
Abstract
New solutions are needed to detect genotype-phenotype associations involved in microbial drug resistance. Herein, we describe a Group Association Model (GAM) that accurately identifies genetic variants linked to drug resistance and mitigates false-positive cross-resistance artifacts without prior knowledge. GAM analysis of 7,179 Mycobacterium tuberculosis (Mtb) isolates identifies gene targets for all analyzed drugs, revealing comparable performance but fewer cross-resistance artifacts than World Health Organization (WHO) mutation catalogue approach, which requires expert rules and precedents. GAM also reveals generalizability, demonstrating high predictive accuracy with 3,942 S. aureus isolates. GAM refinement by machine learning (ML) improves predictive accuracy with small or incomplete datasets. These findings were validated using 427 Mtb isolates from three sites, where GAM inputs are also found to be more suitable in ML prediction models than WHO inputs. GAM + ML could thus address the limitations of current drug resistance prediction methods to improve treatment decisions for drug-resistant microbial infections.
Similar content being viewed by others

Introduction
Microbial drug resistance arising from mutations, horizontal gene transfer, and the overuse of antibiotics is a growing problem that could diminish the utility of many commonly used antibiotics1,2,3,4. Infections caused by drug-resistant microbes are more difficult to treat5, often leading to prolonged treatment, increased healthcare costs, and the development of additional drug resistance1,2,3. Rapid and accurate identification of drug-resistant isolates and their specific drug resistance profiles are thus necessary to guide antibiotic treatment and limit their transmission1,2,6. However, current approaches used to identify drug resistance have significant limitations. Culture-based techniques traditionally require microbial growth at multiple drug concentrations6,7,8; are labour-intensive and time-consuming (requiring days to weeks), especially for slow-growing bacteria7,8,9,10; and may require careful interpretation of susceptible, resistant, or intermediate drug responses5,7,8. Molecular approaches that detect resistance-linked mutations, including polymerase chain reaction and microarray methods, are faster8,11,12 but identify only known and relatively common mutations. DNA sequencing can detect novel and rare mutations but is limited by the accuracy of catalogued mutations associated with phenotypic drug resistance11,13. New methods that can identify drug-resistance phenotypes from an isolate’s genomic information are thus needed to guide treatment decisions.
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), represents a strong case for the value of accurate drug resistance determinations14,15. There were an estimated 10.6 million new TB cases and 1.6 million TB deaths in 202116, including 450,000 rifampicin-resistant TB (RR-TB) cases, the majority of which were also resistant to isoniazid16. Accurate assessment of Mtb drug resistance is essential since the 86% overall TB treatment success rate drops to 57% for RR-TB and multidrug-resistant TB (MDR-TB: RR-TB plus at least isoniazid resistance)16, and underdiagnosis and treatment failures can increase the spread of RR-TB and MDR-TB, as occurred during the COVID-19 pandemic16,17.
Effective TB treatment requires the use of a multi-drug regimen that employs drugs with distinct mechanisms of action. Spontaneous mutations that confer resistance to one of drugs can allow Mtb bacilli to proliferate under selective pressure for mutations that confer resistance to other drugs in the multi-drug regimen18. Reliance of pre-defined drug regimens, including those tailored to resistant strains, can further promote development of multidrug-resistant (MDR-TB) or extensively drug-resistant (XDR-TB) Mtb strains19. This process drives sequential accumulation mutations in genes that directly confer resistance to specific drugs as well as in genes or sets of genes that alter metabolic processes to confer overlapping resistance to multiple drugs18,20. Mutations that produce overlapping resistance phenotypes can thus complicate efforts to genetically disentangle resistance mechanisms and identify specific mutations linked to resistance, particularly when a single mutation alters a pathway, or several pathways, that confers resistance to multiple drugs18. This complexity highlights the need for advanced analytical approaches that can address the dynamic and interconnected nature of the evolution of Mtb drug resistance.
Genome-wide association studies (GWAS) can identify genetic variations associated with specific traits21,22,23,24, including mutations linked to specific Mtb drug resistance phenotypes23,25. However, GWAS has limitations when used to evaluate mutations associated with multiple drug resistance phenotypes, including resistance to second-line TB drugs23,26,27. Most notably, GWAS can detect artificial associations with non-targeted drug resistances28,29, particularly in MDR-TB and XDR-TB30 isolates that can arise sequentially during TB treatment. These artificial cross-resistances can identify false-positive associations between drug resistance and genetic variations31,32,33 by detecting irrelevant genetic markers or incorrectly linking mutations involved in drug resistance that affect different drugs, mechanisms, and gene targets. It is thus important to consider a drug’s mechanism of action, if known, and use functional assays to validate GWAS associations29, which is not always feasible34. Other statistical approaches, such as those employed by the WHO can mitigate artificial cross-resistance but require prior knowledge, including masking to remove neutral mutations and expert rules to resolve interim cases35,36. Novel approaches that identify genetic variations associated with drug resistance should reduce or eliminate cross-resistance artifacts detected by GWAS and require no prior knowledge.
We hypothesized that this could be done using a novel Group Association Model (GAM) approach that identifies sequence variants enriched in isolates grouped by shared drug-resistance profiles and then identifies variants associated with specific drug-resistance by evaluating their enrichment in all isolates that are and are not resistant to a specific drug. We evaluated this hypothesis using sequence and drug-resistance data from the CRyPTIC (Comprehensive Resistance Prediction for Tuberculosis: An International Consortium) database29,37. GAM analysis performed with 7179 Mtb isolates identified single gene associations with eight of the nine first- and second-line drugs, unlike previous GWAS studies that identified multiple erroneous gene targets for each drug29, or untargeted confidence gating approaches that required expert rules or precedents35,36. GAM also demonstrated generalizability when applied to evaluate drug resistance using sequence from 3942 S. aureus isolates. Further, GAM predictive accuracy could be significantly improved by ML (Fig. 1), particularly for datasets with relatively few isolates or incomplete data, and ML inputs supplied by GAM performed better than those from the WHO approach as shown in 427 retrospective and prospective Mtb clinical isolates collected from multiple sites.

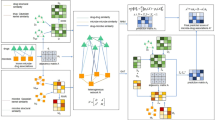
a Genotyping and minimum inhibitory concentration (MIC) culture analysis for drug susceptibility testing (DST) phenotypes of Mtb isolates. b Data filtration via genotype and phenotype information. c Mtb isolate sequence and DST data are fed into GAM to identify mutations associated with drug resistance, after which GAM classification performance is evaluated using statistical metrics. d Machine learning is applied to SNPs that GAM classifies as being associated with drug resistance to predict drug resistance profiles. e Multi-site cross-validation is performed to characterize the utility of this GAM + ML prediction approach. Created in BioRender.
Results
Group characterization of drug-resistant isolates
We screened 12,288 CRyPTIC Mtb genome entries from lineage 1 through 4 (>99% of the database entries)37 and identified 10,228 entries (DS1) that met our criteria for acceptable data quality after excluding those lacking high-quality drug resistance or sequence information or that had contig data that did not meet abundance or length criteria (Supplementary Fig. 1). DS1 entries with incomplete drug resistance profiles were excluded to generate a dataset containing 7179 entries (DS2) that were analyzed to identify mutations associated with specific drug-gene interactions, while all DS1 entries were analyzed to identify mutations associated with resistance to specific drugs (Supplementary Fig. 2). A phylogenetic analysis revealed that DS2 isolates were not disproportionally distributed within or among the four major Mtb lineages (Fig. 2a, b), reducing the potential for lineage-based bias during our screening and GAM processes. All four DS2 lineage groups also revealed similar fractions of drug-sensitive and drug-resistant isolates and drug-resistance category distributions (Monoresistance, MDR/RR, Pre-XDR, XDR, etc.; Supplementary Fig. 3).

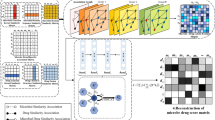
a GAM scheme and phylogenetic tree of DS2 isolates. b DS2 percentages derived from each lineage of all CRyPTIC isolates. Created in BioRender. c Number of isolates in groups containing multiple (Test/Control) or single (Non-Test) isolates. Mono-resistant (Mono), MDR/RR, Pre-XDR, XDR, Poly (RIF susceptible but resistant to ≥2 other drugs), and INH S (RIF + INH susceptible but resistant to ≥2 other drugs). d Group size ranges in size-ranked drug-resistant group quartiles. e Number of DS2 groups resistant to one or more drugs. f Mean number of isolates in groups resistant to one or more drugs. g Specific drug resistance frequencies in all drug-resistant DS2 groups. Source data are provided as a Source Data file.
Segregating the DS2 isolates by their shared drug-resistance profiles to increase data dimensionality generated 126 groups with ≥2 isolates for further analysis, and excluded 132 isolates (1.8%) belonging to single member groups not fit for statistical analysis (Fig. 2c). Most of these DS2 isolates (3997; 54.7%) were susceptible to all 13 analyzed drugs, but 3182 isolates segregated into 126 groups of varying size (2–551 isolates) with distinct drug-resistance profiles. Most drug-resistant isolates (86.6%) belonged to groups containing ≥14 isolates (Fig. 2d), and relatively uniform distribution were observed for the number of groups and mean number of isolates per group when ranking them by their number of drug resistances (Fig. 2e–f). Mtb isolates across the mono- to multi-drug resistance spectrum had multiple drug resistance phenotypes at high frequency when analyzed at the isolate and group level (Supplementary Figs. 4, 5), although specific drug resistance frequencies in these groups varied by drug tier. DS2 group resistance rates were highest for the first-line drugs rifampicin (67%), isoniazid (74%), and ethambutol (49%), and the second-line rifampicin replacement rifabutin (62%); moderately lower for the remaining second-line drugs (24–42%); and lowest for the new and repurposed drugs (6–23%) (Fig. 2g), and mimicked frequencies observed in DS2 isolates (Supplementary Fig. 6). The distribution of DS2 isolates among the four Mtb lineages, and isolates and specific drug resistance among the resistance groups was expected to provide sufficient discriminatory power to identify specific gene-drug associations and attenuate cross associations due to its classification approach.
Identification of genes and mutations associated with drug-resistance phenotypes
To identify variants associated with specific groups for inclusion in future analyses, variant detection rate differences were analyzed between all drug-resistant groups and the drug-susceptible control group by Fisher’s exact test adjusted for multiple comparisons (Fig. 3a). Variant p-values for all GAM Mtb groups are available on Figshare (SF2). All variants enriched in a drug-resistant group (odds ratio ≥1) were assigned to that group and all others were excluded from further analysis, markedly reducing the number of target variants (55.8 × 106 to 31.0 × 103). Variants present in groups resistant to a specific drug (e.g., rifampicin) were then compared to those in groups sensitive to that drug to identify variants associated with a specific drug resistance phenotype, using a -log10p-value > 5.22 threshold to identify variants associated with drug resistance (Fig. 3b). This approach correctly identified variants in six genes associated with the nine first-and second-line drugs, with a single false-positive match between katG and rifampicin38,39. No significant associations were detected for the four new or repurposed drugs (bedaquiline, clofazimine, delamanid, and linezolid), likely due to scarcity of groups/isolates resistant to them (8–29 groups; mean 2.4–8.6 isolates/group) versus the first- and second-line drugs (30–93 groups; mean 9.8–28.7 isolates/group).

a GAM workflow for data grouping and association. b Gene level interpretations of DNA variants associated with specific drug resistance as calculated by Fisher’s exact test, indicating the significance threshold (dashed line; -log10p-value < 5.22) determined after Bonferroni correction for multiple tests. c Gene-drug interactions detected by both LMM and GAM (orange), LMM alone (blue), or neither (white), using associations in the top 20 LMM associations for each drug. d Co-occurrence of DS2 drug-resistant phenotypes, where dark and light green indicates high and low percent overlap, respectively. e True positive, (f) false positive, (g) false negative mutations found by GWAS LMM (blue) and GAM (red). Source data are provided as a Source Data file.
GWAS gene-drug associations previously reported using a LMM accurately detected gene targets for the first- and second-line drugs, but detected multiple false-positive gene associations (≥20) for most of them29. Cross-association was most prominent for rpoB, but all first-and second-line drugs had multiple cross-associated genes (Fig. 3c) due to extensive drug resistance overlaps (Fig. 3d). Such overlaps were highest (72.2–90.1%) for drugs targeting the same genes (rifampicin/rifabutin, amikacin/kanamycin, and levofloxacin/moxifloxacin), but were also substantial for drugs with different gene targets. Spurious associations were also detected with two genes (pncA and rpsL) involved in resistance to drugs not analyzed in this dataset (pyrazinamide, and streptomycin), and with ethA, a reported target for ethionamide14,40,41. GAM results excluded all cross-resistances detected by the LMM, except a single erroneous katG association with rifampicin resistance that was likely caused by the limited number of rifampicin-sensitive but isoniazid-resistant groups (82.5% overlap in these resistance phenotypes).
GAM results primarily identified a single gene variant associated with a specific drug resistance, but multiple variants are often involved in resistance. We therefore next employed a similar confidence grading method as developed by the World Health Organization (WHO)35, and identified 3–20 drug-resistance-associated DS2 variants in each GAM-identified gene per drug (Supplemental Table 1), including variants detected at low frequencies (Supplementary Fig. 7), which had highly variable sensitivity (0.43–76.27%) but uniformly high specificity (98.75–100%; Supplementary Fig. 8).
To evaluate the performance of GAM relative to standard GWAS LMM, true positives, false positives, and false negatives were compared between the two methods (Fig. 3e–g). Raw Mtb LMM results are available on Figshare (SF3). GAM consistently identified substantially fewer false positives than LMM (1-2 vs. 1000~5000) across all drugs in this comparison, highlighting its ability to avoid misleading cross-associations often caused by overlapping drug resistances. Both approaches detected comparable numbers of false negative associations for each drug (Fig. 3g). For new and repurposed drugs, GAM exhibited lower sensitivity (fewer true positives) than LMM, which was expected given the small number of resistant isolates and the stratification required by GAM. However, for drugs with larger sample sizes, GAM not only minimized false positives but also sometimes matched or outperformed LMM in detecting true associations, showcasing its precision when sufficient data is available. Overall, GAM showed more than a 200-fold higher positive prediction value (PPV) than LMM.
To examine GAM’s ability to identify antimicrobial resistance (AMR) variants in other pathogens, we next employed it to analyze an S. aureus dataset containing 3942 isolates with variable resistance signatures for 13 drugs. Variant p-values for all GAM S. aureus groups are available on Figshare (SF4 and SF5). GAM identified SNP resistance-associated mechanisms for three drugs (fusidic acid, ciprofloxacin, and trimethoprim) with a broad-range of resistance-associated isolates (466–3027; Supplementary Table 2), where 23 SNPs linked with five genes (fusA, gyrA, parE, parC, and dfrB) had p-values ranging from >1 × 10−200 to 1.33 × 10−4. GAM also detected horizontal gene transfer mechanisms for four drugs (gentamicin, methicillin, tetracycline, and trimethoprim) with a broad-range of resistance-associated isolates (277–3501; Supplementary Table 2). Six horizontally transferred genes (aacA-aphD, mecA, tetM, tetK, dfrA, and dfrG) that conferred resistance to four drugs (gentamicin, methicillin, tetracycline, and trimethoprim) were detected with p-values ranging from 4.09 × 10−24 to 4.10 × 10−9. GAM detected some SNPs within transferred genes, but these were not directly associated with drug resistance. Four of the remaining seven drugs had few resistant isolates (2–50), and data for two of these four drugs did not appear in the database. The three remaining drugs with horizontal resistance mechanisms did not have these issues, and it is not clear why GAM did not detect gene associations with these drugs, although the SNP-based GAM design was not intended to detect such horizontal gene transfer mechanisms. All significant S. aureus genetic variants found by GAM are available on Figshare (SF6). Additionally, GAM identified a comparable number of true positives while producing fewer false positives than LMM (Supplementary Table 4), demonstrating that GAM outperformed LMM in detecting SNP-based drug resistance. However, for drugs such as penicillin, erythromycin, and clindamycin, where resistance primarily arises from horizontally transferred genes, LMM detected more true positives. Notably, this advantage came at the expense of a significantly higher number of false positives. All raw S. aureus LMM results are available on Figshare (SF7 and SF8).
Optimization of variant detection to improve phenotypic prediction
To evaluate predictive accuracy, DS1 dataset variants identified by GAM were compared to those of the WHO catalogue. GAM drug-specific variants demonstrated variable overlap with the WHO 2023 and 2021 Mtb mutation catalogues generated with the WHO dataset with (1.2–68.8%) and without (9.1–80.0%) interim criteria (Supplementary Figs. 9 and 10). Since GAM minimized the detection of misleading variants and requires no prior knowledge, it was hypothesized that GAM outputs could achieve better predictive accuracy and thus serve as improved inputs for a machine learning (ML) model (Fig. 4a). To assess the best model, nine ML models were screened and optimized for classification performance. Gradient Boosting achieved the highest mean accuracy (81.0%) and lowest overall variance (1.66%) across all nine drugs, (Fig. 4b), and this model was then introduced into a streamlined workflow to evaluate predictive performance (Fig. 4c).

a Schematic comparing prior knowledge requirements and accuracy of different approaches. b Boxplot of GAM + ML classification accuracy across model runs (N = 10), each using a different random test set and seed. Data depict median (center bar), 25th and 75th percentile (lower and upper box bounds), and minimum and maximum values (lower and upper whiskers). P-values were calculated from repeat measure 1-way ANOVAs, followed by Dunnett’s test for multiple comparisons, comparing the results to a Gradient Boosting reference model. c Workflow of the ML model using GAM variants as input. Calculated (d) PPV, (e) specificity, and (f) sensitivity (error bars indicate two-sided 95% confidence intervals) of predictive approaches applied to DS1 for specific drug resistance using variants identified by GAM (blue); 2021 (yellow) and 2023 (green) WHO interim criteria; and a gradient boosting model using GAM variants (red). Sample sizes for these comparisons varied according to the number of Mtb isolates with phenotype data for AMI (n = 10027), EMB (n = 8911), ETH (n = 9356), INH (n = 10025), KAN (n = 10085), LEV (n = 10114), MXF (n = 10139), and RIF (n = 10052). Source data are provided as a Source Data file.
PPV comparisons on AMR strains between the GAM, WHO, and GAM + ML approaches revealed differential predictive performance characteristics for these methods. GAM consistently minimized false-positive drug resistance strains by reducing cross-resistance associations. However, GAM PPV results varied by drug and differed from those of the WHO approach for amikacin, kanamycin, and rifampicin resistance (Fig. 4d). Integrating GAM results into a machine learning (ML) model enhanced the PPV for rifampicin resistance, without major effects on PPVs for other drugs resistances except for slight decreases in ethionamide, isoniazid, and levofloxacin.
Specificity analyses also emphasized the advantages of combining GAM with ML. GAM exhibited high specificity across all drugs, but the addition of ML led to a notable increase in rifampicin specificity (10.9%) (Fig. 4e). Specificity estimates modestly changed for other drugs, with minor decreases (ethionamide, isoniazid, and levofloxacin) and increases (ethambutol) observed for specific drugs. These findings indicate the robust predictive performance of GAM, and the enhancement provided by ML, particularly for rifampicin specificity. Notably, WHO+interim criteria did not markedly contribute to resistance classification since GAM and WHO non-interim variants produced similar PPV and specificity estimates (Supplementary Fig. 11).
Comparison of GAM, WHO, and GAM + ML sensitivity in predicting drug resistance revealed distinct performance patterns. GAM demonstrated variable sensitivity across different drugs, with the highest sensitivity observed for ethionamide resistance (Fig. 4f), while the WHO approach had better sensitivity for isoniazid resistance. Integrating GAM into an ML model improved predictive sensitivity for amikacin, ethionamide, isoniazid, kanamycin, and levofloxacin resistance, without affecting sensitivity estimates for other drugs. The use of ML thus enhances GAM predictive ability, particularly for drugs with complex resistance patterns. Area under the receiver operating characteristic curve (AUC) values ranged from 85.0 ± 1.0% to 97.0 ± 1.0% (Supplementary Fig. 12). Applying optimal cutoff values corresponding to the highest F1 scores increased overall model accuracy by 2.6%, further enhancing prediction of all nine drug phenotypes. These results demonstrate that integrating GAM outputs with ML can improve diagnostic accuracy, particularly for drugs where sensitivity and specificity remain challenging.
Impact of sample size and data completeness on GAM
Sample size and data completeness are expected to influence the ability of GAM to recover known gene resistance associations. To assess the effect of sample size on GAM and LMM performance, samples containing 179–7179 DS2 isolates were randomly generated and assessed for the number of true positive drug-gene pairs identified in ten replicates (Fig. 5a). For GAM, the analysis revealed an asymptotic relationship between sample size and true positive genes, with an inflection near the midpoint reflecting sample size difference required to detect specific drug-gene interactions (Supplementary Fig. 13), and false-positive increases detected in large samples resulting from a cross-association between rpoB and katG.

a Effect of sample size on GAM and LMM true positive (TP) and false positive (FP) gene identifications. Y-axis breaks between 20 and 200. b Heatmap of mean PPV from model runs, each using a different random test set and seed (N = 10), for GAM and LMM for varying sample sizes. c Effect of missing data on GAM performance. a, c Solid and dashed lines represent nonlinear sigmoidal curves and their two-sided 95% confidence intervals, respectively. Data points display mean ± standard error values from model runs, each using a different random test set and seed (N = 10). d ML-GAM workflow for datasets with missing data. e ML training set size effect on GAM accuracy, indicating median (central line) and minimum and maximum range (box boundaries), and p-value from a 1-way ANOVA with Tukey’s multiple comparison test from model runs, each using a different random test set and seed (N = 30). f Effect of missing data on accurate GAM gene identification after adjusting data with ML models trained with different sample sizes, where the remaining samples are analyzed as the GAM test samples. Solid and dashed lines represent nonlinear sigmoidal curves and their two-sided 95% confidence intervals, respectively. Data points display mean ± standard error values from model runs, each using a different random test set and seed (N = 5). Source data are provided as a Source Data file.
Comparing GAM and LMM reveals that while LMM generally identified a slightly higher number of true positives, it exhibited a 100–1000 times higher false positive rate than GAM across all sample sizes (Fig. 5a). Notably, PPV analysis further demonstrated that GAM consistently exhibited a significantly higher PPV than LMM (Fig. 5b), indicating its superior ability to distinguish true positives from false positives in the data. Similar trends were observed at the mutation level, where true positive differences remained minimal, but false positive rates and positive predictive value (PPV) declined more rapidly as sample size increased (Supplementary Fig. 14a, b). When new and repurposed drugs were included in the analysis, LMM showed an increase in true positives, while GAM struggled to capture significant associations due to the limited sample size available for these drugs; however, the LMM false positive rate also increased, while GAM’s rate remained stable (Supplementary Fig. 14c, d).
Randomly varying the drug resistance information drop rate from data entries also detected an inverse sigmoidal relationship between accurate gene identifications and the missing data percentage (Fig. 5c) since removing drug resistance data from an isolate deletes it and its variants from its assigned group and transfers it to a group that has indeterminate resistance data for this drug. In larger groups, this obscures variant frequency differences between it and the control group, but in groups with only two isolates (>25% of DS2 groups) this deletes all this group’s data. Data losses in drug-susceptible isolates diminishes the size and pool of neutral variants of this group. Losses affecting isolates of a drug-resistant group eliminates these isolates and their variants from the group, but in isolates resistant to two or more drugs this can create groups that include variants associated with both drug-susceptible and drug-resistant isolates of the eliminated phenotypes. GAM analyses that included groups with missing data must thus increase the multiple testing correction factor and significance threshold, particularly when isolates have missing data for more than one drug resistance phenotype.
We next explored the potential of ML to attenuate the effect of data omission on GAM predictions by partitioning the dataset into a training set with complete data and a testing set that was resampled with varying data omission rates. ML models generated with training set data were employed to reconstitute the missing testing set data prior to GAM analysis (Fig. 5d), and revealed similar prediction accuracy for missing data (0.767 versus 0.801) even when comparing models that used 1% and 75% of the total sample as their training sets (Fig. 5e). Notably, these training sets included data from single-isolate groups that could not be used in GAM analyses. However, while ML adjustment mitigated the effect of information loss on GAM identifications, irrespective of the amount of data removed from the test samples, GAM gene detections still relied on sample size (Fig. 5f). Thus, ML models built with small but complete training sets can allow the use of large but incomplete datasets that would otherwise exhibit poor performance in GAM analyses.
Relative performance of GAM and WHO variant data in ML phenotype prediction
Subsequent analyses performed using both GAM and 2021 and 2023 WHO interim variants as inputs for Gradient Boosting ML models found that GAM-based models had predictive accuracies greater than (amikacin, isoniazid, kanamycin, and rifampicin) or comparable to those of the WHO+interim models (Supplementary Fig. 15). Similar results were obtained when using WHO non-interim variant inputs, except that GAM models then also had higher predictive accuracy for ethionamide (both WHO datasets) and ethambutol resistance (2023 WHO dataset only) (Supplementary Fig. 16).
This analysis was subsequently replicated using an independent dataset of 428 Mtb isolates (DS3) that produced similar trends, as GAM inputs yielded predictive accuracies that were significantly higher than 2021 and 2023 WHO+interim inputs for all drugs but ethionamide, kanamycin, and levofloxacin (Supplementary Fig. 17). Superior performance of the 2021 and 2023 WHO+interim variants in predicting ethionamide resistance was apparently due to the greater number of inputs (296 and 331 versus 4; 3 shared among all), since GAM variants performed better than WHO non-interim variants for the same analysis (4 and 8 versus 4; 1 shared among all) (Supplementary Fig. 18). Loss of 2021 and 2023 WHO variants identified by interim criteria also decreased the predictive accuracy of the WHO models for kanamycin, isoniazid, and rifampicin, while eliminating the predictive difference of the GAM and 2023 models for levofloxacin. These differences may be due to ML model’s ability to better handle fewer inputs and the relative quality of these inputs.
Similar model performance trends were also obtained when we used GAM and WHO-based ML models to analyze genomic sequence variants of 427 Mtb isolates collected at the West China Hospital (Chengdu), Beijing Chest Hospital, and Shenzhen Third People’s Hospital (Fig. 6a; Supplementary Table 5). GAM + ML performance varied by hospital (Supplementary Table 6), but models that used GAM inputs had consistently better accuracy overall in predicting resistance to ethambutol, ethionamide, isoniazid, kanamycin, moxifloxacin, and rifampicin than those using 2021 or 2023 WHO+interim inputs, while revealing similar results for all other drugs (Fig. 6b–i). GAM and WHO non-interim model results were also comparable to previous results, although the 2021 and 2023 non-interim models had reduced mean predictive accuracies for ethambutol, ethionamide, kanamycin, and moxifloxacin resistance and the 2023 model had a slight increase in accuracy for isoniazid (Supplementary Fig. 19). These results support our hypothesis that GAM inputs are more suitable for ML-based drug resistance prediction models than WHO inputs.

a Mtb isolates from three hospital sites in China were analyzed by drug susceptibility testing and sequenced to identify variant sequences. Created in BioRender. b–i Pair-matched model accuracy for isolates resistant to eight drug targets as assessed across N = 10 random seeds and analyzed by 1-way ANOVAs with Geisser-Greenhouse corrections and Dunnett’s tests for multiple comparisons. The number of isolates used for these comparisons varied according to the number of isolates with phenotype data for (b) amikacin (n = 427), (c) ethambutol (n = 423), (d) ethionamide (n = 421), (e) isoniazid (n = 352), (f) kanamycin (n = 427), (g) levofloxacin (n = 112), (h) moxifloxacin (n = 415), and (i) rifampicin (n = 185) susceptibility tests. Source data are provided as a Source Data file.
Discussion
Cohort size, data characteristics, known resistant mechanisms and other prior knowledge should all be considered when selecting the most appropriate means to analyze drug resistance in populations with complex resistance profiles. GAM has several advantages for streamlined identification of gene-drug interactions from datasets of microbial genome sequence data and drug resistance data, including its ability to remove neutral mutations by discarding those that are not differentially expressed between the control and drug-resistant groups to reduce cross-resistant artifacts observed with other prediction methods. This process also reduces potential lineage-bias. GAM also does not rely on prior knowledge of genes, pathways, or mechanisms involved in drug resistance, enabling it to comprehensively analyze all variants present in a dataset. In contrast, the current WHO approach is specifically tailored to Mtb and relies on identifying resistance-associated mutations through a combination of expert-driven rules, masking strategies to exclude naturally occurring or neutral mutations, pathway knowledge derived from prior genetic studies to pinpoint relevant gene functions, and relaxed statistical thresholds to account for rare mutations35,36. While these strategies provide valuable insights within the constrained scope of Mtb, they inherently limit broader applicability. The ability of GAM to operate independently of such predefined rules or assumptions allows it to provide a more flexible and data-driven framework for identifying drug resistance-associated variants across diverse pathogens. Additionally, GAM utilizes a straightforward computational process, unlike LMMs that must account for random effects and intricate covariance structures. Finally, unlike current drug resistance prediction methods, GAM allows rapid identification of key input features for ML approaches that could ultimately improve treatment of drug-resistant microbial infections.
GAM analyses have specific limitations despite these advantages. First, GAM datasets should contain high-quality phenotype data and a diverse array of resistance profiles, with multiple entries for each profile, to capture the full range of variations associated with all drugs required to avoid incorporating resistance-associated mutations in the control group. Resistance to a specific drug should also be spread across groups with different resistance profiles to enhance statistical power, and most isolates in the dataset should be resistant to multiple drugs outside a single drug family to avoid categorization bias. This can require a substantial numbers of isolates per resistance depending upon the complexity and prevalence of resistance profiles in an isolate population. Alternate approaches may be more effective when it is not possible to meet these criteria, such as when analyzing small datasets with limited resistance group diversity. The WHO model leverages prior knowledge, which can be particularly beneficial for identifying drug-gene associations in limited datasets. While LMMs used in GWAS analyses can partially address missing data through imputation methods, they still require complete phenotype data for each isolate to ensure accurate association analyses. Similarly, standard GAM analyses also exhibit substantial performance losses when applied to datasets that contain incomplete phenotype data. However, our results indicate that ML models trained using small datasets with complete data (1% the size of the test population) can adjust for this missing data to prevent performance losses. These ML models do not reduce the dataset size requirement for GAM analysis, but they can use isolates with complete phenotype data but unique resistance profiles, which are excluded by GAM criteria (4.2% of DS1 isolates) to maximize number of isolates available for GAM analyses. Further, the ability to correct for missing data can permit GAM to analyze isolates with incomplete data (30.2% of DS1 isolates) to improve predictive power gene-drug associations for less frequent drug resistances.
GAM demonstrates limited performance in identifying extra-genomic resistance mechanisms, as shown by its inability to detect horizontal gene transfer mechanisms in isolates resistant to penicillin, erythromycin, and clindamycin within an S. aureus database containing 295 to 3840 isolates with these resistance phenotypes. This performance deficit could potentially be mitigated by developing coding frameworks that facilitate the analysis of gene-level differences across multiple reference genomes and by applying machine learning approaches. However, the current version of GAM is not designed to capture resistance mechanisms that primarily arise from horizontal gene transfer, as occurs in Enterobacterales and many other microbes. This is due to its reliance on SNP-based correlations against a single reference genome, an approach that does not accommodate the complex resistance mechanisms conferred by the expansion of the accessory genome. However, we hypothesize that there are two scenarios where GAM might identify horizontal gene transfer mechanisms. First, GAM could directly detect SNPs within horizontally transferred genes, regardless of their direct association with resistance, leading to association of the entire gene with the resistance phenotype. Second, if a horizontally transferred gene integrates near a polymorphism in the host genome, GAM might detect this polymorphism as being associated with drug resistance due to its linkage equilibrium with an adjacent integrated gene. However, this latter approach would require that a substantial number of isolates contain the same integration site.
ML models that used GAM data had better overall sensitivity and accuracy for drug resistance predictions than the GAM or WHO methods alone or equivalent WHO + ML models, without major specificity differences. This superior performance could be attributed to multiple factors. ML algorithms have the inherent capability to capture complex, nonlinear relationships within data42,43,44, while GAM relies on additive components that may not adequately model intricate interactions and patterns present in antibiotic resistance data. Consequently, ML introduces an additional layer of flexibility, enabling the model to adapt and better capture this underlying complexity to improve predictions. It is also worth noting that the specific ML technique chosen for this study, Gradient Boosting, has demonstrated strong performance for various classification tasks45,46, including successful applications for similar biomedical research topics, making it a suitable candidate approach to improve antibiotic susceptibility prediction. However, future studies are still needed to accurately evaluate the full potential of GAM + ML to enhance antibiotic susceptibility prediction.
Comparison of the GAM and WHO workflows and outputs clearly favor the use of GAM results as the input for such Gradient Boosting models as GAM isolates exhibit less overlap to yield a more distinct set of features, as demonstrated by the relative predictive value of models built with these inputs. Further, GAM requires fewer input features (mutations associated with resistance) to effectively capture drug resistance patterns than the WHO approaches. Classification accuracy differences obtained using GAM and WHO inputs were small but consistent, underscoring GAM’s ability to optimize predictive accuracy within a ML framework.
GAM analyses represent a powerful means to identify genetic variations associated with drug resistance in species with diverse resistance profiles, which is essential for development of new treatments and effective management of drug-resistant infections6,47. GAM analyses could predict specific drug-gene associations in multidrug-resistant Candida auris48, Escherichia coli49, and other infectious diseases with a complex spectrum of drug resistance. GAM could also be applied to identify genetic factors associated with disease resistance in crops, such as maize lethal necrosis disease or downy mildew, which could help inform breeding programs, promote more sustainable agricultural practices, and improve food security50,51. Future studies will incorporate GAM into the analysis of multi-gene mutations and horizontal gene transfer mechanisms to enhance the prediction of resistance phenotypes in Mtb, S. aureus, and other microbes. This includes understanding how mutations in multiple genes or compensatory mutations can significantly increase drug resistance52.
In summary, GAM provides a valuable framework to mitigate neutral mutations, false positives, and cross-resistance artifacts that can hinder accurate identification of gene-drug associations involved in drug resistance in populations with complex resistance profiles, without requiring prior knowledge of resistance mechanisms. Coupling GAM with ML can also improve the sensitivity and accuracy of resistance predictions made with complete data or adjust incomplete data to improve its predictive power. Future studies could therefore benefit from incorporating GAM + ML to predict drug-gene associations in species with complex drug resistance profiles.
Methods
Ethical approval
The use of archived clinical Mtb sample data analyzed in this study25,53 was approved of use by the Beijing Chest Hospital, and West China Hospital, Sichuan University. For all prospective data analyzed, ethical approval was obtained from the Research Ethics Committee of Third People’s Hospital of Shenzhen.
Dataset and exclusion criteria
The CRyPTIC database was selected for its complete and consistent phenotyping across 13 drugs, as well as its extensive global coverage, features that distinguish it from other larger databases. Sequence and drug susceptibility testing (DST) data for 13 antibiotics were obtained from 12,289 Mtb isolates in the CRyPTIC database37. These files were filtered using CRyPTIC criteria to remove entries that lacked high-quality DST results or sequence data29, and to exclude entries with incomplete DST data profiles for any drug (N/A, blank, or indeterminate) prior to their use as input files in a GAM analysis (Supplementary Fig. 1).
GAM analysis procedure in identification of AMR-associated sequence variants
CRyPTIC database entries that did not meet study exclusion criteria (Supplementary Fig. 1) were grouped based on their unique drug resistance profiles to produce a control group that contained the genetic variations present in isolates susceptible to all 13 drugs, and an array of groups with distinct drug resistance profiles and associated genetic variants. Resistance groups analyzed by GAM were defined based on drug susceptibility testing (DST) profiles, where isolates sharing identical drug resistance profiles were grouped together. To ensure statistical robustness, we selected all observed resistance groups that included two or more Mtb strains to maximize both the number and diversity of resistance groups. Non-synonymous genetic variants that differed from the Mtb H37Rv reference genome (NC000962.3) were identified in all groups. Differences in variant detection frequency between the drug-susceptible control group and each drug-resistant group were analyzed using Fisher’s exact tests corrected for multiple tests to identify genetic variants associated with drug resistance without considering specific associations (Supplementary Fig. 2). Variants not significantly enriched in at least one drug-resistant group were eliminated from further analyses. Next, all variants enriched in groups resistant to a specific drug were compared to those present in all groups not resistant to that drug by Fisher’s exact tests corrected for multiple tests to identify variants associated with specific resistance phenotypes.
The GAM procedure identifies DNA variants that are significantly associated with specific single drug AMR phenotypes, while results are interpreted at both the gene and sequence variant level. AMR-associated variants are first linked with their respective genes and then all mutations within these genes are then analyzed to identify those that are significantly associated with the targeted single-drug AMR phenotype.
Significant drug-gene interactions detected by GAM were then further analyzed for variants associated with drug resistance by screening coding region mutations with these genes using WHO-proposed confidence gating criteria35. A variant was considered to have positive predictive value (PPV) if it was the only variant in the target gene of ≥5 isolates (solo mutation), had a resistance-associated solo mutation odds ratio (OR-solo) ≥1, an OR-solo PPV 95% lower confidence interval ≥25%, and an OR-solo false discovery rate-corrected Fisher’s exact test p-value ≤ 0.05. Double mutations were considered when there were no solo mutations in a target gene and one mutation was a neutral mutation.
Correction for multiple testing
Fisher’s exact test results were adjusted using Bonferroni corrections for multiple tests based on the number of unique variants employed in each test. Variants were considered significantly associated with resistance if they were detected at odds ratios ≥1 and had p-values < α/n, where α = 0.05 was employed as the false positive rate and n denoted the number of analyzed genetic variants28,54. There were 8320 mutations across all groups that significantly differed from the control group, resulting in a final p-value threshold of −log10p-value 5.22. The −log10p-value significance thresholds for all 126 group associations are available on Figshare (SF9) along with a full list of Mtb isolates within each group (SF10).
GAM identification of S. aureus AMR variants
A database of 3942 S. aureus isolates resistant to a spectrum of 13 drugs (gentamicin, penicillin, cefoxitin, fusidic acid, teicoplanin, vancomycin, erythromycin, clindamycin, linezolid, ciprofloxacin, rifampicin, tetracycline, and trimethoprim)55 were analyzed using a methodology that mirrored that used to analyze the CRyPTIC Mtb isolates. However, given the smaller size of the overall sample and its control group, the order of GAM analyses was reversed, so that resistance groups, were first compared to identify SNPs that were detected at different frequencies in the resistance groups that were resistant and sensitive to a specific drug, and these SNPs were then compared to the control group to exclude neutral mutations. Groups were also allowed to contain single isolates, and the number of solo mutations required for inclusion was lowered to ≥1. For instances where drug resistance mechanisms stemmed from gene acquisition rather than mutation, a dataset of all genes present within each isolate was used and the analysis terminated at the gene level, as further data extraction was not feasible. S. aureus HO 5096 0412 (HE681097) was used as the reference genome.
Evaluation of GAM performance
To evaluate the accuracy of GAM identification of variants associated with resistance, GAM and GWAS LMM outputs were compared for true positive, false positive, and true negative counts based on known variant-resistance associations summarized in the WHO Mtb mutation catalogue, which is the clinically recognized gold standard reference for resistance-associated Mtb mutations36.
Additionally, we assessed predictive performance by comparing the sensitivity, specificity, and PPV of GAM outputs versus the WHO mutation catalogue as predictors of phenotypic drug susceptibility across different datasets.
Comparison of ML models
Eight machine learning models (Gradient Boosting, Naive Bayes, Random Forest, AdaBoost, Nearest Neighbor, RBF SVC, Decision Tree, and Neural Network) were trained on the same 7129 Mtb isolates using default hyperparameter tuning set by scikit-learn library (Version 1.3.0). Statistical significance of accuracy differences among these models, the primary performance metric, was assessed using one-way analysis of variance (ANOVA) against the Gradient Boosting reference model with Dunnett’s test used to correction for multiple comparisons. Analyses were performed in Python using scikit-learn using a ten-fold cross-validation approach to ensure model robustness.
GAM + ML model generation
A multi-output classification was performed using the scikit-learn library (Version 1.3.0), where input data (X) was GAM-highlighted mutations and labeled data (y) was the drug resistance profiles for individual isolates. To assess the ML model’s performance, the data was analyzed using 10-fold stratified cross-validation (90% and 10% into training and testing sets, respectively) and fold outputs were conjugated for calculation of overall results. This ML classification model used a Gradient Boosting classifier with a learning rate of 0.1 and 950 estimators and a multi-output classifier wrapper to allow it to manage multiple target variables, using the mean of 100 repeats to evaluate the model’s performance.
ML model ROC and AUC analyses
ROC curves were generated for each drug using the scikit-learn library (Version 1.3.0) to assess the ability of the Gradient Boosting models to predict resistance to each analyzed drug. This process was repeated three times using different random seeds to ensure data robustness, after which mean AUC ± standard deviations were computed for each drug. Optimal cutoff value determinations for individual drug resistance predictions were identified as the values that matched the highest F1 scores.
Comparison of ML models generated with GAM and WHO catalog input data
Significant GAM outputs and 2021 and 2023 WHO mutation catalog results were utilized as inputs for a Gradient Boosting model that used consistent model parameters and differed only in their respective input sources. The 7129 Mtb isolate dataset was partitioned into training and testing subsets containing 75% and 25% of these isolates, respectively, and model accuracy for each drug was compared using 10 consecutive random seeds. Statistical significance of accuracy disparities versus the GAM reference model was analyzed using row-matched one-way ANOVAs employing Geisser-Greenhouse corrections and Dunnett’s test for multiple comparisons. These analyzes were performed in Python using the scikit-learn library (Version 1.3.0) using a 10-fold cross-validation approach to improve result reliability.
This process was replicated using held out data from 428 DS3 isolates employed as a validation dataset and with an independent dataset of 427 isolates from a multisite cross validation dataset (DS4) generated from three sites in China. DS4 consisted of 80 samples from West China Hospital (Chengdu), 281 from Beijing Chest Hospital (Beijing), and 102 samples from Shenzhen Third People’s Hospital (Shenzhen), after excluding 36 samples with low genome alignment rates (<50%) or read depths (<10), poor DST quality, or missing phenotypic information.
Clinical sample culturing, phenotyping, and genotyping
Archived data from West China Hospital and Beijing Chest Hospital isolates analyzed in this study was previously collected using published protocols25,53. Data from Shenzhen Third People’s Hospital was generated from isolates cultured between 2015 and 2023. Sputum specimens obtained from patients were inoculated into Bactec MGIT 960 culture tubes (Becton, Dickinson and Co., Sparks, MD, USA, 245122) to verify positive isolates after standard NALC-NaOH decontamination (BaSO, Wuhan Jinhong Biotech Development Co., China, BC1999). Positive isolates were identified using acid-fast staining kit (BaSO, Wuhan Jinhong Biotech Development Co., China, Catalog Catalog BA4092A). Strains isolated from sputum were subcultured on Lowenstein-Jensen (L-J) medium agar (BaSO, Wuhan Jinhong Biotech Development Co., China, CUSTOM0038) for phenotypic drug susceptibility testing (DST) and DNA extraction.
Phenotypic MIC DST was performed according to the manufacturer’s instructions (Trek Diagnostic Systems Ltd., UK, V3020)56,57. Colonies in the log-phase growth stage were suspended in a saline-Tween solution (Trek Diagnostic Systems, T3491), adjusted to a McFarland standard of 0.5, and allowed to settle for 15 min. A 100 µL aliquot was transferred to 11 mL of Middlebrook 7H9 broth (Trek Diagnostic Systems, T3441) and vortex-mixed for 20 s. Another 100 µL of this material was inoculated into each well of the Sensititre MYCOTB MIC plate (Trek Diagnostic Systems, MYCOTBI). Each MYCOTB plate consisted of 2 antibiotic-free positive control wells and 94 antibiotic-containing wells testing 7 antibiotics: amikacin, ethambutol, ethionamide, isoniazid, kanamycin, moxifloxacin, and rifampicin. Plates were covered with permanent plastic seals provided in the test kit and incubated at 37 °C in 5% CO2. Growth was monitored at days 7, 10, 14, and 21 using a mirrored viewer. The lowest concentration with no visible growth was recorded as the MIC. Unless otherwise specified, MYCOTB test results were based on the first time point with adequate growth in the drug-free control wells.
Genomic DNA was extracted using the CTAB method with reagents from Sangon Biotech (Shanghai) Co., Ltd. Colonies from Lowenstein-Jensen (L-J) slants were collected into 500 µL of Tris-EDTA buffer (pH 8.0 Sangon Biotech, Catalog #B540625) and heated at 80 °C for 20 min. Lysozyme (50 mg/mL, Sangon Biotech, Catalog #B541002) was added (10 µL per tube), followed by vortex mixing and incubation at 37 °C for 2 h. Proteinase K (2 mg/mL, Sangon Biotech, Catalog #A414170) and 10% sodium dodecyl sulfate (SDS, Sangon Biotech, Catalog #A425678) were then added (50 µL each), vortexed gently, and incubated at 65 °C for 20 min. A 150 µL mixture of N-acetyl-N,N,N-trimethyl ammonium bromide (CTAB, Sangon Biotech, Catalog #A600108) and NaCl was added, followed by the addition of NaCl alone. The suspension was vortexed until milky and incubated at 65 °C for 10 min. Chloroform-isoamyl alcohol (24:1, Sangon Biotech, Catalog #A610278) was added (700 µL), vortexed, and centrifuged at 13,000 rpm for 5 min at room temperature using a microcentrifuge (Thermo Fisher Scientific Inc., USA, ModelST40R). The genomic DNA in the aqueous phase was then isolated by ethanol precipitation and resuspended in 30 µL of nuclease-free water (Sangon Biotech, Catalog #B300591).
DNA quality control was performed using an Agilent 5400 system. DNA shearing was conducted using a Covaris instrument (Covaris, USA, model ML230) to generate ~350 bp fragments. End repair was performed using T4 DNA polymerase (New England Biolabs, M0203L), and 3’ adenylation was performed with Klenow fragment (New England Biolabs, Catalog #M0212). DNA adaptors (Illumina, Catalog #FC-121-1031) were ligated using T4 DNA ligase (New England Biolabs, Catalog #M0202). Size selection was performed using SPRIselect beads (Beckman Coulter, USA, Catalog #2358413). The DNA library was amplified using high-fidelity polymerase (New England Biolabs, Catalog #M0530), and sequencing was performed using the Illumina NovaSeq platform (Illumina, USA, model NovaSeqX) with 150 bp paired-end reads (PE150).
Sample size and data completeness analyses
The effect of sample size on the ability for GAM to distinguish true-positive from false-positive associations was assessed by systematically reducing the dataset to contain 179, 529, 879, 1579, 2279, 2979, 4329, 5779, and 7179 samples. Each subset was analyzed using GAM to identify true- and false-positive drug-gene associations.
A comparative analysis of the effect of sample size on the ability of GAM and LMM to identify drug-gene associations was performed using the same datasets used to evaluate sample size effects on GAM performance. The LMM model employed in this analysis used FaST-LMM58 with adjustments for kinship, lineage, geographic region of sample collection, and the testing site of each sample, where the kinship matrix was assessed using the Jaccard similarity index, and lineage, geographic region, and test site were included as covariates.
The effect of missing data was assessed by eliminating random drug resistance data fields from random isolates to generate data sets with missing DST data rates of 1, 5, 7.5 10, 12.5, 15, 17.5, 20, and 25%. Sample and data exclusion processes were repeated 10 times, and the resulting data was analyzed to determine the mean and standard error of the number of true- and false-positive drug-gene associations detected at each sample size or data drop rate. This analysis was not performed using the LMM approach, as FaST-LMM inherently excludes isolates with missing phenotype information, and thus the effects for missing DST data can be drawn from the effects of reduced sample size.
Both studies analyzed and/or dropped data from the three first-line drugs and six second-line drugs (amikacin, ethambutol, ethionamide, isoniazid, kanamycin, levofloxacin, moxifloxacin, rifabutin, and rifampicin), since the four new/reproposed drugs analyzed in the CRyPTIC dataset lacked significant drug-gene associations and thus did not contribute to GAM performance.
ML adjustment for missing data
To assess the impact of ML on GAM analyses performed with incomplete DST data, a multi-output classification task was performed using the scikit-learn library (Version 1.3.0). In this analysis, the 7179 CRyPTIC isolates suitable for GAM analyses were systematically divided into ML training sets containing 1, 25, 50, and 75% of these entries, with the remaining samples employed for the corresponding GAM testing dataset. These ML training datasets were supplemented with 428 entries (DS3) with unique drug resistance profiles that could not be grouped for GAM (Supplementary Fig. 1) but provided valuable information to train the ML model. These training datasets were used to train Gradient Boosting classifiers, configured with a learning rate of 0.1 and 100 estimators, and later applied to predict missing values in the testing datasets employed for GAM analyses. Missing data in these testing datasets was produced by randomly excluding a drug resistance data field from a randomly selected isolate to generate testing datasets 0, 5, 20, 40, 60, 80, and 99% missing DST data rates, and repeating this procedure five times for each missing data rate value. Missing DST data values were then imputed by the ML models and the adjusted data was subjected to GAM evaluation. All GAM results were analyzed to determine mean and standard error values.
Data visualization and statistics
Data visualization and statistical analysis were carried out using various software tools. Figures were created using GraphPad Prism (Version 10.0.2), with p-values obtained through a one-way analysis of variance (ANOVA) and Tukey’s multiple comparison test. Additionally, Matplotlib (Version 3.8.0) and Seaborn (Version 0.13.0) were used for generating other data plots to provide comprehensive visual representations of the study’s findings. The performance of machine learning models was assessed using the scikit-learn library (Version 1.3.0), enabling rigorous evaluation of their predictive capabilities. In investigating associations between categorical variables and addressing the need for multiple comparisons, we conducted Fisher’s exact tests and applied Bonferroni correction, making use of the statsmodel library (Version 0.14.0) to maintain statistical rigor. For the visualization of phylogenetic trees and their associated data, we utilized the phylobase (Version 0.8.10) and ggtree (Version 3.2.1) packages, providing a comprehensive representation of evolutionary relationships within the dataset. The GAM analysis code was developed and tested in Python (version 3.8.1) using Jupyter Notebook (version 7.3.2). Key libraries included Pandas (version 1.3.5), NumPy (version 1.18.2), SciPy (version 1.6.2), PySnpTools (version 0.0.2), FastLMM (version 0.0.1), and scikit-learn (version 1.6.1).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated in this study are provided in the Supplementary Information and Source Data within this paper. Significant variants, SNPs, genes, and group thresholds for GAM and LMM analyses of Mtb and S. aureus, along with large input files for code execution, are accessible on Figshare [https://doi.org/10.6084/m9.figshare.28191398]59. Gene variant and DST data for the 12,289 CRyPTIC database Mtb isolates (DS1-DS3) used in this study are available at the European Bioinformatics Institute (EBI) FTP site [https://ftp.ebi.ac.uk/pub/databases/cryptic/release_june2022/]. The 427 DS4 isolates from China with linked DST profiles (DS4) are available under restricted access for research purposes only, access can be obtained by request from the corresponding authors, subject to institutional approval. Raw whole genome sequencing data for Mtb isolates collected by the Third People’s Hospital of Shenzhen were deposited in Sequence Read Archive (SRP567794) [https://www.ncbi.nlm.nih.gov/sra/?term=SRP567794]. Whole genome sequencing raw data from the Beijing Chest Hospital were deposited in Sequence Read Archive (SRP134826) [https://www.ncbi.nlm.nih.gov/sra/?term=SRP134826] and Genome Sequence Archive (CRA000786) [https://ngdc.cncb.ac.cn/gsa/browse/CRA000786]. S. aureus gene variants and linked DST data for the 3,942 isolates used in this study are available at GitHub [https://github.com/nwheeler443/staph_gwas]. Source data are provided with this paper.
Code availability
The GAM pipeline code is available on Zenodo [https://doi.org/10.5281/zenodo.14975407]60.
References
Cohen, M. L. Epidemiology of drug resistance: implications for a post-antimicrobial era. Science 257, 1050–1055 (1992).
Alanis, A. J. Resistance to antibiotics: are we in the post-antibiotic era? Arch. Med. Res. 36, 697–705 (2005).
Michael, C. A., Dominey-Howes, D. & Labbate, M. The antimicrobial resistance crisis: causes, consequences, and management. Front. Public. Health 2, 145 (2014).
Mazel, D. & Davies, J. Antibiotic resistance in microbes. Cell. Mol. Life Sci. 56, 742–754 (1999).
Rowneki, M. et al. Detection of drug resistant Mycobacterium tuberculosis by high-throughput sequencing of DNA isolated from acid fast bacilli smears. PLoS One 15, e0232343 (2020).
Bergeron, M. G. & Ouellette, M. Preventing antibiotic resistance through rapid genotypic identification of bacteria and of their antibiotic resistance genes in the clinical microbiology laboratory. J. Clin. Microbiol. 36, 2169–2172 (1998).
Didelot, X., Bowden, R., Wilson, D. J., Peto, T. E. A. & Crook, D. W. Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 13, 601–612 (2012).
Coll, F. et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 7, 51 (2015).
Cohen, K. A., Manson, A. L., Desjardins, C. A., Abeel, T. & Earl, A. M. Deciphering drug resistance in Mycobacterium tuberculosis using whole-genome sequencing: progress, promise, and challenges. Genome Med. 11, 45 (2019).
World Health Organization. Technical Report on Critical Concentrations for TB Drug Susceptibility Testing of Medicines Used in the Treatment of Drug-Resistant TB (World Health Organization, 2018).
Witney, A. A. et al. Clinical application of whole-genome sequencing to inform treatment for multidrug-resistant tuberculosis cases. J. Clin. Microbiol. 53, 1473–1483 (2015).
Frickmann, H., Masanta, W. O. & Zautner, A. E. Emerging rapid resistance testing methods for clinical microbiology laboratories and their potential impact on patient management. Biomed. Res. Int. 2014, 375681 (2014).
Yang, S. & Rothman, R. E. PCR-based diagnostics for infectious diseases: uses, limitations, and future applications in acute-care settings. Lancet Infect. Dis. 4, 337–348 (2004).
Palomino, J. C. & Martin, A. Drug Resistance Mechanisms in Mycobacterium tuberculosis. Antibiotics 3, 317–340 (2014).
Muller, B., Borrell, S., Rose, G. & Gagneux, S. The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis. Trends Genet. 29, 160–169 (2013).
World Health Organization. Global Tuberculosis Report 2022 (World Health Organization, 2022).
Di Gennaro, F. et al. Increase in Tuberculosis Diagnostic Delay during First Wave of the COVID-19 Pandemic: Data from an Italian Infectious Disease Referral Hospital. Antibiotics 10, 272 (2021).
Dookie, N., Rambaran, S., Padayatchi, N., Mahomed, S. & Naidoo, K. Evolution of drug resistance in Mycobacterium tuberculosis: a review on the molecular determinants of resistance and implications for personalized care. J. Antimicrob. Chemother. 73, 1138–1151 (2018).
Rattan, A., Kalia, A. & Ahmad, N. Multidrug-resistant Mycobacterium tuberculosis: molecular perspectives. Emerg. Infect. Dis. 4, 195–209 (1998).
Waller, N. J. E., Cheung, C. Y., Cook, G. M. & McNeil, M. B. The evolution of antibiotic resistance is associated with collateral drug phenotypes in Mycobacterium tuberculosis. Nat. Commun. 14, 1517 (2023).
Earle, S. G. et al. Identifying lineage effects when controlling for population structure improves power in bacterial association studies. Nat. Microbiol. 1, 16041 (2016).
Coll, F. et al. Genome-wide analysis of multi- and extensively drug-resistant Mycobacterium tuberculosis. Nat. Genet. 50, 307–316 (2018).
Farhat, M. R. et al. GWAS for quantitative resistance phenotypes in Mycobacterium tuberculosis reveals resistance genes and regulatory regions. Nat. Commun. 10, 2128 (2019).
Oppong, Y. E. A. et al. Genome-wide analysis of Mycobacterium tuberculosis polymorphisms reveals lineage-specific associations with drug resistance. BMC Genomics 20, 252 (2019).
Huang, H. et al. Cross-sectional Whole-genome Sequencing and Epidemiological Study of Multidrug-resistant Mycobacterium tuberculosis in China. Clin. Infect. Dis. 69, 405–413 (2019).
Farhat, M. R. et al. Genetic Determinants of Drug Resistance in Mycobacterium tuberculosis and Their Diagnostic Value. Am. J. Respir. Crit. Care Med 194, 621–630 (2016).
Walker, T. M. et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect. Dis. 15, 1193–1202 (2015).
Rao, S., Yao, Y. & Bauer, D. E. Editing GWAS: experimental approaches to dissect and exploit disease-associated genetic variation. Genome Med. 13, 41 (2021).
The CRyPTIC Consortium. Genome-wide association studies of global Mycobacterium tuberculosis resistance to 13 antimicrobials in 10,228 genomes identify new resistance mechanisms. PLoS Biol. 20, e3001755 (2022).
WHO. WHO announces updated definitons of extensively drug-resistant tuberculosis (WHO, 2021).
Marigorta, U. M., Rodriguez, J. A., Gibson, G. & Navarro, A. Replicability and Prediction: Lessons and Challenges from GWAS. Trends Genet. 34, 504–517 (2018).
Ishigaki, K. Beyond GWAS: from simple associations to functional insights. Semin. Immunopathol. 44, 3–14 (2022).
Shen, X. & Carlborg, O. Beware of risk for increased false positive rates in genome-wide association studies for phenotypic variability. Front. Genet. 4, 93 (2013).
Dick, T. & Dartois, V. TB drug susceptibility is more than MIC. Nat. Microbiol. 3, 971–972 (2018).
World Health Organization. Catalogue of Mutations in Mycobacterium Tuberculosis Complex and Their Association with Drug Resistance (World Health Organization, 2021).
World Health Organization. Catalogue of Mutations in Mycobacterium Tuberculosis Complex and Their Association with Drug Resistance Second Edition (World Health Organization, 2023).
The CRyPTIC Consortium. A data compendium associating the genomes of 12,289 Mycobacterium tuberculosis isolates with quantitative resistance phenotypes to 13 antibiotics. PLoS Biol 20, e3001721 (2022).
Isakova, J. et al. Mutations of rpoB, katG, inhA and ahp genes in rifampicin and isoniazid-resistant Mycobacterium tuberculosis in Kyrgyz Republic. BMC Microbiol. 18, 22 (2018).
Muthaiah, M. et al. Prevalence of mutations in genes associated with rifampicin and isoniazid resistance in Mycobacterium tuberculosis clinical isolates. J. Clin. Tuberc. Other Mycobact. Dis. 8, 19–25 (2017).
Miotto, P., Zhang, Y., Cirillo, D. M. & Yam, W. C. Drug resistance mechanisms and drug susceptibility testing for tuberculosis. Respirology 23, 1098–1113 (2018).
Morlock, G. P., Metchock, B., Sikes, D., Crawford, J. T. & Cooksey, R. C. ethA, inhA, and katG loci of ethionamide-resistant clinical Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 47, 3799–3805 (2003).
Medina-Ortiz, D., Contreras, S., Quiroz, C. & Olivera-Nappa, A. Development of Supervised Learning Predictive Models for Highly Non-linear Biological, Biomedical, and General Datasets. Front. Mol. Biosci. 7, 13 (2020).
Almeida, J. S. Predictive non-linear modeling of complex data by artificial neural networks. Curr. Opin. Biotechnol. 13, 72–76 (2002).
Yang, Y. et al. DeepAMR for predicting co-occurrent resistance of Mycobacterium tuberculosis. Bioinformatics 35, 3240–3249 (2019).
Ma, B. et al. Diagnostic classification of cancers using extreme gradient boosting algorithm and multi-omics data. Comput. Biol. Med. 121, 103761 (2020).
Moradigaravand, D. et al. Prediction of antibiotic resistance in Escherichia coli from large-scale pan-genome data. PLoS Comput. Biol. 14, e1006258 (2018).
Miotto, P. et al. A standardised method for interpreting the association between mutations and phenotypic drug resistance in Mycobacterium tuberculosis. Eur. Respir. J. 50, 1701354 (2017).
Carolus, H. et al. Genome-Wide Analysis of Experimentally Evolved Candida auris Reveals Multiple Novel Mechanisms of Multidrug Resistance. mBio 12, e03333-20 (2021).
Van den Bergh, B. et al. Frequency of antibiotic application drives rapid evolutionary adaptation of Escherichia coli persistence. Nat. Microbiol. 1, 16020 (2016).
Liu, X. et al. Identification of Novel Loci and Candidate Genes for Cucumber Downy Mildew Resistance Using GWAS. Plants 9, 1659 (2020).
Gowda, M. et al. Genome-wide association and genomic prediction of resistance to maize lethal necrosis disease in tropical maize germplasm. Theor. Appl. Genet. 128, 1957–1968 (2015).
Safi, H. et al. Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-beta-D-arabinose biosynthetic and utilization pathway genes. Nat. Genet. 45, 1190–1197 (2013).
Lin, Y. et al. Comparative performance of microbiological methods for the detection of tuberculous meningitis pathogens in cerebrospinal fluid. Diagn. Microbiol. Infect. Dis. 107, 116025 (2023).
Kaler, A. S. & Purcell, L. C. Estimation of a significance threshold for genome-wide association studies. BMC Genomics 20, 618 (2019).
Wheeler, N. E., et al. Contrasting approaches to genome-wide association studies impact the detection of resistance mechanisms in Staphylococcus aureus. bioRxiv, https://www.biorxiv.org/content/10.1101/758144v1 (2019).
Lee, J. et al. Sensititre MYCOTB MIC plate for testing Mycobacterium tuberculosis susceptibility to first- and second-line drugs. Antimicrob. Agents Chemother. 58, 11–18 (2014).
Somerville, W., Thibert, L., Schwartzman, K. & Behr, M. A. Extraction of Mycobacterium tuberculosis DNA: a question of containment. J. Clin. Microbiol. 43, 2996–2997 (2005).
Lippert, C. et al. FaST linear mixed models for genome-wide association studies. Nat. Methods 8, 833–835 (2011).
Saliba, J. Enhanced diagnosis of multi-drug-resistant microbes using group association modeling and machine learning. Figshare, https://doi.org/10.6084/m9.figshare.28191398 (2025).
Saliba, J. Enhanced diagnosis of multi-drug-resistant microbes using group association modeling and machine learning. Zenodo, https://doi.org/10.5281/zenodo.14975407 (2025).
Acknowledgements
The work was primarily supported by research funding provided by the National Institutes of Health U01CA252965 (T.Y.H.), R01AI144168 (T.Y.H.), R01AI175618 (T.Y.H.), R01HD090927 (T.Y.H.), R01HD103511 (T.Y.H.), R21NS130542 (T.Y.H.), the United States Department of Defense W81XWH1910026 (T.Y.H.), and Lavin-Bernick Award of Tulane University (W.Z.). We thank Dr. Robin Forman for his inspiring discussion on this project.
Author information
Authors and Affiliations
Contributions
J.G.S. and Q.S. conceptualized the study. J.G.S. performed methodology, validation, and investigation, and wrote the original draft. W.Z. contributed to investigation and writing. L.L., C.W., J.Q., Y.X., B.Y. and H.H. conducted clinical data analysis. C.J.L. contributed to investigation and writing. T.Y.H. provided funding acquisition and investigation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Saliba, J.G., Zheng, W., Shu, Q. et al. Enhanced diagnosis of multi-drug-resistant microbes using group association modeling and machine learning. Nat Commun 16, 2933 (2025). https://doi.org/10.1038/s41467-025-58214-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58214-6
