
Abstract
The persistent monkeypox outbreaks intensify the demand for monkeypox vaccines. Based on the mRNA vaccine platform, we conduct a systematic screening of monkeypox virus (MPXV) surface proteins from two types of viral particles, extracellular enveloped viruses (EVs) and intracellular mature viruses (MVs). This screening unveils 12 important antigens with diverse levels of neutralizing immunogenicity. Further assessment reveals that the combinations of 4, 8, and 12 of these antigens, namely Mix-4, Mix-8, and Mix-12, induce varying degrees of immune protection, with Mix-12 being the most potent. This finding demonstrates the significance of not only the level but also the diversity of the neutralizing antibodies in providing potent immune protection. Additionally, we utilize a T cell-epitope enrichment strategy, analyzing the complete proteome sequence of the MPXV to predict antigenic epitope-rich regions. Integration of these epitope-rich regions into a cellular immune-targeting antigen, named MPX-EPs, showcases that a cellular immune-targeting mRNA vaccine can independently confer immune protection. Furthermore, co-immunization with Mix-12 and MPX-EPs achieves complete protection against MPXV challenge. Overall, these results suggest an effective approach to enhance the immune protection of mRNA vaccines through the specific coordination of humoral and cellular immune responses.
Similar content being viewed by others
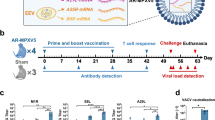

Introduction
Monkeypox, a rare but potentially severe zoonotic disease, is caused by the monkeypox virus (MPXV), a member of the orthopoxvirus family. MPXV has been recognized as a significant public health concern due to its similarity to a related deadly virus, the variola virus responsible for smallpox, and its sporadic outbreaks across 110 countries in 2022–20241, rapidly spreading among men who have sex with men (MSM)2,3. The persistence and development of monkeypox outbreaks underscore the importance of vaccine development to combat this emerging threat4. Notably, the JYNNEOS vaccine and the ACAM2000 vaccine designed against smallpox have received conditional approval from the United States Food and Drug Administration for the prevention of monkeypox5,6. Further vaccine development against MPXV has been based on the research conducted earlier on the smallpox virus vaccines7,8, especially in terms of identifying antigens that evoke protective immune responses against the vaccinia virus (VACV)9,10,11. Such insights provide a roadmap for monkeypox vaccine exploration. However, at present, systematic research on the key neutralizing antigens of the monkeypox virus is insufficient. Additionally, there have been no reports on the mechanisms of cellular immunity in mediating anti-MPXV immunity and the characterization of the related antigen design12,13,14,15.
In response to the growing demand for effective monkeypox vaccines, mRNA vaccine technology has emerged as a promising avenue. Inspired by the success of COVID-19 mRNA vaccines16,17, much attention has been dedicated to the development of mRNA-based monkeypox vaccines. While several monkeypox mRNA vaccines have already been reported18,19,20,21,22,23,24, there exists a critical gap in fully accounting for the polymorphism of monkeypox-neutralizing antibodies and the protective cellular immune response. The cellular immunity, often overlooked in the antigen designs for mRNA vaccines25,26,27,28, is essential for viral clearance and the establishment of a protective immune response during primary infections. T-cell immunity plays a pivotal role in combating VACV29, and hundreds of VACV-specific CD8+ T-cell epitopes have been identified in vaccine recipients30,31, monkeys32, and mice33,34,35,36,37. We presented here a significant step forward in the quest to develop a monkeypox vaccine by comprehensively screening the surface proteins of two types of monkeypox virus particles, named extracellular enveloped virus (EVs) and intracellular mature virus (MVs), identifying critical neutralizing antigens. Additionally, our study demonstrated the superior protective potency of a complex combination of antigens over a simple combination or a single antigen, highlighting the importance of diversified humoral immunity in vaccine efficacy. Further, we adopted an epitope-enrichment strategy to analyze the sequence of the complete MPXV proteome, leading to the design of a cellular immune-targeting antigen, MPX-EPs. The efficacy of this innovation highlights the potential of cellular immunity as an independent source of immune protection. As the findings of our study demonstrated, a coordinated approach that leverages both humoral and cellular immune responses holds promise for enhancing the efficacy of mRNA vaccines against monkeypox. In the face of persistent outbreaks and the evolving landscape of infectious diseases, the development of effective vaccines remains essential for global public health preparedness and response.
Results
Systematic screening of MPXV surface proteins for critical neutralizing antigens
MPXV displays distinct surface proteins in EVs and MVs, its two infectious mature forms. Surface proteins play a crucial role in inducing neutralizing antibodies against viruses, making them vital in vaccine designs. While previous studies on MPXV mRNA vaccines focused on counterparts of VACV antigens within MPXV18,19,20, we comprehensively screened all reported membrane proteins from both MVs and EVs for potential antigens (Fig. 1a).

a Schematic diagram of experimental design. Created in BioRender. Tian, C. (2025) https://BioRender.com/x66i748. 27 surface proteins of MPXV MVs and 6 surface proteins of MPXV EVs were selected and designed as mRNAs for vaccination (12 μg per dose). Collected sera after boosting vaccination (3 weeks interval) were subjected to MPXV-based neutralization assay. b Neutralizing potency of MPXV surface antigens. The neutralizing potency against MPXV in the sera (1:50 dilution, n = 5 mice per group) was evaluated with plaque reduction neutralization tests. The neutralizing ratios are indicated as mean ± SEM. The horizontal lines denoted the threshold for selecting the critical neutralization antigens. c Neutralizing antibodies titers (NT50) induced by MPXV surface antigens. The neutralization potencies of selected antigens (neutralizing ratio > 60%) were further confirmed by NT50. PBS-immunized groups were included as controls (n = 5). NT50 is indicated as mean ± SEM from the indicated number of biological repeats. Data are representative of two independent experiments with similar results (b, c). Source data are provided as a Source Data file.
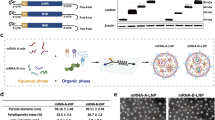
To evaluate the immunogenicity of these proteins, we constructed mRNAs expressing each of the surface proteins (Supplementary Fig. 1). The mRNAs were encapsulated into lipid nanoparticles (LNPs) to formalize into vaccines. LNPs were a classic four-component formulation that consisted of ionizable lipid (ALC-0315), DSPC, cholesterol, and PEGylated lipid (ALC-0159). The characterization of the LNPs was determined by transmission electron microscopy (TEM) (Supplementary Fig. 2a), dynamic light scattering (DLS) analysis (Supplementary Fig. 2b), and the zeta potential analysis (Supplementary Fig. 2c). These mRNA antigens were then used to immunize BALB/c mice at a dose of 12 μg per mouse, with a booster administered 3 weeks later. Two weeks after the booster, blood samples were collected from the mice to evaluate the levels of neutralizing antibodies against MPXV (hMpxV/China/GZ8H-01/2023). An over 60% inhibition of the MPXV infection by 1:50 diluted sera from the blood samples deemed the corresponding protein potent in inducing neutralizing antibodies. Based on this criterion, we identified 12 candidate antigens: A21, A29, M1, H3, A17, A30, A28, H2, I2, and G2 in MVs, and A35 and B6 in EVs (Fig. 1b and Supplementary Table 1). The neutralizing potency of anti-sera induced by the 12 candidate antigens was further characterized by measuring the 50% neutralization titer (NT50, Fig. 1c). Notably, several candidate antigens, including M1, H3, A29, A35, and B6, were previously reported as potential vaccine targets and have been employed in the development of MPXV mRNA vaccines by earlier studies38,39,40,41.
Enhancement of the protectivity of mRNA vaccine by multiple surface antigens
Similar to other poxviruses, MPXV possesses a large genome consisting of numerous genes42,43,44. Multiple surface proteins encoded by the MPXV genome play synergistic roles in infection and replication. Relying on a single surface protein as the vaccine target often fails to provide complete protection. Consequently, the mainstream approach has shifted towards developing multivalent vaccines, such as tetravalent or hexavalent multi-component vaccines45,46,47. Based upon the aforementioned screening of neutralizing antigens, we aimed to enhance the protective immune responses by incorporating multiple candidate antigens. We designed 4-valent, 8-valent, and 12-valent mRNA vaccines. The 4-valent vaccine (Mix-4) comprised 4 antigens (A29, M1, B6, and A35) that exhibited the highest neutralizing potential when evaluated individually. The 8-valent vaccine (Mix-8) included 8 candidate antigens (A29, M1, B6, A35, H3, A17, A30 and H2) with high and moderate neutralizing potential, and the 12-valent vaccine (Mix-12) incorporated all 12 antigens (A29, M1, B6, A35, H3, A17, A30, H2, A28, A21, G2 and I2) (Fig. 2a). These vaccines were formulated by an equal-ratio mixture of mRNAs encoding each component. Following formulation, BALB/c mice were immunized with a 12 μg dose of the mRNA mixture and received a booster vaccination 3 weeks later. Blood samples collected 2 weeks after the booster was used to harvest sera, of which NT50 of the sera against MPXV was measured. The group immunized with Mix-12 exhibited the highest titer of neutralizing antibodies, followed by the group receiving Mix-8, while the group immunized with Mix-4 had the lowest (Fig. 2b). Notably, all multi-component vaccines induced higher neutralizing antibody titers compared to those generated by single-antigen vaccines. We then examined the MPXV-specific IgG subclass responses elicited by these mRNA mixtures (Fig. 2c) and evaluated their protective efficacy using a passive immunization model. Young BALB/c mice (3 weeks old) received 200 µl of plasma from immunized mice via retro-orbital injection 2 h before being challenged with MPXV at a viral load of 1.0 × 106 PFU. The protective efficacy was characterized by the viral loads in the lungs, quantitated by a plaque assay at 4 days post-infection (Fig. 2d). Of note, the Mix-12 group demonstrated significantly lower viral loads in the lungs compared to the other groups. The association between antibody subtypes and the viral loads indicated that the IgG1 and IgG2a were associated with higher protective potency compared to other subtypes (Supplementary Fig. 3). Regarding the cellular immune response and associated protection potency, we observed strong IFN-γ induction by Elispot assays (Fig. 2e), along with notable immune protection conferred by CD8+ T cell infusion. Specifically, 2.0 × 107 CD8+ or CD4+ T cells sorted from immunized mice were transfused into naive BALB/c mice (3 weeks old), which were challenged with MPXV at a viral load of 1.0 × 106 PFU 6 h later. The viral loads in the lungs were quantitated by a plaque assay at 4 days post-infection. Immune protection against MPXV challenge was evident in the CD8+ T cell infusion group, with reduced viral loads in the lungs (Fig. 2f), while CD4+ T cell infusion did not show similar effects. The group receiving CD8+ T cell infusion from Mix-12 vaccinated mice demonstrated superior immune protection. Overall, plasma transfusion was more potent in inhibiting MPXV infection than T cell transfer, suggesting that more efficient cellular immune protection necessitates further antigen design efforts.

a Schematic diagram of the immunization and challenge experiments for multicomponent mRNA vaccines. Created in BioRender. Tian, C. (2025) https://BioRender.com/s30e801. The designed 4-valent, 8-valent, and 12-valent mRNA vaccines, comprising 4, 8, and 12 antigens, respectively. These vaccines were formulated by an equal-ratio mixture of mRNAs for each component and named as Mix-4, Mix-8, and Mix-12, accordingly. b Neutralizing antibody titers (NT50) induced by different mRNA vaccines. The neutralizing antibodies titers (NT50) induced by Mix-4, Mix-8, and Mix-12 were measured by plaque reduction neutralization tests. PBS-immunized groups were included as controls. NT50 is indicated as mean ± SEM. (n = 6 per group). c Heatmaps demonstrating IgG subclass levels in the serum from immunized mice (n = 6 per group). Heatmaps were created using the hiplot online platform (https://hiplot.com.cn/). d Protective potency of plasma passive transfusion. Three-week-old BALB/c mice (n = 6) were transfused with 200 μl of plasma from immunized mice and challenged with MPXV (1.0 × 106 PFU) via intranasal inoculation. Viral titers in the lungs were measured using a standard plaque assay. e Proliferation of epitope-specific splenocytes producing IFN-γ following stimulation. IFN-γ-producing epitope-specific splenocytes were assessed by ELISpot. Each point represents the mean of 3 technical replicates, with a limit of detection (LOD) of 10, and representative spots are shown. f Protective potency of CD8+ T cells and CD4+ T cells transfusion. (Created in BioRender. Tian, C. (2025) https://BioRender.com/h28r862). Three-week-old BALB/c mice (n = 6) were transfused with 2.0 × 107 CD8+ or CD4+ T cells sorted from immunized mice and intranasally inoculated with MPXV at the dose of 1.0 × 106 PFU. Mice were sacrificed 4 days post-infection, and lungs were harvested for viral load detection. Data are shown as mean ± SEM from individual mice (b, d, e, f). Two independent experiments were performed with 2 technical replicates (b–f). P values were determined by one-way ANOVA with Tukey’s multiple comparison post-hoc test. Source data are provided as a Source Data file.
Characterization of the immune response of MPX-EPs, a CTL specific antigen
Given the substantial role played by cytotoxic T-lymphocyte (CTL)-mediated cellular immune responses in clearing infected cells and its reported correlation between cellular immunity and protection against VACV12,48,49, we aimed to further optimize our vaccine design by enhancing the CTL-mediated immune response. To achieve this, we constructed MPX-EPs, an mRNA vaccine specifically designed to activate cellular immunity (Fig. 3a). MPX-EPs were designed by combining our previously established method for identifying CTL-epitope-enriched peptides from the viral proteome (Supplementary Fig. 4 and Supplementary Table 2) and leveraging mRNA-based antigen activation of CTLs50. In order to enhance the formation of CTL antigens, antigen presentation to CTLs, and activation of CD8+ T lymphocytes, we introduced a sequence encoding ubiquitin (Ub) at the N-terminus of the peptide encoded by MPX-EPs (Supplementary Fig. 5a)51. This sequence served to direct the peptide to proteasome degradation (Supplementary Fig. 5b). To characterize the immune response within the background of human HLA, we evaluated this mRNA vaccine in humanized HLA-A*02:01/DR1 mice52,53. The mice were immunized with 3 μg of each MPX-EPs mRNA and a booster vaccination 3 weeks apart. The activation of cellular immunity was characterized by analyzing splenocytes from immunized mice using flow cytometry, ELISpot, and intracellular cytokine staining (ICS) 3 weeks post-booster vaccination. Flow cytometry analysis showed an increase in CD45+CD8+ T lymphocytes, but no significant difference in CD45+CD4+ T lymphocytes (Fig. 3b). ELISpot experiments revealed elevated numbers of IFN-γ secreting cells in the spleens of MPX-EPs-immunized mice compared to the control group (Fig. 3c). ICS indicated higher frequencies of CD8+IFN-γ+TNF-α+ T cells in the spleens of MPX-EPs-immunized mice (Fig. 3d, e). The immune activation by individual CTL-epitope-enriched peptides from each MPXV protein was assessed using a reporter cell-based epitope expression system54,55. Predicted HLA-I epitope-enriched peptides were ectopically expressed in human 293 reporter cells respectively, which harbor a modified infrared fluorescence protein (IFP) with a granzyme B (GzB) cleavage sequence. Upon GzB cleavage, the IFP emits a fluorescent signal56. When co-cultured with CD8+ T cells isolated from immunized humanized HLA-A02:01/DR1 mice, all the CTL-epitope-enriched peptides successfully presented antigens via HLA-A02:01 in the reporter cells and activated cognate CD8+ T cells. The activation resulted in GzB-dependent fluorescence and CD8+ T cell-mediated apoptosis (Supplementary Fig. 6). In contrast, the ectopic expression of an unrelated MPXV protein did not activate CD8+ T cells from the immunized mice. Further, the immunogenicity of the specific epitopes predicted by our HLA-I-epitope identification method was evaluated with an HLA-A*02:01 tetramer assay. HLA-A*02:01-specific epitopes with the highest predicted affinity were synthesized and assembled into HLA-A*02:01 tetramers (Supplementary Table 3), which were used to stain CD8+ T cells from MPX-EPs-immunized transgenic mice. The HLA-A*02:01 tetramers containing predicted epitopes showed enhanced binding to CD8+ T cells compared to those containing an unrelated peptide (Supplementary Fig. 7). Results from the three assays indicated a successful activation of CD8+ T lymphocytes in the spleens of MPX-EPs immunized mice by the designed epitopes. The protective efficacy of the CTL-specific mRNA vaccine was further assessed by comparing mice that received a CD8+ T-cell depleting antibody or an isotype IgG2b control prior to the MPXV challenge. The absence of protective effects and the restoration of viral titers in the lungs upon CD8+ T cell depletion suggested a CD8+ T cell dependency of the protection (Fig. 3i). Analysis of viral load and histopathological changes indicated partial protection conferred by the cellular immunity-specific vaccine (Fig. 3i–k). Collectively, these results demonstrated that immunization with MPX-EPs elicited a robust CD8+ T cell-mediated immune response, leading to CD8+ T cell proliferation and cytokine secretion against MPXV, demonstrating its potential as a cellular immunity-targeting vaccine.

a Schematic diagram of the MPX-EPs mRNA design and evaluation. Created in BioRender. Tian, C. (2025) https://BioRender.com/b59j557. Predicted CTL epitopes enriched peptides were linked with A-A-Y motifs, with a sequence encoding ubiquitin (Ub) added to the N-terminus of the peptides. b–e Evaluation of the cellular immune response. The percentages of the lymphocyte subsets of CD45+CD4+ T cells (b, left) and CD45+CD8+ T cells (b, right) in the spleens of immunized HLA-A*02:01/DR1 transgenic mice were assessed by flow cytometry. The proliferation of MPX-EPs-specific, IFN-γ producing splenocytes was measured by ELISpot (c). Each point represents the mean of 3 technical replicates, with a lower limit of detection (LLOD) of 5, and representative spots were shown (c). The percentage of TNF-α and IFN-γ positive CD8+ T cells was measured (d, e). Representative flow cytometry plots are presented, with symbols representing the means of two technical replicates per individual mouse. f–h MPX-EPs-induced protection against MPXV. HLA-A*02:01/DR1 transgenic mice were intramuscularly vaccinated with two doses of MPX-EPs (3 μg/dose). At 20 days post-booster immunization, the immunized mice were injected (i.p.) with an anti-CD8-α antibody (to deplete CD8+ T cells) or an IgG2b isotype control antibody (without CD8+ T-cells depletion) (200 mg/mouse) twice with a 24 h interval. The mice were then infected with the MPXV (1.0 × 107 PFU/mouse). Viral titers after 4 days post-inoculation in lung tissues from HLA-A*02:01/DR1 transgenic mice, with and without CD8+ T cell depletion (n = 6), were determined by a plaque assay with two technical replicates (f). The LLOD was 20 PFU per gram of tissue. Lung tissue pathology was evaluated by HE staining (g) and pathological scoring (h), Scale bar is 200 μm. Statistical significance was calculated via two-sided unpaired t-test (b, c, e) or one-way ANOVA with Tukey’s multiple comparisons post-hoc tests (f, h). Experiments were repeated twice independently, and similar results were obtained. Both independent experiments contain 3 technical repeats (b–h). Data are presented as mean ± SEM (b, c, e, f, h). Source data are provided as a Source Data file.
Synergistic activation of humoral and cellular immunity to protect humanized HLA-transgenic mice from infection with MPXV
Having observed that the two types of mRNA vaccines we designed effectively induced protective humoral and cellular immune responses respectively, we next investigated whether the combination of the mRNA vaccines could synergistically enhance the protective efficacy and confer full protection. To test this hypothesis, we vaccinated humanized HLA-A*02:01/DR1 mice with two formulations: MPX-m-Mix and MPX-p-Mix, respectively, and assayed both humoral and cellular immune responses (Fig. 4a). MPX-m-Mix contained 12 μg of Mix-12 mRNA and 3 μg of MPX-EPs mRNA, while MPX-p-Mix consisted 12 μg of recombinant proteins corresponding to those encoded by Mix-12 mRNAs and 3 μg of synthesized peptides derived from the sequence of MPX-EPs, with PBS as the control. The same reagents were administered again 3 weeks later as a booster. To evaluate the humoral immune responses, lymph nodes, and blood samples were collected 7 days post-booster. First, we examined the frequencies of T follicular helper (Tfh) cells (CD4+CD185+PD-1+) and germinal center B (GC B) cells (B220+CD95+GL-7+) in the lymph nodes using flow cytometry. Both cell types were more frequent in mice that received MPX-m-Mix or MPX-p-Mix compared to PBS-treated control mice, indicating robust activation of B cells in the lymph nodes (Supplementary Fig. 8a, b). The activation of the humoral immune response was further characterized by measuring viral-specific and neutralizing antibodies in sera harvested from the immunized mice. Compared with the control group, MPX-m-Mix immunized mice had a higher titer of virus-specific antibodies in the sera, whereas MPX-p-Mix immunized mice showed slightly lower antibody levels (Fig. 4b). This demonstrated the robust immunogenicity of the humoral-targeting component of the combinational vaccine. Considering that MPXV infection was introduced intranasally in this study, we analyzed antibody subclasses specific to respiratory tissues by ELISA. Results indicated that IgA and IgG2a titers are associated most strongly with the reduction in viral loads in the lungs (Supplementary Fig. 9a, b). Consistent with virus-specific antibody titers, the sera from MPX-m-Mix immunized mice exhibited the highest titer of neutralizing antibodies against MPXV (Fig. 4c). Longitudinal analysis at 40, 90, and 150 days post-boost showed that MPX-m-Mix-immunized mice maintained stably higher levels of virus-specific and neutralizing antibodies compared to MPX-p-Mix-immunized mice (Supplementary Fig. 12a, b). Regarding the cellular immune response, splenocytes were isolated 21 days post-booster and analyzed. Both MPX-m-Mix and MPX-p-Mix groups showed significant increases in the ratios of CD4+ and CD8+ T cells to total T lymphocytes (Supplementary Fig. 10a, b). Furthermore, ELISpot assays revealed increases in IFN-γ-secreting splenocytes (Fig. 4d) and IL-4-secreting splenocytes (Fig. 4e) in mice immunized with the synergistic stimulation strategy compared to control mice. Notably, the MPX-m-Mix group showed a higher number of IFN-γ specific spots and fewer IL-4 specific spots compared to the MPX-p-Mix group. Long-term assessment at 150 days post-boost vaccination demonstrated a similar pattern, with MPX-p-Mix showing prolonged cellular immune response (Supplementary Fig. 12c). In addition, ICS confirmed that the frequencies of the viral peptide-stimulated splenocytes from the MPX-m-Mix-immunized mice capable of releasing the cytokines TNF-α and IFN-γ were higher than those from MPX-p-Mix-immunized or PBS inoculated transgenic mice (Supplementary Fig. 10c, d). Taken together, these results indicated that immunization with MPX-m-Mix induced robust antigen-specific cellular immune responses in the humanized animals. Both MPX-m-Mix and MPX-p-Mix groups displayed a Th1-biased immune response (Fig. 4f). To assess memory immune responses, we examined MPXV-specific memory immune cells. Notably, MPX-m-Mix immunization significantly expanded the CD44+CD62L-CD8+ effector memory T cells (Tem) population (Supplementary Fig. 11a, b) suggesting strong memory cell activation. Finally, lung tissue-resident immune responses were evaluated by isolating CD8+CD69+CD103+ T cells from the lungs of immunized mice. In vitro stimulation assays revealed that MPX-m-Mix induced the highest level of antigen-specific cytokines, including IFN-γ, TNF-α, GM-CSF, IL-2, and Granzyme B, indicating strong CTL activation (Fig. 4g, h and Supplementary Fig. 13).

a Schematic diagram of the dual immunization design using Mix-12 and MPX-EPs antigens. HLA-A*02:01/DR transgenic mice were immunized with MPX-m-Mix (containing 3 μg MPX-EPs and 12 μg Mix-12 per mouse), MPX-p-Mix (containing 12 μg proteins from Mix-12 and 3 μg peptides from MPX-EPs per mouse), or PBS with 8.7% sucrose as a control. A booster vaccination was administered 3 weeks later. Blood and lymph nodes were collected 1-week post-booster for humoral immune response analysis, while cells from lungs and spleens were collected 3 weeks post-booster for cellular immune response evaluation. Immunized mice (n = 5 per group) were challenged with MPXV virus (1.0 × 107 PFU) 3 weeks after the booster vaccination. b, c Antibody production following immunization against MPXV. MPXV-specific IgG antibodies in the sera collected 21 days after the booster vaccination were detected with ELISA. Neutralizing antibody titers (NT50) were determined by plaque reduction neutralization test. d–f Th1-biased cellular immune response induced by MPX-Mix. Splenocytes from immunized HLA-A*02:01/DR1 transgenic mice (n = 5) collected 21 days post-booster immunization were re-stimulated ex vivo and subjected to IFN-γ ELISpot (d) (lower limit of detection (LLOD) = 20) and IL-4 ELISpot (e) (LLOD = 2), with the correlation of IL-4- and IFN-γ-secreting cells shown in the scatter plot (f). g–h Analysis of lung-resident CD8+ T cells from immunized mice (n = 5 per group). Lung cells were stained with anti-CD45-Alexa Fluor™ 700, CD8-PerCP-Cyanine5.5, CD69-FITC, and CD103-AF594. The frequencies of CD45+CD8+CD69+CD103+ T cells were measured by flow cytometry, with the representative flow cytometry plots presented. i–k Protection conferred by MPX-m-Mix or MPX-p-Mix against MPXV. Viral loads in the lungs (i) and lung pathology (j, k) were evaluated at 4 days post MPXV inoculation (i.n., 1.0 × 107 PFU, n = 5 mice per group), Scale bar is 200 μm. Data are representative of two independent experiments with three technical replicates (b–k). Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons (n = 5). Data are presented as mean ± SEM (b–e, h, I, k). Source data are provided as a Source Data file.
To evaluate the protective efficacy of the vaccines using the synergistic stimulation strategy, another set of mice was challenged with MPXV 3 weeks after the booster. Viral loads in the lungs were evaluated at 4 days post-infection by a plaque assay. Compared to the control treatment, immunization with MPX-m-Mix fully prevented MPXV infection in the lungs (Fig. 4i). Consistently, immunized mice did not exhibit any significant pathological changes (Fig. 4j, k). In contrast, while the MPX-p-Mix immunized mice showed reduced viral loads and fewer pathological changes in the lungs, they did not achieve the same level of protection as those immunized with MPX-m-Mix. Overall, the dual mRNA vaccination strategy effectively prevented MPXV infection in HLA transgenic mice. MPX-m-Mix achieved complete protection and outperformed the individual immune activation strategies targeting either humoral or cellular responses alone, as well as the recombinant protein vaccine MPX-p-Mix.
Discussion
The successful application of mRNA vaccine technology in COVID-19 vaccines has opened new avenues for the development of vaccines against other viral diseases, including monkeypox (MPXV)23,57,58. In this study, we proposed a multicomponent mRNA vaccine strategy for the development of a monkeypox vaccine. Recent studies have shown that mRNA vaccine technology platform has been explored for multivalent MPXV antigens and can induce potent MPXV-specific antibody and T-cell responses, suggesting that it is a promising strategy for controlling the monkeypox pandemic18,19,20,21,59,60,61,62,63,64,65. Inspired by this success, we systematically evaluated MPXV surface proteins and identified 12 candidate antigens: A21, A29, M1, H3, A17, A30, A28, H2, I2, and G2 in MVs, and A35 and B6 in EVs. These candidate antigens were categorized into three groups based on their potency to induce neutralizing antibodies: high, moderate, and low neutralizing-potential antigens. In order to enhance the diversity of neutralizing antibodies induced by the mRNA vaccine, rather than solely increasing the levels of neutralizing antibodies within the immunized individuals, we explored combinations of the candidate antigens with different neutralizing potentials. Our findings suggest that increasing antigen diversity, even by including low-neutralization-potential antigens, enhanced the overall protective efficacy of the humoral immune response.
In addition to neutralizing antibodies, robust and broad cellular immune responses could provide potent protection against orthopoxviruses66. CTLs play a crucial role in clearing virus-infected cells, preventing further viral invasion, and mitigating disease progression. While the importance of CTLs has been validated in immune defense against various viral infections67,68,69,70, their contribution to the immune protection against MPXV, especially their role in clearing MPXV-infected cells, remains unclear. Currently, the consensus is that both neutralizing antibodies and virus-specific CTLs are essential for preventing viral infections25,71,72,73. Although significant progress has been made in identifying CTL epitopes for MPXV, the application of these epitopes in antigen design, particularly in the design of mRNA vaccine antigens, remains underexplored28,74,75,76. In our previous research on the Zika virus (ZIKV), we designed a candidate DNA vaccine targeting the viral NS3 protein, intended to induce a predominately CD8+ CTL response51. This vaccine, incorporating Ub and rearranged ZIKV NS3 protein, successfully elicited CTLs without generating NS3-specific antibodies. Similarly, in the context of SARS-CoV-2, a strong and comprehensive cellular response—beyond neutralizing antibodies—plays a critical role in conferring effective immunity against COVID-1977,78,79. In the development of the COVID-19 mRNA vaccine, we proposed the conception of the CTL epitope enrichment region for the design of mRNA antigen targeting cellular immunity50. Building upon this approach, we constructed MPX-EPs, an mRNA vaccine antigen specifically designed to activate CTL-associated protective immunity against MPXV, demonstrating its potency in protecting humanized mice from MPXV challenge. This antigen was designed by incorporating the identified CTL epitope enrichment region from the MPXV proteome. In order to enhance the formation of CTL-specific antigens, antigen presentation to CTLs, and activation of CD8+ T lymphocytes, we introduced a Ub at the N-terminus of the peptide encoded by MPX-EPs. This sequence facilitates proteasome degradation and functional epitope formation. To characterize the immune responses in a human-relevant setting, we evaluated this mRNA vaccine in humanized HLA-A*02:01/DR1 mice. Of note, by stimulating lymphocytes in mice with functional epitopes identified through the predictive analysis, we detected specific CD8+ T cell responses by intracellular staining and ELISpot assays. Among the validated CD8+ T cell epitopes, one had been previously reported80, while the others were characterized in this study. These findings not only validate our antigen design strategy but also provide different insights into MPXV immunity, demonstrating the potential of leveraging CTL-targeting antigens for enhanced protection against orthopoxviruses.
Moreover, we successfully synergized the activation of both humoral and cellular immune responses to achieve superior protection by co-administering surface antigen mRNAs with the CTL-targeting MPX-EPs. In this study, mice immunized with the dual-mRNA vaccine (MPX-m-Mix) were compared to those receiving a mixture of the corresponding recombinant protein antigens and peptide antigens (MPX-p-Mix). The results demonstrated that mRNA immunization offered significant advantages in activating both humoral and cellular immune responses, and the effect was long-lasting. Over a 150-day observation period, the MPX-m-Mix group exhibited higher levels of humoral responses, particularly tissue-resident MPXV-specific antibody responses, while also eliciting stronger memory cellular immune responses. The superior immune protection observed in MPXV challenge experiments, the epitope-specificity of lung tissue-resident CTLs, and the more pronounced Th1 bias of the cellular response suggest that our mRNA vaccine design provides a dual advantage in both safety and efficacy.
In conclusion, our vaccine strategy had two major advantages over previous monkeypox vaccine designs: the diversification of neutralizing antibodies and the induction of potent cellular immunity. Contemporary vaccine research tends to emphasize the augmentation of neutralizing antibody levels as the primary mechanism of protection. In contrast, our findings indicate that augmenting the diversity of neutralizing antibodies instead of merely increasing their titers, significantly enhances the protective efficacy of the humoral immune response. Furthermore, our mRNA vaccine activated robust cellular immunity, demonstrating not only the protective potential of an independent cellular immune response but also its synergy with the humoral immune responses. By incorporating a CTL-targeting antigen alongside neutralizing antigen components, our approach led to a more durable and comprehensive adaptive immune response, effectively protecting against high-titer MPXV challenges. Beyond MPXV, our findings have broader implications for the design of next-generation vaccines against emerging and re-emerging infectious diseases. This study underscores the importance of rational antigen selection, balanced immune activation, and a strategic combination of humoral and cellular immune targets in vaccine development. By shifting the focus from simply increasing antibody levels to designing vaccines that leverage immune diversity and synergy, our approach provides a framework for improving vaccine efficacy, particularly for pathogens where cellular immunity plays a crucial role. These insights could be instrumental in guiding future vaccine strategies against orthopoxviruses and other viral infections with pandemic potential.
Methods
Ethics statement
All animal care and viral-related experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Shenzhen Bay Laboratory (BACG202101) and the ethics committee of Changchun Veterinary Research Institute (Approval number: IACUC of AMMS-11-2024-028).
Cells, viruses, and plasmids
Vero E6 cells (Cat. No# CRL-1586) and HEK293T cells (Cat. No# CRL-3216) were obtained from the American Type Culture Collection (ATCC). All cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with GlutaMAX (Gibco, Cat. No# 10566016) supplemented with 10% fetal bovine serum (FBS) (PAN-Biotech, Cat. No# ST220622) and 100 units/mL of penicillin-streptomycin. The MPXV virus (Genbank: PP778666.1) used in this study was isolated from a patient in Guangzhou, China. The virus was propagated in Vero E6 cells that were cultured in DMEM (Sigma-Aldrich) containing 10% fetal bovine serum (Invitrogen), 50 U/ml of penicillin, and 50 µg/ml of streptomycin. All experiments with infectious MPXV virus were conducted in a biosafety level 3 laboratory. The plasmids encoding MPXV surface proteins or CTL-specific antigens were constructed and managed within our laboratory.
Animals
BALB/c mice and HLA-A*02:01/DR1 transgenic mice were chosen for vaccination and/or challenge experiments. All mice were housed in standard ventilated cages (5–6 mice per cage) under specific pathogen-free (SPF) conditions. The animal room was maintained on a 12-h light/dark cycle (lights on at 7:00 A.M.) at an ambient temperature of 22 ± 2 °C and a relative humidity of 50–60%. Food and water were provided ad libitum, and bedding was replaced twice weekly. All animal care and experimental procedures were conducted following the approval of the Institutional Animal Care and Use Committee (IACUC) of Shenzhen Bay Laboratory and the ethics committee of Changchun Veterinary Research Institute.
Analysis of functional MPXV HLA-I epitopes and identification of epitope-enriched fragments
The NetMHCpan 4.1 server (http://www.cbs.dtu.dk/services/NetMHCpan/) was employed to predict potential HLA-I epitope locations within all structural and nonstructural proteins of the MPXV strain (GenBank: ON563414.2). All potential epitopes with predicted affinity values (IC50) less than 20 nM were included for further analysis. Epitope-enriched fragments were characterized as fragments of MPXV proteins containing over 20 such epitopes per 100 amino acids.
Generation of modified mRNA
The mRNAs were synthesized in vitro through T7 polymerase-mediated DNA-dependent RNA transcription, incorporating 1-methylpseudoUTPs in place of UTPs. To enhance the stability and translation efficiency of the mRNAs, modifications including a 5′-UTP cap and a 3′ poly-A tail were introduced using the ScriptCap™ Cap 1 Capping System and A-Plus™ Poly (A) Polymerase Tailing Kit. For surface antigens, the mRNA templates were constructed so that the mRNA sequences comprised a signal peptide from tissue plasminogen activator (MDAMKRGLCCVLLLCGAVFVSAS), the viral extracellular region of the surface protein, and a His tag (HHHHHH) inserted before the stop codon in sequential order. In the case of MPX-EPs, epitope-enriched peptides were linked using A-A-Y motif linkers, with a ubiquitin sequence featuring a G76A amino acid modification at the N-terminus and a V5 tag sequence (GKPIPNPLLGLDST) at the C-terminus.
Validation of naked mRNA expression
To verify the in vitro expression from naked mRNA, HEK293T cells were transfected with each mRNA using TransIT®-mRNA Reagent (Mirus Bio, Cat. No# MIR-2250). HEK293T cells were seeded in 12-well plates 24 h prior to transfection, and the medium was changed to Opti-MEM (Gibco, Cat. No# 31985062) 2 h before transfection. Each well was transfected with 0.5 μg of mRNA mixed with TransIT®-mRNA Reagent. For surface antigens, 72 h post-transfection, supernatants were collected for Western blot analysis. The antibodies used in the Western blot analysis included a horseradish peroxidase (HRP)-conjugated anti-His antibody (Sino Biological, Cat. No# 105327-MM02T-H, 1:2000).
For MPX-EPs, MG132 (APEXBIO, Cat. No# A2585) was added to the cell culture medium 48 h post-transfection. Six hours later, the cells were collected, and treated with trypsin-EDTA (Gibco, Cat. No# 25300120) to create single cells, fixed, and permeabilized using Cytofix/Cytoperm reagent (BD Biosciences, Cat. No# BDB554714) following the manufacturer’s instructions. The cells were stained with anti-V5 tag mouse mAb (Cell Signaling Technology, Cat. No# 80076S, 1:1000) and subsequently with goat anti-mouse IgG H&L (Cy3®) (Abcam, Cat. No# ab97035, 1:1000). Mock-transfected cells and cells not treated with MG132 served as two different controls. The stained cells were analyzed using flow cytometry (BD FACSAria III cell analyzer).
Preparation and characterization of LNP
LNP (Lipid Nanoparticle) formulations were prepared following a modified protocol derived from Precision Nanosystems. Initially, ionizable lipid (ALC-0315), DSPC, cholesterol, and PEGylated lipid (ALC-0159) were dissolved in ethanol with a weight ratio of 48%/10%/40%/2%. This lipid mixture was combined with citrate buffer (100 mM, pH 5.0) containing mRNA at a concentration of 174 μg/mL using microfluidic cartridges. The aqueous solution and the ethanol solution were mixed rapidly at an aqueous-to-ethanol ratio of 3/1 by volume with a weight ratio of 40/1 (total lipids/mRNA). The resulting formulations were concentrated using 30-kDa Amicon Ultra Centrifugal Filters, passed through a 0.22-μm filter, and stored at 4 °C until needed. The concentrations of all mRNA formulations were verified using the Quant-iT RiboGreen RNA Kit (Invitrogen, Cat. No# R11490). Each formulation exhibited an endotoxin level below 1 EU/ml. For the transmission electron microscope test, the prepared LNPs@mRNA was dialyzed against Milli-Q water for 2 h. Then the dialyzed LNPs@mRNA was diluted 100 times using Milli-Q water and 10 μL of the sample was dropped onto a carbon-coated copper grid. Samples were dried under a vacuum to remove water. Tests were carried out on a Hitachi HT-7700. For dynamic light scattering analysis and zeta potential analysis, dialyzed LNPs@mRNA was diluted 10 times using Milli-Q water, and tests were carried out on Zetasizer Nano-ZS90 (Malvern Instruments, UK).
Animal immunization
-
a.
BALB/c mice (female, 6–8 weeks) were randomly allocated into groups (n = 5). First, mice were intramuscularly (I.M.) immunized with mRNA-LNP for each MPXV surface protein (12 μg/100 μl/mouse), or PBS (control), and boosted at 3 weeks with the same immunogens (I.M.). 14 days after the 2nd immunization, sera were collected to detect neutralizing antibodies of MPXV.
-
b.
BALB/c mice (female, 6–8 weeks) were randomly allocated into groups (n = 12). First, mice were intramuscularly (I.M.) immunized with mRNA-LNP mixtures of MPXV surface antigens (12 μg/100 μl/mouse), or PBS (control), and boosted at 3 weeks with the same immunogens (I.M.). 14 days after the 2nd immunization, sera were collected from 6 mice per group to detect neutralizing antibodies. The rest immunized mice were then subjected to challenge experiments, described below.
-
c.
HLA-A*02:01/DR1 transgenic mice (female, 4–5 weeks) were randomly allocated into groups (n = 11). MPX-EPs diluted in PBS with 8.7% sucrose were injected intramuscularly (I.M., 3 μg per mouse) following a prime and boost regimen with an interval of 3 weeks, PBS with 8.7% sucrose served as the control. The immunized mice were further processed for subsequent experiments as described below.
-
d.
HLA-A*02:01/DR1 transgenic mice (female, 4–5 weeks) were randomly allocated into groups (n = 25) and vaccinated via the intramuscular route with either the MPX-m-Mix, which contained MPX-EPs (3 μg per mouse) and a surface antigen mRNA mix (12 μg per mouse). The MPX-p-Mix, which was formulated with an equal-ratio mixture of 12 recombinant surface antigens (A29, M1, B6, A35, H3, A17, A30, H2, A28, A21, G2, and I2, 12 μg per mouse), the MPX-EPs-derived synthetic peptide pool (3 μg per mouse) and Alum (Alhydrogel® adjuvant 2%, 150 μg/mouse, InvivoGen) or PBS with 8.7% sucrose as buffer controls. The same doses were administered again for a booster immunization at 3 weeks post-priming vaccination. Serum samples were collected from 5 mice per group at 21 days, 40 days, 90 days, and 150 days after the boost vaccination and analyzed as described below. Lymph nodes were collected from 5 mice per group at 7 days after the boost vaccination and analyzed as described below. Another 5 mice per group were sacrificed at 21 days and 150 days post-booster vaccination to analyze T cell immune responses as described below. The remaining mice were further processed for subsequent challenge experiments as described below.
ELISA analysis of MPXV-specific IgG and subtypes
To detect MPXV-specific antibodies in immunized sera or BALF collected from immunized mice, plates were coated with purified MPXV at a concentration of 1 μg/ml in PBS, followed by sequential addition of serially diluted serum samples and HRP-conjugated anti-mouse IgG (1:5000) antibodies (Invitrogen, Cat. No# A16066), HRP-conjugated anti-mouse IgG1 (1:5000) antibodies (Invitrogen, Cat. No# PA1-74421), HRP-conjugated anti-mouse IgG2a (1:5000) antibodies (Invitrogen, Cat. No# A-10685), HRP-conjugated anti-mouse IgG2c (1:5000) antibodies (Abcam, Cat. No# ab97255), HRP-conjugated anti-mouse IgG2b (1:5000) antibodies (Invitrogen, Cat. No# SA5-10266), HRP-conjugated anti-mouse IgG3 (1:2000) antibodies (Invitrogen, Cat. No# M32607), or HRP-conjugated anti-mouse IgA (1:1000) antibodies (Proteintech, Cat. No#SA00012-7) for 1 h at 37 °C. The plates were sequentially incubated with the substrate TMB (3,3′,5,5′-tetramethylbenzidine) (Sigma, Cat. No# T0440) and then H2SO4 (1N) to stop the reaction. The absorbance at 450 nm was measured on a microplate reader.
Plaque reduction neutralization test (PRNT)
The neutralizing activity of serum samples from immunized mice against the authentic MPXV was evaluated using a plaque formation-based neutralization assay. The Vero E6 cells were seeded at a density of 1.0 × 105 cells per well and cultured for 2 h at 37 °C. Serial dilutions of the serum samples were prepared in 96-well plates by a factor of two and then incubated with a viral solution containing ~00 PFU of MPXV at 37 °C for 2 h. The serum-virus mixture was added to the Vero E6 cells. Plaque formation was examined 3 or 4 days post-infection, and neutralizing titers were calculated from the serial dilutions of sera that exhibited plaque formation reduction.
ELISpot assays of IFN-γ and IL-4
ELISpot assays were conducted utilizing mouse IFN-γ (Abcam, Cat. No# ab64029) and IL-4 ELISpot kits (Mabtech, Cat. No# 3311-4HPW-10) following the manufacturer’s instructions. The enumerated splenocytes were ex vivo restimulated with a mixture of 15-amino-acid overlapping peptide pool (at 2 μg/ml per peptide) or the DMSO control. Subsequently, spots were counted using an ELISpot reader (iSpot).
Profiling of lymphocytes in the spleens, the lungs, and the lymph nodes
The spleens and lungs from immunized HLA-A*02:01/DR1 transgenic mice (n = 5 under animal immunization c. and n = 5 under animal immunization d.) were harvested on Day 21 post-second immunization. The spleens and lungs were briefly lysed, and red blood cells were removed by suspension in ammonium-chloride-potassium buffer. The splenocytes were then washed and resuspended in RPMI medium 1640 supplemented with 10% fetal bovine serum. Cells were first stained with Ghost Dye™ Red 780 (TONBO Biosciences, Cat. No# 13-0865-T100) to exclude dead cells, and then stained with a cocktail of the following fluorescently labeled antibodies: anti-CD45-Alexa Fluor™ 700 (Invitrogen, Cat. No# 56-0451-82), anti-CD8-PerCP-Cyanine5.5 (TONBO Biosciences, Cat. No# 65-0081-U100), anti-CD44-APC (Biolegend, Cat.No# 103012), anti-CD62L-BV421 (Biolegend, Cat.No# 104436), and anti-CD107a-BV711 (BioLegend, Cat. No# 121631). For ICS, the cells underwent additional fixation and permeabilization using the Cytofix/Cytoperm reagent (BD Biosciences, Cat. No# BDB554714) according to the manufacturer’s instructions and staining with the anti-IFN-γ-PE (TONBO Biosciences, Cat. No# 50-7311-U100), anti-TNF-α-BV421 (BioLegend, Cat. No# 506328), anti-Granzyme B-FITC (BioLegend, Cat. No# 372206), and anti-CD107a-BV711 (BioLegend, Cat. No# 121631). The lung cells were washed and resuspended in RPMI medium 1640 supplemented with 10% fetal bovine serum. Cells were first stained with Ghost Dye™ Red 780 (TONBO Biosciences, Cat. No# 13-0865-T100) to exclude dead cells, and then stained with a cocktail of the following fluorescently labeled antibodies: anti-CD45-Alexa Fluor™ 700 (Invitrogen, Cat. No# 56-0451-82), anti-CD8-PerCP-Cyanine5.5 (TONBO Biosciences, Cat. No# 65-0081-U100), anti-CD69-FITC (Biolegend, Cat.No# 104506), anti-CD103-AF594 (Biolegend, Cat.No# 121428). The inguinal lymph nodes from immunized mice (n = 5, HLA-A*02:01/DR1 transgenic mouse under animal immunization d.) were harvested at day 7 post-second immunization and pooled. The nodes were homogenized into single-cell suspensions using a syringe plunger and passed through a 70 µm cell strainer in complete RPMI 1640 media containing 10% fetal bovine serum. Cells were washed and resuspended in fresh RPMI-10% FBS media in a 96-well round-bottomed plate for staining. Cells were first stained with Ghost Dye™ Red 780 (TONBO Biosciences, Cat. No# 13-0865-T100) for dead cells, and then stained with a cocktail of the following fluorescently labeled antibodies: anti-CD45-Alexa Fluor™ 700 (Invitrogen, Cat. No# 56-0451-82), anti-CD4-FITC (Tonbo Biosciences, Cat. No# 35-0042-U100), anti-CD185-Brilliant Violet 605™ (BioLegend, Cat. No# 145513), anti-PD-1-Brilliant Violet 421™ (BioLegend, Cat. No# 135218), anti-B220-PerCP-Cyanine5.5 (TONBO Biosciences, Cat. No# 65-0452-U100), anti-CD95-PE (BioLegend, Cat. No# 152608), anti-GL-7-APC (BioLegend, Cat. No# 144618) in the cell staining buffer (BioLegend, Cat. No# 420201) and incubated for 20 min in the dark at room temperature. Cells were then washed and resuspended in cell stain buffer and were analyzed using BD Aria III cell analyzer. The resulting data were processed using FlowJo software V_10.
Profiling of lung tissue-resident immune responses
HLA-A*02:01/DR1 transgenic mice were vaccinated intramuscularly with two doses of MPX-p-Mix or MPX-m-Mix at a 3-week interval. Twenty-one days post boost vaccination, lung cells were stained with anti-CD45-Alexa FluorTM 700, CD8-PerCP-Cyanine5.5, CD69-FITC and CD103-AF594. The population of CD45+CD8+CD69+CD103+ T cells was sorted by flow cytometry and stimulated in vitro with a control peptide pool (Con-Pool) from unrelated antigens or a mixture of 15-amino-acid overlapping peptides (Pep-Pool, at 2 μg/ml per peptide) for 48 h. The supernatant was collected for the cytokine detection using a Mul-Analyte Flow Assay Kit (BioLegend, Cat. No# 740150, for INF-γ, TNF-α and GM-CSF), and ELISA kits (Invitrogen, Cat. No# BMS601 for IL-2; Invitrogen, Cat. No# BMS6029 for granzyme B), according to the manufacturer’s instructions.
HLA tetramer preparation and staining assay
Predicted functional peptides (Supplementary Table 3) matched with HLA-A*02:01were synthesized and used for peptide-specific tetramer preparation following the instructions of the HLA-A*02:01 Tetramer Kit-PE (MBL International Corporation, Cat. No# TB-7300-K1). Cells of spleens or lungs isolated from immunized HLA-A*02:01/DR1 transgenic mice were washed and stained with Ghost Dye™ Red 780 (TONBO Biosciences, Cat. No# 13-0865-T100) for dead cells. Surface markers were stained with CD45-Alexa Fluor™ 700 (Invitrogen, Cat. No# 56-0451-82), CD8-PerCP-Cyanine5.5 (TONBO Biosciences, Cat. No# 65-0081-U100) and Peptide-tetramer-PE mixture, for 20 min at room temperature. The stained cells were pelleted and washed three times before being analyzed by flow cytometry (BD Aria III cell analyzer).
Validation of epitope-enriched regions
HEK293T cells stably expressing the pHAGE_EF1a_ICADCR, pHAGE_EF1a_IFPGZB, and pHAGE_EF1aHLA vectors (HLA-A*02:01 were separately overexpressed in HEK293T cells)56 were transfected with plasmids encoding the computationally predicted epitope-enriched regions of each MPXV protein. At 36 h post-transfection, 1 million HEK293T cells were cocultured with 0.2 million CD8+ T cells from MPX-EPs immunized mice for 12 h, after which IFP-positive target cells were sorted and analyzed with FlowJo V_10 software.
MPXV challenge studies
Immunized, plasma, or T cells transfused mice were challenged with MPXV in the following experiments to evaluate immune protection efficacy. (1). BALB/c mice (male and female, 3 weeks) randomly allocated into groups (n = 6) were transfused with 200 μl of the plasma collected from Mix-12, Mix-8, Mix-4, or PBS vaccinated mice (under animal immunization b.). Two hours later, the transfused mice were intranasally inoculated with MPXV at the dose of 1.0 × 106 PFU. Mice were sacrificed at 4 days post-infection to collect lungs for viral load quantification by a plaque assay. (2). BALB/c mice (male and female, 3 weeks) randomly allocated into groups (n = 6) were transfused with 2.0 × 107 CD8+ or CD4+ T cells sorted from immunized mice collected from Mix-12, Mix-8, Mix-4, or PBS vaccinated mice (under animal immunization b.). Six hours later, the transfused mice were intranasally inoculated with MPXV at the dose of 1.0 × 106 PFU. Mice were sacrificed at 4 days post-infection to collect lungs for viral load quantification by a plaque assay. (3). MPX-EPs (3 μg) or PBS immunized HLA-A*02:01/DR1 transgenic mice (n = 6 under animal immunization c.) were injected intraperitoneally with an anti-mouse anti-CD8-α (IgG2b) mAb (Bio X Cell, Cat. No# BE0061, 100 μg/mouse) or IgG2b isotype control mAb (Bio X Cell, Cat. No# BE0090, 100 μg/mouse) on Days-1 and 0 of the challenge. Peripheral blood cells were collected for the evaluation of CD8+ T-cell depletion by flow cytometry on the day of the challenge. The mice were intranasally inoculated with MPXV (1.0 × 107 PFU), and half of the mice were sacrificed at 4 days post-infection. The lung tissues were collected for viral load detection by a plaque assay and pathological analysis. (4). Mice immunized using the synergistic strategy (n = 5, HLA-A*02:01/DR1 transgenic mouse under animal immunization d.) were intranasally inoculated with MPXV (1.0 × 107 PFU). The challenged mice per group were sacrificed at 4 days post-infection to collect lung tissues for viral loads and pathological analysis. All procedures adhered to institutional guidelines for animal use and care.
Histopathological assay
Lung tissues collected from challenged mice were immediately fixed in 10% neutral-buffered formalin without inflation, and embedded in paraffin. Approximately 5-μm sections were cut and mounted on slides. Histopathological changes caused by MPXV infection were examined by standard hematoxylin and eosin (H&E) staining and viewed under a light microscope. H&E-stained lung tissue sections were blindly examined and scored by trained histo-pathologists.
Statistics
Statistical analyzes were conducted using Prism 8.0 (GraphPad software). Student’s t-test was used for pairwise comparisons. Group comparisons were executed using one-way ANOVA, followed by Tukey’s multiple comparison post-hoc tests. Specific details regarding the statistical methods and results were provided in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors affirm that all data supporting the findings in this study are accessible in the paper and supplementary information. Source data are provided in this paper.
References
Moss, B. Understanding the biology of monkeypox virus to prevent future outbreaks. Nat. Microbiol. 9, 1408–1416 (2024).
Poland, G. A., Kennedy, R. B. & Tosh, P. K. Prevention of monkeypox with vaccines: a rapid review. Lancet Infect. Dis. 22, e349–e358 (2022).
Kupferschmidt, K. Why monkeypox is mostly hitting men who have sex with men. Science 376, 1364–1365 (2022).
Huang, Y., Mu, L. & Wang, W. Monkeypox: epidemiology, pathogenesis, treatment and prevention. Signal. Transduct. Target Ther. 7, 373 (2022).
Rao, A. K. et al. Use of JYNNEOS (smallpox and monkeypox vaccine, live, nonreplicating) for preexposure vaccination of persons at risk for occupational exposure to orthopoxviruses: recommendations of the advisory committee on immunization practices - United States, 2022. MMWR Morb. Mortal. Wkly Rep. 71, 734–742 (2022).
Rizk, J. G., Lippi, G., Henry, B. M., Forthal, D. N. & Rizk, Y. Prevention and treatment of monkeypox. Drugs 82, 957–963 (2022).
Paran, N. & Sutter, G. Smallpox vaccines: new formulations and revised strategies for vaccination. Hum. Vaccin 5, 824–831 (2009).
Melamed, S., Israely, T. & Paran, N. Challenges and achievements in prevention and treatment of smallpox. Vaccines 6, 8 (2018).
Xiao, Y. et al. Short-term and longer-term protective immune responses generated by subunit vaccination with smallpox A33, B5, L1, or A27 proteins adjuvanted with aluminum hydroxide and CpG in mice challenged with vaccinia virus. Vaccine 38, 6007–6018 (2020).
Gilchuk, I. et al. Cross-neutralizing and protective human antibody specificities to poxvirus infections. Cell 167, 684–694.e689 (2016).
Jacobs, B. L. et al. Vaccinia virus vaccines: past, present and future. Antivir. Res. 84, 1–13 (2009).
Gilchuk, P. et al. Discovering naturally processed antigenic determinants that confer protective T cell immunity. J. Clin. Investig. 123, 1976–1987 (2013).
Terajima, M. et al. Vaccinia virus-specific CD8(+) T-cell responses target a group of epitopes without a strong immunodominance hierarchy in humans. Hum. Immunol. 69, 815–825 (2008).
Sette, A. et al. Definition of epitopes and antigens recognized by vaccinia specific immune responses: their conservation in variola virus sequences, and use as a model system to study complex pathogens. Vaccine 27, G21–G26 (2009).
Terajima, M. et al. Quantitation of CD8+ T cell responses to newly identified HLA-A*0201-restricted T cell epitopes conserved among vaccinia and variola (smallpox) viruses. J. Exp. Med. 197, 927–932 (2003).
Szabó, G. T., Mahiny, A. J. & Vlatkovic, I. COVID-19 mRNA vaccines: platforms and current developments. Mol. Ther. 30, 1850–1868 (2022).
Verbeke, R., Lentacker, I., De Smedt, S. C. & Dewitte, H. The dawn of mRNA vaccines: the COVID-19 case. J. Control Release 333, 511–520 (2021).
Sang, Y. et al. Monkeypox virus quadrivalent mRNA vaccine induces immune response and protects against vaccinia virus. Signal. Transduct. Target Ther. 8, 172 (2023).
Hou, F. et al. mRNA vaccines encoding fusion proteins of monkeypox virus antigens protect mice from vaccinia virus challenge. Nat. Commun. 14, 5925 (2023).
Zhang, R. R. et al. Rational development of multicomponent mRNA vaccine candidates against mpox. Emerg. Microbes Infect. 12, 2192815 (2023).
Zeng, J. et al. Mpox multi-antigen mRNA vaccine candidates by a simplified manufacturing strategy afford efficient protection against lethal orthopoxvirus challenge. Emerg. Microbes Infect. 12, 2204151 (2023).
Yang, X. et al. Evaluation and comparison of immune responses induced by two Mpox mRNA vaccine candidates in mice. J. Med. Virol. 95, e29140 (2023).
Freyn, A. W. et al. An mpox virus mRNA-lipid nanoparticle vaccine confers protection against lethal orthopoxviral challenge. Sci. Transl. Med. 15, eadg3540 (2023).
Zuiani, A. et al. A multivalent mRNA monkeypox virus vaccine (BNT166) protects mice and macaques from orthopoxvirus disease. Cell 187, 1363–1373.e1312 (2024).
Grifoni, A. et al. Defining antigen targets to dissect vaccinia virus and monkeypox virus-specific T cell responses in humans. Cell Host Microbe 30, 1662–1670.e1664 (2022).
Hammarlund, E. et al. Monkeypox virus evades antiviral CD4+ and CD8+ T cell responses by suppressing cognate T cell activation. Proc. Natl. Acad. Sci. USA 105, 14567–14572 (2008).
Aziz, S. et al. Contriving multi-epitope vaccine ensemble for monkeypox disease using an immunoinformatics approach. Front. Immunol. 13, 1004804 (2022).
Song, H. et al. Characterizing monkeypox virus specific CD8+ T cell epitopes in rhesus macaques. Virology 447, 181–186 (2013).
Goulding, J., Bogue, R., Tahiliani, V., Croft, M. & Salek-Ardakani, S. CD8 T cells are essential for recovery from a respiratory vaccinia virus infection. J. Immunol. 189, 2432–2440 (2012).
Oseroff, C. et al. HLA class I-restricted responses to vaccinia recognize a broad array of proteins mainly involved in virulence and viral gene regulation. Proc. Natl. Acad. Sci. USA 102, 13980–13985 (2005).
Smith, C. L. et al. Immunodominance of poxviral-specific CTL in a human trial of recombinant-modified vaccinia Ankara. J. Immunol. 175, 8431–8437 (2005).
Walsh, S. R. et al. Diverse recognition of conserved orthopoxvirus CD8+ T cell epitopes in vaccinated rhesus macaques. Vaccine 27, 4990–5000 (2009).
Tscharke, D. C. et al. Identification of poxvirus CD8+ T cell determinants to enable rational design and characterization of smallpox vaccines. J. Exp. Med. 201, 95–104 (2005).
Pasquetto, V. et al. HLA-A*0201, HLA-A*1101, and HLA-B*0702 transgenic mice recognize numerous poxvirus determinants from a wide variety of viral gene products. J. Immunol. 175, 5504–5515 (2005).
Tscharke, D. C. et al. Poxvirus CD8+ T-cell determinants and cross-reactivity in BALB/c mice. J. Virol. 80, 6318–6323 (2006).
Moutaftsi, M. et al. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nat. Biotechnol. 24, 817–819 (2006).
Moutaftsi, M. et al. Correlates of protection efficacy induced by vaccinia virus-specific CD8+ T-cell epitopes in the murine intranasal challenge model. Eur. J. Immunol. 39, 717–722 (2009).
Xia, H., He, Y. R., Zhan, X. Y. & Zha, G. F. Mpox virus mRNA-lipid nanoparticle vaccine candidates evoke antibody responses and drive protection against the Vaccinia virus challenge in mice. Antivir. Res. 216, 105668 (2023).
Boulanger, D. et al. Identification and characterization of three immunodominant structural proteins of fowlpox virus. J. Virol. 76, 9844–9855 (2002).
Xia, A. et al. Cross-reactive antibody response to monkeypox virus surface proteins in a small proportion of individuals with and without Chinese smallpox vaccination history. BMC Biol. 21, 205 (2023).
Golden, J. W. & Hooper, J. W. Heterogeneity in the A33 protein impacts the cross-protective efficacy of a candidate smallpox DNA vaccine. Virology 377, 19–29 (2008).
Kugelman, J. R. et al. Genomic variability of monkeypox virus among humans, Democratic Republic of the Congo. Emerg. Infect. Dis. 20, 232–239 (2014).
Shchelkunov, S. N. et al. Human monkeypox and smallpox viruses: genomic comparison. FEBS Lett. 509, 66–70 (2001).
Shchelkunov, S. N. et al. Analysis of the monkeypox virus genome. Virology 297, 172–194 (2002).
Heraud, J. M. et al. Subunit recombinant vaccine protects against monkeypox. J. Immunol. 177, 2552–2564 (2006).
Sakhatskyy, P. et al. Immunogenicity and protection efficacy of subunit-based smallpox vaccines using variola major antigens. Virology 371, 98–107 (2008).
Golden, J. W., Josleyn, M. D. & Hooper, J. W. Targeting the vaccinia virus L1 protein to the cell surface enhances production of neutralizing antibodies. Vaccine 26, 3507–3515 (2008).
Panchanathan, V., Chaudhri, G. & Karupiah, G. Correlates of protective immunity in poxvirus infection: where does antibody stand? Immunol. Cell Biol. 86, 80–86 (2008).
Xu, R. H., Remakus, S., Ma, X., Roscoe, F. & Sigal, L. J. Direct presentation is sufficient for an efficient anti-viral CD8+ T cell response. PLoS Pathog. 6, e1000768 (2010).
Tai, W. et al. An mRNA-based T-cell-inducing antigen strengthens COVID-19 vaccine against SARS-CoV-2 variants. Nat. Commun. 14, 2962 (2023).
Gambino, F. Jr. et al. A vaccine inducing solely cytotoxic T lymphocytes fully prevents Zika virus infection and fetal damage. Cell Rep. 35, 109107 (2021).
Jin, X. et al. Screening HLA-A-restricted T cell epitopes of SARS-CoV-2 and the induction of CD8(+) T cell responses in HLA-A transgenic mice. Cell Mol. Immunol. 18, 2588–2608 (2021).
Shen, A. R. et al. Exosomal vaccine loading T cell epitope peptides of SARS-CoV-2 induces robust CD8(+) T cell response in HLA-A transgenic mice. Int. J. Nanomed. 17, 3325–3341 (2022).
Ferretti, A. P. et al. Unbiased screens show CD8(+) T Cells of COVID-19 patients recognize shared epitopes in SARS-CoV-2 that largely reside outside the spike protein. Immunity 53, 1095–1107.e1093 (2020).
Kula, T. et al. T-Scan: a genome-wide method for the systematic discovery of T cell epitopes. Cell 178, 1016–1028.e1013 (2019).
Zhang, H. et al. Profiling CD8(+) T cell epitopes of COVID-19 convalescents reveals reduced cellular immune responses to SARS-CoV-2 variants. Cell Rep. 36, 109708 (2021).
Thompson, M. G. et al. Prevention and attenuation of Covid-19 with the BNT162b2 and mRNA-1273 Vaccines. N. Engl. J. Med. 385, 320–329 (2021).
Mucker, E. M. et al. Comparison of protection against mpox following mRNA or modified vaccinia Ankara vaccination in nonhuman primates. Cell 187, 5540–5553.e5510 (2024).
Moss, B. Smallpox vaccines: targets of protective immunity. Immunol. Rev. 239, 8–26 (2011).
Ye, Q. et al. A penta-component mpox mRNA vaccine induces protective immunity in nonhuman primates. Nat. Commun. 15, 10611 (2024).
Ye, T. et al. Polyvalent mpox mRNA vaccines elicit robust immune responses and confer potent protection against vaccinia virus. Cell Rep. 43, 114269 (2024).
Zhou, J. et al. Circular RNA vaccines against monkeypox virus provide potent protection against vaccinia virus infection in mice. Mol. Ther. 32, 1779–1789 (2024).
Kong, T. et al. Single-chain A35R-M1R-B6R trivalent mRNA vaccines protect mice against both mpox virus and vaccinia virus. EBioMedicine 109, 105392 (2024).
Li, E. et al. An mpox quadrivalent mRNA vaccine elicits sustained and protective immunity in mice against lethal vaccinia virus challenge. Emerg. Microbes Infect. 14, 2447619 (2025).
Zhang, N. et al. Multi-valent mRNA vaccines against monkeypox enveloped or mature viron surface antigens demonstrate robust immune response and neutralizing activity. Sci. China Life Sci. 66, 2329–2341 (2023).
Traut, C. C. et al. Orthopoxvirus-specific T cell responses in convalescent MPOX patients. J. Infect. Dis. https://doi.org/10.1093/infdis/jiad245 (2023).
Ogura, H. et al. Dysfunctional Sars-CoV-2-M protein-specific cytotoxic T lymphocytes in patients recovering from severe COVID-19. Nat. Commun. 13, 7063 (2022).
Wodarz, D. & Nowak, M. A. Correlates of cytotoxic T-lymphocyte-mediated virus control: implications for immunosuppressive infections and their treatment. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355, 1059–1070 (2000).
Wilson, J. A. & Hart, M. K. Protection from Ebola virus mediated by cytotoxic T lymphocytes specific for the viral nucleoprotein. J. Virol. 75, 2660–2664 (2001).
Iborra, S. et al. The DC receptor DNGR-1 mediates cross-priming of CTLs during vaccinia virus infection in mice. J. Clin. Investig. 122, 1628–1643 (2012).
Li, M. et al. Long-lasting humoral and cellular memory immunity to vaccinia virus Tiantan provides pre-existing immunity against mpox virus in Chinese population. Cell Rep. 43, 113609 (2024).
Xu, R., Johnson, A. J., Liggitt, D. & Bevan, M. J. Cellular and humoral immunity against vaccinia virus infection of mice. J. Immunol. 172, 6265–6271 (2004).
Hobbs, S. J., Osborn, J. F. & Nolz, J. C. Activation and trafficking of CD8(+) T cells during viral skin infection: immunological lessons learned from vaccinia virus. Curr. Opin. Virol. 28, 12–19 (2018).
Adamo, S. et al. Memory profiles distinguish cross-reactive and virus-specific T cell immunity to mpox. Cell Host Microbe 31, 928–936.e924 (2023).
Ezzemani, W. et al. Identification of novel T-cell epitopes on monkeypox virus and development of multi-epitopes vaccine using immunoinformatics approaches. J. Biomol. Struct. Dyn. 42, 5349–5364 (2024).
Yu, C. et al. Designing a smallpox B-cell and T-cell multi-epitope subunit vaccine using a comprehensive immunoinformatics approach. Microbiol Spectr. 12, e0046524 (2024).
Keeton, R. et al. T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature 603, 488–492 (2022).
Tarke, A. et al. SARS-CoV-2 vaccination induces immunological T cell memory able to cross-recognize variants from Alpha to Omicron. Cell 185, 847–859.e811 (2022).
Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 23, 186–193 (2022).
Farzan, M. et al. Immunoinformatics-based multi-epitope vaccine design for the re-emerging monkeypox virus. Int. Immunopharmacol. 123, 110725 (2023).
Acknowledgements
This work was funded by grants from the Shenzhen Medical Research Fund (E24010010, E24010014) to W.T.; the National Natural Science Foundation of China (82271872, 82341046 and 32100755) to W.T.; the Shenzhen Bay Laboratory Startup Fund (21330111) to W.T.; the National Key Research and Development Plan of China (2021YFC2300200, 2022YFC2303200, 2021YFC2302405, and 2022YFC2303400) to G.C.; the National Natural Science Foundation of China (32188101) to G.C.; Vanke Special Fund for Public Health and Health Discipline Development, Tsinghua University (2022Z82WKJ005, 2022Z82WKJ013) to G.Y.; the National Natural Science Foundation of China (82241082, 32270182) to Y.L.; the National Natural Science Foundation of China (82372254) to J.L.; the National Natural Science Foundation of China (82372254) to L.Z.; Shenzhen San-Ming Project for Prevention and Research on Vector-borne Diseases (SZSM202211023) to G.C.; Shenzhen Medical Research Fund (B2404002) to G.C.; Yunnan Provincial Science and Technology Project at Southwest United Graduate School (202302AO370010) to G.C.; The New Cornerstone Science Foundation through the New Cornerstone Investigator Program and the Xplorer Prize from Tencent Foundation to G.C.
Author information
Authors and Affiliations
Contributions
W.T., G.Y., and G.C. conceived and designed the experiments. W.T., C.T., H.S., B.C., X.Y., X.Z., P.D., M.L., Q.Y., S.F., W.W., O.Z., Y.L., J.L., L.Z., and G.Z. performed the experiments. W.T., C.T., H.S., B.C., S.L., G.Z., M.T., G.Y., and G.C. analyzed the data. W.T., H.S., and G.C. wrote the manuscript. All authors have given approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
A patent application has been filed for the antigens presented here. G.C., W.T., H.S., C.T., and B.C. are inventors. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tai, W., Tian, C., Shi, H. et al. An mRNA vaccine against monkeypox virus inhibits infection by co-activation of humoral and cellular immune responses. Nat Commun 16, 2971 (2025). https://doi.org/10.1038/s41467-025-58328-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58328-x
