
Abstract
Proton exchange membrane fuel cells show great potential as power source for automobiles, yet are now facing technological challenges of low efficiency in the cathodic oxygen reduction reaction and severe degradation from Nafion ionomers. Herein, we report the design and construction of a core/shell nanoparticle, composing of Pt3Fe intermetallic nanoparticle as core and atomically-thin porous N-doped carbon layer as shell, to alleviate Nafion ionomer poisoning and local oxygen transport at the interfaces, thereby improving the performance of membrane electrode assemblies. Combining electrochemical, spectroscopic and calculation results verify that the sandwiching carbon layer can effectively prevent surface Pt active sites from poisoning by ionomers. Moreover, this deliberate design facilitates a more homogeneous distribution of ionomers in catalyst layer, and drives a H2-air fuel cell peak power density up to 1.0 W cm-2. Due to the configuration-induced strong Fe-N coordination, our unique catalyst efficiently preserves transition metals and consequently delivers a notable fuel cell durability at a constant potential of 0.5 V for 100 h.
Similar content being viewed by others


Introduction
Proton exchange membrane fuel cells (PEMFCs) have attracted considerable attention from the scientific community because of their exceptional energy conversion efficiency1,2, limited environmental impact3,4,5,6 and reliable product performance. However, one of the biggest challenges is that the performance of Pt-based membrane electrode assemblies (MEAs), such as short-term power density and long-term stability, is still far from satisfactory7,8,9,10,11. Extensive researches have been conducted to identify the impediments that hinder the efficacy of Pt-based materials in PEMFCs4,9,12,13. Ionomers are commonly used as essential additives in the catalyst layer of PEMFCs to provide protons and enhance the dispersion of catalytic slurries during the film-forming procedure9,14,15. However, the unmodified sulfonate groups of these incorporated ionomers inevitably interact with Pt sites at the generated Pt-ionomer interface, leading to distinct adsorption and a significant reduction in mass activity by 2 to 4 times, resulting in only 20%-50% usable Pt surface area16,17,18. Furthermore, uneven dispersion of ionomers on the catalyst surface also increases resistance to O2 diffusion across the micro-interface, causing undesirable voltage drop losses at the region of high current density17. Various strategies have been implemented to address these issues, including the use of modified ionomers with improved oxygen permeability14,19, integration of Pt nanoparticles (NPs) into porous carbon17, and introduction of ionomer shielding agents9, among others. However, an optimal balance between ionomer toxicity, O2 transfer, and catalyst stability remains a big challenge, which calls for abrupt innovations of interface engineering to unlock the full potential of Pt-based materials in MEAs.
Herein, we report an interfacing approach to manipulate the adsorption and distribution of ionomers within the cathodic catalyst layer, which notably improves the activity and stability of Pt-based materials in MEAs (Fig. 1). We designed a Pt3Fe NPs/porous N-doped atomically-thin carbon layer core/shell structure (PNAC-o-Pt3Fe/C) through the carbonization of oleylamine-captured nanocrystals, and then constructed a unique Pt3Fe/porous carbon layer/ionomer sandwiching interface. When integrated in the cathodic catalyst layers of MEAs, this design efficiently eliminates ionomer absorptive pathway by physically separating sulfonate groups from the Pt surface. Furthermore, atomically-thin porous N-doped carbon layer facilitates mass transfer and homogeneous distribution of ionomers on catalysts surface for enhancing O2 accessibility. As a result, the coverage of sulfonate groups on the Pt in the sandwich-structured catalyst is merely 10.1%, notably lower than the catalyst without carbon layer (21.2%). The PNAC-o-Pt3Fe/C catalyst exhibits superior activity in kinetic regions and improved mass transport properties (a lower O2 transport resistance RO2PI value of 0.03 s cm-1) in the region of high current density areas, surpassing those previously-reported PtFe-based catalysts. Furthermore, PNAC-o-Pt3Fe/C catalyst demonstrates high durability in both rotating disc electrode (RDE) and MEA tests, remaining stable even after a 60 K accelerated decay test (ADT) and a 100 h MEA constant potential test.

N-doped atomically-thin carbon layer in PNAC-o-Pt3Fe/C shields Pt atoms from ionomer poisoning, increases the number of active sites, and improves O2 permeability and long-term stability. Note: This model is schematic and a relatively reasonable structure has been chosen and this image was created using 3D Max and PS software.
Results and discussion
Synthesis and structural characterizations
The PNAC-o-Pt3Fe/C was synthesized using oleylamine carbonization strategy, in which platinum acetylacetonate (Pt(acac)2) and iron acetylacetonate (Fe(acac)3) were used as metal precursors, molybdenum carbonyl was used as a reducing agent, and oleylamine was utilized as a solvent and protective agent, and also as a carbon source to facilitate the encapsulation of Pt3Fe NPs within the carbon layer during the high-temperature pyrolysis process6 (details in Methods and Supplementary Fig. 1).
The transmission electron microscopy (TEM) image of the as-synthesized PNAC-o-Pt3Fe/C shows a uniform dispersion of Pt3Fe NPs on the carbon support with an average size of 4.0 ± 1.0 nm (Supplementary Fig. 2a). The combined high-resolution transmission electron microscopy (HRTEM) (Fig. 2a and Supplementary Figs. 3, 4, 5) with electron energy loss spectroscopy (EELS) mapping (Supplementary Fig. 6) and Raman spectroscopy20,21 (Supplementary Figs. 7, 8 and Supplementary Table 1) of the exhaustive regions reveals that the Pt3Fe NPs are covered with a single layer (or a few layers) of carbon featured with numerous micro-pore (caused from the fracture of the carbon layer) structures, and forming the highly cross-linked structure with the carbon support at the margin. Thermogravimetric (TGA-DSC) analysis of the resulting PNAC-o-Pt3Fe/C illustrates a distinct weight loss and an exothermic reaction at 503 K under an air atmosphere (Fig. 2b), which is consistent with the results obtained from the PNAC-o-Pt3Fe catalyst without Ketjenblack support (Supplementary Fig. 9). These findings imply that oleylamine is present on the surface of Pt3Fe NPs and can be effectively removed at lower temperatures. Fourier-transform infrared spectroscopy (FT-IR) analysis of the original Pt3Fe/C before thermal annealing reveals four characteristic peaks at 2920.6 cm-1, 1580.9 cm-1, 1464.3 cm-1, and 940.8 cm-1 (Supplementary Fig. 10), also indicating the presence of functional groups of oleylamine molecules22. This suggests that the pyrolysis of these organic ligands that remain on the surface of Pt3Fe NPs during the high-temperature treatment likely contributes to the formation of this carbon. This oleylamine-derived carbon layer, being atomically-thin and porous, not only separates the internal active sites from the surrounding environment but also retains their electrochemical activity. This is evidenced by the competitive electrochemically active surface area (ECSA) of 60.3 m2 gPt-1 observed in PNAC-o-Pt3Fe/C, which is only slightly lower than its theoretical value of 72.6 m2 gPt-1 (Supplementary Table 2).

a HRTEM image of PNAC-o-Pt3Fe/C showing o-Pt3Fe/C NPs enclosed by a thin and porous N-doped carbon layer. b TGA-DSC curve of PNAC-o-Pt3Fe/C. c XRD patterns of PNAC-o-Pt3Fe/C, d-Pt3Fe/C and original Pt3Fe/C tested with Cu Kα radiation. d Atomically-resolved HAADF-STEM image of PNAC-o-Pt3Fe/C. e, f Enlarged dark (f) and bright (e) field STEM images of PNAC-o-Pt3Fe/C of the rectangular region in (d). g Intensity profile of PNAC-o-Pt3Fe/C extracted from the yellow dashed area in (e) showing the alternating Pt and Fe atoms. h EDS elemental mapping of PNAC-o-Pt3Fe/C. i N 1 s XPS spectrum of PNAC-o-Pt3Fe/C. j, k Pt L3-edge (j) and Fe K-edge (k) FT-XAFS spectra of PNAC-o-Pt3Fe/C compared with respective oxide and metallic foil references at R space; X-axis abbreviation R represents the radial distance. Source data for (b, c, g) and (i–k) were provided as a Source Data file.
X-ray diffraction (XRD), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), energy dispersive spectrometer (EDS), X-ray photoelectron spectroscopy (XPS) and inductively coupled plasma mass spectrometry (ICP-MS) techniques were employed to examine the atomic structure, elemental distribution and crystalline phase of Pt3Fe NPs in PNAC-o-Pt3Fe/C. XRD patterns indicate an ordered crystal structure in PNAC-o-Pt3Fe/C, with a distinct superlattice reflection that closely aligns with the Joint Committee on Powder Diffraction Standards (JCPDS) cards (Fig. 2c and Supplementary Fig. 11). Moreover, HAADF-STEM images display a periodic square arrangement of Fe (appearing darker) columns enclosed by Pt dots (appearing brighter) within the unit cell along the [001] zone axis (Fig. 2d–g and Supplementary Fig. 12), inferring the presence of an L12-type, face-centered cubic intermetallic structure in PNAC-o-Pt3Fe/C. EDS elemental analysis (Fig. 2h and Supplementary Fig. 13), together with the XPS and ICP-MS measurements (Supplementary Table 3), confirm the uniform dispersion of Pt and Fe elements from the interior to the periphery within each Pt3Fe NP, with atomic percentages of 69% Pt and 31% Fe, closely matching the theoretical value (Supplementary Fig. 14).
The interaction of electrons between the carbon layer and its inner Pt3Fe intermetallic NPs was verified using EDS elemental analysis, XPS and X-ray absorption fine structure (XAFS) techniques. EDS elemental analysis indicates that the carbon layer obtained from oleylamine in the PNAC-o-Pt3Fe/C contains a small amount of N heteroatom (Fig. 2h), which tends to coordinate with neighboring atoms23. XPS analysis of N element in PNAC-o-Pt3Fe/C reveals the presence of five coordination configurations17,20,23 (Fig. 2i and Supplementary Table 4), along with Fe-N24,25 and Pt-N26 formations, indicating chemical interaction between Pt3Fe NPs and the N atoms on the carbon layer. The positively shifted XPS adsorption peak of Pt 4 f (Supplementary Fig. 15a) and elevated Pt L3-edge white line absorption intensity in the X-ray absorption near-edge structure (XANES) (Supplementary Fig. 15b) of PNAC-o-Pt3Fe/C compared to those of o-Pt3Fe/C further confirm this Pt/Fe-N interaction and suggest a direction of electron transfer from metal to N. The Fe K-edge and Pt L3-edge XANES spectroscopy analysis in the R space reveal clear peaks at 1.91 Å and 2.03 Å (Fig. 2j, k), providing the coexistence of Pt-N/O and Fe-N/O bonds in PNAC-o-Pt3Fe/C. Additionally, the model-based extended X-ray absorption fine structure (EXAFS) fitting of PNAC-o-Pt3Fe/C reveals that the coordination numbers of Pt-N/O and Fe-N/O are 0.5 and 2.2 (Supplementary Tables 5, 6), respectively, corresponding to the bonding circumstances observed in the wavelet transform (WT) (Supplementary Figs. 15, 16), again validating this Pt-N and Fe-N coordination interaction in PNAC-o-Pt3Fe/C. These findings (EDS, XPS, XAFS) highlight that the carbon layer produced by carbonizing small oleylamine molecules not only physically encapsulates Pt3Fe NPs, but also creates a distinctive coordination structure through N heteroatoms doping.
In order to assess the scalability and potential of this approach, we performed experiments with a feedstock that was amplified by a factor of 25 (Supplementary Fig. 17). We find that this method of carbonization allows for large-scale production, rendering it a feasible and scalable synthesis technology for Pt-based catalysts.
Performance evaluations
To assess the impact of a carbon layer on the electrochemical behavior of PNAC-o-Pt3Fe/C, we conducted initial electrocatalytic measurements using a RDE in an O2-saturated 0.1 M HClO4 solution (pH was 1.0 ± 0.1). For fairness and accuracy, the Nafion content in the catalyst slurry and the amount of Pt catalyst on the electrode surface were standardized at 5 μL mL-1 and 2 μg, respectively. Under these identical conditions, o-Pt3Fe/C displays a high half-wave potential (E) of 0.917 V vs. reversible hydrogen electrode (RHE) compared to commercial Pt/C (0.847 V vs. RHE) (Fig. 3a); moreover, the carbon layer encapsulated PNAC-o-Pt3Fe/C exhibits a further boost in the half-wave potential (0.926 V vs. RHE), indicating the higher oxygen reduction reaction (ORR) activity of the carbon layer encapsulation configuration. When normalized for Pt loading (Fig. 3b), PNAC-o-Pt3Fe/C exhibits the highest mass activity (MA) of 1.66 A mgPt-1 at 0.9 V, surpassing the values obtained for o-Pt3Fe/C (1.15 A mgPt-1) and commercial Pt/C (0.11 A mgPt-1), further demonstrating its notable activity towards ORR. In terms of specific activities (SAs) normalized based on the electrochemical active surface areas (Fig. 3b and Supplementary Fig. 18, Table 2), PNAC-o-Pt3Fe/C achieves the highest SA of 2.75 mA cm-2, followed by o-Pt3Fe/C (1.73 mA cm-2), and commercial Pt/C (0.16 mA cm-2), signifying a 1.6- and 17.1-fold increase in SAs compared to o-Pt3Fe/C and commercial Pt/C, respectively. Additionally, the Tafel slope of PNAC-o-Pt3Fe/C is determined to be 53.0 mV dec-1, the smallest value among the three catalysts (Fig. 3c), indicating that the catalytic reaction kinetics on the surface of PNAC-o-Pt3Fe/C occur at a faster rate.

a iR-corrected (95%, R was 30 ± 2 Ω) ORR polarization curves of various electrocatalysts; X-axis abbreviation E represents the potential, Y-axis abbreviation j represents the current density. b Histograms showing mass and specific activities at 0.9 V vs. RHE; error bars represent the standard deviation obtained from two separate experiments. c Tafel slopes of various electrocatalysts; X-axis abbreviation jk represents the kinetic current density, Y-axis abbreviation E represents the potential. d Activity decay after ADT cycling of above-mentioned Pt-based catalysts; error bars represent the standard deviation obtained from two separate experiments. Electrochemical evaluations were conducted under ambient conditions in a 0.1 M HClO4 solution with a pH value of 1.0 ± 0.1. The Pt loading was maintained at 10 μg cm−2, the scan rate was 20 mV s−1, the rotation rate was constant at 1600 rpm, and the gas flow was sustained at 0.1 L min−1. Source data for (a–d) were provided as a Source Data file.
Such an increase in ORR activity of PNAC-o-Pt3Fe/C can be attributed to: (1) the carbon layer presenting on the active sites of Pt alters their electronic structure; (2) the N-containing groups (especially pyridinic N) in the carbon layer facilitate the dissociation of O2 by generating Lewis basic sites, thus enhancing the ORR activity20,27; (3) the carbon layer serves as a barrier, impeding the sulfonate groups in the ionomers from poisoning the Pt active sites. Computational simulations of PNAC-o-Pt3Fe confirm that the coordination of N atoms in the carbon layer with Pt causes a down shift of the d-band center in the Pt sites (Supplementary Fig. 19), which actually hampers its ORR activities28,29. Meanwhile, the literatures indicates that these N-containing groups typically demonstrate excellent ORR performance in alkaline solutions but are less effective in acidic electrolytes30,31. Therefore, the unique carbon layer in PNAC-o-Pt3Fe/C, which physically separates the Nafion ionomers from the Pt to release more active sites15,18,32, is the most probable reason for this enhanced catalytic activity in the acidic conditions.
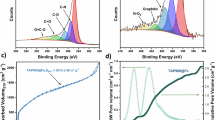
To quantify the Pt active sites released by carbon layer in encapsulated PNAC-o-Pt3Fe/C, CO displacement measurements were conducted33,34. Both PNAC-o-Pt3Fe/C, o-Pt3Fe/C and commercial Pt/C exhibit a distinct inverse peak in CO displacement upon applying a potential of 0.4 V vs. RHE (Fig. 4a and Supplementary Fig. 20), indicating the successful substitution of absorbed sulfonate groups on the Pt surface with CO; notably, the peak area of PNAC-o-Pt3Fe/C is apparently smaller than that of o-Pt3Fe/C and commercial Pt/C, suggesting a lower occupancy of sulfonate groups on the Pt sites within PNAC-o-Pt3Fe/C. To quantitatively compare the coverage of sulfonate groups on these catalyst layers, we divided CO replacement charge by CO reduction charge (calculated according to Eq. 1), as shown in Fig. 4b, c and Supplementary Fig. 18. The calculated coverage of sulfonate of PNAC-o-Pt3Fe/C was found to be 10.1%, which is substantially lower than that of o-Pt3Fe/C (21.2%) and commercial Pt/C (23.3%) (Fig. 4c), thereby further revealing the inhibition of poisoning due to the presence of the carbon layer. Further, we analyzed the XPS spectra of the electronic interaction between Pt and S, with or without carbon layer, for both catalysts when incorporated with ionomers. Pt 4 f XPS peak of PNAC-o-Pt3Fe/C exhibits a smaller shift than that of o-Pt3Fe/C (Fig. 4d, e), which aligns with prior research9 and is corroborated by the XPS findings of S 2p presented in Fig. 4f. This feeble charge transfer from Pt to S implies a restricted interaction between Pt and ionomers in the catalyst layer of PNAC-o-Pt3Fe/C, which can be attributed to the unique structure of its carbon layer.

a, b CO displacement (a) and CO stripping (b) curves of PNAC-o-Pt3Fe/C, o-Pt3Fe/C and commercial Pt/C; X-axis abbreviation E represents the potential. c ECSA and sulfonate group coverage obtained from CO stripping experiments of those Pt-based catalysts; error bars represent the standard deviation obtained from two separate experiments. d–f XPS spectra of Pt 4 f (d, e) and S 2p (f) for PNAC-o-Pt3Fe/C and o-Pt3Fe/C before or after ionomer adsorption. Source data for (a–f) were provided as a Source Data file.
The performance of PNAC-o-Pt3Fe/C, o-Pt3Fe/C and commercial Pt/C in PEMFC was evaluated using a MEA test system under H2-O2 and H2-air conditions. The H2-O2 fuel cell using commercial Pt/C as the cathode demonstrates a current density of 1.1 A cm-2 at 0.6 V and a peak power density of 1.1 W cm-2 (Fig. 5a). When utilizing PNAC-o-Pt3Fe/C, the H2-O2 fuel cell shows notably improved performance, achieving a current density of 1.8 A cm-2 at 0.6 V and a peak power density of 1.6 W cm-2. Furthermore, PNAC-o-Pt3Fe/C-based H2-air fuel cells outperforms both o-Pt3Fe/C and commercial Pt/C-based ones across the entire current density range (Fig. 5b), and it achieves a high peak power density of 1.0 W cm-2, 1.7- and 1.3-folds higher than those of o-Pt3Fe/C (0.6 W cm-2) and commercial Pt/C (0.8 W cm-2). The performance of PNAC-o-Pt3Fe/C-based H2-air fuel cell not only surpasses that of PtFe-based catalysts reported in literature (Supplementary Table 7) but also is comparable to most Pt-based catalysts, including some PtCo-based catalysts (Supplementary Table 8).

a H2-O2 polarization curves and power density plots of the MEAs with PNAC-o-Pt3Fe/C and commercial Pt/C as cathode catalysts, respectively. b, c H2-air polarization curves (b) and power density plots (c) of MEAs with PNAC-o-Pt3Fe/C, o-Pt3Fe/C, and commercial Pt/C as cathode, respectively. d Activity comparisons of PNAC-o-Pt3Fe/C, o-Pt3Fe/C and commercial Pt/C-based fuel cells in the regions of low and high current densities. e H2-air fuel cells stability of PNAC-o-Pt3Fe/C and o-Pt3Fe/C tested at 0.5 V. In all MEAs, the anode catalyst was Pt/C with a loading of 0.2 mgPt cm−2, while the cathode Pt loading was 0.15 mgPt cm−2. The temperature was set at 353.15 K, relative humidity at 100%, and back pressure at 0.2 MPa. In the H2-O2 fuel cells, flow rates for H2 and O2 were fixed at 0.35 L min−1 and 0.8 L min−1, respectively. In the H2-air fuel cells, flow rates for H2 and air were fixed at 0.35 L min−1 and 1.2 L min−1, respectively. Source data for (a–e) were provided as a Source Data file.
This marked enhancement in PNAC-o-Pt3Fe/C-based fuel cell efficiency results from the heightened catalytic activity detected at low (0.85 V) and high (0.5 V) current densities (Fig. 5c, d). The catalytic material’s inherent activity governs the low current density performance, whereas O2 transfer influences the high current density performance4. Specifically, at 0.85 V (Fig. 5d), H2-air fuel cell based on PNAC-o-Pt3Fe/C further boosts this mass activity to 0.67 A mgPt-1 and achieves even higher mass activity in H2-O2, reaching 0.85 A mgPt-1, higher than those of H2-air fuel cell-based on o-Pt3Fe/C (0.59 A mgPt-1) and commercial Pt/C (H2-air, 0.35 A mgPt-1; H2-O2, 0.39 A mgPt-1). In the region of high current density (0.5 V) (Fig. 5d), PNAC-o-Pt3Fe/C demonstrates unexpectedly improved performance in mass transfer (H2-air, 2.00 A cm-2; H2-O2, 2.81 A cm-2) relative to o-Pt3Fe/C (H2-air, 1.13 A cm-2) and commercial Pt/C (H2-air, 1.56 A cm-2; H2-O2, 1.79 A cm-2), indicating the existence of a rapid O2 transport channel in the PNAC-o-Pt3Fe/C catalyst layer. Typically, carbon layers on catalyst surface lead to increased resistance to O2 diffusion35, thus resulting in undesirable pressure drop losses in high current density regions. However, the PNAC-o-Pt3Fe/C catalyst exhibits markedly improved oxygen transport properties (lowest RO2PI value of 0.03 s cm-1 relative to 0.09 s cm-1 for o-Pt3Fe/C and 0.13 s cm-1 for commercial Pt/C) due to the thin and N-doped porous structure of carbon layer (Supplementary Fig. 21). Here, the atomically-thin and porous character of carbon layer enables efficient transport of oxygen during catalytic reactions; moreover, N atom doping of the carbon layer improves the uniform distribution of ionomers on catalyst surface17, which further enhances the O2 availability. Therefore, by incorporating an N-doped ultrathin porous carbon layer onto the surface of Pt3Fe NPs, we achieve simultaneous enhancement in catalyst performance in both low and high current density regions, a finding that has not been reported previously.
To further evaluate the potential of PNAC-o-Pt3Fe/C under real operating conditions, a comparison of MEA polarization curves at 6 cm2 under a constant differential flow and 25 cm2 under stoichiometric flow of 1.5 for H2 and 2.0 for air at 353.15 K was performed (Supplementary Fig. 22). The result shows a consistent pattern of performance variation across all current density regions for both assemblies (6 cm2 and 25 cm2), and only a slight performance degradation observed in the 25 cm2 MEA relative to the 6 cm2. This suggests that optimizing the structure of PNAC-o-Pt3Fe/C can also improve the mass activity and O2 transport even in a larger MEA assembly under stoichiometric flow, similar to the effects observed in a smaller MEA under differential flow.
The stability analysis of PNAC-o-Pt3Fe/C was performed under both RDE and MEA operating conditions. It is noteworthy that during the initial RDE examination, PNAC-o-Pt3Fe/C shows only a 2 mV shift in its half-wave potential, even after undergoing 60 K ADT cycles, which is in contrast with the notable negative shifts observed in both o-Pt3Fe/C (9 mV) and Pt/C (30 mV) (Supplementary Fig. 23). Furthermore, PNAC-o-Pt3Fe/C demonstrates considerably lower degradation rates of 7.2% (60 K, MA) and 4.8% (60 K, SA), much lower than those of o-Pt3Fe/C (50 K, MA, 27.5%; SA, 27.4%) and Pt/C (30 K, MA, 45.0%; SA, 27.9%) (Fig. 3d and Supplementary Fig. 24). This outstanding durability of PNAC-o-Pt3Fe/C is at the forefront of the top-performing PtFe alloy electrocatalysts tested under RDE conditions (Supplementary Table 9), and also exceeds those of Pt-based electrocatalysts (Supplementary Table 10). Additionally, the durability of PNAC-o-Pt3Fe/C was evaluated using an MEA evaluation system with an abundant H2-air feedstock. Impressively, PNAC-o-Pt3Fe/C-based fuel cell demonstrates an impressive durability with only 19.1% activity degradation during a 100 h constant potential test at 0.5 V (Fig. 5e). Such a high durability is not only notably outperforming that of o-Pt3Fe/C (42.4% after 57 h), but also competitive with the majority of Pt-based catalysts (Supplementary Table 11), highlighting the greatly improved stability of PNAC-o-Pt3Fe/C and demonstrating the potential of this interface engineering strategy.
The morphology and structure of PNAC-o-Pt3Fe/C were analyzed after 60 K cycles of ADT using TEM, HAADF-STEM, EDS and density functional theory (DFT) to determine the factors responsible for enhanced stability. During the ADT process, the NPs on the surface of commercial Pt/C experience severe agglomeration, leading to their size increasing from 3.3 ± 0.6 nm to 6.0 ± 1.9 nm (Supplementary Figs. 2, 25). In this context, the carbon layer enclosure has the capability to prevent the movement and coalescence of NPs36, and also slow down their oxidative dissolution and diffusion through physical barriers. Actually, the average size of the Pt3Fe NPs remains almost the same at 4.1 ± 1.0 nm in PNAC-o-Pt3Fe/C (Supplementary Figs. 2, 25), indicating that the carbon layer has effectively prevented the size increase of the Pt3Fe nanocatalysts under these electrochemically challenging conditions.
Another important factor that affects the stability of the catalyst is the loss of transition metal Fe, which causes interference with the ligand effect and initiates the Fenton reaction37. Fig. 6b–f demonstrate that PNAC-o-Pt3Fe/C maintains a well-organized crystalline structure for its NPs, and the corresponding Pt/Fe elemental composition is 69/31 at.%, which is similar to the original PNAC-o-Pt3Fe/C without ADT cycling (Supplementary Fig. 14). We constructed a representative DFT computational model based on the Pt3Fe (111) crystal plane and the porous carbon layer by considering the unique coordination structure of the carbon layer with Fe atoms (based on the EXAFS fitting result in Supplementary Table 6). DFT calculations show that the formation energy of Fe vacancies in the encapsulated nano-structure (PNAC-o-Pt3Fe) increases obvious from 1.78 eV (o-Pt3Fe) to 2.10 eV (Fig. 6g, h and Supplementary Fig. 26). This is due to the Fe-N coordination structure, originating from carbon layer, effectively prevents the spillover of Fe atoms. Therefore, the high durability of PNAC-o-Pt3Fe/C is believed to be attributed to the well-preserved carbon layer6,36, which effectively inhibits the leaching of transition metal elements during the catalytic process.

a HRTEM image of PNAC-o-Pt3Fe/C after 60 K ADT cycling showing o-Pt3Fe/C NPs remain enclosed by a thin and porous N-doped carbon layer. b Atomically-resolved HAADF-STEM image of PNAC-o-Pt3Fe/C after ADT cycling. c, d Enlarged STEM images (c) of PNAC-o-Pt3Fe/C and the corresponding FFT pattern (d) taken from the yellow dashed area in (b). e, f STEM-EDS spectrum (e) and elemental mapping (f) of the tested PNAC-o-Pt3Fe/C catalyst after ADT cycling; Y-axis abbreviation Cps represents counts per second. g, h DFT model (g) of PNAC-o-Pt3Fe and the calculated Fe hole formation energy (h) in the model of PNAC-o-Pt3Fe and o-Pt3Fe; Y-axis abbreviation EV-Fe represents the hole formation energy of Fe atom. Source data for (g) was provided as a Supplementary Data 1 file. Source data for (e, h) were provided as a Source Data file.
We have successfully implemented a sandwiching strategy to enhance the catalytic efficiency and stability of Pt-based materials in MEAs. This was achieved by creating a N-doped porous carbon layer on the surface of a Pt3Fe intermetallic NPs through in-situ carbonization of oleylamine surfactants. CO-displacement and XPS spectroscopy analysis demonstrated that this approach efficiently alleviates the poisoning of Nafion sulphonate groups to surface Pt sites and thus releases more active sites. Meanwhile, the porous carbon layer, along with the N-heteroatom doping character, enhances the O2 mass transfer, leading to a distinct activity enhancement in the region of high current density. This atomically-thin porous N-doped carbon layer encapsulated PNAC-o-Pt3Fe/C shows outstanding peak power densities in fuel cells, with 1.6 W cm-2 in H2-O2 fuel cell and 1.0 W cm-2 in H2-air fuel cell. Furthermore, PNAC-o-Pt3Fe/C exhibited prominent durability in both RDE and MEAs testing conditions, due to the effective coordination between transition metal Fe atoms and N atoms within the carbon layer. The present work provides a concept for balancing the Pt-ionomer interactions and lays the foundation for the development of fuel cell technology.
Methods
Preparation of PNAC-o-Pt3Fe/C catalyst
(a) Synthesis of Pt3Fe NPs: 10 mg of Pt(acac)2, 4.49 mg of Fe(acac)3, 33 mg of Mo(CO)6 and 5 mL of oleylamine were subjected to sonication at 293 K for 1 h to achieve a homogeneous solution. Subsequently, the solution was transferred to an oil bath and maintained at 493 K for 2 h. After cooling to room temperature, the product was obtained by centrifugation and washed with a mixture of ethanol and cyclohexane (v:v, 1:4) to remove any excess impurities. Finally, the resulting product was dispersed in cyclohexane for future use.
(b) Pt3Fe NPs loaded onto carbon: Following the synthesis of Pt3Fe NPs, Ketjenblack EC-300J carbon was mixed with hexane and subjected to sonication at 293 K for 1 h. Subsequently, a precise quantity of Pt3Fe NPs was added to the suspension and sonicated for an additional 3 h to achieve a Pt3Fe/C catalyst. The product was then washed with alcohol once and dried under vacuum conditions, resulting in the formation of Pt3Fe/C.
(c) Annealing of Pt3Fe/C to obtain the PNAC-o-Pt3Fe/C catalyst: After being cleaned with alcohol and dried in a vacuum, Pt3Fe/C material underwent annealing in a mixture of 5% H2 and 95% Ar at a temperature of 923 K for 6 h with a ramp rate of 2 K min-1 and a gas flow rate of 100 mL min-1. Throughout the entire heating procedure, the Pt3Fe/C was kept in the center and allowed to cool naturally to room temperature in the furnace after the heat treatment. This annealing process resulted in the formation of our PNAC-o-Pt3Fe/C catalyst. During this process, Pt3Fe NPs underwent a transformation into an intermetallic structure, while the oleylamine present on the surface of the NPs was in-situ carbonized. The main loading of Pt was obtained in the range of 6–10 wt.%.
Preparation of o-Pt3Fe/C catalyst
The experimental procedure for this process was similar to that for the synthesis of PNAC-o-Pt3Fe/C, with the only difference being that Pt3Fe/C was pretreated at 503 K in air for 2 h to remove the oleylamine molecule prior to annealing at 923 K for 6 h. Here, the main Pt loading was obtained in the range of 6-10 wt.%.
Preparation of d-Pt3Fe/C catalyst
The experimental procedure for this process was similar to the synthesis of o-Pt3Fe/C, with the exception that the annealing condition was modified from 923 K for 6 h to 823 K for 2 h.
Preparation of PNAC-o-Pt3Fe catalyst
The experimental procedure for this process was similar to the synthesis of PNAC-o-Pt3Fe/C, with the exception of drying and annealing the obtained Pt3Fe NPs directly, without loading onto a carbon support.
Preparation of PNAC-o-Pt3Fe/SiO2 catalyst
The experimental procedure for this process was analogous to the impregnation method. Specifically, the newly synthesized Pt3Fe NPs were added to an ethanol solution containing SiO2 and stirred for an extended period of time until the solution was completed dried. Subsequently, the resulting Pt3Fe/SiO2 precursor was subjected to annealing under the identical conditions employed for the preparation of PNAC-o-Pt3Fe/C.
Batch preparation of PNAC-o-Pt3Fe/C catalyst
The batch preparation experiments were conducted using a similar method as the preparation of PNAC-o-Pt3Fe/C. However, the added amount of feedstock was increased by a factor of 25. In a typical synthesis, 250 mg of Pt(acac)2, 112.25 mg of Fe(acac)3, 825 mg of Mo(CO)6 and 40 mL of oleylamine were dispersed in a 250 mL blue-capped glass bottle using ultrasonic treatment (KQ2200DE CNC ultrasonic cleaner, Kunshan Ultrasonic Instrument Co., Ltd.). The resulting solution was then heated in an oil bath at 493 K for 2 h (IKAC-MAG HS 7 digital display constant temperature magnetic heated stirrer, Germany IKA Instruments & Equipment Co., Ltd.), resulting in the formation of a black colloidal solution. This sol-gel dispersion was subsequently washed by centrifuging it with a mixture of cyclohexane and ethanol (v:v, 1:4) using 50 mL centrifuge tubes (TG16-WS desktop high-speed centrifuge, Hunan Xiangyi Instrument Development Co., Ltd.), and then it was dispersed in 40 mL of cyclohexane using a 250 mL blue-capped glass bottle and left to stand. The resulting cyclohexane solution was mixed with a carbon support that was evenly distributed in a 2500 mL glass bottle with 2000 mL of cyclohexane via ultrasonication for a duration of 3 h. The mixture was then washed with ethanol, dried and prepared for use. Finally, the dried power was placed into an aluminum boat measuring 50 mm × 40 mm × 20 mm and subjected to calcination at 923 K (H2/Ar) for a period of 6 h (OTF-1200X tube furnace, Hefei Kejing Material Technology Co., Ltd.).
MEA performance measurements
The MEAs activity and stability of those catalysts were evaluated using the AL-CS-150 fuel cell test system provided by Darkstream Technology Co., Ltd. The catalyst inks were first prepared by mixing 5 mg of catalyst, 1 mL of isopropanol, and 38.8 μL of Nafion through ultrasonic to achieve a homogeneous slurry solution. The resulting PNAC-o-Pt3Fe/C slurry was then sprayed directly onto a 25.4 μm thick proton exchange membrane (DuPont 211, prior to use the membrane was first treated at 353.15 K in 5 wt.% H2O2 for 2 h and then at 353.15 K in 0.5 M H2SO4 for a further 2 h) to create a 6 cm2 cathode catalyst layer. Following the drying of the cathode layer, a commercial Pt/C (20 wt.%, Johnson-Matthey) catalyst slurry was applied to the opposite side of the proton exchange membrane as the anode. The cathode loading was set at 0.15 mg cm-2 and the anode loading was set at 0.2 mg cm-2, respectively. To ensure comparability, the cathode and anode catalyst loadings in the reference samples (o-Pt3Fe/C; commercial Pt/C,10 wt.%, Johnson-Matthey) were maintained at the same level as the PNAC-o-Pt3Fe/C sample. To assemble the MEAs, gas diffusion layers were placed on both sides of the proton exchange membrane with catalysts layers, and then subjected to a hot-pressing process at a temperature of 403 K and a pressure of 3 MPa for 4 min. Before testing, the electrode material underwent a 4 h activation process to achieve a stable performance. This activation procedure involved a steady current of 4.5 A for 1 h, a constant voltage of 0.65 V for 0.5 h, a constant voltage of 0.45 V for 0.5 h, and was repeated for a total of 3 cycles. After activation, polarization current experiments were carried out under controlled conditions of 353.15 K, 100% relative humidity, a pressure of 0.2 MPa, and H2 flow of 0.35 L min-1, O2 flow rate of 0.8 L min-1 (or an air flow rate of 1.2 L min-1). Meanwhile, the stability tests were conducted under a constant pressure of 0.5 V at a temperature of 353.15 K, relative humidity of 100%, a pressure of 0.2 MPa, H2 flow rate of 0.35 L min-1, and air flow rate of 1.2 L min-1.
Measurements of sulphonate group coverage
To determine the coverage of sulphonate group on the Pt surface, the CO displacement and CO stripping experiments were conducted. CO displacement experiment measured the amount of Pt active sites absorbed by sulfonate groups, while the CO stripping experiment determined the overall number of Pt active sites on the catalyst surface. Both experiments were carried out in a 0.1 M HClO4 electrolyte using a three-electrode system. To ensure accurate results, a quantity of catalyst (Pt loading of 20 μg) was applied to the carbon paper surface, and used as the working electrode. Prior to the experiments, catalytic material was activated by performing 100 cycles in an electrolyte saturated with Ar to remove impurities. This activation was performed within the voltage range of 0.05-1.1 V vs. RHE, at a scan rate of 500 mV s-1.
Detailed CO displacement process was as follows: First, the catalyst material was immersed in an electrolyte saturated with N2 for at least 30 min to ensure complete removal of O2 in the solution. Then, a potential of 0.4 V vs. RHE was applied until a stable current was obtained. Without delay, CO was immediately introduced into the electrolyte system to measure the reduction current associated with the replacement of sulphonate groups. Upon successful displacement, the CO was immediately removed from the system. To ensure reproducibility, each displacement sequence was repeated twice.
Detailed CO stripping process was as follows: First, the catalyst material was immersed in a CO saturated solution for 10 min at a constant potential of 0.1 V vs. RHE to ensure complete monolayer adsorption of CO on the metal surface. Following this, the CO gas was substituted with Ar gas and kept for an additional 10 min to eliminate any remaining CO in the electrolyte. Subsequently, the CO stripping curve was generated by performing CV twice within a potential range of 0.05 V vs. RHE to 1.1 V vs. RHE at a scan rate of 20 mV s-1. Here, each stripping process was repeated twice for the sake of reproducibility.
The rate of sulphonate adsorption (θdis) was determined by calculating the integrated CO displacement charge (qdis) and the CO stripping charge (qstrip) using Eq. 1.
O2 transport resistance measurements
The investigation of O2 transport resistance was performed by limiting current measurements at 353.15 K and 100% RH, using an H2 flow rate of 1 L min-1 and an O2 flow rate of 2 L min-1. The experimental setup included a dry oxygen mole fraction of 5% O2 in N2, with the cell potential controlled at 0.3, 0.15, and 0.1 V for a duration of 2 min, and the current density calculated based on the average of the last 30 s. To distinguish between pressure-independent and pressure-dependent oxygen transport resistance, the assessments were performed at pressures of 150, 175, 200, 225 KPaabs.
Here, the total O2 transport resistance (RO2total) can be calculated from the following Eqs. 2, 3 using ilim as the limiting current, Pin as the total pressure, PH2Oin as the partial pressure of H2O, and Xdry,O2 as the dry oxygen mole fraction.
Notably, RO2total can be divided into pressure dependent (RO2PD) and pressure independent (RO2PI) components. The former represents the resistance to intermolecular gas diffusion, while the latter represents the resistance to Knudsen diffusion and oxygen transport through the ionomer/liquid water layer. The pressure-independent resistance to O2 transport (RO2PI) could be determined by analyzing the Y-intercept of the linear relationship between RO2total and the total pressure.
DFT calculation
All the calculations were assessed using the DFT with the projector augmented plane-wave method, which was implemented in the Vienna ab initio simulation package38. Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation was used to determine the exchange-correlation potential39. A cut-off energy of 450 eV was set for the plane wave. The Kohn-Sham equation was solved iteratively with an energy criterion of 10-5 eV. The structures were relaxed until the residual forces on the atoms decreased to < 0.02 eV/Å. To prevent interlaminar interactions, a vacuum spacing of 20 Å was applied perpendicular to the slab. The formation energy (Eform) was calculated using Eq. 4, which takes into account the total energy of the defect (Edefect), the total energy of the perfect model (Eperfect), and the chemical potential of the Fe atom (EFe).
Data availability
The data that support the conclusions of this study are available within the paper and Supporting information. Source data are provided with this paper.
References
Cullen, D. A. et al. New roads and challenges for fuel cells in heavy-duty transportation. Nat. Energy 6, 462–474 (2021).
Sun, Y. Y. et al. Advancements in cathode catalyst and cathode layer design for proton exchange membrane fuel cells. Nat. Commun. 12, 5984 (2021).
Islam, M. N. et al. Designing fuel cell catalyst support for superior catalytic activity and low mass-transport resistance. Nat. Commun. 13, 6157 (2022).
Kodama, K., Nagai, T., Kuwaki, A., Jinnouchi, R. & Morimoto, Y. Challenges in applying highly active Pt-based nanostructured catalysts for oxygen reduction reactions to fuel cell vehicles. Nat. Nanotechnol. 16, 140–147 (2021).
Xiao, F. et al. Atomically dispersed Pt and Fe sites and Pt-Fe nanoparticles for durable proton exchange membrane fuel cells. Nat. Catal. 5, 503–512 (2022).
Zhao, Z. P. et al. Graphene-nanopocket-encaged PtCo nanocatalysts for highly durable fuel cell operation under demanding ultralow-Pt-loading conditions. Nat. Nanotechnol. 17, 968–975 (2022).
Stephens, I. E. L., Rossmeisl, J. & Chorkendorff, I. Toward sustainable fuel cells. Science 354, 1378–1379 (2016).
Lazaridis, T., Stühmeier, B. M., Gasteiger, H. A. & El-Sayed, H. A. Capabilities and limitations of rotating disk electrodes versus membrane electrode assemblies in the investigation of electrocatalysts. Nat. Catal. 5, 363–373 (2022).
Chen, F. D. et al. Blocking the sulfonate group in Nafion to unlock platinum’s activity in membrane electrode assemblies. Nat. Catal. 6, 392–401 (2023).
Jiao, K. et al. Designing the next generation of proton-exchange membrane fuel cells. Nature 595, 361–369 (2021).
Zaman, S. et al. Oxygen reduction electrocatalysts toward practical fuel cells: progress and perspectives. Angew. Chem. Int. Ed. 60, 17832–17852 (2021).
Huang, L. et al. An integrated platinum-nanocarbon electrocatalyst for efficient oxygen reduction. Nat. Commun. 13, 6703 (2022).
Tang, M. H., Zhang, S. M. & Chen, S. L. Pt utilization in proton exchange membrane fuel cells: structure impacting factors and mechanistic insights. Chem. Soc. Rev. 51, 1529–1546 (2022).
Jinnouchi, R. et al. The role of oxygen-permeable ionomer for polymer electrolyte fuel cells. Nat. Commun. 12, 4956 (2021).
Zhang, Q. et al. Covalent organic framework-based porous ionomers for high-performance fuel cells. Science 378, 181–186 (2022).
Kodama, K. et al. Effect of the side-chain structure of perfluoro-sulfonic acid ionomers on the oxygen reduction reaction on the surface of Pt. ACS Catal 8, 694–700 (2018).
Ott, S. et al. Ionomer distribution control in porous carbon-supported catalyst layers for high-power and low Pt-loaded proton exchange membrane fuel cells. Nat. Mater. 19, 77–85 (2020).
Shinozaki, K., Morimoto, Y., Pivovar, B. S. & Kocha, S. S. Suppression of oxygen reduction reaction activity on Pt-based electrocatalysts from ionomer incorporation. J. Power Sources 325, 745–751 (2016).
Katzenberg, A. et al. Highly permeable perfluorinated sulfonic acid ionomers for improved electrochemical devices: insights into structure-property relationships. J. Am. Chem. Soc. 142, 3742–3752 (2020).
Kawai, R., Yamada, Y., Gohda, S. & Sato, S. Bottom-up synthesis of carbon materials with high pyridinic-nitrogen content from dibenzacridine isomers with zigzag and armchair edges. J. Mater. Sci. 57, 7503–7530 (2022).
Yamada, Y. et al. Toward strategical bottom-up synthesis of carbon materials with exceptionally high pyridinic-nitrogen content: development of screening techniques. Carbon 198, 411–434 (2022).
Zhan, W. C. et al. Surfactant-assisted stabilization of Au colloids on solids for heterogeneous catalysis. Angew. Chem. Int. Ed. 56, 4494–4498 (2017).
Liu, J. et al. High performance platinum single atom electrocatalyst for oxygen reduction reaction. Nat. Commun. 8, 15938 (2017).
Peng, H. L. et al. High performance Fe- and N- doped carbon catalyst with graphene structure for oxygen reduction. Sci. Rep. 3, 1765 (2013).
Song, P., Zhang, Y. W., Pan, J., Zhuang, L. & Xu, W. L. Cheap carbon black-based high-performance electrocatalysts for oxygen reduction reaction. Chem. Commun. 51, 1972–1975 (2015).
Veith, G. M. et al. Evidence for the formation of nitrogen-rich platinum and palladium nitride nanoparticles. Chem. Mater. 25, 4936–4945 (2013).
Guo, D. H. et al. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 351, 361–365 (2016).
Zhao, Z. P. et al. Pt-based nanocrystal for electrocatalytic oxygen reduction. Adv. Mater. 31, 1808115 (2019).
Wang, Z. C. et al. Tailored lattice compressive strain of Pt-skins by the L12-Pt3M intermetallic core for highly efficient oxygen reduction. Adv. Mater. 35, 2301310 (2023).
Dai, L. M., Xue, Y. H., Qu, L. T., Choi, H. J. & Baek, J. B. Metal-free catalysts for oxygen reduction reaction. Chem. Rev. 115, 4823–4892 (2015).
Shui, J. L., Wang, M., Du, F. & Dai, L. M. N-doped carbon nanomaterials are durable catalysts for oxygen reduction reaction in acidic fuel cells. Sci. Adv. 1, e1400129 (2015).
Liu, C. et al. Operando X-ray absorption spectroscopic study on the influence of specific adsorption of the sulfo group in the perfluorosulfonic acid ionomer on the oxygen reduction reaction activity of the Pt/C catalyst. ACS Appl. Energy Mater. 4, 1143–1149 (2021).
Takeshita, T., Kamitaka, Y., Shinozaki, K., Kodama, K. & Morimoto, Y. Evaluation of ionomer coverage on Pt catalysts in polymer electrolyte membrane fuel cells by CO stripping voltammetry and its effect on oxygen reduction reaction activity. J. Electroanal. Chem. 871, 114250 (2020).
Garrick, T. R., Moylan, T. E., Yarlagadda, V. & Kongkanand, A. Characterizing electrolyte and platinum interface in PEM fuel cells using CO displacement. J. Electrochem. Soc. 164, F60–F64 (2017).
Yamada, H., Kato, H. & Kodama, K. Cell performance and durability of Pt/C cathode catalyst covered by dopamine derived carbon thin layer for polymer electrolyte fuel cells. J. Electrochem. Soc. 167, 084508 (2020).
Khedekar, K. et al. Revealing in-plane movement of platinum in polymer electrolyte fuel cells after heavy-duty vehicle lifetime. Nat. Catal. 6, 676–686 (2023).
Tsuneda, T. Fenton reaction mechanism generating no OH radicals in Nafion membrane decomposition. Sci. Rep. 10, 18144 (2020).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Physical Review B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Acknowledgements
S.J.G. acknowledges funding from the National Key R&D Program of China (No. 2021YF1501001), National Science Fund for Distinguished Young Scholars (No. 52025133), and National Natural Science Foundation of China (No. 52261135633). X.Q.C. expresses gratitude to the National Natural Science Foundation of China (No. 52201197) and the China Postdoctoral Science Foundation (No. 2021M700187). C.S.S. extends thanks to the National Natural Science Foundation of China (No. 22105007). The authors gratefully acknowledge the support of the Shanghai Synchrotron Radiation Facility (BSRF) for XAFS analysis.
Author information
Authors and Affiliations
Contributions
S.J.G. supervised the entire project and conceived the idea. M.C.L. and X.Q.C. designed the study. X.Q.C. carried out the synthesis, general characterization, and electrochemical tests. H.Y.G., Y.H. and M.G.L. contributed to the electrochemical tests. C.S.S., R.Z. and Q.Z.H. performed the TEM characterization. Q.H.Z. conducted the HAADF-STEM experiments. M.L., F.L., H.T. and Z.Y.Q. provided constructive feedback on the manuscript. S.J.G., M.C.L. and X.Q.C. wrote the paper. All authors discussed the results and provided comments on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Frédéric Jaouen and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, X., Guo, H., Han, Y. et al. Sandwiching intermetallic Pt3Fe and ionomer with porous N-doped carbon layers for oxygen reduction reaction. Nat Commun 16, 2851 (2025). https://doi.org/10.1038/s41467-025-58116-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58116-7
