nedd4-1(NE), but not Nedd4-1 (Fig. 4A). A major binding partner of VPS33B is VPS16B (also known as VIPAS39 or SPE39)20,21,22. Like VPS33B, VPS16B is involved in protein trafficking22,23. VPS33B and VPS16B form a unique bidirectional Sec1-Munc18 protein complex, with VPS16B-VPS33B implicated in SNARE-mediated vesicular trafficking21. Consistent with this, we found that VPS16B also interacts with Nedd4-1(NE), but not with Nedd4-1 (Fig. 4B, C), suggesting the existence of a complex involving Nedd4-1(NE) and VPS16B-VPS33B. Moreover, we also validated binding (by co-IP) of Nedd4-1(NE), but not canonical Nedd4-1, to the miniTurbo hit VPS16A (Fig. 3A, Figure S8), which is part of the HOPS and CORVET complexes; VPS16A was previously shown to regulate endocytic trafficking to lysosomes and endosome/autophagosome maturation24,25.

HEK293T cells were transfected with the indicated constructs. Cell lysates were (A–C) subjected to Flag immunoprecipitation (IP), or (D, E, G) incubated in 1% SDS and boiled to dissociate non-covalent protein complexes prior to Flag IP, and the indicated proteins were detected by immunoblotting (Ub: ubiquitin). The ubiquitination signal was normalized to both VPS16B (or VPS33B) IP levels and Nedd4-1(NE) (or Nedd4-1) lysate levels. F Protein levels of Flag-VPS16B were quantified and normalized to cellular actin levels. In (A), Flag blots were imaged at the same time and images were corrected using the same parameters (MW: molecular weight). Constructs labeled “CS” correspond to full-length, catalytically inactive (Cys → Ser) mutants, and the construct labeled E70Q in (G) represents the lymphangiectasia patient mutation. All data are means ± SD of (A–E) 3 or (F, G) 4 independent experiments. Statistical significance was determined using (A–C) Student’s t-test, or (D–G) one-way ANOVA (N.S.: not significant; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001). The markers beside each blot indicate molecular weight (measured in kDa).
We recently demonstrated that Nedd4-1(NE) is phosphorylated at 14-3-3 binding sites, Thr16 and Ser458, in the NE region, and that phosphorylation-ablating mutations (T16A/S458A; “2A”) stabilize or enhance global substrate binding12. In accord, we found that VPS16B exhibits increased binding to the 2A mutant compared to the wild-type (WT) NE region (Figure S9B, C). However, these mutations did not affect the punctate localization of Nedd4-1(NE) (Figure S9A), suggesting that 14-3-3 binding and phosphorylation at these sites do not regulate Nedd4-1(NE) subcellular localization.
Nedd4-1(NE) ubiquitinates and colocalizes with VPS16B
The main function of ubiquitin ligases, including Nedd4-1(NE), is to catalyze the transfer of ubiquitin onto their specific substrates. Thus, we investigated whether VPS33B and/or VPS16B are substrates of Nedd4-1(NE) by analyzing their ubiquitination by Nedd4-1(NE). We found no significant difference between VPS33B ubiquitination mediated by Nedd4-1(NE) WT or by Nedd4-1(NE) CS, a catalytically inactive mutant that displays similar subcellular distribution to the WT protein (Figure S9A), suggesting that VPS33B is not a substrate of Nedd4-1(NE) (Fig. 4D). On the other hand, we observed Nedd4-1(NE)-mediated ubiquitination of VPS16B, ubiquitination that was significantly reduced in the presence of Nedd4-1(NE) CS (Fig. 4D). Specifically, we show that Nedd4-1(NE) catalyzes the attachment of K63-linked ubiquitin chains onto VPS16B (Figure S10A). Consistent with the above interaction data, we found that unlike Nedd4-1(NE), Nedd4-1 was unable to ubiquitinate VPS16B (Fig. 4E). Moreover, in line with the known consequence of ubiquitination often leading to protein degradation, we found that VPS16B (but not VPS33B) steady-state levels were decreased in the presence of Nedd4-1(NE) compared to Nedd4-1(NE) CS (Fig. 4F, Figure S10B). (We could not determine the regulation of VPS16B half-life by Nedd4-1(NE) using cycloheximide-chase because Nedd4-1(NE) is much less stable than VPS16B). Taken together, these data suggest that although VPS16B and VPS33B both exist in a complex with Nedd4-1(NE), only VPS16B is a substrate of Nedd4-1(NE).
In search for pathogenic variants associated with protein-losing enteropathy (due to lymphangiectasia), a homozygous variant of unknown significance (chr15:g.56208822 C > G, NM_001285338.2:c.208 G > C, p.Glu70Gln) located in the NE region of Nedd4-1(NE) (hereafter called Nedd4-1(NE)-E70Q), was identified in a patient with consanguineous parents by whole exome sequencing (WES). This variant was relatively rare in the population (minor allele frequency of 0.001807) with no homozygotes reported in gnomAD v3 and with potential deleterious computational predictions (i.e., CADD score 15.0) (Table S4). Analysis of catalytic activity of this mutant revealed a significant reduction in its ability to ubiquitinate VPS16B (Fig. 4G). As expected, and much like the catalytically inactive Nedd4-1(NE) CS control, such a reduction in ubiquitination did not affect the subcellular localization of Nedd4-1(NE) E70Q relative to WT (Figure S9A). Interestingly, ubiquitination of the model substrate YY1, which binds the Nedd4-1 (and Nedd4-2) WW domains via its PY motifs26, did not exhibit reduced ubiquitination by the Nedd4-1(NE) E70Q mutant (Figure S9D). This suggests that it is not the catalytic activity per se of Nedd4-1(NE) that is affected by the E70Q mutation, but rather, ubiquitination of specific substrates that bind the NE region, such as VPS16B. Curiously, the reduced ubiquitination of VPS16B by the Nedd4-1(NE) E70Q mutant was not associated with reduced binding to this substrate, which was actually stronger than binding to the WT Nedd4-1(NE) (Figure S9E); the latter may suggest that tighter binding between Nedd4-1(NE) E70Q and VPS16B may hinder access of the HECT domain to VPS16B, or occlude access to some Lys residues that serve as ubiquitination sites.
Given the notable and dynamic localization of Nedd4-1(NE) at late endosomes, we next assessed the potential colocalization of Nedd4-1(NE) with its substrate VPS16B, a protein that has been reported to localize to late endosomes as well as the Golgi27. To do so, we co-transfected HeLa cells with VPS16B and Nedd4-1(NE) and assessed colocalization by immunofluorescence. When cells were incubated in DMEM, VPS16B displayed a significant colocalization with Nedd4-1(NE) in endosomes (Fig. 5A, B; Pearson’s r = 0.78 ± 0.08 SD). However, the co-localization between Nedd4-1(NE) and VPS16B was reduced when cells were incubated in HBSS (Fig. 5A, B; Pearson’s r = 0.55 ± 0.17 SD), consistent with our earlier finding that Nedd4-1(NE) localization is dependent on amino acid abundance. Collectively, these data suggest that when amino acids are abundant, Nedd4-1(NE) and VPS16B are both localized at late endosomes, where Nedd4-1(NE) can bind, ubiquitinate, and degrade VPS16B. In contrast, under nutrient deprivation, Nedd4-1(NE) re-localizes to the cytosol and is separated from VPS16B.

A, C, D HeLa cells were transfected with the indicated constructs. In (A), cells were incubated for 1 hr in DMEM or HBSS. Cells were fixed, permeabilized, and immunostained with anti-Flag (green) and/or anti-V5 (red) antibodies. Nuclei were stained with DAPI (blue). All scale bars are 10 μm. B, E Quantification of the Pearson correlation coefficient between (B) VPS16B and Nedd4-1(NE) or (E) GFP-LC3 and either Nedd4-1(NE) or Nedd4-1. Each data point represents an individual cell (B) n = 68 DMEM cells, 67 HBSS cells, across 3 independent experiments; (E) n = 86 cells/condition, across 3 independent experiments). Horizontal bars represent the means ± SD. Statistical significance was determined using Student’s t-test (****: p < 0.0001).
Notably, we observed some variability in the size of Nedd4-1(NE)-positive puncta between different cells (Figure S11A), which may be attributed to differences in the expression levels of Nedd4-1(NE). Because larger size, “swollen” late endosome may indicate impaired cargo traffic between late endosomes and lysosomes19, we analyzed distribution of endogenous Rab7 in the absence or presence of ectopically expressed Nedd4-1(NE). We found that cells ectopically expressing Nedd4-1(NE) contain a significantly greater percentage of large late endosomes (i.e., Rab7-positive puncta greater than 3 μm3) than untransfected cells (Figure S11B). Thus, we propose that Nedd4-1(NE) may perturb cargo traffic by disrupting endo-lysosomal maturation.
Nedd4-1(NE) suppresses autophagy
To delineate a functional role for Nedd4-1(NE), we focused on autophagy, a process influenced by both its substrate VPS16B and its kinase IKKβ. Indeed, VPS16B has been implicated in endo-lysosomal maturation22, an important precursor to autolysosome formation during autophagy28. Moreover, IKKβ was previously shown to stimulate autophagy in response to amino acid deprivation18. Consistent with the known colocalization between Rab7 and LC3 at autophagic vesicles29,30, we found that Nedd4-1(NE) displayed a strong colocalization with LC3 (Fig. 5C, E; Pearson’s r = 0.71 ± 0.10 SD). On the other hand, the diffusely localized Nedd4-1 displayed significantly less colocalization with LC3 (Fig. 5D-E; Pearson’s r = 0.53 ± 0.13 SD). Notably, our results show that LC3 is not a substrate of Nedd4-1(NE) (Figure S12), suggesting that although Nedd4-1(NE) is localized to autophagosomes, the potential role for Nedd4-1(NE) in the regulation of autophagy is not due to the degradation of LC3.
In support of the potential role of Nedd4-1(NE) in autophagy, our aforementioned miniTurbo screen identified multiple components of autophagy machinery (Fig. 3A, Table S3), including WDR45 (also known as WIPI4; mammalian homolog of Atg18), which we validated as an interactor of Nedd4-1(NE), but not Nedd4-1 (Figure S13A). Furthermore, we show that Nedd4-1(NE) binds ATG2B (Figure S13B), a known binding partner of WDR4531 and a paralog of ATG2A, another hit from our miniTurbo screen. Together, WDR45 and ATG2A/B control nascent autophagosome expansion32; thus, they represent potential targets through which Nedd4-1(NE) may regulate autophagy.
To test if Nedd4-1(NE) indeed regulates autophagy, we expressed Nedd4-1(NE) in HeLa cells and assessed autophagic flux by measuring the stabilization of endogenous LC3B-II protein levels in cells treated with bafilomycin A, a lysosome inhibitor, relative to untreated cells. Our results show that when amino acids are abundant, autophagic flux was significantly decreased in cells expressing Nedd4-1(NE) compared to control cells (Fig. 6A-B), consistent with a role for Nedd4-1(NE) in suppressing autophagy. In accord, we show that Nedd4-1(NE) reduces the number of GFP-LC3-positive puncta (i.e., autophagosomes) under conditions known to stimulate their increase (HBSS), a result that was blunted when cells were instead transfected with the catalytically inactive Nedd4-1(NE) CS (Figure S13C-F). Taken together, these results suggest that Nedd4-1(NE) inhibits autophagy, possibly through the dysregulation of endosome/autophagosome maturation.

A HeLa cells were transfected with 3xFlag-Nedd4-1(NE) as indicated. Cells were incubated in either DMEM or HBSS for 4 hrs in the presence or absence of bafilomycin A (100 nM). The markers beside each blot indicate molecular weight (measured in kDa). B Autophagic flux was quantified by measuring endogenous LC3B-II (normalized to total LC3B) stabilization upon bafilomycin A treatment. Data are means ± SD of 5 independent experiments. Statistical significance was determined using Student’s t-test (***: p < 0.001). C Model of the proposed mechanism. When amino acids are abundant, Nedd4-1(NE) localizes to late endosomes (or autophagosomes), resulting in the suppression of autophagy. However, under starvation conditions, IKKβ is activated and phosphorylates Nedd4-1(NE), leading to Nedd4-1(NE) re-localization to the cytosol, thus allowing autolysosomal maturation and autophagy to proceed. Model created in BioRender. Rotin, D. (2025). https://BioRender.com/x91o950.
Discussion
Our studies here distinguish Nedd4-1(NE) from the canonical (Nedd4-1) isoform by demonstrating differences in their subcellular localization and their cellular substrates and functions. While Nedd4-1 is primarily dispersed throughout the cytosol, Nedd4-1(NE) localizes to late endosomes when amino acids are abundant. Once there, it can interact with and ubiquitinate substrates such as VPS16B, and suppress autophagy in a mTORC1-independent manner (Fig. 6C), likely via interaction with the IKK complex that is known to regulate autophagy18. This work provides evidence of alternative splicing regulating the subcellular localization of Nedd4-1 isoforms, which we propose contributes to the regulation of autophagy in primates; while canonical Nedd4-1 was previously shown to promote autophagy (see below), we show here that Nedd4-1(NE) inhibits it.
Members of the Nedd4 family typically contain an N-terminal C2 domain that regulates plasma membrane localization, through interactions with calcium and phospholipids3,33, and catalytic activity, through an intramolecular interaction with the HECT domain4,5,6. Structural studies of Nedd4 family members suggest that a common interface exists on the C2 domain to which HECT domains, calcium, and phospholipids can all bind4,33. The residues that form this interface are all absent in Nedd4-1(NE), resulting in the lack of a functional C2 domain and associated forms of regulation12. Instead, Nedd4-1(NE) utilizes its unique NE region, an N-terminal segment not found in other Nedd4 family members or any other proteins, as a regulatory domain. We previously demonstrated that phosphorylation of the NE region at 14-3-3 binding sites regulates strength of substrate binding12. In our current study, we identify important biological functions of the NE region: regulation of the subcellular localization of Nedd4-1(NE), complex formation with VPS16B and IKKβ (novel substrates of Nedd4-1(NE)), and suppression of autophagy.
Through immunofluorescence experiments, we demonstrated the dynamic subcellular localization of Nedd4-1(NE). In addition to displaying a unique, isoform-specific localization to late endosomes, Nedd4-1(NE) re-localizes primarily to the cytosol under conditions of amino acid starvation. Notably, we found that mTORC1 does not regulate Nedd4-1(NE) localization, suggesting that a different amino acid sensing pathway is involved. Indeed, we show that IKKβ, which was previously shown to promote autophagy in response to nutrient deprivation (independent of NF-κB activation18), phosphorylates Nedd4-1(NE) and prevents its late endosomal localization, thus providing an additional mechanism to allow autophagy to proceed.
Another unique role of the NE region of Nedd4-1(NE), compared to the C2 domain of Nedd4-1, is its ability to mediate the formation of a complex between Nedd4-1(NE), VPS16B, and VPS33B, resulting in VPS16B ubiquitination and degradation. VPS16B was previously reported to be ubiquitinated in response to EGF stimulation34, however no ubiquitin ligase has been implicated in this process. Here, we report that Nedd4-1(NE), but not Nedd4-1, colocalizes with VPS16B at late endosomes, where it can catalyze its ubiquitination and degradation. This represents a novel mechanism by which the VPS16B-VPS33B complex can be regulated in higher order mammals in which Nedd4-1(NE) is expressed. This has notable implications in humans, where VPS16B and VPS33B are necessary for α-granule maturation in platelets; loss of function of either of these proteins causes Arthrogryposis, Renal dysfunction, and Cholestasis (ARC) syndrome, a multi-system disorder associated with defects in α-granule formation in megakaryocytes and platelets27,35,36,37. Notably, we found late endosome localization of Nedd4-1(NE) in the megakaryocytic Dami cell line, which expresses Nedd4-1(NE) endogenously.
In addition to VPS33B, our miniTurbo screen identified other proteins involved in protein trafficking that are proximal or binding to Nedd4-1(NE), but not Nedd4-1. Interestingly, while VPS16B was not identified as a hit in our screen, we did identify (and validate) VPS16A, a protein that shares both structural similarity and a common ancestor with VPS16B27. VPS16A is a core component of the HOPS and CORVET complexes, which are involved in endo-lysosomal membrane fusion events, and endosome and autophagosome maturation24,25. Other notable hits included VPS18, another core component of the HOPS and CORVET complexes22, and VPS37B, a subunit of the ESCRT-I complex, which is involved in sorting cargo into intraluminal vesicles38. Collectively, these proteins represent additional targets that may be regulated by Nedd4-1(NE) to control endosomal maturation, protein trafficking, and/or autophagy.
The process of endosomal maturation is paralleled in autophagy, where autophagosomes mature and eventually fuse with lysosomes to form autolysosomes28. These two pathways share common machinery, including certain Rab proteins and ESCRT complexes. In this study, we suggest a role for Nedd4-1(NE) in both processes. Interestingly, the canonical Nedd4-1 isoform (which is mainly localized to the cytosol) has been reported to positively regulate autophagy through multiple different mechanisms, such as stabilizing Beclin-139, stabilizing VPS3440, and promoting SQSTM1-mediated inclusion body autophagy41. In contrast, in our current study we show that Nedd4-1(NE) negatively regulates autophagy.
Our studies here mostly utilized ectopic expression of tagged Nedd4-1(NE) due to its low abundance of protein expression. Nevertheless, we were also able to detect punctate localization of endogenous Nedd4-1(NE) by immunofluorescence in cells using NE-specific antibodies, consistent with earlier proteomic analyses identifying endogenous Nedd4-1(NE) protein in human tissues13,14. Moreover, the ectopically tagged Nedd4-1(NE) protein was also detected in puncta with our NE-specific antibodies. Further, ectopic expression of tagged Nedd4-1(NE) did not affect its subcellular localization, since the same batch of transfected cells exhibited a different localization pattern depending on amino acids availability. Thus, lower abundance proteins, especially those selected for in evolution (such as Nedd4-1(NE) that is expressed in primates), cannot be overlooked simply because they are harder to detect with existing technology, as they are likely to have important biological functions. Indeed, several lines of evidence underscore the physiological/pathological importance of Nedd4-1(NE) despite being normally expressed at low levels: (i) We describe here a significant reduction in ubiquitination of the bona fide Nedd4-1(NE) substrate, VPS16B, by Nedd4-1(NE) in a lymphangiectasia and protein-losing enteropathy (PLE) patient with a homozygous mutation (E70Q) in the NE region. This syndrome is characterized by loss of plasma proteins in the intestine resulting in hypoproteinemia, edema, infections, and intestinal disease such as diarrhea42. Numerous diseases, both genetic or acquired, are manifested as PLE, including those caused by block in intestinal lymphatic flow42, as observed in the patient with E70Q variant described here (see Table S4). (ii) a SNP in the non-coding region of the NEDD4 gene (rs80321580; C/C alleles), identified previously as a risk allele for keloids43, specifically enhances the expression of Nedd4-1(NE) (also called Nedd4-TV3), promoting keloid development44. (iii) our recent experiments show a significantly elevated mRNA expression of Nedd4-1(NE), but not canonical Nedd4-1, following stimulation with specific cytokines, such as TNFα (Persaud, Kefalas and Rotin, manuscript in preparation), suggesting that Nedd4-1(NE) may have a role in protecting cells from pathogenic infection in primates.
In summary, we propose a model (Fig. 6C) whereby the splice isoform Nedd4-1(NE) exhibits a unique, amino acid-dependent localization to late endosomes/autophagosomes, where it inhibits endosome-to-lysosome/autophagosome maturation and hence autophagy, by reducing the levels of VPS16B and possibly other LE proteins. Under starvation conditions, IKKβ phosphorylates Nedd4-1(NE), re-localizing it to the cytosol and allowing autophagy to proceed. Nedd4-1(NE) may also have additional suppressive effects on autophagy by disrupting the autophagy complex (demarcated by the presence of lipidated LC3) via interactions with specific autophagy proteins, such as ATG2B and WDR45. Importantly, these findings are unique to the Nedd4-1(NE) isoform, suggesting species-specific evolution of Nedd4-1 to provide a unique mechanism to regulate endosomal maturation and autophagy in primates.
Methods
Reagents used in this study are listed in the Supplementary Information (Table S1).
Cell culture
HEK293T, HeLa, and 293 T-REx cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and antibiotic-antimycotic solution. Dami cells were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM), supplemented with 10% horse serum (HS). 293 T-REx and Dami stable cell lines were maintained in media containing 100 µg/ml hygromycin B. Transfections were performed using PolyJet or PolyEZ reagents. All experiments were performed using cells incubated in media supplemented with 10% serum (FBS or HS) unless otherwise indicated.
Dami stable cell line generation and induction
Tetracycline-inducible, 3xFlag-tagged Nedd4-1(NE) was stably expressed in Dami cells using the Sleeping Beauty (SB) system45 with the SB100X transposase46. To generate this cell line, Dami cells were co-transfected with pCMV(CAT)T7-SB100 (transposase expression vector) and a pSB plasmid encoding Nedd4-1(NE) as well as GFP and hygromycin resistance. Selection was initiated by incubation with 200 µg/ml hygromycin B and single cell clones were generated using fluorescence activated cell sorting (FACS). Expression of 3xFlag-Nedd4-1(NE) in individual clones was validated by immunoblotting (Figure S14A). To induce differentiation, cells were treated with 1 μM phorbol 12-myristate 13-acetate (PMA) for 3 days. To induce Nedd4-1(NE) expression, cells were treated with 100 μg/ml tetracycline for 1 day.
Generation of Nedd4-1(NE) knockout line using CRISPR-Cas9
HeLa cells were transfected with a PX458 plasmid containing Cas9, GFP, and a gRNA targeting the NE region (GCTATCCAAGTCATCTCCGG). Transfected (GFP + ) cells were sorted by FACS into 96-well plates to generate single cell clones. After expansion, Nedd4-1(NE) knockout was confirmed by Sanger sequencing of genomic DNA (Figure S3A).
Constructs
Cloning of recombinant DNA into various expression vectors was performed using Gibson assembly, Gateway cloning, and/or restriction digestion and ligation. The NE region cDNA was derived from HeLa cells and was cloned into expression vectors as previously described12. Catalytically inactive (CS), 14-3-3 binding-deficient (T16A/S458A; “2 A”), and the lymphangiectasia patient (E70Q) Nedd4-1(NE) mutants were generated via site-directed mutagenesis. Recombinant DNA constructs used in this study are listed in Table S1.
Immunofluorescence and microscopy
HeLa cells seeded on glass cover slips were transfected as indicated. One day after transfection, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and stained with the indicated primary antibodies, fluorophore-conjugated secondary antibodies, and 4’,6-diamidino-2-phenylindole (DAPI) to label the nucleus. Samples were mounted on glass microscope slides using Dako mounting medium. Image acquisition was performed using an IX81 confocal microscope (Olympus) equipped with a 60X/1.35 oil immersion objective. Image processing and quantification were performed using Volocity version 7.0.0 (Quorum Technologies). Higher resolution imaging was performed using a LSM880 confocal microscope (Zeiss) equipped with a 63X/1.40 oil immersion Plan Apochromat objective and an Airyscan detector. Images were acquired using Airyscan fast sequential mode with an optical magnification of 1.8X. Z stacks were acquired with optimal Airyscan settings (144 nm Z-steps) for each individual channel using lasers 405 nm, 488 nm (argon), 552 nm and 642 nm. The Z-stack setting for each individual channel was set to maximize focus at the top and bottom of the imaging stack to cover the dynamic range of the detector. Scan average and digital gain were set to 1. Image (Airyscan) processing was performed in 3D-batch mode using Zen 2.3 SP1 FP3 (black) version 14.01.19.201 (Zeiss). Puncta scoring was performed by a blinded observer.
Immunoprecipitation (IP), ubiquitination assays, and immunoblotting
Cells were lysed in lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1.5 mM MgCl2, 1 mM EGTA) freshly supplemented with 1 mM PMSF, 10 µg/ml leupeptin, 10 µg/ml aprotinin, and 10 µg/ml pepstatin. Flag IPs were performed by incubating 1 mg clarified cell lysate with anti-Flag M2 affinity gel for 2 hrs at 4 °C. For ubiquitination assays, lysates were boiled in 1% sodium dodecyl sulfate (SDS) for 5 min at 95 °C prior to IP. Proteins from cell lysates and/or IPs were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. Antibodies used in this study are listed in Table S1. Affinity-purified antibodies against the NE region of Nedd4-1(NE) were purified from egg yolks following the immunization of chickens with the KLH-conjugated synthetic peptide CLQISLQPTRYSGYLQSSNVLA, corresponding to amino acids 86-107 of the Nedd4-1(NE) full-length protein (Capralogics Inc., Hardwick, MA). Immunoblots were imaged using a ChemiDoc MP imaging system (Bio-Rad) and analyzed using Image Lab version 6.1.0 (Bio-Rad). Uncropped immunoblot images are provided in the Source Data file.
BioID (miniTurbo) sample preparation
Tetracycline-inducible, miniTurbo-tagged proteins were stably expressed in 293 T-REx cells using the Flp-In T-REx site-specific recombination system. To generate these cell lines, cells were co-transfected with pOG44 (Flp-recombinase expression vector) and a pcDNA5-FRT-TO plasmid containing the coding sequence for Flag-miniTurbo (FmT)16 alone, or the FmT-tagged fusion protein. Nedd4-1 and Nedd4-1(NE) fusion proteins were C-terminally tagged, rendering them catalytically inactive, reducing the possibility of false negative hits due to protein degradation. Cell lines were generated by selection with 200 µg/ml hygromycin B. Expression of fusion proteins in the resulting isogenic cell pools was validated by immunoblotting (Figure S14B). Two independent pools (biological replicates) were created for each “bait” protein of interest. Stable isogenic cell pools expressing FmT alone (i.e., “tag”-only controls) or the FmT-fused bait proteins were expanded to five 15-cm plates. Expression of the fusion protein was induced by the addition of 1 μg/ml tetracycline to the culture media for 24 hrs. Cells were treated with 50 μM biotin for 30 min and were subsequently collected by scraping and centrifugation. Cell pellets were washed twice with PBS and stored at −80 °C until lysis. Biotin-streptavidin affinity purification was performed as previously described47.
Mass spectrometry
High performance liquid chromatography was conducted using a 2 cm pre-column (Acclaim PepMap; 50 mm × 100 μm inner diameter (ID)), and 50 cm analytical column (Acclaim PepMap; 500 mm × 75 um ID; C18; 2 μm; 100 Å; Thermo Fisher Scientific, Waltham, MA), running a 120 min reversed-phase buffer gradient at 225 nl/minute on a Proxeon EASY-nLC 1000 pump in-line with a Thermo Q-Exactive HF quadrupole-Orbitrap mass spectrometer. A parent ion scan was performed using a resolving power of 60,000, then up to the twenty most intense peaks were selected for MS/MS (minimum ion count of 1000 for activation) using higher energy collision induced dissociation (HCD) fragmentation. Dynamic exclusion was activated such that MS/MS of the same m/z (within a range of 10 ppm; exclusion list size = 500) detected twice within 5 sec were excluded from analysis for 15 sec. For protein identification, Thermo.RAW files were converted to the.mzXML format using Proteowizard48, then searched using X!Tandem49 and COMET50 against the Human RefSeq Version 45 database (containing 36,113 entries). Data were analyzed using the trans-proteomic pipeline (TPP)51 via the ProHits software suite (v3.3)52. Search parameters specified a parent ion mass tolerance of 10 ppm, and an MS/MS fragment ion tolerance of 0.4 Da, with up to two missed cleavages allowed for trypsin. Variable modifications of +16@M and W, +32@M and W, +42@N-terminus, and +1@N and Q were allowed. Proteins identified with an iProphet cut-off of 0.9 (corresponding to ≤ 1% FDR) and at least two unique peptides were analyzed with SAINT Express v.3.653. Twenty control runs (from cells expressing the FmT tag alone) were collapsed to the two highest spectral counts for each prey and compared to the experimental data, consisting of two biological replicates (each analyzed with two technical replicates). High confidence interactors were defined as those with Bayesian false discovery rate (BFDR) ≤ 0.01.
Human study subject and genetics evaluation
The patient, a male child, carried a diagnosis of intestinal lymphangiectasia with hypogammaglobulinemia and recurrent infections of the respiratory tract and middle ear since infancy. His parent provided written informed consent for his enrollment and participation in research protocol (06-I-0015), which was approved by the Institutional Review Board of the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH). Trio whole exome sequencing was performed on the patient and his unaffected parents using genomic DNA extracted from whole blood and sequenced in-kind by Regeneron Genetics Center using an Illumina HiSeq sequencing system (Illumina). An in-house custom analysis pipeline at NIAID was then used to filter and prioritize candidates according to an autosomal recessive model consistent with the patient’s consanguineous background. WES data for the patient and his parents has been deposited to dbGaP (accession number phs003940.v1.p1 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003940.v1.p1]).
Statistics and reproducibility
Statistical significance was assessed using Student’s t-test, one-way ANOVA, or a chi-squared test, as indicated in each figure legend. In Figs. 2D–G, 3H–I, and S9A, data from three replicate experiments were pooled together prior to performing a chi-squared test (Table S2). All Student’s t-tests were two-tailed. For one-way ANOVA, significance between sample means was assessed using Tukey’s multiple comparisons test. Exact p values are indicated in the Source Data file. All statistical analyses were performed using GraphPad Prism version 8.4.3. All experiments were reproducible, and no data were excluded from the analyses. Quantification of immunofluorescence and immunoblot data are found in each figure, and individual experiment-level data are found in the Source Data file. In Figs. 1B-E and 3D, unquantified immunofluorescence images are representative of at least 3 independent experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The source data underlying all results in the manuscript and the Supplementary Information, including raw quantification data and uncropped immunoblot images, are provided as a Source Data file. All raw mass spectrometry data files have been deposited in the MassIVE archive with accession ID MSV000093801. Sequencing data has been deposited in dbGaP with accession number phs003940.v1.p1 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003940.v1.p1]. Source data are provided with this paper.
References
Hershko, A. & Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 (1998).
Rotin, D. & Prag, G. Physiological functions of the ubiquitin ligases Nedd4-1 and Nedd4-2. Physiol. (Bethesda) 39, 18–29 (2024).
Plant, P. J., Yeger, H., Staub, O., Howard, P. & Rotin, D. The C2 domain of the ubiquitin protein ligase Nedd4 mediates Ca2+-dependent plasma membrane localization. J. Biol. Chem. 272, 32329–32336 (1997).
Wiesner, S. et al. Autoinhibition of the HECT-type ubiquitin ligase Smurf2 through its C2 domain. Cell 130, 651–662 (2007).
Mari, S. et al. Structural and functional framework for the autoinhibition of Nedd4-family ubiquitin ligases. Structure 22, 1639–1649 (2014).
Persaud, A. et al. Tyrosine phosphorylation of NEDD4 activates its ubiquitin ligase activity. Sci. Signal 7, ra95 (2014).
Staub, O. et al. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 15, 2371–2380 (1996).
Kanelis, V., Rotin, D. & Forman-Kay, J. D. Solution structure of a Nedd4 WW domain-ENaC peptide complex. Nat. Struct. Biol. 8, 407–412 (2001).
Huibregtse, J. M., Scheffner, M., Beaudenon, S. & Howley, P. M. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl Acad. Sci. USA. 92, 2563–2567 (1995).
Garrone, N. F., Blazer-Yost, B. L., Weiss, R. B., Lalouel, J. M. & Rohrwasser, A. A human polymorphism affects NEDD4L subcellular targeting by leading to two isoforms that contain or lack a C2 domain. BMC Cell Biol. 10, 26 (2009).
Araki, N. et al. Expression, transcription, and possible antagonistic interaction of the human Nedd4L gene variant: implications for essential hypertension. Hypertension 51, 773–777 (2008).
Kefalas, G. & Rotin, D. Primate-specific isoform of Nedd4-1 regulates substrate binding via Ser/Thr phosphorylation and 14-3-3 binding. Sci. Rep. 13, 17903 (2023).
Drake, J. M. et al. Phosphoproteome integration reveals patient-specific networks in prostate cancer. Cell 166, 1041–1054 (2016).
Sharma, K. et al. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 8, 1583–1594 (2014).
Sancak, Y. et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 (2008).
Branon, T. C. et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887 (2018).
Roux, K. J., Kim, D. I., Raida, M. & Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 (2012).
Criollo, A. et al. The IKK complex contributes to the induction of autophagy. EMBO J. 29, 619–631 (2010).
Galmes, R. et al. Vps33B is required for delivery of endocytosed cargo to lysosomes. Traffic 16, 1288–1305 (2015).
Pulipparacharuvil, S. et al. Drosophila Vps16A is required for trafficking to lysosomes and biogenesis of pigment granules. J. Cell Sci. 118, 3663–3673 (2005).
Liu, R. J. Y. et al. The Sec1-Munc18 protein VPS33B forms a uniquely bidirectional complex with VPS16B. J. Biol. Chem. 299, 104718 (2023).
Zhu, G. D. et al. SPE-39 family proteins interact with the HOPS complex and function in lysosomal delivery. Mol. Biol. Cell 20, 1223–1240 (2009).
Zhu, G. D. & L’Hernault, S. W. The Caenorhabditis elegans spe-39 gene is required for intracellular membrane reorganization during spermatogenesis. Genetics 165, 145–157 (2003).
Solinger, J. A. & Spang, A. Tethering complexes in the endocytic pathway: CORVET and HOPS. FEBS J. 280, 2743–2757 (2013).
Liang, C. et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 10, 776–787 (2008).
Persaud, A. et al. Comparison of substrate specificity of the ubiquitin ligases Nedd4 and Nedd4-2 using proteome arrays. Mol. Syst. Biol. 5, 333 (2009).
Urban, D. et al. The VPS33B-binding protein VPS16B is required in megakaryocyte and platelet α-granule biogenesis. Blood 120, 5032–5040 (2012).
Hyttinen, J. M., Niittykoski, M., Salminen, A. & Kaarniranta, K. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochimica et. Biophysica Acta (BBA)-Mol. Cell Res. 1833, 503–510 (2013).
Gutierrez, M. G., Munafó, D. B., Berón, W. & Colombo, M. I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 117, 2687–2697 (2004).
Jager, S. et al. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 117, 4837–4848 (2004).
Behrends, C., Sowa, M. E., Gygi, S. P. & Harper, J. W. Network organization of the human autophagy system. Nature 466, 68–76 (2010).
Bakula, D. et al. WIPI3 and WIPI4 β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat. Commun. 8, 15637 (2017).
Escobedo, A. et al. Structural basis of the activation and degradation mechanisms of the E3 ubiquitin ligase Nedd4L. Structure 22, 1446–1457 (2014).
Ishii, A. et al. Inhibitory effect of SPE-39 due to tyrosine phosphorylation and ubiquitination on the function of Vps33B in the EGF-stimulated cells. FEBS Lett. 586, 2245–2250 (2012).
Lo, B. et al. Requirement of VPS33B, a member of the Sec1/Munc18 protein family, in megakaryocyte and platelet alpha-granule biogenesis. Blood 106, 4159–4166 (2005).
Gissen, P. et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat. Genet. 36, 400–404 (2004).
Cullinane, A. R. et al. Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization. Nat. Genet. 42, 303–312 (2010).
Stuchell, M. D. et al. The human endosomal sorting complex required for transport (ESCRT-I) and Its role in HIV-1 budding*♦. J. Biol. Chem. 279, 36059–36071 (2004).
Pei, G. et al. The E3 ubiquitin ligase NEDD4 enhances killing of membrane-perturbing intracellular bacteria by promoting autophagy. Autophagy 13, 2041–2055 (2017).
Xie, W. et al. Auto-ubiquitination of NEDD4-1 Recruits USP13 to Facilitate Autophagy through Deubiquitinating VPS34. Cell Rep. 30, 2807–2819.e2804 (2020).
Lin, Q. et al. The HECT E3 ubiquitin ligase NEDD4 interacts with and ubiquitylates SQSTM1 for inclusion body autophagy. J. Cell Sci. 130, 3839–3850 (2017).
Ozen, A. & Lenardo, M. J. Protein-Losing Enteropathy. N. Engl. J. Med. 389, 733–748 (2023).
Nakashima, M. et al. A genome-wide association study identifies four susceptibility loci for keloid in the Japanese population. Nat. Genet. 42, 768–771 (2010).
Fujita, M. et al. NEDD4 is involved in inflammation development during keloid formation. J. Invest. Dermatol. 139, 333–341 (2019).
Kowarz, E., Löscher, D. & Marschalek, R. Optimized sleeping beauty transposons rapidly generate stable transgenic cell lines. Biotechnol. J. 10, 647–653 (2015).
Mátés, L. et al. Molecular evolution of a novel hyperactive sleeping beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 41, 753–761 (2009).
Coyaud, E. et al. BioID-based identification of Skp Cullin F-box (SCF)β-TrCP1/2 E3 ligase substrates. Mol. Cell. Proteom. 14, 1781–1795 (2015).
Kessner, D., Chambers, M., Burke, R., Agus, D. & Mallick, P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 (2008).
Craig, R. & Beavis, R. C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 (2004).
Eng, J. K., Jahan, T. A. & Hoopmann, M. R. Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24 (2013).
Deutsch, E. W. et al. A guided tour of the trans-proteomic pipeline. Proteomics 10, 1150–1159 (2010).
Liu, G. et al. ProHits: integrated software for mass spectrometry-based interaction proteomics. Nat. Biotechnol. 28, 1015–1017 (2010).
Teo, G. et al. SAINTexpress: improvements and additional features in significance analysis of INTeractome software. J. Proteom. 100, 37–43 (2014).
Knight, J. D. et al. ProHits-viz: a suite of web tools for visualizing interaction proteomics data. Nat. methods 14, 645–646 (2017).
Acknowledgements
We thank SPARC-Drug Discovery (SickKids) for reagents and the SickKids Imaging Facility for assistance with microscopy. This work was supported by Canadian Institutes of Health Research (CIHR), FRQS (Quebec) and RESTRACOMP (SickKids) studentships to G.K., a CIHR Foundation grant (FDN-387969) to D.R., and in part by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health (1ZIAAI000769 to M.J.L.; 1ZIAAI001059 to H.C.S. and Y.Z.). CIHR funding is also acknowledged for W.H.A.K., J.H.B., A.M.M., and B.R.
Author information
Authors and Affiliations
Contributions
G.K. and D.R. conceived the study and wrote the manuscript. G.K., A.P., A.A., A.Pe., J.L., A.M.S., and H.H.Y.Y. performed experiments and analyzed data. N.W., Y.Z., A.M.M., S.S., H.C.S., and M.J.L. contributed to clinical studies. J.H.B., W.H.A.K., and B.R. contributed to study design and manuscript editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zan Chen, Peter McPherson, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kefalas, G., Priya, A., Astori, A. et al. The primate-specific Nedd4-1(NE) localizes to late endosomes in response to amino acids to suppress autophagy. Nat Commun 16, 2682 (2025). https://doi.org/10.1038/s41467-025-57944-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57944-x
