
Abstract
Heart injury has been characterized by the irreversible loss of cardiomyocytes comprising the contractile tissues of the heart and thus strategies enabling adult cardiomyocyte proliferation are highly desired for treating various heart diseases. Here, we test the ability of human induced pluripotent stem cell-derived primitive macrophages (hiPMs) and their conditioned medium (hiPM-cm) to promote human cardiomyocyte proliferation and enhance cardiac regeneration in adult mice. We find that hiPMs promote human cardiomyocyte proliferation, which is recapitulated by hiPM-cm through the activation of multiple pro-proliferative pathways, and a secreted proteome analysis identifies five proteins participating in this activation. Subsequent in vivo experiments show that hiPM-cm promotes adult cardiomyocyte proliferation in mice. Lastly, hiPM-cm enhances cardiac regeneration and improves contractile function in injured adult mouse hearts. Together, our study demonstrates the efficacy of using hiPM-cm in promoting adult cardiomyocyte proliferation and cardiac regeneration to serve as an innovative treatment for heart disease.
Similar content being viewed by others

Introduction
Heart diseases, characterized by their high incidence and leading cause of death, pose a significant challenge to global health1. These conditions often share a common pathology regarding the loss of contractile myocardial tissue, which has been considered irreversible due to the inability of adult cardiomyocytes to regenerate, leading to the progression of heart failure and other debilitating complications2,3. The field has been cognizant of the limitations inherent in current treatments and therapies. For instance, pharmacological interventions such as beta-blockers and ACE inhibitors4,5,6,7, while effective in managing cardiac symptoms, do not address the fundamental issue of cardiomyocyte loss. Similarly, mechanical assist devices and surgical interventions, although life-saving in some cases8,9, are palliative rather than regenerative. These treatments are recognized as inadequate in compensating for the loss of contractile myocardial tissue that occurs following injury. Accordingly, there has been a surge of research efforts directed towards achieving a ‘regenerative’ goal in adult hearts.
Much of the inspiration for the regenerative approach comes from neonatal rodent hearts, with an inherent capacity of self-renewal during the initial stages of life10. In this context, macrophages have been identified as key contributors to the heart’s regenerative potential. The clearance of macrophages in neonatal mice impedes the heart’s regenerative capacity11, while transplantation of macrophages derived from injured neonatal mouse hearts stimulates cardiomyocyte proliferation in infarcted adult mouse hearts12. In more detail, cardiac macrophages contain two subpopulations based on their divergent origins, namely monocyte-derived macrophages and embryonic-derived cardiac-resident macrophages13,14,15,16. Between them, monocyte-derived macrophages originating from bone marrow hematopoietic progenitors lack reparative activities, whereas embryonic-derived cardiac-resident macrophages originating from primitive macrophages, as well as their conditioned medium can stimulate neonatal rat cardiomyocyte proliferation in vitro17. Thus, we hypothesize that primitive macrophages hold the key to unlocking the heart’s innate regenerative potential.
Although primitive macrophages hold promise in the context of heart regeneration, both biologically for their inherent properties and therapeutically for their potential to repair damaged myocardium18,19, they are difficult to obtain in sufficient quantities for clinical applications. We realized that having a reliable source of these cells would enable for exploring the use of primitive macrophages (and related materials) as interventions for cardiac diseases and ideally, to induce regeneration in adult hearts. Accordingly, in this study, we generated primitive macrophages (hiPMs) from human induced pluripotent stem cells (hiPSCs) by optimizing a cocktail of cytokines and growth factors for directed differentiation and discovered that these cells promoted human cardiomyocyte proliferation. Further, we found that hiPM conditioned medium (hiPM-cm) recapitulated the pro-proliferative ability of hiPMs on human cardiomyocytes. Proteomics analysis of hiPM-cm revealed protein components partially mediated the proliferative effects via the activation of multiple pro-proliferative signaling pathways. Subsequently, we found that hiPM-cm promoted cardiomyocyte proliferation in non-injured mouse hearts via the activation of the aforementioned pro-proliferative signaling pathways. Lastly, we found that hiPM-cm resulted in enhanced cardiomyocyte proliferation and improved cardiac function in infarcted mouse hearts, thus demonstrating the efficacy of hiPM-cm in promoting adult cardiomyocyte proliferation and cardiac regeneration.
Results
Generation of hiPSC-derived primitive macrophages
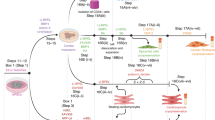
To evaluate the potential of primitive macrophages to reignite human cardiomyocyte proliferation, we first generated hiPMs from hiPSCs by optimizing a cocktail of cytokines and growth factors for directing the differentiation towards mesoderm and the specification of primitive hematopoietic progenitors. This optimization facilitates the efficient differentiation of hiPSCs into primitive macrophages, mirroring the developmental process of the generation of these cells within the yolk sac (Fig. 1a). Accordingly, hiPSC-derived embryoid bodies (EBs) were subjected to BMP4, bFGF, and Activin signaling activation for mesoderm formation followed by BMP4, bFGF, and VEGF signaling activation for hemogenic endothelium (HE) specification to generate CD235+KDR+ cells (Fig. 1b). Subsequently, CD45+ myeloid progenitors were generated upon the induction with M-CSF and IL-3 (Fig. 1c) and gave rise to hiPM precursors (hiPMpres). Eventually, hiPMpres were matured to hiPMs positive for CD11b and CD14 upon the induction with M-CSF and bFGF (Fig. 1d, e).

a Schematic diagram of the protocols to generate hiPMs from human iPSCs. Relevant growth factors and duration of differentiation steps are as indicated. Cartoon elements used in the schematic diagram were created using BioRender. b Flow cytometry analysis of cells double positive for KDR and CD235 during hemogenic epithelium (HE) specification at day 5 after the start of hiPM differentiation. c Flow cytometry analysis of cells positive for CD45 during myeloid progenitor generation at day 11 after the start of hiPM differentiation. d Flow cytometry analysis of hiPMs positive for CD11b and CD14 at day 32 after the start of hiPM differentiation. e Immunofluorescence images for CD11b and CD14 expression in hiPMs. iPSCs were used as a negative control. Cells were stained with CD11b (red) and CD14 (green); nuclei were counterstained with Hoechst (blue). Scale bar, 10 µm. The experiment was repeated three times independently with similar results. f Phagocytosis assessment of hiPMs with FITC-conjugated Dextran (green). Human iPSC-derived cardiac fibroblasts (hiCFs) were used as a negative control. Scale bar, 50 μm. g Flow cytometry analysis of CCR2 expression in hiPMs. THP-1Ms were used as a positive control. The experiment was repeated three times independently with similar results. h Principal component analysis of RNA sequencing data revealing in-group clusters with minimal overlap between hiPMs and THP-1Ms. n = 4 biological repeats per group. i Barplot of differentially expressed genes (hiPMs vs THP-1Ms) specific to primitive macrophages. Differentially expressed genes were identified with a cutoff of |Fold change | > 2 and adjusted p-value < 0.05. j Measurement of LYVE1, FOLR2, CD163, and MAF mRNA levels in hiPMs and THP-1Ms via qPCR analysis. β-actin was used as a control. n = 3 biological repeats per group. k Representative immunofluorescence images for LYVE1 expression in hiPMs. THP-1Ms were used as a negative control. Cells were stained with LYVE1 (green); nuclei were counterstained with Hoechst (blue). Scale bar, 20 μm. Quantitative data are presented as the mean ± SEM. Groups were compared using a two-tailed unpaired Student’s t test (j) or ANOVA (i). Source data are provided as a Source Data file.
The functionality of hiPMs was first shown by the engulfment of FITC-labeled dextran, as compared to cardiac fibroblasts (hiCFs) derived from hiPSCs (Fig. 1f), demonstrating their phagocytotic ability as macrophages. Next, we performed a comparative analysis of the expression of CCR2, a well-established marker for differentiating monocyte-derived macrophages, between hiPMs and human macrophages derived from monocytes collected from human peripheral blood (THP-1Ms) that were double positive for CD11b and CD1420 (Supplementary Fig. 1a, b). While THP-1M cells exhibited strong CCR2 expression, the expression in hiPMs was significantly lower (Fig. 1g).
Upon conducting an RNA sequencing (RNA-seq) analysis to compare hiPMs and THP-1Ms, we observed a distinctive gene expression profile in hiPMs from THP-1Ms (Fig. 1h). Notably, out of 25 previously identified genes specific to primitive macrophages15,21, 20 were significantly upregulated in hiPMs compared to the THP-1Ms (Fig. 1i). To examine the specificity and homogeneity of hiPMs, we performed qPCR analysis and immunostaining for LYVE1, a marker specifically expressed by primitive macrophages15,21. Both methods revealed the high expression of LYVE1 in hiPMs over THP-1Ms (Fig. 1j, k), reinforcing the identity of hiPMs. Likewise, qPCR analysis of a few other markers expressed by primitive macrophages, including FOLR2, CD163, and MAF15,21, revealed high expression only in hiPMs (Fig. 1j). These findings establish that hiPMs can be generated from hiPSCs.
HiPMs and their conditioned medium promote human cardiomyocyte proliferation
Previous studies have reported that cardiac macrophages are implicated in the regeneration of neonatal mouse heart11. To test whether hiPMs promote the proliferation of human cardiomyocytes, we used both 2D and 3D culture systems to co-culture hiPMs and human cardiomyocytes derived from hiPSCs (positive for α-actinin and cardiac troponin T (cTnT))22 (Fig. 2a and Supplementary Fig. 2a, b). Co-culture with hiPMs promoted human cardiomyocyte proliferation in the 2D system, evidenced by a significantly higher percentage of Ki67-positive cardiomyocytes compared to the control group without hiPMs (Fig. 2b, c). Conversely, a significant reduction was observed in the proportion of Ki67-positive cardiomyocytes when they were co-cultured with THP-1Ms (Supplementary Fig. 2c, d). Similarly, when human cardiomyocytes were substituted with hiCFs, co-culture with hiPMs did not elicit a significant promotion in their proliferation (Supplementary Fig. 2e). These results support that co-culture with hiPMs promotes the proliferation of human cardiomyocytes.

a Schematic diagram depicting the co-culture of human cardiomyocytes and hiPMs in 2D and 3D culture systems. Cartoon elements used in the schematic diagram were created using BioRender. b Images showing co-culturing of human cardiomyocytes and hiPMs by brightfield microscopy and immunofluorescence microscopy. Human cardiomyocytes co-cultured with hiPMs (+hiPM) were compared with human cardiomyocytes alone (-hiPM). For immunofluorescence images, human cardiomyocytes were labeled with cardiac Troponin T (cTnT) (red), hiPMs were labeled with CD14 (green), and all the nuclei were counterstained with Hoechst (blue). Scale bars, 100 μm for brightfield images and 10 μm for immunofluorescence images. The experiment was repeated three times independently with similar results. c Representative immunofluorescence images and quantification of Ki67-positive human cardiomyocytes co-cultured with hiPMs in a 2D culture system. Human cardiomyocytes were labeled with cTnT (red) and Ki67 (green); cardiomyocyte nuclei were counterstained with Hoechst (blue). Scale bar, 10 μm. n = 3 biological repeats per group. d Brightfield images showing the co-culturing of human cardiomyocytes and hiPMs in 3D culture system. Human cardiomyocytes co-cultured with hiPMs (+hiPM) were compared with human cardiomyocytes alone (-hiPM) at 10 days after the start of 3D culturing in Ultralow attachment 96 U-well plates. Scale bar, 100 μm. The experiment was repeated three times independently with similar results. e Representative immunofluorescence images and quantification of Ki67-positive human cardiomyocytes co-cultured with hiPMs in the 3D culture system. Human cardiomyocytes were labeled with cTnT (red) and Ki67 (green); cardiomyocyte nuclei were counterstained with Hoechst (blue). Scale bar, 15 μm. n = 3 biological repeats per group. Quantitative data are presented as the mean ± SEM. Groups were compared using a two-tailed unpaired Student’s t test (c and e). Source data are provided as a Source Data file.
For the 3D system, we again observed a significantly increased proliferation of human cardiomyocytes upon co-culture with hiPMs (Fig. 2d, e). Notably, most of the Ki67-positive human cardiomyocytes were not in direct contact with hiPMs (Supplementary Fig. 2f), suggesting that the stimulatory effects of hiPMs on cardiomyocyte proliferation were not solely mediated by cell-to-cell contact. These findings from the 2D and 3D co-culture experiments support that hiPMs promote cardiomyocyte proliferation, apparently through the secretion of bioactive molecules.
We subsequently examined whether the conditioned medium from hiPM cultures (hiPM-cm) can promote human cardiomyocyte proliferation (Fig. 3a). Specifically, we performed daily cell counts of cardiomyocytes for a period of four days following the initiation of hiPM-cm treatment. Our results demonstrated a significant increase in the number of cardiomyocytes over time (Fig. 3b), providing direct evidence of the proliferative effect of hiPM-cm on these cells. Likewise, we used an IncuCyte instrument to monitor the proliferation of human cardiomyocytes cultured in either hiPM-cm or fresh B27-supplemented RPMI medium (FM) across a 4-day period. While the FM-treated samples maintained a relatively constant cell confluence, a notable escalation in cell confluence was detected in hiPM-cm-treated samples over time (Fig. 3c), indicative of the proliferative effect of hiPM-cm on cardiomyocytes. Immunostaining analysis also revealed that a significantly elevated percentage of cardiomyocytes positive for Ki67 was detected in the hiPM-cm group than those in the FM group, with comparable percentages of cTnT-positive cardiomyocytes between the two groups (Fig. 3d and Supplementary Fig. 3a). Likewise, a significantly larger size of 3D spheres, along with a significantly higher percentage of Ki67-positive cardiomyocytes, was observed in the hiPM-cm group as compared to those in the FM group, both starting with the same numbers of human cardiomyocytes (Fig. 3e, f).

a Schematic diagram depicting the incubation of human cardiomyocytes with fresh culture medium (FM group) or conditioned medium collected from hiPMs (hiPM group). Cartoon elements used in the schematic diagram were created using BioRender. b Daily cell count in human cardiomyocytes treated with FM or hiPM-cm for 4 days. n = 4 per group. c Cardiomyocyte proliferation monitored by IncuCyte instrument at a 12 h interval for 4 days. n = 4 per group. Scale bar, 50 μm. d Immunofluorescence staining of Ki67-positive human cardiomyocytes treated with FM and hiPM-cm in 2D culture system. Human cardiomyocytes were labeled with cTnT (red) and Ki67 (green); nuclei were counterstained with Hoechst (blue). Scale bar, 10 μm. n = 3 biological repeats per group. e Cross-sectional areas of cardiac spheres composed of human cardiomyocytes treated with FM and hiPM-cm at 10 days after the start of 3D culturing. Scale bar, 100 μm. n = 8 spheres for FM group, n = 7 spheres for hiPM-cm group. f Immunofluorescence staining of Ki67-positive human cardiomyocytes treated with FM and hiPM-cm in the 3D culture system. Human cardiomyocytes were labeled with cTnT (red) and Ki67 (green); nuclei were counterstained with Hoechst (blue). Scale bar, 15 μm. n = 3 biological repeats per group. g Immunofluorescence staining of αSMA-positive human cardiomyocytes treated with FM and hiPM-cm. Human cardiomyocytes were labeled with cTnT (red) and αSMA (green); nuclei were counterstained with Hoechst (blue). Scale bar, 10 μm. n = 3 biological repeats per group. h Extracellular acidification rate (ECAR) analysis in human cardiomyocytes treated with FM and hiPM-cm. n = 4 biological repeats per group. i Oxygen consumption rate (OCR) analysis in human cardiomyocytes treated with FM and hiPM-cm. n = 3 biological repeats per group. j ATP levels in human cardiomyocytes treated with FM and hiPM-cm. n = 6 biological repeats per group. k Calcium handling analysis in human cardiomyocytes treated with FM and hiPM-cm. n = 12 cardiomyocytes per group. Quantitative data are presented as the mean ± SEM. Groups were compared using a two-tailed unpaired Student’s t test (b–k). Source data are provided as a Source Data file.
Immunofluorescence staining revealed a significantly increased percentage of cardiomyocytes that were positive for αSMA, a marker for cardiomyocyte dedifferentiation23,24, in human cardiomyocytes in the hiPM-cm group versus those in the FM group (Fig. 3g). Consistent with active proliferation25,26,27, we found that the human cardiomyocytes in the hiPM-cm group had increased glycolysis, decreased oxidative phosphorylation and ATP production, along with remodeled calcium handling, when compared to human cardiomyocytes in the FM group (Fig. 3h–k and Supplementary Fig. 3b). These results collectively support that hiPM-cm promotes the proliferation of human cardiomyocytes.
Protein components in hiPM-cm partially mediated the proliferative effects through the activation of multiple pro-proliferative signaling pathways
To identify the components underlying the pro-proliferative effects of hiPM-cm, we fractionated hiPM-cm and found that only the recovered fractions with relatively large-molecule weight molecules retained by 3-kDa filters were able to promote cardiomyocyte proliferation whereas the supernatants through 3-kDa filters were not (Fig. 4a). Subsequently, we focused on the protein components of hiPM-cm, which were mostly large-molecule weight molecules. As the conditioned medium from hiPMpres (hiPMpre-cm), the precursors of hiPMs, were unable to promote cardiomyocyte proliferation, they were used as a negative control group in liquid chromatography-mass spectrometry (LC-MS) analysis (Fig. 4b and Supplementary Fig. 4a).

a Immunofluorescence staining of Ki67-positive human cardiomyocytes treated with FM, hiPM-cm, hiPM-cm components retained by or passing through 3-kDa filters. Human cardiomyocytes were labeled with cTnT (red) and Ki67 (green); nuclei were counterstained with Hoechst (blue). Scale bar, 10 μm. n = 3 biological repeats per group. b Experimental strategy for analyzing protein components from hiPMpre-cm or hiPM-cm collected in RPMI medium by LC/MS. Cartoon elements used in the schematic diagram were created using BioRender. c Venn diagram showing the overlap between upregulated proteins identified in hiPM-cm versus hiPMpre-cm by LC/MS and upregulated genes encoding secreted proteins in CCR2- human cardiac tissue-resident macrophages versus CCR2+ human monocyte-derived macrophages in the heart. d Unsupervised clustering analysis of relative pro-proliferative potentials of pairwise protein combinations evaluated by the percentages of Ki67-positive human cardiomyocytes. e Immunofluorescence staining of Ki67-positive human cardiomyocytes treated with a five-protein combination including C1QB, NRP1, PLTP, FUCA1, and SERPING1 (5-P combo). Scale bar, 10 μm. n = 3 biological repeats per group. f Principal component analysis of RNA sequencing data from human cardiomyocytes treated with FM and hiPM-cm. n = 3 biological repeats per group. g Volcano plot illustrating DEGs between human cardiomyocytes treated with FM and hiPM-cm. h Representative KEGG terms of cell proliferation-related genes upregulated in cardiomyocytes incubated with hiPM-cm versus FM. i Immunoblotting analysis showing the phosphorylated and total protein levels of AKT, ERK, and STAT3 in human cardiomyocytes treated with FM and hiPM-cm. GAPDH was used as a loading control. n = 3 biological repeats per group for phosphorylated and total ERK; n = 6 biological repeats per group for the other proteins. j Quantification of Ki67-positive human cardiomyocytes treated with hiPM-cm and inhibitors for PI3K-AKT (A-674563), ERK (PD0325901), and JAK-STAT (Stattic) pathways in single or in combination. n = 3 biological repeats per group. Quantitative data are presented as the mean ± SEM. Groups were compared using a two-tailed unpaired Student’s t test (i) or one-way ANOVA followed by post hoc Tukey test (a, e, and j). Source data are provided as a Source Data file.
We identified 220 secreted proteins significantly enriched in hiPM-cm versus hiPMpre-cm that were both collected in RPMI-1640 medium (Supplementary Data 1), 9 (SERPING1, TF, NRP2, C1QC, GAS6, C1QB, FUCA1, NRP1, and PLTP) out of which have been found upregulated in CCR2- human cardiac-resident macrophages versus CCR2+ human monocyte-derived macrophages in the heart15 (Fig. 4c and Supplementary Data 2). After treating human cardiomyocytes with each of these 9 candidate proteins individually (or with FM as a control), an immunostaining analysis showed that none of them resulted in a significantly increased percentage of Ki67-positive cardiomyocytes (Supplementary Fig. 4b). These results suggest that no single protein component in hiPM-cm was sufficient to induce the observed promotion of cardiomyocyte proliferation as hiPM-cm did. Further, when we examined the combinatory pro-proliferative effects of any two given proteins, we found several combinations involving C1QB, NRP1, PLTP, FUCA1, and SERPING1 resulted in a significantly increased percentage of Ki67-positive cardiomyocytes (Fig. 4d). Indeed, treatment with the five-protein combination (5-P combo) resulted in a significantly higher percentage of Ki67-positive human cardiomyocytes compared to those treated with PBS. However, this increase was not as pronounced as that observed with hiPM-cm treatment (Fig. 4e). These results suggest that this five-protein combination can partially recapitulate the pro-proliferative effects induced by hiPM-cm. Moreover, to pinpoint potential receptors that these five proteins might engage to stimulate cardiomyocyte proliferation, we employed NicheNet (version 2.2.0), a bioinformatic tool for predicting ligand-receptor interactions from RNA sequencing data, to compile a list of candidate receptors for these five proteins (Supplementary Data 3). We then proceeded to validate these candidates by using siRNAs to assess their impact on cardiomyocyte proliferation and found that the siRNA-mediated knockdown of individual candidate receptors did not markedly affect the proliferative response to the five-protein combination (Supplementary Fig. 4c). Next, we focused on the protein pair NRP1 and C1QB, which exhibited the most significant pro-proliferative effects among all the aforementioned tested protein pairs. We found that the synergistic proliferative effect of NRP1 and C1QB could be reduced by about 80% through the concurrent use of siFLT1 and siLRP1 (Supplementary Fig. 4d), suggesting that the activities of NRP1 and C1QB are predominantly mediated through their respective receptors, FLT1 and LRP1.
Next, we used a transcriptomics approach to identify potential pro-proliferative signaling pathways that contributed to the observed cardiomyocyte proliferation induced by hiPM-cm (Fig. 4f). RNA sequencing analysis identified 214 downregulated genes and 407 upregulated genes in human cardiomyocytes exposed to hiPM-cm when compared to those exposed to FM (Fig. 4g and Supplementary Data 4). KEGG analysis of the differentially expressed proliferation-related genes indicated that the hiPM-cm group had a signature showing the enrichment of genes related to PI3K/AKT, JAK/STAT, and MAPK pathways28,29,30 in the hiPM-cm group (Fig. 4h), aligning with preceding research that have implicated multiple proteins from the aforementioned five-protein combination in the regulation of (cancer) cell proliferation, with their proliferative impacts often mediated through the PI3K/AKT, ERK, and JAK/STAT pathways31,32,33,34,35,36. Accordingly, we conducted immunoblotting and inhibitors studies to investigate whether these signaling pathways contributed to the observed cardiomyocyte proliferation induced by hiPM-cm. Immunoblotting with antibodies to measure the phosphorylation status of pathway component proteins28,29,30 revealed significant increases in human cardiomyocytes incubated with hiPM-cm compared to those incubated with FM (Fig. 4i), supporting that hiPM-cm induces the activation of the PI3K/AKT, JAK/STAT, and MAPK pathways in human cardiomyocytes. In addition, treatment of hiPM-cm with individual inhibitors targeting the PI3K/AKT, JAK/STAT, and MAPK pathways37,38,39 resulted in significant decreases in the extent of cardiomyocyte proliferation. Notably, treatment with all three inhibitors simultaneously led to the most pronounced reduction in proliferation (Fig. 4j). Thus, beyond demonstrating that a combination of five proteins partially recapitulates hiPM-cm’s capacity to induce the proliferation of human cardiomyocytes, our findings have revealed PI3K/AKT, JAK/STAT, and MAPK pathways that mediate these effects.
HiPM-cm promotes cardiomyocyte proliferation in adult mouse hearts
We next investigated whether hiPM-cm promotes cardiomyocyte proliferation in vivo in C57BL/6J mice that received a single intramyocardial injection of PBS, FM, or hiPM-cm at 8 weeks old (Fig. 5a). A significant increase in heart weight to tibial length ratio was observed at one week after hiPM-cm injection, but not FM injection (Supplementary Fig. 5a). Immunostaining for proliferation markers (Ki67, pHH3, and Aurora B) showed significantly increased percentages of cardiomyocytes positive for these markers at one week after hiPM-cm injection; no increases were detected in those that received FM injection (Fig. 5b–d). In contrast, hiPM-cm injection did not promote the proliferation of cardiac fibroblasts (Supplementary Fig. 5b). In addition, we conducted experiments using a previously reported genetic model known as MADM (mosaic analysis with double markers), in which non-dividing cardiomyocytes are labeled with dual fluorescence markers whereas cardiomyocytes labeled with either red or green fluorescence alone indicate bona fide cardiomyocyte division40. Briefly, a single intramyocardial injection of hiPM-cm, but not of PBS or FM, led to a significant increase in the number of single-labeled cardiomyocytes of MADM adult mouse hearts (Fig. 5e), supporting that hiPM-cm induces cardiomyocyte proliferation in adult hearts. Further, in MADM mice that underwent a single intramyocardial injection of either PBS or the five-protein combination—comprising C1QB, NRP1, PLTP, FUCA1, and SERPING1 that partially recapitulated the pro-proliferative effects on human cardiomyocytes by hiPM-cm in vitro—we observed that the five-protein combination significantly enhanced adult cardiomyocyte proliferation at one week after injection (Supplementary Fig. 5c), suggesting that the five-protein combination partially recapitulates the pro-proliferative effects of hiPM-cm within the adult heart.

a Schematic diagram depicting the experiment to evaluate the pro-proliferative potential of hiPM-cm on cardiomyocytes in adult mouse hearts intramyocardially injected with PBS (PBS group), FM (FM group), and hiPM-cm (hiPM-cm group). Cartoon elements used in the schematic diagram were created using BioRender. b–d Representative immunofluorescence images and quantification of the percentages of cardiomyocytes positive for Ki67 (b), pHH3 (c), and Aurora B (d) one week after the injection. Ki67/pHH3/Aurora B (green), PCM1 (red), and Hoechst (blue). Scale bar, 20 μm. n = 5 mice per group. e Representative immunofluorescence images and quantification of single-colored (red and green) cardiomyocytes using MADM mice one week after the injection. Scale bar, 50 μm. n = 9 mice per group. f–h Representative immunofluorescence images and quantification of the percentages of cardiomyocytes positive for phosphorylated AKT (f), ERK (g), and STAT3 (h) three days after the injection. pAKT/pERK/pSTAT3 (green), PCM1 (red), and Hoechst (blue). Scale bar, 20 μm. n = 5 mice per group. i Representative immunofluorescence images and quantification of Ki67-positive human cardiomyocytes treated with hiPM-cm and three inhibitors in combination (hiPM-cm + 3i). Inhibitors used were AKT inhibitor A-674563, ERK inhibitor PD0325901, and STAT3 inhibitor Stattic. Scale bar, 20 μm. Ki67 (green), PCM1 (red), and Hoechst (blue). White arrows denote Ki67-positive cells and yellow arrows denote Ki67-negative cells. n = 5 mice per group. Quantitative data are presented as the mean ± SEM. Groups were compared using a two-tailed unpaired Student’s t test (e and i) or one-way ANOVA followed by post hoc Tukey test (b–d and f–h). Source data are provided as a Source Data file.
We next determined whether hiPM-cm induces adult cardiomyocyte proliferation in vivo through the PI3K/AKT, JAK/STAT, and MAPK pathways, as observed in vitro with human cardiomyocytes. We conducted immunostaining for the pro-proliferative phosphorylation signals associated with these pathways and found that all of the three pathways were activated in cardiomyocytes three days after hiPM-cm injection, a response not seen with FM injection (Fig. 5f–h). Further, we conducted experiments that combined the intramyocardial injection of hiPM-cm with intraperitoneal administration of inhibitors for the PI3K/AKT, JAK/STAT, and MAPK pathways (as 4 doses across the first 4 days). Immunostaining of hearts sampled at one week showed that the combinatory use of these inhibitors led to a significant reduction in the extent of cardiomyocyte proliferation induced by hiPM-cm (Fig. 5i). Taken together, these in vivo results establish that hiPM-cm induces cardiomyocyte proliferation in adult hearts and specifically implicate the PI3K/AKT, JAK/STAT, and MAPK pathways in this induction.
HiPM-cm enhances cardiac regeneration in mouse hearts after I/R
To explore the potential therapeutic application of hiPM-cm in promoting cardiac regeneration upon injury, 8-week-old C57BL/6J mice were subjected to Sham surgery (Sham group) or myocardial ischemia/reperfusion injury (I/R) followed by a single intramyocardial injection of FM (I/R + FM group) or hiPM-cm (I/R + hiPM-cm group) at 5 min before the reperfusion (Fig. 6a). Consistently, serum cTnT, an established biomarker of cardiac injury, significantly increased in I/R + FM group and it was partially reversed in the I/R + hiPM-cm group at three days after the surgery (Fig. 6b). At one week after the surgery, a significant hypertrophic response in the heart of mice from I/R + FM group was observed when compared to the sham group as reflected by a significant increase in heart weight to tibial length ratio, which was attenuated in mice from I/R + hiPM-cm group (Fig. 6c). Further, Masson’s trichrome staining revealed that the I/R + hiPM-cm group had significantly less fibrotic tissues than I/R + FM group did at one week after the surgery (Fig. 6d), indicative of a protective effect exerted by the hiPM-cm against I/R.

a Schematic diagram depicting the experiment to evaluate the pro-proliferative potential of hiPM-cm on cardiomyocytes in adult mouse hearts that received sham surgery (Sham group), or I/R surgery followed by intramyocardial injection with FM (I/R + FM group) and hiPM-cm (I/R + hiPM-cm group) at five minutes before reperfusion. Cartoon elements used in the schematic diagram were created using BioRender. b Measurement of serum cTnT levels by ELISA at three days after the surgery. n = 8 mice per group. c Ratios of heart weight to tibial length (HW/TL) at one week after the surgery. n = 8 mice per group. d Representative images and quantification of Masson’s trichrome staining of heart sections from Sham, I/R + FM, and I/R + hiPM-cm groups at one week after the surgery. Scale bar, 50 μm. n = 5 mice per group. e–g Representative immunofluorescence images and quantification of the percentages of cardiomyocytes positive for Ki67 (e), pHH3 (f), and Aurora B (g) one week after the surgery. Scale bar, 20 μm. n = 4 mice per group for Ki67 staining and Aurora B staining; n = 5 mice per group for pHH3 staining. Ki67/pHH3/Aurora B (green), PCM1 (red), and Hoechst (blue). h Representative images of wheat germ agglutinin (WGA) staining and quantification of cardiomyocyte cross-sectional areas one week after the surgery. Scale bar, 10 μm. n = 5 mice per group. Quantitative data are presented as the mean ± SEM. Groups were compared using one-way ANOVA followed by post hoc Tukey test (b-h). Source data are provided as a Source Data file.
Given that hiPM-cm was collected from induced primitive macrophages, we wondered whether the cardioprotective effects may be a result of immunomodulation. We pursued this possibility by performing a flow cytometry analysis of the immune cells isolated from mouse hearts at three days after I/R (Supplementary Fig. 6a, b). While the proportions of CD11b+ macrophages (CD45+CD11b+F4/80+), neutrophils (CD45+CD11b+Ly6Ghigh), T lymphocytes (CD45+CD11b-TCRβ+B220-) and B lymphocytes (CD45+CD11b-TCRβ-B220+) significantly increased after I/R, no changes were detected between the two I/R groups treated with either FM or hiPM-cm (Supplementary Fig. 6c), suggesting that hiPM-cm confers cardioprotection against I/R independent of immunomodulation.
To investigate whether hiPM-cm improves cardiac regeneration specifically by promoting cardiomyocyte proliferation in adult mouse hearts, we evaluated cardiomyocyte proliferation in mice from Sham, I/R + FM, and I/R + hiPM-cm groups at one week after I/R. Immunostaining showed that only hiPM-cm increased the percentages of cardiomyocytes positive for Ki67, pHH3, and Aurora B (Fig. 6e–g), each indicative of enhanced adult cardiomyocyte proliferation by hiPM-cm. Consistently, wheat germ agglutinin (WGA) staining revealed significantly smaller cardiomyocyte cross-sectional areas in the hearts of the I/R + hiPM-cm group when compared to those from the I/R + FM group (Fig. 6h). By contrast, triphenyltetrazolium chloride (TTC) staining that the extent of cardiac injury was comparable between the I/R + FM and I/R + hiPM-cm groups at 1 day after I/R (Supplementary Fig. 7a), suggesting that the beneficial effects of hiPM-cm were not immediately linked to a reduction in reperfusion injury during the acute phase of I/R. However, a significant decrease in cardiac injury was observed in the I/R + hiPM-cm group relative to the I/R + FM group at both 3 and 7 days after I/R (Supplementary Fig. 7a), likely attributable to cardiomyocyte proliferation at later stages after I/R. Together, these results indicate that hiPM-cm promotes adult cardiomyocyte proliferation, contributing to improved heart regeneration after I/R.
HiPM-cm improves cardiac function in mouse hearts after I/R
To evaluate whether hiPM-cm can improve heart function after I/R, we performed serial echocardiography on mice from Sham, I/R + FM, and I/R + hiPM-cm groups one week after the surgery (Fig. 7a). The ultrasound scans (H.Z.) and their interpretations (J.B.) were performed by investigators who were blinded to the treatment to prevent bias. Compared with the values of the Sham group, the I/R + FM group displayed decreased left ventricular ejection fraction (LVEF) and fractional shortening (LVFS) at one week after I/R, indicating a progression toward heart failure. By contrast, the I/R + hiPM-cm group showed a significant improvement in LVEF and LVFS as compared to the I/R + FM group (Fig. 7b). Consistently, compared with the values of the Sham group, the I/R + FM group displayed significantly increased left ventricular end-diastolic dimension (LVEDD) and end-systolic dimension (LVESD) at one week after I/R. By comparison, the I/R + hiPM-cm group showed a significant attenuation of the increases in LVEDD and LVESD as compared to the I/R + FM group (Fig. 7b). Taken together, these data show that hiPM-cm improves cardiac function in mouse hearts at one week after I/R.

a Schematic diagram depicting the experiment to evaluate cardiac function in adult mouse from the Sham group, I/R + FM group, and I/R + hiPM-cm group. Cartoon elements used in the schematic diagram were created using BioRender. b, c Representative echocardiographic images and quantitative data of left ventricular ejection fraction (LVEF), fractional shortening (LVFS), end-diastolic dimension (LVEDD), and end-systolic dimension (LVESD) at one week (b) and 12 weeks (c) after the surgery. n = 8 mice per group. d Representative images and quantification of Masson’s trichrome staining of heart sections from Sham, I/R + FM, and I/R + hiPM-cm groups at 12 weeks after the surgery. Scale bar, 2 mm. n = 8 mice per group. Quantitative data are presented as the mean ± SEM. Groups were compared using one-way ANOVA followed by post hoc Tukey test (b–d). Source data are provided as a Source Data file.
To evaluate whether the improved cardiac function by hiPM-cm can be long-lasting, we performed functional and histological evaluations for up to 12 weeks. Echocardiogram examinations demonstrated improved cardiac function in the I/R + hiPM-cm group at two weeks, four weeks, and 12 weeks after I/R, as evidenced by significantly higher percentages of LVEF and LVFS, along with significantly lower LVEDD and LVESD when compared to the I/R + FM group at the same time points (Fig. 7c and Supplementary Fig. 7b, c). In addition, assessment of heart failure using running exercise test revealed that mice from the I/R + hiPM-cm group ran significantly longer duration and total distances compared to mice from the I/R + FM group at 12 weeks after I/R (Supplementary Fig. 7d), indicating that hiPM-cm confers long-term cardioprotection against heart failure in mouse hearts after I/R. Consistently, a significant hypertrophic response in the heart of mice from the I/R + FM group was observed when compared to the sham group, as reflected by a significant increase in heart weight to tibial length ratio, which was attenuated in mice from I/R + hiPM-cm group at 12 weeks after the surgery (Supplementary Fig. 7e). Histological assessment of mouse hearts at 12 weeks after I/R surgery also revealed significantly less fibrotic tissues in I/R + hiPM-cm group as compared to I/R + FM group (Fig. 7d). Taken together, our data suggest that hiPM-cm ameliorates cardiac dysfunction in mouse hearts after I/R in both short and long terms.
Discussion
Our study shows that hiPMs and hiPM-cm can promote adult cardiomyocyte proliferation and cardiac regeneration. By identifying several secreted proteins in hiPM-cm, we show that this approach can activate multiple pro-proliferative signaling pathways in human and mouse cardiomyocytes, thereby stimulating their proliferation in both in vitro and in vivo settings. Importantly, our findings demonstrate that hiPM-cm can effectively promote adult cardiomyocyte proliferation, enhance cardiac regeneration, and improve contractile function in adult mouse hearts after I/R, demonstrating its therapeutic potential for the regenerative treatment of various heart diseases.
Cardiovascular regenerative treatment has been pursued at both basic and translational levels41,42,43,44. Despite progress in harnessing adult stem and progenitor cells for cardiac tissue regeneration45,46, strategies to efficiently generate fully functional new cardiomyocytes have yet to yield definitive successes in clinical settings47,48. Alternatively, the discovery of residual cardiomyocyte turnover in adult mammals has raised hope for therapeutic intervention to reinvigorate the endogenous regenerative potential of the heart by promoting pre-existing cardiomyocyte proliferation49,50. Our study reveals the efficacy of hiPMs and hiPM-cm in promoting human cardiomyocyte proliferation, highlighting its potential as an innovative regenerative treatment of heart diseases.
A prerequisite underlying our study was the capacity to readily generate hiPMs. A recent study reported a human embryonic stem cell (hESC)-based approach for generating hiPMs that differs from our approach in several ways51. While both approaches involve the critical steps of embryonic body formation, the collection of suspended cells, and the maturation of macrophages, we streamlined the induction process by simplifying the steps leading to the generation of primitive hematopoietic progenitor cells. While the use of growth factors such as Activin A, BMP4, and VEGF is common to both approaches51,52, we omitted the addition of cytokines such as IL-6, IL-1, and IGF1, as well as the sorting of primitive hematopoietic progenitor cells. This tailored approach not only highlights the adaptability of hiPM generation protocols but also underscores the potential for fine-tuning macrophage properties to suit specific therapeutic objectives.
The use of conditioned medium has garnered considerable attention in the field of regenerative medicine, particularly in the context of mesenchymal stem cell therapies, where it has been shown to exert paracrine effects that ameliorate cardiac dysfunction53. As a therapeutic alternative to direct cell therapy, conditioned medium possesses several benefits for being safer, avoiding immune reactions, rejection, and tumor risks due to its acellular nature. In addition, its preparation is more straightforward and cost-effective, facilitating scalability, and its ease of use allows for direct patient administration, bypassing complex surgeries53,54,55,56. Given the demonstrated pro-proliferative effects of hiPM-cm in our study, it is conceivable that hiPM-cm itself can serve as the foundation for the translational application of our findings.
Alternatively, further investigations into the specific medium components (and potentially cofactors, etc) or specific forms (such as extracellular vesicles) concerning the pro-proliferative effect could lay the groundwork for formulating a targeted biological therapeutic cocktail. Specifically, our study identified that a five-protein combination, including C1QB, NRP1, PLTP, FUCA1, and SERPING1, partially recapitulates the pro-proliferative effects of hiPM-cm, both in enhancing human cardiomyocyte proliferation in vitro and in promoting adult mouse cardiomyocyte proliferation in vivo. Our study also revealed a subset of this cocktail, consisting of NRP1 and C1QB, which exhibited the most significant pro-proliferative effects on human cardiomyocytes in vitro among all tested protein pairs, mainly through their respective receptors FLT1 and LRP1. Given the documented involvement of these proteins and receptors in the activation of PI3K/AKT, JAK/STAT, and MAPK pathways33,57,58,59,60,61,62,63, which were also triggered by hiPM-cm, it is plausible that these pathways mediated the pro-proliferative effects of NRP1 and C1QB on human cardiomyocytes in vitro. Future studies are needed to investigate the potential pro-proliferative effects of NRP1 and C1QB in adult hearts. Notably, considering the complexity of hiPM-cm, it may encompass additional components that could exert benefits via alternative pathways, warranting further investigation to fully elucidate their contributions and potential applications64,65,66.
In conclusion, our study has demonstrated the therapeutic potential of hiPM-cm in promoting cardiomyocyte proliferation and mitigating myocardial I/R injury. Through the facilitation of adult cardiomyocyte proliferation and the promotion of heart regeneration, hiPM-cm serves as a promising candidate for regenerative therapy. While the current findings are encouraging in the context of I/R-induced acute cardiac injury, the application of hiPM-cm to a diverse array of cardiac disease models characterized by “irreversible” cardiomyocyte loss presents a compelling avenue for future research.
Methods
Animals
All the experiments and protocols were approved by the Institutional Animal Care and Use Committee of the Institute of Zoology, Chinese Academy of Sciences (permit number: IOZ-IACUC-2023-249). All mice were housed in SPF animal facilities under a 12 h light/dark cycle. All the 8-week-old C57BL/6J mice were purchased from SPF Biotechnology, China. MADM-ML-11TG/GT mouse lines40 were generously provided by Dr. Chong Liu at the College of Medicine, Zhejiang University (Hangzhou, China). Cardiomyocyte-specific MADM mice were created by crossing homozygous MADM-ML-11TG/GT with αMHC-MerCreMer strain67.
Culture and maintenance of human induced pluripotent stem cells (iPSCs)
The human induced pluripotent stem cell (hiPSC) line utilized in this investigation for cardiomyocyte induction, human iPSC-derived primitive macrophages (hiPMs), and human iPSC-derived cardiac fibroblasts (hiCFs) originated from amniotic mesenchymal cells (Ethics Committee Approval: Institute of Zoology, Chinese Academy of Sciences, China; Cell Line: Q-iPS-6, permit number: DYSL[2023]004). The hiPSCs were maintained in chemically defined PSCeasyII medium (Cellapy, CA1014500) and passaged using 0.5 mM EDTA at a split ratio ranging from 1:10 to 1:20. Cells were replated onto plates coated with 0.5% Matrigel (Corning, 356230) in medium supplemented with 10 μM ROCK inhibitor Y-27632 (Selleck, S1049).
For cryopreservation, the cells were frozen in a solution containing 90% PSCeasyII medium and 10% DMSO. For thawing, the cells were resuspended in PSCeasyII medium supplemented with 10 μM Y-27632. The fresh medium was changed daily to maintain optimal culture conditions. All the cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2.
Generation of hiPSC-derived primitive macrophage precursors (hiPMpres) and primitive macrophages (hiPMs)
The hiPMs were generated by differentiation of hiPSCs as follows: On day 0, a single well of 80%-confluence iPSC cultured in a 12-well plate was harvested by incubation in 0.5 mM EDTA for 8 min at room temperature. The cells were resuspended in PSCeasyII medium containing 10 μM Y-27632 and replated in one well of a 6-well ultra-low adherence plate (Corning, 3471) to facilitate the formation of EBs. On day 1, EBs were incubated in PSCeasyII medium supplemented with 2 ng/mL Activin A (STEMCELL Technologies, 78001), 5 ng/mL bFGF (STEMCELL Technologies, 78003), 50 ng/mL BMP4 (STEMCELL Technologies, 78211), 20 ng/mL SCF (STEMCELL Technologies, 78062), and 5 μM Y-27632 for mesoderm formation. On day 3, the medium was replaced with PSCeasyII supplemented with 100 ng/mL VEGF (Novoprotein, C083), 5 ng/mL bFGF, 50 ng/mL BMP4, and 20 ng/mL SCF for hemogenic endothelium (HE) specification. On day 5, 10–20 EBs were plated into one well of a 0.2% gelatin-coated 6-well plate and cultured in X-VIVO 15 medium (Lonza, 04-418Q) supplemented with 100 ng/mL M-CSF (Novoprotein, C417), 25 ng/mL IL-3 (Novoprotein, CX90), 1% GlutaMax (Gibco, 35050061), and 0.055 mM 2-mercaptoethanol (Thermo Fisher Scientific, 31350010) with the medium changed every 3 days for myeloid progenitor generation and macrophage differentiation. From day 17 onwards, the non-adherent hiPM precursors (hiPMpres) were collected when the medium was refreshed every 5 days for about 2 months until a discernible decline in their production rate was observed. The collected hiPMpres were centrifuged at 600 g for 3 min, resuspended in X-VIVO 15 medium supplemented with 100 ng/mL M-CSF, 5 ng/mL bFGF, and 1% GlutaMax (Gibco, 35050061), and seeded at a density of 5-6 × 105 cells per well in a 6-well plate (NEST, 703001) for them to become adherent and mature to hiPMs, with the medium changed every 5 days. After 10 days, mature hiPMs were harvested for downstream analyses.
Generation of hiPSC-derived cardiomyocytes
Human cardiomyocytes were induced from hiPSCs using a slightly modified previously published protocol22. Briefly, hiPSCs cultured to 80% confluence in PSCeasyII medium were first treated with RPMI + B27 medium (RPMI-1640 (Gibco, 11875-093) supplemented with B27, minus insulin (Gibco, A1895601)) and 4 μM CHIR99021 (Selleck, S2924) for 48 hr. Following this initial treatment, CHIR99021 was removed, and the cells were cultured in RPMI + B27 medium for an additional 24 hr. On day 3, 5 μM Wnt pathway inhibitor IWR-1 (Selleck, S7086) was added to the medium. Starting from day 5, the cells were cultured in RPMI + B27 medium, with medium changes every other day. On day 11, the medium was switched to glucose-free RPMI + B27 medium for 4 days, with medium changes every other day, to induce glucose starvation and selectively eliminate non-cardiomyocyte cell types. From day 15, the resulting purified cardiomyocytes were maintained in RPMI + B27 medium, with medium changes every other day, until subsequent experiments at around day 30.
Generation of hiPSC-derived cardiac fibroblasts (hiCFs)
The hiCFs were induced according to a previously published protocol68. A culture of hiPSCs grown to 80% confluence in PSCeasyII medium was first exposed to RPMI + B27 medium supplemented with 4 μM CHIR99021 (Selleck, S2924) for 48 hr. After the initial treatment, CHIR99021 was removed, and the cells were cultured in RPMI + B27 medium for another 24 hr. On day 3, 5 μM Wnt pathway inhibitor IWR-1 (Selleck, S7086) was added to the medium. On day 5, the cells were cultured in the RPMI + B27 medium. On day 6, the cells were dissociated using Accutase (Thermo Fisher Scientific, A1110501) and centrifuged at 200 × g for 3 min. The cells were then resuspended in advanced DMEM/F-12 medium (Thermo Fisher Scientific, 12634010) supplemented with 1% GlutaMax (Thermo Fisher Scientific, 35050061), 5 μM CHIR99021, 2 μM retinoid acid (Sigma, R2625), 5 μM Y-27632 (ROCK inhibitor), and 1% FBS (Gibco). Subsequently, the cells were plated onto Matrigel-coated cell culture dishes at a density of 2 × 104 per cm². On day 7, the medium was replaced with advanced DMEM/F-12 medium supplemented with 1% GlutaMax, 5 μM CHIR99021, and 2 μM retinoid acid. On day 9, the medium was replaced with advanced DMEM/F-12 medium supplemented with 1% GlutaMax. On day 11, the cells were dissociated using Accutase, centrifuged at 200 × g for 3 min, resuspended in the medium with advanced DMEM/F-12 medium supplemented with 1% GlutaMax and 2 μM SB431542 (Selleck, S1067), and seeded onto Matrigel-coated cell culture dishes at a split ratio ranging from 1:3 to 1:6, with the medium changed every other day. Upon 90–100% confluence, the cells were dissociated using Accutase, centrifuged, resuspended in PromoCell Fibroblast Growth Medium (PromoCell, C-23025) supplemented with 20 ng/mL of bFGF (STEMCELL Technologies, 78003) and 10 μM SB431542, and then seeded onto a Matrigel-coated cell culture dish at a density of 1 × 104 cells per cm² for culturing for 4 days, with the medium changed every other day. Subsequently, the resultant hiCFs were passaged and seeded again onto a Matrigel-coated cell culture dish at a density of 1 × 104 cells per cm² and cultured in PromoCell Fibroblast Growth Medium (PromoCell, C-23025) containing 20 ng/mL of bFGF and 10 μM SB431542, for two days. The resultant hiCFs were kept in PromoCell Fibroblast Growth Medium (PromoCell, C-23025) containing 10 μM SB431542, with the medium changed every other day until used for the experiment.
THP-1 maintenance and macrophage differentiation
THP-1 cell line, originally obtained from ATCC (catalog number TIB-202), was maintained in RPMI-1640 plus 10% FBS. For macrophage differentiation69, 5 × 105 THP-1 cells were seeded into a well of a 6-well plate (NEST, 703001). Following 48 hr of stimulation with 500 nM Phorbol 12-myristate 13-acetate (PMA) (Abcam, ab120297), the resultant THP-1-derived macrophages (THP-1Ms) became adherent and were collected for downstream analyses.
Cell coculturing
For 2D coculture experiments with cardiomyocytes, cardiomyocytes were dissociated using 2.5% TryPLE (Gibco, 12605010) at 37 °C for 8 min and seeded at a density of 1 × 105 cells per well in a 24-well plate (NEST, 702001), with the culture medium consisting of RPMI + B27 medium supplemented with 10 μM Y-27632. After 24 hr, hiPMs or THP-1Ms were added to the coculture system at a ratio of 1:4 relative to the cardiomyocytes. The medium was replaced with RPMI + B27 medium, changed every other day, and the colcuture was maintained for 4 days prior to immunofluorescence analysis of cardiomyocyte proliferation.
For 2D coculture experiments with hiCFs, hiCFs were dissociated using Accutase and seeded into 24-well plates at a density of 5 × 104 per well. The hiPMs were then added at a ratio of 1:1 relative to hiCFs and cocultured in PromoCell Fibroblast Growth Medium (PromoCell, C-23025) containing 10 μM SB431542. The medium was changed every other day, and the coculture was maintained for 4 days prior to immunofluorescence analysis of fibroblast proliferation.
For 3D coculture experiments with cardiomyocytes, cardiomyocytes were dissociated, counted, resuspended at a concentration of 2.5 × 105/mL in RPMI + B27 medium containing 10 μM Y-27632, and mixed with hiPMs at a number ratio of 1:4 relative to the cardiomyocytes. Then the cells were seeded into a 96-well ultra-low adherence plate (Corning, 7007) with 200 μL per well and aggregated to the bottom of the wells by centrifugation at 100 × g for 3 min. After 24 hr, the medium was replaced with RPMI + B27 medium, with the medium changed every other day, and the coculture was maintained for 10 days prior to immunofluorescence analysis of cardiomyocyte proliferation.
Acquisition of conditioned medium
To obtain conditioned medium from hiPMpres (hiPMpre-cm) for incubation with cardiomyocytes, hiPMpres were harvested, counted, and resuspended in RPMI + B27 medium. The cells were then seeded into a single well of a 6-well plate at a density of 5-6 × 105 cells. After 12 hr, the cells adhered to the plate and were incubated for an additional 48 hr. Following this incubation, the supernatant was collected, and cell debris was removed through an initial centrifugation at 600 × g for 3 min, followed by a second centrifugation at 2000 × g for another 3 min. The hiPMpre-cm was then obtained from the resultant supernatant for subsequent experiments. For proteomics analysis, hiPMpre-cm was collected in RPMI-1640 medium (Gibco, 11875-093) alone, following the same procedure.
To obtain conditioned medium from hiPMs (hiPM-cm) for incubating with cardiomyocytes), hiPMs were harvested, counted, and resuspended in RPMI + B27 medium. The cells were then seeded into a single well of a 6-well plate at a density of 5-6 × 105 cells and incubated for an additional 48 hr. The supernatant was collected and subjected to a two-step centrifugation to remove any residual cellular debris. The resultant hiPM-cm was used for subsequent experiments. For proteomics analysis, hiPM-cm was collected in RPMI-1640 medium (Gibco, 11875-093) alone, following the same procedure.
To analyze the various components within hiPM-cm, a 3 kDa Amicon® Ultra-2 Centrifugal Filter Device (Merck, UFC200324) was employed for centrifugation, enabling the efficient separation of molecules based on size, specifically those with molecular weights larger than 3 kDa. The medium was concentrated to approximately one-tenth of its original volume. The concentrated medium, enriched with molecules larger than 3 kDa, was then diluted tenfold with RPMI medium to achieve a suitable concentration for cell culture applications. Both the diluted solution, containing concentrated molecules larger than 3 kDa, and the filtrate, comprising molecules smaller than 3 kDa, were supplemented with B27 prior to incubation with cardiomyocytes.
Incubation of cardiomyocytes with conditioned medium
For 2D cultures, cardiomyocytes were counted and replated at a density of 1 × 105 cells per well in a 24-well plate, with the culture medium consisting of RPMI + B27 supplemented with 10 μM Y-27632. The cardiomyocytes were then incubated in a conditioned medium for 4 days, with the medium changed every other day, before being harvested for downstream analyses.
For 3D cultures, cardiomyocytes were counted and resuspended at a concentration of 2.5 × 105/mL in RPMI + B27 containing 10 μM Y-27632 and seeded into a 96-well ultra-low-adherence plate (Corning, 7007) with 200 μL per well. After 24 hr, the medium was replaced with either RPMI + B27 or hiPM-cm, and the 3D organoids were maintained for 10 days, with the medium changed every other day, before being harvested for downstream analyses.
Immunofluorescence staining and flow cytometry analysis of cells
For immunofluorescence staining, the cells were initially fixed with 4% paraformaldehyde (PFA) (Servicebio, G1101) at room temperature for 30 min. Subsequently, they were permeabilized and blocked using QuickBlock™ Blocking Buffer for Immunol Staining (Beyotime, P0260) at room temperature for 1 hr. The cells were then incubated with primary antibodies at 4 °C overnight. After three washes with PBS to eliminate any unbounded primary antibodies, second antibodies and Hoechst 33342 (Beyotime, C1022) were added, and the cells were incubated at room temperature for 1 hr. Immunofluorescence images were captured with a Nikon A1R MP microscope, and the intensity of the immunofluorescent signals were analyzed with NIS-Elements 5.20.00 from at least 2 visual fields and more than 500 cells per well for at least three experimental repeats for each group.
Primary antibodies included: rabbit anti-CD14 (Proteintech, 17000-1-AP), rabbit anti-CD11b (Beyotime, AF6396), rabbit anti-Ki67 (Abcam, ab15580), mouse anti-Ki67 (Abcam, Ab279653), goat anti-cTnT (Abcam, Ab64623), rabbit anti-α-actinin (Proteintech, 11313-2-AP), mouse anti-cTnT (Abcam, Ab10214), rabbit anti-vimentin (Abmart, T55134F) and rabbit anti-αSMA (Proteintech, 14395-1-AP). All the primary antibodies were diluted with QuickBlock™ Primary Antibody Dilution Buffer for Immunol Staining (Beyotime, P0262) at a ratio of 1:200.
Secondary antibodies included: goat anti-rabbit conjugated with Alexa Fluor™ 488 (Thermo Fisher Scientific, A11008), goat anti-mouse conjugated with Alexa Fluor™ 568 (Thermo Fisher Scientific, A11004) and donkey anti-goat conjugated with Alexa Fluor™ 647 (Thermo Fisher Scientific, A21447). All the secondary antibodies were diluted by QuickBlock™ Primary Antibody Dilution Buffer for Immunol Staining (Beyotime, P0262) at 1:500.
For flow cytometry analysis, the cells were first digested into single cells and then stained, either through direct labeled primary antibodies or using a two-step immunofluorescence method. For the direct labeling approach, the cells were incubated with primary antibodies for 30 min at 4 °C, followed by washing with PBS to eliminate any unbounded antibodies. Alternatively, the cells were stained through a process that encompassed fixing, permeabilizing, and blocking, followed by incubation with both primary and secondary antibodies. All samples were analyzed on a LSRFortessa X-20 analyzer (BD Biosciences), and the acquired data was performed with the FlowJo software (FlowJo 10.8.1).
Direct labeling primary antibodies included: ABflo® 647 rabbit anti-human VEGFR2/CD309 (Abclonal, A23357), Abflo® 488 rabbit anti-human CD235a/Glycophorin A (Abclonal, A23014), mouse CCR2 APC-conjugated Antibody (R&D, FAB5538A), and FITC anti-human CD45 Antibody [HI30] (Elabscience, E-AB-F1137C). All the antibodies were diluted at a ratio of 1:50.
Immunofluorescence staining of cardiac organoids
Cardiac organoids were fixed in 4% PFA at 4 °C overnight. Subsequently, they underwent gradient dehydration using 15% and 30% sucrose solutions and were embedded in 20% gelatin to enable frozen sectioning at a thickness of 5 μm. A microwave oven was used to retrieve antigens with citric acid buffer. The sections were then permeabilized and blocked with QuickBlock™ Blocking Buffer for Immunol Staining (Beyotime, P0260) for 1 hr at room temperature. Next, the sections were incubated with primary antibodies overnight at 4 °C. On a subsequent day, the sections were rinsed with PBS and incubated with Alexa-Fluor conjugated secondary antibodies and Hoechst 33342 for 1 hr at room temperature. Finally, immunofluorescence images were captured using a Nikon A1R MP microscope. Immunofluorescence signals were analyzed using NIS-Elements 5.20.00 software from at least 2 visual fields and more than 500 cells for each organoid for at least 6 organoids for each group.
Phagocytosis assay
FITC-conjugated dextran (molecular mass 40 kDa, Sigma-Aldrich, FD40S) was used to determine the phagocytic ability of hiPMs70. Both hiCFs and hiPMs were cultured in RPMI-1640 supplemented with 1% BSA (Solarbio, A8020) and incubated with 1 mg/mL FITC-conjugated dextran for 2 hr. The phagocytosis was then detected by FITC fluorescence signals within the cells using Cytation (BioTek, Cytation5).
IncuCyte monitoring of cardiomyocyte proliferation
Cardiomyocyte proliferation was monitored using an IncuCyte S3 real-time imaging platform. Briefly, cardiomyocytes were seeded into a 96-well plate at a density of 500 cells per well and subsequently maintained in a continuous culture environment for 4 days. Throughout this duration, imaging was carried out using a 10x objective lens, with images captured at a 12 hr interval.
Seahorse mitochondrial and glycolysis stress test
The energy metabolism of cardiomyocytes was evaluated with the Seahorse XF Cell Mitochondrial Stress Test and Glycolysis Stress Test (Agilent, XF9637) using a slightly modified previously published protocol23. Prior to seeding the cardiomyocytes, Matrigel was applied to coat the wells of the Seahorse XF 96 microplate. Both FM and hiPM-cm group cardiomyocytes were plated into the prepared wells. Subsequently, Y-27632 was removed after 24 hr (day 0), and the cells were then cultured in the Seahorse XF 96 for another 4 days, with the medium changed every other day. To ensure comparable cell counts on day 4, 7 × 103 cardiomyocytes were seeded for the FM group, while 5 × 103 cardiomyocytes were seeded for the hiPM-cm group on day 0.
For the glycolysis stress test, the culture medium was replaced with Agilent Seahorse XF DMEM Basal Medium enriched with 2 mM glutamine and 1 mM sodium pyruvate, 1 hr before the assay commenced. During the measurement phase, the following inhibitors were added: 10 mM glucose (Diamond, A100188), 1 μM oligomycin (Merck Millipore, 495455), and 50 mM 2-DG (SIGMA, D8375-1G). The extracellular acidification rate (ECAR) values obtained were normalized based on the result of the cell viability assay using a Cell Counting Kit-8 (CCK8) (Beyotime, C0037).
For the mitochondrial stress test, the assay medium was supplemented with the following drugs: 2.5 μM Oligomycin (Merck Millipore, 495455), 2.5 μM FCCP (Selleck, S8276), 2.5 μM Antimycin A (AA) (ENZO, ALX-380-075-M010), and 2.5 μM Rotenone (Selleck, S2348). The resulting O2 consumption rate (OCR) values were normalized based on the result of CCK8 (APExBIO, K1018-5).
Measurement of ATP production
Cardiomyocytes were seeded in a 96-well plate with 2500 cells per well and cultured in either FM or hiPM-cm for 4 days. Cardiomyocytes were then lysed, and ATP production was detected using an ATP Assay Kit (Beyotime, S0026) and normalized to protein content determined by the BCA Protein Assay Kit (Beyotime, P0012).
Detection of calcium handling
Cardiomyocytes were dissociated into single cells and seeded onto a glass-bottom 96-well plate at a density of 500 cells per well. The cells were then cultured for 4 days in FM or hiPM-cm. To evaluate Ca2+ handling, the cardiomyocytes were incubated with Fluo-4, AM (Abcam, ab241082) in RPMI + B27 medium for 30 min at 37 °C. Ca2+ imaging was taken every 600 ms with a Nikon inverted microscope (Eclipse TiE) with a 40x magnification oil immersion objective by excitation at 488 nm. Single-cell Fluo-4, AM images were captured and processed by Metamorph software (version 7.0). Quantitative analyses of fluorescence signal intensity, frequency, and slope of the generated curve were performed with GraphPad Prism 8.4 software (Prism Inc)71.
RNA sequencing and data analysis
Total RNA was used as input material for library preparation. Sequencing libraries were generated using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, E7530L), and index codes were added to attribute sequences to each sample. Qualified libraries were pooled and sequenced on Illumina platforms with PE150 strategy (Novogene Bioinformatics Technology), based on the effective library concentration and the desired data volume. The differential expression genes (DEGs) were analyzed with the DESeq2 package (version 1.40.2) and identified with an absolute fold change ≥ 2 and an adjusted p-value < 0.05 (Benjamini-Hochberg method)72. The full set of processed detected genes were first examined using principal component analysis (PCA). With variance stabilizing transformation of the read counts, PCA was performed using the prcomp function of the R Stats Package (version 3.6.2). DEGs were subjected to volcano plots, heatmaps, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses. Volcano plots and heatmaps were visualized with ggplot2 (version 3.4.2) and pheatmap (version 1.0.12) R packages, respectively. KEGG pathway enrichment analysis was conducted using clusterProfiler (version 4.8) R package73 and visualized with ComplexHeatmap (version 2.16.0) R package74. NicheNet (version 2.2.0) was employed to identify receptors for NRP1, SERPING1, FUCA1, C1QB, and PLTP in cardiomyocytes using bulk RNA sequencing data of human cardiomyocytes treated with FM and hiPM-cm as the input, with a threshold set for receptor expression at an average log2 value greater than 475.
qPCR analysis
Total RNA was extracted from cells with the FastPure Cell/Tissue Total RNA Isolation Kit V2 (Vazyme, RC112-01) and reversely transcribed with the RevertAid™ First Strand cDNA Synthesis Kit (Thermo Scientific, M1632). qPCR analysis was then carried out in the Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Q712-03). Actb gene was used as a reference gene for normalization, and data were processed using the 2-(ΔΔCt) method76. Sequences of primers used for qPCR are as below:
LYVE1 Fw: 5’-TCTGAAGGGGTAGGCACGAT-3’;
LYVE1 Re: 5’-CTGGATGGAAAGCTCTTCTGC-3’;
CD163 Fw: 5’-AGGCCATTCTGAGCCACACT-3’;
CD163 Re: 5’-AAGGTATCTTAAAGGCTGAACTCAC-3’;
MAF Fw: 5’-TCTCCCGAGTTTTTCATAACTGAGC-3’;
MAF Re: 5’-TCCAAAATGTGGCGTATCCC-3’;
FOLR2 Fw: 5’-CGAAGACAGAGGCTGTGGG-3’;
FOLR2 Re: 5’- TGCAGGCGGAGCTGAACC-3’;
ACTB Fw: 5’-AGAGCTACGAGCTGCCTGAC-3’;
ACTB Re: 5’-AGCACTGTGTTGGCGTACAG-3’.
Mass spectrometry-based proteomic analysis of conditioned medium
For mass spectrometry analysis, hiPM-cm and hiPMpre-cm were collected in RPMI-1640 without phenol red (Gibco, 11835-030) from the incubation with the cells for 48 hr followed by a two-step centrifugation to remove cellular debris. Mass spectrometry analysis was then performed as previously described77. The conditioned medium samples were then reduced with 25 mM dithiothreitol for 1 hr at 37 °C and alkylated with 50 mM iodoacetamide for 30 min in the dark at room temperature. Digestion was performed with trypsin with a 1:50 enzyme-to-protein ratio at 37 °C overnight. Digested samples were resuspended with 50 mM NH4HCO3 solution to wash the membrane, and the collected components were vacuum-dried. The dried peptide samples were resuspended in a solvent of 0.1% formic acid (v/v) and subjected to an EASY-nLC 1200 interfaced via a Nanospray Flex ion source to an Orbitrap Exploris 240 mass spectrometer (Thermo Fisher Scientific). Samples were loaded onto a C18 trap column (3 μm particles, 150 μm ID, 3 cm length, Dr. Maisch GmbH) and separated using a C18 analytical column (1.9 μm particles, 150 μm ID, 20 cm length, Dr. Maisch GmbH) at a flow rate of 0.5 μL/min with a 110 min gradient composed of water containing 0.1% formic acid (solvent A) and 80% acetonitrile containing 0.1% formic acid (solvent B). The gradient was 8–12% B for 5 min, 12–30% B for 75 min, 30–45% B for 15 min, 45–100% B for 5 min, and finally 100% B for 10 min. MS settings included the MS1 scan (300-1500 m/z, 6 × 104 resolution, AGC target 3e6) and MS2 scans (110-1500 m/z, 1.5 × 104 resolution, AGC target 7.5e4). Peptides and proteins were identified by comparing the raw data to the Maxquant. Statistical analysis was carried out using Perseus software. Further quantitative data analyses were qualified on unique proteins with at least two unique peptides and an FDR < 0.01. The significantly differentially expressed proteins in hiPM-cm versus hiPMpre-cm were identified with a cutoff of |Fold change | >2 and adjusted p-value < 0.05. Venn plot showing the intersection between significantly upregulated proteins in hiPM-cm versus hiPMpre-cm and significantly upregulated genes encoding secreted proteins78 in CCR2- macrophages versus CCR2+ macrophages (p-value < 0.05) of human hearts15 downloaded from the GEO database (GSE112630) were visualized using ggVenn R package (version 0.1.10).
Evaluation of pro-proliferative effects of candidate proteins
Cardiomyocytes were seeded into 384-well plates (NEST, 761002) at a density of 5 × 103 cells per well in RPMI + B27 medium containing 10 μM Y-27632. After 24 hr, the culture medium was aspirated and replaced with fresh RPMI + B27 medium containing candidate proteins identified by proteomics analysis, or their combinations, at a concentration of 100 ng/mL for each protein for 4 days, with the medium containing indicated protein(s) refreshed every other day. Following this incubation period, immunofluorescence analysis was conducted to assess cardiomyocyte proliferation.
The proteins screened included: Transferrin (Novoprotein, CJ41), NRP2 (Sino Biological, 10695-H08H), SERPING1 (Novoprotein, C539), C1QC (Abclonal, RP01865), GAS6 (Novoprotein, C01W), C1QB (MCE, HY-P75464), FUCA1 (Sino Biological, 13893-H08H), NRP1 (Novoprotein, C11K), and PLTP (Sino Biological, 11171-H08H).
Validation of potential receptors for identified pro-proliferative proteins
To validate the potential receptors for the identified pro-proliferative proteins, we employed siRNA technology. The siRNAs were procured from JTSBIO Co. and used to knock down target genes in hiPSC-derived cardiomyocytes using Lipofectamine™ RNAiMAX Transfection Reagent (Thermo Fisher Scientific, 13778075) following manufacturer’s instructions.
siNC: 5’-GCUUGUUGCAAAUAUGCUAUC-3’;
PLXNA1 siRNA-1: 5’-GGUUCACCUUCCUCUUGUAUA-3’;
PLXNA1 siRNA-2: 5’-GGCAGCAGAUUGACUACAAGA-3’;
FLT1 siRNA: 5’-GCAACAGUCAAUGGGCAUU-3’;
KDR siRNA: 5’-CGGAGAAGAAUGUGGUUAA-3’;
SELE siRNA-1: 5’-GGUUGAAUGCACCACUCAAGG-3’;
SELE siRNA-2: 5’-GCUGGAGAGUUCACCUUCAAA-3’;
LRP1 siRNA: 5’-GGCCGUGGAUUAUCACAAUTT-3’;
APOA1 siRNA-1: 5’-GAAGCUGCACGAGCUGCAAGA-3’;
APOA1 siRNA-2: 5’-UGGACGACUUCCAGAAGAAGU-3’;
EGFR siRNA: 5’-AGGAAUUAAGAGAAGCAACAUTT-3’.
Immunoblotting analysis
Samples for immunoblotting analysis were lysed by RIPA (Beyotime, P3001) with 1x protease inhibitor mixture (LabLead, C0101) and 5 mM NaF. Samples were separated by 10% SDS-PAGE and transferred to 0.22-μm PVDF membranes. The PVDF membranes were blocked with 5% skim milk and incubated with primary antibody at 4 °C overnight. After being washed three times with PBS supplemented with 0.1% Tween 20 (Solarbio, T8220), the PVDF membranes were incubated with HRP-conjugated secondary antibodies. The signals were detected by NcmECL Ultra (NCM Biotech, P10300).
Primary antibodies included: rabbit anti-phospho-AKT (Abclonal, AP0637, 1:2000), rabbit anti-AKT (Abclonal, A18120, 1:2000), rabbit anti-phospho-ERK1/2 (CST, 4370, 1:2000), rabbit anti-ERK1/2 (CST, 4695, 1:2000), rabbit anti-phospho-STAT3 (Abclonal, AP0715, 1:2000), rabbit anti-STAT3 (Abclonal, A16975), and mouse anti-GAPDH (Abclonal, AC002, 1:5000).
In vitro treatment with inhibitors for pro-proliferative pathways
To assess the effect of AKT, STAT3, and MAPK signaling pathways on the promotion of cardiomyocyte proliferation by hiPM-cm, cardiomyocytes were treated with hiPM-cm alone, or hiPM-cm supplemented with 0.2 μM AKT inhibitor A-674563 HCl (Selleck, S2670), 2 μM ERK inhibitor PD0325901 (Selleck, S1036), 5 μM STAT3 inhibitor Stattic (MCE, HY-13818) in single or in combination for 4 days, with the medium changed every other day, before immunofluorescence analysis of cardiomyocyte proliferation.
Myocardial ischemia/reperfusion injury
Mouse heart ischemia/reperfusion injury was operated as previously described79. C57BL/6J mice were anesthetized with 2% isoflurane and their chest cavities were then opened at the fourth intercostal space. Upon the removal of pericardium, the left anterior descending coronary artery was ligated for 40 min using silk ligature and then released to repercuss the ischemic area. Thoracic wall and skin were then sutured, and the mice were placed under a heat lamp and warmed until recovery.
Intramyocardial injection of conditioned medium
For I/R-injured mice, a total of 40 μL FM or hiPM-cm was injected into 3 points of the left ventricle, one at the left border zone of the ligation point, one at the right border zone of the ligation point, and one at the apex region right below the ligation point, using a 30-gauge syringe at 2 min after reperfusion in an open-chest surgery80,81. For Sham mice, a total of 40 μL PBS was injected into the same 3 points of the left ventricle as did for I/R-injured mice.
For those that were not subjected to Sham or I/R surgery, C57BL/6J or MADM mice were anesthetized by 2% isoflurane, and a thoracotomy was performed at the fourth intercostal space. After the removal of the pericardium, a total volume of 40 μL containing PBS, FM, hiPM-cm, or the five-protein combination was injected into the same three points within the left ventricle of the heart as did for I/R-injured mice. The five proteins were diluted in PBS to a final concentration and administered at a dosage of 1 μg per protein per mouse. The thoracic wall and skin were then sutured, and the mice were placed under a heat lamp until they recovered from anesthesia.
In vivo treatment with inhibitors for pro-proliferative pathways
AKT inhibitor A-674563 HCl, ERK inhibitor PD0325901, and STAT3 inhibitor Stattic 30,38,39were diluted by DMSO or peanut oil as appropriate. Mice were gavaged with A-674563 HCl and PD0325901 at a dose of 10 mg/kg/day and intraperitoneally injected with and Stattic at a dose of 10 mg/kg/day. The inhibitors were administered in combination at 6 hr before the intramyocardial injection of the conditioned medium, and this administration was repeated once per day for another three consecutive days.
Measurement of cardiomyocyte proliferation in vivo using MADM mice
To induce MADM expression, tamoxifen (Selleck, S1238) was administered via intraperitoneal injection at a dosage of 40 mg/kg/day for a period of five consecutive days40,82. Following this, MADM mice received intramyocardially injection with conditional medium or five-protein combination at three distinct points across the left ventricles as described above, one week post the commencement of tamoxifen treatment. One week later, hearts were harvested, and each ventricle was divided into three segments from apex to base, fixed with 4% PFA, dehydrated in a sucrose gradient up to 30%, and embedded in OCT for cryosectioning. The 10 μm-thick sections were stained with Hoechst 33342 for 15 min at room temperature before being mounted using a neutral balsam mounting medium (Aladdin, N116470). For imaging, the PerkinElmer Vectra Polaris system was used to perform full scans on the sections at 40x magnification. For each mouse, quantification of cardiomyocyte proliferation was performed based on a total of 8–10 fields across three distinct sections (one from each of the three ventricle segments) by calculating the ratio of single-labeled (either green or red) to double-labeled cells using ImageJ (Image J2, Fiji).
Measurement of serum cTnT levels
For the measurement of cardiac Troponin T (cTnT) levels in the serum, blood was collected from mouse facial vein83, left undisturbed at 4 °C for 4 hr to clot, and centrifuged at 1000 × g for 10 min. Serum from the resultant supernatant was collected and used for the measurement of serum cTnT levels by an ELISA kit (Yutong, F7649B) following the manufacturer’s instructions.
Flow cytometry analysis of immune cells in the heart
A single-cell suspension of the mouse heart was prepared as previously described84,85. The mouse heart was initially injected with EDTA buffer (130 mM NaCl, 5 mM KCl, 0.5 mM NaH2PO4, 10 mM HEPES, 10 mM Glucose, 10 mM BDM, 10 mM Taurine, 5 mM EDTA), with the inferior vena cava open in the cavity. Subsequently, the aorta was clamped, and the heart was isolated and perfused with EDTA buffer, perfusion buffer (130 mM NaCl, 5 mM KCl, 0.5 mM NaH2PO4, 10 mM HEPES, 10 mM Glucose, 10 mM BDM, 10 mM Taurine, 1 mM MgCl2), and collagenase buffer (0.5 mg/mL Collagenase 2, (Worthington, LS004176) 0.5 mg/mL Collagenase 4 (Worthington, LS004188)). After removing the clamp, heart tissues were gently pulled into 1 mm3 pieces using forceps and dissociated by gentle pipetting in a dish with the forementioned collagenase buffer. Following 1 hr of digestion, enzymes were deactivated by the addition of stop buffer (perfusion buffer with the addition of FBS). The resulting cell suspensions were filtered through a 100 μm cell strainer. Cardiomyocytes were gravity-settled for 20 min, and the supernatant containing other types of cells was then collected and centrifuged at 300 × g for 5 min at 4 °C. The resultant supernatant was discarded, and the cell pellet was resuspended for downstream analysis. Cells were stained with monoclonal antibodies for 30 min at room temperature and treated with Fcg-blocking antibody anti-mouse CD16/32 (Biolegend, 101302) for 10 min at 4 °C. The CD45+ gate was used for all immune cells. Neutrophils were gated as CD11b+Ly6G+. Macrophages (Mφ) were gated as CD11b+F4/80+ and divided by CD11b+F4/80+Ly6Chigh and CD11b+F4/80+Ly6Clow. B cells and T cells were gated by CD11b-B220+ and CD11b-TCR-β+, respectively. All samples were analyzed on a FACS Canto II Cell analyzer (BD Biosciences). Analysis of acquired data was performed with the FlowJo software (FlowJo 10.8.1).
The antibodies used were: BV421 anti-mouse CD45 (Biolegend, 103134, 1:50), BV510 anti-mouse CD11b (Biolegend, 101263, 1:100), APC anti-mouse B220 (Biolegend, 103212, 1:100), PE/Cy7 anti-mouse TCR-β (Biolegend, 109222, 1:100), BV605 anti-mouse Ly6C (Biolegend, 128036, 1:100), FITC anti-mouse Ly6G (Biolegend, 127605, 1:100), and PE anti-mouse F4/80 (Biolegend, 123109, 1:100).
Immunofluorescence staining of mouse hearts
For immunofluorescence staining, mouse hearts were fixed in 4% PFA at 4 °C overnight, embedded in OCT, and frozen sectioned at 10 μm thickness. A microwave oven was used to retrieve antigens with citric acid buffer. Then, all sections were permeabilized and blocked by a QuickBlock™ Blocking Buffer for Immunol Staining (Beyotime, P0260) for 1 hr at room temperature and incubated with primary antibodies at 4 °C overnight. The next day, sections were washed and incubated with Alexa-Fluor conjugated secondary antibodies and Hoechst 33342 for 1 hr at room temperature.
Multiplex immunofluorescence (mIF) analysis was performed according to a previous protocol using tyramide signal amplification (TSA)86. In brief, heart sections were treated with 3% H2O2 and underwent antigen retrieval as immunohistochemistry. Then incubated with primary antibodies and corresponding HRP-conjugated secondary antibodies (Servicebio, GB23303) and TSA-diluted FITC-tyramide (Servicebio, G1222).
Immunofluorescence images were captured using a Nikon A1R MP microscope. Immunofluorescence signals were analyzed using NIS-Elements 5.20.00 software from at least 5 visual fields and more than 2000 cells for each heart.
The primary antibodies included: rabbit anti-PCM1 (Abclonal, A16637, 1:500), rabbit anti-Ki67 (Abclonal, A23722, 1:1000), mouse anti-pHH3 (Proteintech, 66863-1-Ig, :1000), rabbit anti-Aurora B (Abclonal, A19539, 1:500), rabbit anti-phospho-AKT (Abmart, T40067, 1:200), rabbit anti-phospho-ERK1/2 (Abmart, TA1015, 1:200), rabbit anti-phospho-STAT3 (Abclonal, AP0715, 1:200) and rabbit anti-Vimentin (Abmart, T55134F, 1:200).
Wheat germ agglutinin staining
To measure cardiomyocyte cross-sectional area, heart sections were stained with 5 μg/mL Alexa Fluor 488-conjugated Wheat germ agglutinin (WGA) (Thermo Fisher Scientific, W11261) for 10 min at room temperature. The sections were washed with PBS, and the nuclei were counterstained with Hoechst 33342. The cross-sectional area of cardiomyocytes was measured by ImageJ software (Image J2, Fiji).
Masson’s trichrome staining
To measure cardiac fibrosis, heart sections were performed Masson’s trichrome staining as previously described76,79. Heart sections were rehydrated and immersed in a Weigert iron hematoxylin (ZSGB-BIO, ZLI-9610) solution for 10 min, followed by immersion in Biebrich Scarlet-Acid Fuchsin (Trichrome Stain Kit, TRM-1) for 15 min at room temperature. The sections were then incubated in a phosphotungstic-phosphomolybdic acid solution (Trichrome Stain Kit, TRM-1) for 5 min at room temperature. After immersion in aniline blue solution (Trichrome Stain Kit, TRM-1) for 10 min at room temperature and 1% acetic acid (Trichrome Stain Kit, TRM-1) for 1-2 min, the sections were dehydrated in an ethanol series and xylene and mounted using a neutral balsam mounting medium (Aladdin, N116470). With an Axio Observer Z1 widefield microscope, images of stained sections were acquired, and Blue-colored areas indicative of cardiac fibrosis were analyzed with ImageJ software (Image J2, Fiji).
TTC staining
Mouse hearts were quickly excised and perfused with ice-cold saline to remove any residual blood. Subsequently, the hearts were sectioned into transverse slices at a thickness of 1-2 mm. These slices were then incubated in 1% 2,3,5-triphenyltetrazolium chloride (TTC) solution (Sigma-Aldrich, T8877) at 37 °C for 30 min. The TTC solution was freshly prepared in phosphate-buffered saline (PBS) and adjusted to a pH of 7.4. The infarcted area (white) and the viable myocardium (red) were quantified using a digital camera and analyzed with ImageJ (Image J2, Fiji). The infarct size was calculated as the percentage of the infarct area relative to the total left ventricular area.
Echocardiography
Visual Sonics Vevo 3100 imaging system (Visual Sonics) was used to perform transthoracic echocardiography with a 30 MHz transducer (MX400) as previously described79,87. Two-dimensional targeted M-mode traces were obtained at the level of the papillary muscle in all mice anesthetized with 1.5% isoflurane in 100% oxygen. Left ventricular ejection fraction (LVEF), left ventricular fractional shortening (LVFS), left ventricular end-diastolic dimension (LVEDD), and left ventricular end-systolic dimension (LVESD) were measured using the cardiac echocardiography software and calculated by Vevo LAB (Vevo LAB 5.7.1) from three consecutive cardiac cycles.
Treadmill exhaustion test
A treadmill exhaustion test was performed88 on mice at 12 weeks after I/R. The running protocol started with an adaptation phase of 5 m/min for 5 min, followed by a test phase of 14 m/min with an increase of 2 m/min every 2 min until signs of fatigue appeared. The treadmill was set at an elevation angle of 20 degrees. The test was terminated when the mice failed to respond to consecutive fatigue stimuli (1.5 mA at 3 Hz) for 10 sec. Once fatigue was observed, the mice were promptly removed from the treadmill. Treadmill running time and distance were recorded and analyzed for all the mice tested.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8.4 software (Prism Inc., San Diego, CA, USA). Two groups were compared using a two-tailed unpaired Student’s t test as appropriate. When comparing more than two groups, a one-way analysis of variance (ANOVA) was used, followed by the post hoc Tukey test as appropriate. Individual data points or mean ± SEM are presented.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the RNA sequencing data have been deposited to the National Center for Biotechnology Information Sequence Read Archive (SRA) under accession number PRJNA1154450 and PRJNA1154419. Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD057702. Source data are provided in this paper.
References
Braunwald, E. The war against heart failure: the Lancet lecture. Lancet 385, 812–824 (2015).
Chen, Y. et al. Reversible reprogramming of cardiomyocytes to a fetal state drives heart regeneration in mice. Science 373, 1537–1540 (2021).
Xia, B. et al. The alternative splicing landscape of infarcted mouse heart identifies isoform level therapeutic targets. Sci. Data 11, 1154 (2024).
Humphries, S., Mars, K., Hofmann, R., Held, C. & Olsson, E. M. G. Randomized evaluation of routine beta-blocker therapy after myocardial infarction quality of life (RQoL): design and rationale of a multicentre, prospective, randomized, open, blinded endpoint study. Eur. Heart J. Open 3, oead036 (2023).
Steg, P. G. Routine beta-blockers in secondary prevention - on injured reserve. N. Engl. J. Med. 390, 1434–1436 (2024).
Wolfel, E. E. Effects of ACE inhibitor therapy on quality of life in patients with heart failure. Pharmacotherapy 18, 1323–1334 (1998).
Leong, D. P., McMurray, J. J. V., Joseph, P. G. & Yusuf, S. From ACE inhibitors/ARBs to ARNIs in coronary artery disease and heart failure (Part 2/5). J. Am. Coll. Cardiol. 74, 683–698 (2019).
Doenst, T. et al. PCI and CABG for Treating stable coronary artery disease: JACC review topic of the week. J. Am. Coll. Cardiol. 73, 964–976 (2019).
Sabatine, M. S. & Braunwald, E. Thrombolysis in myocardial infarction (TIMI) study group: JACC Focus seminar 2/8. J. Am. Coll. Cardiol. 77, 2822–2845 (2021).
Porrello, E. R. et al. Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080 (2011).
Aurora, A. B. et al. Macrophages are required for neonatal heart regeneration. J. Clin. Invest. 124, 1382–1392 (2014).
Li, Y., Li, H., Pei, J., Hu, S. & Nie, Y. Transplantation of murine neonatal cardiac macrophage improves adult cardiac repair. Cell. Mol. Immunol. 18, 492–494 (2020).
Liu, K. et al. Lineage tracing clarifies the cellular origin of tissue-resident macrophages in the developing heart. J. Cell Biol. 221, e202108093 (2022).
Ma, Y., Mouton, A. J. & Lindsey, M. L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 191, 15–28 (2018).
Bajpai, G. et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 24, 1234–1245 (2018).
Epelman, S. et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104 (2014).
Lavine, K. J. et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 111, 16029–16034 (2014).
Wong, N. R. et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity 54, 2072–2088.e2077 (2021).
Jia, D. et al. Cardiac resident macrophage-derived legumain improves cardiac repair by promoting clearance and degradation of apoptotic cardiomyocytes after myocardial infarction. Circulation 145, 1542–1556 (2022).
Giambelluca, S., Ochs, M. & Lopez-Rodriguez, E. Resting time after phorbol 12-myristate 13-acetate in THP-1 derived macrophages provides a non-biased model for the study of NLRP3 inflammasome. Front. Immunol. 13, 958098 (2022).
Zaman, R. et al. Selective loss of resident macrophage-derived insulin-like growth factor-1 abolishes adaptive cardiac growth to stress. Immunity 54, 2057–2071.e2056 (2021).
Gundry, R. L. et al. High efficiency differentiation of human pluripotent stem cells to cardiomyocytes and characterization by flow cytometry. J. Vis. Exp. 91, e52010 (2014).
Du, J. et al. A small-molecule cocktail promotes mammalian cardiomyocyte proliferation and heart regeneration. Cell Stem Cell 29, 545–558 (2022).
Li, Y. et al. gp130 Controls cardiomyocyte proliferation and heart regeneration. Circulation 142, 967–982 (2020).
Mohamed, T. M. A., Abouleisa, R. & Hill, B. G. Metabolic determinants of cardiomyocyte proliferation. Stem Cells 40, 458–467 (2022).
Li, X. et al. Inhibition of fatty acid oxidation enables heart regeneration in adult mice. Nature 622, 619–626 (2023).
Nguyen, P. D. et al. Interplay between calcium and sarcomeres directs cardiomyocyte maturation during regeneration. Science 380, 758–764 (2023).
Yu, J. S. & Cui, W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 143, 3050–3060 (2016).
Strash, N. et al. Human Erbb2-induced Erk activity robustly stimulates cycling and functional remodeling of rat and human cardiomyocytes. Elife 10, e65512 (2021).
Fang, Y. et al. Translational profiling of cardiomyocytes identifies an early Jak1/Stat3 injury response required for zebrafish heart regeneration. Proc. Natl. Acad. Sci. USA 110, 13416–13421 (2013).
Dong, W., Albers, J. J. & Vuletic, S. Phospholipid transfer protein reduces phosphorylation of tau in human neuronal cells. J. Neurosci. Res. 87, 3176–3185 (2009).
Lu, Y.-Z. et al. CGRP sensory neurons promote tissue healing via neutrophils and macrophages. Nature 628, 604–611 (2024).
Zhang, P. et al. NRP1 promotes prostate cancer progression via modulating EGFR-dependent AKT pathway activation. Cell Death Dis. 14, 159 (2023).
Dong, Y. et al. Solasonineinduces apoptosis and inhibits proliferation of bladder cancer cells by suppressing NRP1 expression. J. Oncol. 2022, 7261486 (2022).
Vuletic, S., Dong, W., Wolfbauer, G., Tang, C. & Albers, J. J. PLTP regulates STAT3 and NFkappaB in differentiated THP1 cells and human monocyte-derived macrophages. Biochim. Biophys. Acta 1813, 1917–1924 (2011).
Kumar, A., Yadav, R. P., Chatterjee, S., Das, M. & Pal, D. K. Integration of bioinformatics analysis to identify possible hub genes and important pathways associated with clear cell renal cell carcinoma. Urologia 91, 261–269 (2024).
Zou, Y., Fan, G. & Wang, X. Pre-clinical assessment of A-674563 as an anti-melanoma agent. Biochem. Biophys. Res. Commun. 477, 1–8 (2016).
Luo, M. et al. PD0325901, an ERK inhibitor, enhances the efficacy of PD-1 inhibitor in non-small cell lung carcinoma. Acta Pharm. Sin. B 11, 3120–3133 (2021).
Schust, J., Sperl, B., Hollis, A., Mayer, T. U. & Berg, T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Bio.l 13, 1235–1242 (2006).
Ali, S. R. et al. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc. Natl. Acad. Sci. USA 111, 8850–8855 (2014).
Tan, J. et al. Moderate heart rate reduction promotes cardiac regeneration through stimulation of the metabolic pattern switch. Cell Rep. 38, 110468 (2022).
Marchiano, S. et al. Gene editing to prevent ventricular arrhythmias associated with cardiomyocyte cell therapy. Cell Stem Cell 30, 396–414 (2023).
Hashimoto, H., Olson, E. N. & Bassel-Duby, R. Therapeutic approaches for cardiac regeneration and repair. Nat. Rev. Cardiol. 15, 585–600 (2018).
Consortium, A. B. et al. A biomarker framework for cardiac aging: the Aging Biomarker Consortium consensus statement. Life Med. 2, lnad035 (2023).
Kobayashi, H. et al. Regeneration of nonhuman primate hearts with human induced pluripotent stem cell-derived cardiac spheroids. Circulation, 150, 611–621 (2024).
Yap, L. et al. Pluripotent stem cell-derived committed cardiac progenitors remuscularize damaged ischemic hearts and improve their function in pigs. NPJ Regen. Med. 8, 26 (2023).
Peinkofer, G. et al. From early embryonic to adult stage: Comparative study of action potentials of native and pluripotent stem cell-derived cardiomyocytes. Stem Cells Dev. 25, 1397–1406 (2016).
Liu, Y. W. et al. Human embryonic stem cell-derived cardiomyocytes restore function in infarcted hearts of non-human primates. Nat. Biotechnol. 36, 597–605 (2018).
Feng, J. et al. Versican promotes cardiomyocyte proliferation and cardiac repair. Circulation 149, 1004–1015 (2024).
Gao, F. et al. Reduced mitochondrial protein translation promotes cardiomyocyte proliferation and heart regeneration. Circulation 148, 1887–1906 (2023).
Atkins, M. H. et al. Modeling human yolk sac hematopoiesis with pluripotent stem cells. J. Exp. Med. 219, e20211924 (2022).
Sturgeon, C. M., Ditadi, A., Awong, G., Kennedy, M. & Keller, G. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat. Biotechnol. 32, 554–561 (2014).
Nasseri Maleki, S. et al. Conditioned medium obtained from human amniotic membrane-derived mesenchymal stem cell attenuates heart failure injury in rats. Iran J. Basic Med. Sci. 22, 1253–1258 (2019).
Smolinska, V., Bohac, M. & Danisovic, L. Current status of the applications of conditioned media derived from mesenchymal stem cells for regenerative medicine. Physiol. Res. 72, S233–S245 (2023).
Kim, Y. J. et al. Increasing angiogenic efficacy of conditioned medium using light stimulation of human adipose-derived stem cells. Commun. Biol. 5, 957 (2022).
Lin, H. et al. Advances in mesenchymal stem cell conditioned medium-mediated periodontal tissue regeneration. J. Transl. Med. 19, 456 (2021).
Hayashi, H. et al. Apolipoprotein E-containing lipoproteins and LRP1 protect from NMDA-induced excitotoxicity associated with reducing alpha2-macroglobulin in muller glia. Invest. Ophthalmol. Vis. Sci. 62, 23 (2021).
Qu, H. et al. HIF‑3alpha affects preeclampsia development by regulating EVT growth via activation of the Flt‑1/JAK/STAT signaling pathway in hypoxia. Mol. Med. Rep. 23, 68 (2021).
Romeo, R., Boden-El Mourabit, D., Scheller, A., Mark, M. D. & Faissner, A. Low-density lipoprotein receptor-related protein 1 (LRP1) as a novel regulator of early astroglial differentiation. Front. Cell Neurosci. 15, 642521 (2021).
Tian, Y. et al. Extracellular Hsp90alpha and clusterin synergistically promote breast cancer epithelial-to-mesenchymal transition and metastasis via LRP1. J. Cell Sci. 132, jcs228213 (2019).
Verma, M. et al. Endothelial cell signature in muscle stem cells validated by VEGFA-FLT1-AKT1 axis promoting survival of muscle stem cell. Elife 13, e73592 (2024).
Wei, S. C., Tsao, P. N., Weng, M. T., Cao, Z. & Wong, J. M. Flt-1 in colorectal cancer cells is required for the tumor invasive effect of placental growth factor through a p38-MMP9 pathway. J. Biomed. Sci. 20, 39 (2013).
Yu, Y. et al. Neuropilin 1 (NRP1) positively regulates adipogenic differentiation in C3H10T1/2 cells. Int. J. Mol. Sci. 24, 7394 (2023).
Carew, N. T. et al. Loss of cardiomyocyte CYB5R3 impairs redox equilibrium and causes sudden cardiac death. J. Clin. Invest. 132, e147120 (2022).
Xie, S. Y. et al. USP28 Serves as a key suppressor of mitochondrial morphofunctional defects and cardiac dysfunction in the diabetic heart. Circulation 149, 684–706 (2024).
Paulus, W. J. & Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 62, 263–271 (2013).
Mohamed, T. M. A. et al. Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration. Cell 173, 104–116 (2018).
Zhang, H. et al. Generation of quiescent cardiac fibroblasts from human induced pluripotent stem cells for in vitro modeling of cardiac fibrosis. Circ. Res. 125, 552–566 (2019).
Feng, Y. H. et al. Differential regulation of resveratrol on lipopolysacchride-stimulated human macrophages with or without IFN-gamma pre-priming. Int. Immunopharmacol. 4, 713–720 (2004).
Sun, Y. et al. Myeloid dendritic cells in severe aplastic anemia patients exhibit stronger phagocytosis. J Clin. Lab Anal. 35, e24063 (2021).
Bargehr, J. et al. Epicardial cells derived from human embryonic stem cells augment cardiomyocyte-driven heart regeneration. Nat. Biotechnol. 37, 895–906 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Gu, Z. Complex heatmap visualization. iMeta 1, e43 (2022).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Wang, J. et al. CAR-Macrophage therapy alleviates myocardial ischemia-reperfusion injury. Circ. Res. 135, 1161–1174 (2024).
Qiu, S. et al. Building a highly efficient Streptomyces super-chassis for secondary metabolite production by reprogramming naturally-evolved multifaceted shifts. Metab. Eng. 81, 210–226 (2024).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Zhang, H. et al. Prophylactic supplementation with Bifidobacterium infantis or its metabolite inosine attenuates cardiac ischemia/reperfusion injury. Imeta 3, e220 (2024).
Lei, J. et al. FOXO3-engineered human mesenchymal progenitor cells efficiently promote cardiac repair after myocardial infarction. Protein Cell 12, 145–151 (2021).
Liu, X. et al. Transplantation of SIRT1-engineered aged mesenchymal stem cells improves cardiac function in a rat myocardial infarction model. J. Heart Lung Transplant. 33, 1083–1092 (2014).
Liu, C. et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221 (2011).
Wang, J. et al. Deficiency of flavin-containing monooxygenase 3 protects kidney function after ischemia-reperfusion in mice. Commun. Biol. 7, 1054 (2024).
Ackers-Johnson, M. et al. A simplified, langendorff-free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circ. Res. 119, 909–920 (2016).
Wang, J. et al. Prophylactic supplementation with lactobacillus reuteri or its metabolite GABA protects against acute ischemic cardiac injury. Adv. Sci. 11, e2307233 (2024).
Zhang, W. et al. Fully automated 5-plex fluorescent immunohistochemistry with tyramide signal amplification and same species antibodies. Laboratory Invest. 97, 873–885 (2017).
Lindsey, M. L., Kassiri, Z., Virag, J. A. I., de Castro Bras, L. E. & Scherrer-Crosbie, M. Guidelines for measuring cardiac physiology in mice. Am. J. Physiol. Heart Circ. Physiol. 314, H733–h752 (2018).
Schiattarella, G. G. et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 568, 351–356 (2019).
Acknowledgements
We would like to extend our sincere gratitude to the National Stem Cell Resource Center for providing the iPSC cell line. We are deeply appreciative of Prof. Chong Liu from Zhejiang University for supplying the MADM mouse line, Prof. Shiqiang Wang from Peking University for valuable suggestions on the detection of calcium handling, and Prof. Feng Liu and Dr. Guixian Liang from the Institute of Zoology of the Chinese Academy of Sciences for their valuable advice on macrophage differentiation. Our sincere thanks also go to Ms. Hua Qin, Ms. Qing Meng, and Ms. Guoli Hou from the Institute of Zoology of the Chinese Academy of Sciences for their technical support. This study was supported by the National Natural Science Foundation of China (92368112), the National Key Research and Development Program of China (2024YFA1802600, 2020YFA0113400), CAS Project for Young Scientists in Basic Research (YSBR-076), Initiative Scientific Research Program of the Institute of Zoology, Chinese Academy of Sciences (2024IOZ0103 and 2023IOZ0202), and State Key Laboratory of Membrane Biology and State Key Laboratory of Organ Regeneration and Reconstruction of the Chinese Academy of Sciences. Cartoon elements used in schematic diagrams in Figs. 1a, 2a, 3a, 4b, 5a, 6a, 7a, and Supplementary Fig. S7d were created using BioRender (https://BioRender.com/y79o016).
Author information
Authors and Affiliations
Contributions
Y.X. conducted all the cell experiments and assisted part of the mouse experiments, analyzed the data, and wrote the original draft. H.Z. performed mouse surgery and assessed cardiac function. X.L. executed immunofluorescence staining and quantification of tissue sections. P.X. analyzed RNA-seq and proteomic data. H.D. conducted multiplex immunofluorescence (mIF) analysis. Jiawan.W. performed flow cytometry analysis on immune cells. J.S. designed siRNA targeting specific genes. Y.L. carried out the IncuCyte test. Y.W. conducted and analyzed the treadmill exhaustion test. C.H. processed and interpreted Masson’s trichrome and wheat germ agglutinin staining of heart sections. H.F. conducted a calcium handling analysis. J.L. performed mass spectrometry analysis. Y.Z. and S.L. collected heart tissues and prepared sections. Z.G. and J.Z. managed project administration. J.B. measured ATP production. T.T., Q.G., T.W., and Jun.W. provided guidance on experimental details and manuscript preparation. M.S. supervised the project and wrote the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiao, Y., Zhang, H., Liu, X. et al. Medium from human iPSC-derived primitive macrophages promotes adult cardiomyocyte proliferation and cardiac regeneration. Nat Commun 16, 3012 (2025). https://doi.org/10.1038/s41467-025-58301-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58301-8
This article is cited by
-
Metabolic cardiomyopathies: untangling clinical heterogeneity with human stem-cell derived models
EMBO Molecular Medicine (2025)
