
Abstract
Long-circulating, transfection-competent lipid nanoparticle (LNP)-mRNA delivery systems are critical for achieving efficient transfection in extrahepatic tissues. Here we investigate the properties of LNP mRNA systems containing high proportions of bilayer forming lipids, using equimolar egg sphingomyelin and cholesterol as the bilayer-forming components. We show that LNP mRNA systems prepared at bilayer lipid to ionizable lipid molar ratios of 4-0.67 exhibit high mRNA encapsulation efficiencies (90–100%) and excellent transfection potencies in vitro. Systems with bilayer lipid to ionizable lipid molar ratios equating to 4 exhibit a liposomal morphology with a solid core suspended in an aqueous interior surrounded by a lipid bilayer. These liposomal LNPs exhibit longer circulation lifetimes than LNP systems with Onpattro-like lipid compositions and have enhanced extrahepatic transfection properties. The prolonged blood circulation lifetime is attributed to reduced plasma protein adsorption. The transfection competency of liposomal LNP systems is attributed to export of the solid core containing mRNA from the LNP as the endosomal pH is lowered.
Similar content being viewed by others
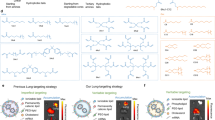


Introduction
Standard lipid nanoparticle (LNP) formulations, such as the clinically approved Onpattro LNP-siRNA formulation that can effectively transfect hepatocytes following intravenous (i.v.) administration1,2, have a lipid composition consisting of ionizable lipid/helper lipid/cholesterol/PEG-lipid in the approximate molar ratios 50:10:38.5:1.5. (This lipid composition is referred to as the Onpattro-like formulation in this study). These formulations exhibit a nanostructured core surrounded, at least in part, by a lipid monolayer3. ~90% of the injected dose of LNP systems with this lipid composition is cleared to the liver within an hour of i.v. administration4. While these formulations have shown considerable utility for vaccine and liver applications, their wider use for gene therapy is hindered by limited access to extrahepatic tissues. In order to transfect extrahepatic tissues, LNP systems with significantly extended circulation lifetimes are necessary to deliver sufficient amounts to these tissues. Such circulation lifetimes can be achieved by incorporating PEG-lipids that reside with the LNP for extended times, however, the presence of the PEG-lipid coating inhibits uptake into and transfection of target cells5.
Extensive efforts have been made to develop liposomal systems containing anticancer drugs with extended circulation lifetimes6,7,8,9,10,11. These efforts followed the observation that long-circulating liposomal systems preferentially accumulate at extrahepatic disease sites such as tumours12. Previous work has focused on using PEG-lipids as agents to achieve long circulation lifetimes13, however, extended circulation lifetimes in the absence of PEG-lipids can be achieved for liposomes composed of lipid mixtures such as distearoylphosphatidylcholine (DSPC) or sphingomyelin (SM) with approximately equimolar levels of cholesterol10. Interestingly, recent work from our laboratory has shown that LNP mRNA formulations containing higher proportions of helper lipids such as DSPC or egg SM (ESM) (molar ratios ionizable lipid/DSPC or ESM/cholesterol/PEG-lipid 33:40:25.5:1.5) can exhibit liposome-like morphology. In these systems the oil droplet formed by the ionizable lipid in the neutral form is suspended in an aqueous medium surrounded by a lipid bilayer. This morphology is interpreted as consisting of an oil droplet surrounded by a monolayer of the bilayer-forming lipids and further surrounded in turn by a lipid bilayer consisting solely of the bilayer-forming lipids14. These systems are highly transfection competent both in vitro and in vivo. The presence of an exterior lipid bilayer potentially allows for some of the design principles that have evolved for liposomal delivery systems to be applied to LNP mRNA systems.
Previous work has shown that hydration of equimolar mixtures of cholesterol with phospholipids such as DSPC or ESM results in robust bilayer structures15,16. Furthermore, other investigations have shown that cholesterol has a limited solubility in the oil (neutral) form of ionizable cationic lipids17. In addition, studies have shown that LNP formed by ethanol dilution/rapid mixing with aqueous media give rise to structures that can be modelled according to the lipid composition18. At low proportions of bilayer lipids such as phosphatidylcholine and cholesterol, the bilayer lipid forms a monolayer around the hydrophobic core. At higher proportions of bilayer lipids, hybrid structures form, where a bilayer protrusion is associated with the hydrophobic core18,19. Sufficiently high proportions of bilayer lipid can result in a solid core surrounded by a lipid bilayer18. Therefore, a rational design approach would suggest that in order to generate long-circulating LNP mRNA systems, a high proportion of bilayer lipid is required to provide a lipid bilayer of DSPC-cholesterol or ESM-cholesterol surrounding the oil droplet in the aqueous interior where the oil droplet consists of the neutral form of the ionizable lipid surrounded by a monolayer of the bilayer preferring lipid.
In this work, we formulate LNP mRNA systems using equimolar proportions of ESM and cholesterol and vary the molar ratio of bilayer lipid (ESM and cholesterol) to ionizable lipid from 9 to 0.43, holding the PEG-lipid content constant at 1.5 mol/mol. We show that LNP mRNA systems containing bilayer to ionizable lipid molar ratios of 4 exhibit liposomal morphology while demonstrating excellent transfection properties both in vitro and in vivo. Further, these liposomal LNPs exhibit longer circulation lifetimes and improved extrahepatic transfection properties compared to formulations with the Onpattro-like composition. Prolonged circulation lifetime of the liposomal LNPs is attributed to reduced opsonisation by plasma proteins. The ability of these systems to maintain transfection competency in the presence of high proportions of bilayer lipid is ascribed to an ionizable lipid-dependent process. This process encourages export of the mRNA complexed with ionizable lipid as the LNP experiences the lower pH environment of the endosome as it matures.
Results and discussion
High proportions of bilayer forming lipids in LNP mRNA systems lead to liposome morphology and enhanced transfection potency in vitro
LNP mRNA systems were prepared containing a range of molar ratios of bilayer lipid (equimolar ESM and cholesterol) to ionizable lipid (RB/I) in four-component systems consisting of ionizable lipid, ESM, cholesterol and PEG-lipid, maintaining the PEG-lipid content constant at 1.5 mol ratio. The molar ratios used in the formulations were 10/45/45/1.5 (RB/I = 9); 20/40/40/1.5 (RB/I = 4); 30/35/35/1.5 (RB/I = 2.3); 40/30/30/1.5 (RB/I = 1.5); 50/25/25/1.5 (RB/I = 1); 60/20/20/1.5 (RB/I = 0.67) and 70/15/15/1.5 (RB/I = 0.43). The ionizable lipid used in each formulation was nor-MC3, a lipid that has a similar structure as DLin-MC3-DMA (MC3; see Fig. 1A, Fig. S1, Table S1)20. The encapsulated mRNA encodes for nano-luciferase (NanoLuc; 808 nt) and was formulated into the LNP at an N/P ratio of 6 (Table S1). As shown in Fig. 1B, excellent encapsulation efficiencies were achieved for RB/I ratios as high as 9 but were reduced at RB/I ratios of 0.67 and lower. LNP sizes were in the range of 40–60 nm in diameter over the whole range of lipid compositions (Fig. 1C).

LNP mRNA systems were formulated as described in Methods containing NanoLuc mRNA (808 nt; N/P = 6) with lipid compositions nor-MC3/ ESM: cholesterol/ PEG-DMG at varying ratios of bilayer lipid (ESM and equimolar cholesterol) to ionizable lipid (RB/I). These formulations correspond to RB/I of 9, 4, 2.3, 1.5, 1, 0.67 and 0.43. The Onpattro-like formulation consists of MC3/DSPC/cholesterol/PEG-DMG (50/10/38.5/1.5 mol/mol). If it is assumed that the cholesterol and DSPC reside exclusively in the monolayer or bilayer environments, the RB/I for this formulation is 1.03. A Lipids in ethanol are mixed with mRNA in 25 mM NaOAc (pH 4) to give rise to LNP mRNA systems that are either liposomal LNP systems or solid core systems where at least part of the exterior surface consists of a lipid monolayer. B Encapsulation efficiencies of LNP NanoLuc mRNA systems with varying RB/I values (mean ± S.D., n = 3). C Hydrodynamic diameter (DLS measurement, number mean) for various RB/I ratios (mean ± S.D., n = 3). D The classification of LNP morphologies within the formulated population. LNP morphology was defined by its oil core or lamellar/multilamellar liposomal features as determined from cryo-TEM micrographs. E Cryo-TEM micrographs of LNP mRNA systems prepared using various RB/I values, micrograph has been reproduced twice. F Luminescence of Huh7 cells incubated for 24 h with LNP NanoLuc mRNA systems (0.1–1 μg mRNA/mL), decrease in RB/I indicated from beige to dark red, with Onpattro-like formulation (indicated as gray) over a range of RB/I values (mean ± S.E.M., n = 6). Source data are provided as a Source Data file. Figure 1A was partially created in BioRender. Cheng, M. (2025) https://BioRender.com/o07c413. All LNP illustrations are original and were created using Inkscape.
Cryo-TEM studies (Fig. 1D, E) revealed a regular progression in morphology as the RB/I ratios were decreased. At high RB/I values (9-2.3), a bilayer liposomal structure is observed containing an aqueous region and an electron dense solid core structure. The proportions of the solid core increase as the ESM/cholesterol levels are decreased below RB/I = 2. In previous work characterizing LNP composed of equimolar DSPC and cholesterol and varying amounts of triolein19 or ionizable lipid18, we have shown that the proportion of LNP that are solid core can be modelled by assuming that the triolein or (neutral) ionizable lipid exists in a hydrophobic environment surrounded by a lipid monolayer with a lipid bilayer protruding from it. Applying the same analysis to the results obtained here, where we assume the neutral form of nor-MC3 exists exclusively in the hydrophobic oil core and ESM and cholesterol reside in the monolayer and bilayer, shows results in close agreement with experimental data (see Table S2).
The properties of the system containing bilayer lipid to ionizable lipid ratios RB/I = 4 are of particular interest as a large proportion (~ 84%) exhibit a bilayer structure containing a solid core component that occupies ~30% of the interior (Fig. 1D). It may be noted that the oil droplet is associated with the membrane bilayer either through a bulge in the bilayer or possibly connected by a tether. The morphology of this liposomal LNP system is similar to that observed previously for LNP systems (in the absence of RNA) containing DSPC and cholesterol (equimolar) and the ionizable lipid DLin-KC2-DMA at proportions corresponding to RB/I = 4.218.
The in vitro transfection potencies of the LNP mRNA systems prepared over the range RB/I 9 to 0.43 were investigated following incubation with Huh7 cells. As shown in Fig. 1F, potent transfection was observed for formulations with RB/I = 4-0.67, in most cases exceeding that achieved using the Onpattro-like lipid composition. This shows that the LNP mRNA systems with a solid core encapsulated in a lipid bilayer are transfection competent, consistent with the results of Chander et al. 14 Further analyses of LNP and mRNA stability stored at 4 °C for 63 weeks, showed LNP formulated with RB/I = 9-2.3 increased in size by <20% and maintained high levels of mRNA encapsulation (> 80%). In contrast, the sizes of LNP formulations with RB/I = 1.5-1 increased by up to 300% with encapsulation levels to as little as 40% (Fig. S2A, B). We further assessed the translatability and mRNA integrity of LNPs stored for 63 weeks. While gel electrophoresis showed that mRNA extracted from all LNPs was significantly degraded upon long term storage, the mRNA extracted from the RB/I = 4 formulation displayed the highest mRNA integrity and translatability compared to the other formulations (Fig. S2C–E). This suggests that LNPs with external bilayer structures exhibit substantially improved protection of mRNA cargo. Finally, cryo-TEM images revealed formation of larger vesicular structures at low RB/I (Fig. S2F), consistent with fusion of LNPs and loss of mRNA encapsulation during storage.
Liposomal LNP mRNA systems are transfection competent in vivo and exhibit enhanced extrahepatic transfection properties
It is of obvious interest to characterize the in vivo protein expression and biodistributions following i.v. administration of the RB/I = 4 (nor-MC3/ESM/cholesterol/PEG-DMG; 20/40/40/1.5 mol/mol) liposomal LNP system in comparison to LNP mRNA systems with the Onpattro-like lipid composition. Both systems contained NanoLuc mRNA (N/P = 6). As shown in Fig. 2A, markedly different transfection profiles were observed for the liposomal LNP compared to the formulation with the Onpattro-like composition. The luminescence signal for the liposomal LNP originated from the liver, spleen and pancreas rather than predominantly in the liver as shown in Fig. 2B and S3. This result was consistent with the data from the homogenized organs (Fig. 2B–C and S4). Liposomal LNP displayed higher luminescence than LNP with the Onpattro-like lipid composition in the heart, lungs, spleen, kidney, muscle, inguinal lymph node and pancreas. In particular, 50-fold and 150-fold increase in luminescence in the spleen and inguinal lymph node, respectively, were observed for the liposomal LNP mRNA systems (Fig. 2C and S4).

Liposomal LNP (nor-MC3/ ESM/cholesterol/ PEG-DMG; 20/40/40/1.5 mol/mol; RB/I = 4) and LNP with the Onpattro-like composition (MC3/DSPC/cholesterol/DMG-PEG: 50/10/38.5/1.5 mol/mol) containing NanoLuc (N/P = 6) were prepared and administered i.v. to CD-1 female mice. A IVIS bioluminescence images expressed as radiance (photons/second/cm2/steradian) at various times following injection of LNP mRNA systems (0.5 mg mRNA/kg). B Ex vivo bioluminescence (24 hpi) in heart, lung, liver, spleen, kidney, skin, fat, muscle (left flank), inguinal lymph node and pancreas. C Ex vivo transfection profile of homogenized tissue measured in relative luminescence units/gram of tissue (Grey: Liver; Pink: Spleen; Green: Lymph node; Blue: Pancreas). [More detailed statistical analyses can be found in Fig. S4]. D Representative 3D rendered SPECT/CT reconstructed images of CD-1 mice injected i.v. with either the 111In-labelled Onpattro-like LNP formulation or the liposomal LNP NanoLuc mRNA system (∼0.4-0.5 mCi/mouse [111In] and ∼15 µg mRNA/mouse corresponding to 0.5 mg mRNA/kg) as a function of time post-injection. E LNP biodistribution as detected by gamma counting of liposomal LNP containing NanoLuc compared to the Onpattro-like composition ex vivo, measured in relative % injected dose/gram (%ID/g) (Grey: Liver; Red: Spleen; Green: Lymph node; Turquoise: Pancreas) [More detailed statistical analyses can be found in Fig. S9 and Table S3]. Source data are provided as a Source Data file.
In order to understand factors leading to enhanced extrahepatic transfection, we characterized the LNP circulation lifetimes and biodistribution using SPECT/CT imaging. A 3% wt/wt ratio of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-diethylenetriaminepentaacetic acid (DTPA-PE) was incorporated into NanoLuc-containing LNPs with the Onpattro-like lipid composition and the liposomal lipid composition (RB/I = 4). The addition of DTPA-PE did not affect morphology and size (Fig. S5). These LNPs were then radiolabelled with the gamma emitter 111InCl3 to achieve radiochemical yields of 97.6% and 100%, respectively, for Onpattro-like and liposomal LNP (Fig. S6). The biodistributions of the LNPs in CD-1 mice following systemic administration (∼0.4-0.5 mCi/mouse- [111In], ∼15 µg mRNA/mouse corresponding to 0.5 mg mRNA/kg) were ascertained. LNPs with the Onpattro-like composition were rapidly cleared within 2 h to liver and spleen as shown in Fig. 2D. The liposomal LNP exhibited a much longer circulation half-life (~ 15-fold increase; Fig. S7–8) and increased accumulation in the heart, thoracic mammary glands (Fig. 2D), blood, inguinal lymph nodes, pancreas and bone compared to LNP with the Onpattro-like lipid composition (Figs. 2E, S9 and Table S3). LNPs formulated at RB/I = 4 with ALC-0315 and SM-102 as the ionizable lipid component also showed liposomal LNP morphologies as observed through cryo-TEM images (Fig. S10) and these LNPs again exhibit similar radiopharmaceutical profiles as the liposomal LNP formulated with nor-MC3 (Figs. S11, Table S3).
In order to determine LNP delivery and protein expression at a tissue and cellular level, mRNA encoding for mCherry was formulated in liposomal LNP containing the fluorescent marker lipid DiD-C18 to determine tissue/cell biodistribution. Tissue sections of the lymphoid organs showed DiD-C18 and mCherry expression at the peripheral regions of both the spleen and inguinal lymph node (Fig. S12), indicating these LNP are preferentially delivered to cells such as macrophages at the marginal zone (spleen) and subcapsular and medullary sinus (lymph node). Immunofluorescence imaging demonstrated colocalization between the peripheral mCherry expression and macrophage (F4/80) staining as shown in Fig. S13.
Liposomal LNP systems adsorb lower levels of corona proteins compared to LNP with the Onpattro-like lipid composition
To elucidate why liposomal LNP mRNA systems exhibit longer circulation lifetimes, we characterized the biomolecular corona (BMC) adsorbing to the surface of the different LNP systems following incubation with mouse plasma. Using size-exclusion column chromatography (SEC), isolation of the plasma protein-bound LNPs was performed (Fig. S14). A ~ 2-fold higher (wt/wt) adsorption of plasma proteins was found for LNP with the Onpattro-like lipid composition as compared to the liposomal LNP system (Fig. 3A). Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) analysis suggested both formulations exhibited a broadly similar plasma protein profile (Fig. 3B, Table S4). Liposomal LNP formulations with other ionizable lipids such as ALC-0315 and SM-102 also showed a decrease in protein adsorption after SEC (Fig. S15). Mass spectrometry (MS) proteomic analysis detected over 1000 different proteins in the BMC of both formulations (Fig. 3C). Volcano plot analysis showed 98 proteins that are statistically different (P value = 0.01) between the two formulations. Notably, the liposomal LNP exhibit significantly lower immunoglobulin heavy variable 10-1 (Ighv10-1) and fibrinogen (Fgb) levels (Fig. S16). The classes of proteins adsorbed by liposomal LNP and Onpattro-like systems were similar but with a slight reduction in immunoproteins (~ 4.4% less) and a slight increase in apolipoprotein (~ 3.6% more) for the liposomal LNP as shown in Fig. 3D and Table S421,22,23. While the dominant contribution to prolonged circulation lifetimes correlates with the overall reduction in adsorbed plasma proteins, it is possible that the reduced levels of immune proteins and increased levels of apolipoproteins could also contribute to this effect.

Liposomal LNP (nor-MC3/ ESM: cholesterol/ PEG-DMG; 20/40:40/1.5 mol/mol; RB/I = 4) and LNP with the Onpattro-like composition (MC3/DSPC/cholesterol/DMG-PEG: 50/10/38.5/1.5 mol/mol), N/P ratio of 6, were formulated, incubated with plasma and isolated as indicated in Methods. A Protein to lipid ratio (wt/wt, µg/µg), total protein measured from a BCA assay, total lipid calculated from DiD-C18 absorbance measurement (mean ± S.D., n = 3). Data were analyzed through a two-tailed unpaired t-test, *p = 0.0296. as indicated in Methods. B SDS–PAGE of the plasma proteins adsorbed to the surface of Onpattro-like and liposomal LNP at 1 hour post incubation (mean ± S.D., n = 3). The plasma protein profile is also shown. C Heatmap analysis of the > 1000 proteins identified (iBAQ intensity log) (White-orange indicates the highest abundance while purple-black indicates lower abundance). D The proportions of proteins with distinct biological functions in the protein coronas of LNP mRNA systems with the Onpattro-like lipid composition as compared to the liposomal LNP (Grey: Others; Yellow: Immuno Proteins; Blue: Apolipoprotein; Green: Complement Proteins; Pink: Albumin; Purple: Coagulation Factors). Source data are provided as a Source Data file.
Mechanism of action for mRNA loading and intracellular delivery by liposomal LNPs
As shown, liposomal LNP systems exhibiting a solid core surrounded by a lipid bilayer can exhibit efficient mRNA loading properties and are transfection competent in vitro and in vivo. In order to characterize the loading and transfection mechanisms, we investigated the cryo-TEM properties of the systems with RB/I values of 4 and 2.3. As shown in Fig. 4A, following T-tube mixing at pH 4, electron dense structures associated with bilayer protrusions are observed. The electron dense structures likely reflect mRNA complexed with positively charged ionizable lipid as observed elsewhere24,25. These structures appear to serve as nucleation points for subsequent deposition of ESM/cholesterol/ionizable lipid, resulting in large bilayer structures emanating from the electron dense core. As the pH is raised to 7.4, the electron dense structure moves inside the bilayer and becomes larger, an event that can be ascribed to properties of the ionizable lipid. As the pH is raised, ionizable lipids in the outer monolayer of the bilayer structure will adopt the neutral form and move to the inner monolayer, becoming re-protonated in the lower pH environment of the LNP interior and thus adding to the interior surface area. This would create an imbalance in transmembrane surface areas, driving invagination26,27,28 of the electron dense structures as observed. As the pH approaches pH 7.4 the ionizable lipids will adopt the neutral oil form and phase separate out of the lipid bilayer into the hydrophobic core structure causing it to become larger, as also observed.

Liposomal LNP with RB/I = 4 (nor-MC3/ ESM: cholesterol/ PEG-DMG; 20/40:40/1.5 mol/mol) or RB/I = 2.3 (nor-MC3/ESM/cholesterol/PEG-lipid; 30/35/35/1.5 mol/mol) were formulated containing Firefly Luciferase mRNA (N/P = 6), n = 3. A Cryo-TEM micrographs generated following the mixing stage at pH 4 (after dialysis against 25 mM NaOAc pH 4 to remove ethanol) and after dialysis against PBS to raise the pH to 7.4, micrograph has been reproduced twice. The white arrows in the pH 4 preparations indicate the presence of electron dense structures interpreted as complexes of mRNA with ionizable lipid. The white arrows in the pH 7.4 micrograph indicate the electron dense oil droplet structures formed by the ionizable lipid in the neutral form. B Cryo-TEM micrographs of LNP mRNA systems (originally in PBS pH 7.4 buffer) that were subsequently dialyzed against lower pH buffers to mimic endosomal pH environments (pH 6.4: early endosomes, pH 5.6: late endosomes, pH 5.0: lysosomes), micrograph has been reproduced twice. The white arrows in the pH 5.6 and pH 5 micrographs show that the electron dense regions decrease in size quickly as the pH is lowered, consistent with a partitioning of the charged form into the lipid bilayer.
In an attempt to simulate the morphological behaviour of the liposomal LNP in the lower pH endosomal environment, the behaviour of the RB/I = 4 and RB/I = 2.3 systems following dialysis against pH 6.4 (early endosome), pH 5.6 (late endosome) and pH 5 (lysosome) buffers was investigated. Remarkably, the interior solid core progressively migrates from the LNP interior to presenting as a protrusion from the surface of the LNP (Fig. 4B). Further, the volume of the solid core decreases in size as the pH is lowered.
This behaviour offers insight into the mechanism whereby LNP mRNA systems containing high levels of bilayer lipid could be transfection competent. First, given the apparent rapid exchange of lipids between the bilayer and the solid core, it is likely that the solid core is tethered to the interior monolayer even in situations where the solid core appears to be independent of the surrounding membrane as visualized by cryo-TEM. As the pH is lowered the ionizable lipid is progressively converted to the positively charged, bilayer-preferring form. Given that the interior of the aqueous region encapsulated by the bilayer is buffered at pH 7.4, it is probable that this lipid will be preferentially located on the outer monolayer of the LNP experiencing the low pH of the endosome. This would lead to asymmetric transbilayer surface areas and extrusion of the remaining solid core to the exterior of the LNP to compensate28,29. One can imagine this as a localized warhead that interacts with the endosomal membrane to achieve fusion and cytosolic delivery. Note, it is likely that this warhead will contain positively charged ionizable lipids complexed with mRNA cargo. The mechanism is illustrated in Fig. 5A, B.

A On mixing an ethanol solution of a lipid mixture containing a high proportion of bilayer-forming lipids and an ionizable lipid with an aqueous buffer (pH 4) containing mRNA, an mRNA cationic lipid complex forms and acts as a nucleation point for subsequent deposition of bilayer lipid that includes residual positively charged ionizable lipid. As the pH is raised to 7.4, the complex moves to the interior of the bilayer protrusion and enlarges due to partitioning of the neutral form of the ionizable lipid into an oil droplet. The mRNA likely dissociates from the oil core at pH 7.4 and resides in the aqueous interior. B Proposed mechanism of transfection of liposomal LNP mRNA systems. Following uptake into an endosome and as the pH of the environment decreases, the ionizable lipid in the oil core moves to the outer monolayer of the LNP bilayer resulting in extrusion of an electron dense region and a positive surface charge on the exterior of the LNP. This encourages close association with the negatively charged endosomal membrane, fusion and release of the mRNA into the cytoplasm. Figure 5 is completely original and was illustrated using Inkscape. All LNP illustrations are original and were created using Inkscape.
Rational design for LNP formulation development
In this study, we demonstrate that LNP mRNA systems can be rationally designed to form liposomal structures with a bilayer surrounding an interior solid core and that these systems are transfection competent both in vitro and in vivo. Points of interest concern the mechanism whereby these liposomal LNPs are transfection potent, the implications of the higher lipid doses inherent in LNP formulations containing high proportions of bilayer lipids, the stability of these systems and the markedly different transfection profile observed following systemic administration.
In previous work, we suggested that upon exposure of LNP RNA systems to the low pH in the endosome, the ionizable lipid is converted to the positively charged form and combines with the negatively charged lipids in the endosomal membrane to produce non-bilayer membrane disruptive structures resulting in cytoplasmic delivery of the nucleic acid cargo30. However, the presence of high levels of bilayer forming lipids such as ESM/cholesterol would be expected to mitigate against such events. The results presented here suggest that transfection competence involves presentation of a small solid core on the exterior of the liposome bilayer as the pH is lowered. The nature of this solid core is of obvious interest. At pH values of 5.6 and 5, which are well below the pKa of the ionizable lipid, the residual solid core likely consists of ionizable lipid in the positively charged form complexed with the mRNA cargo.
LNP mRNA systems prepared with high RB/I ratios contain significantly more lipid per mRNA dose than LNP mRNA systems containing 50 mol% ionizable lipids. For example, for an RB/I of 1 and an mRNA dose of 1 mg/kg, the lipid dose is 22 mg/kg for an N/P of 6. The equivalent dose for LNPs with an RB/I of 9 or 4 is 107 and 54 mg lipid/kg respectively. Two things should be noted. First, these lipid doses are lower than what is commonly used for liposomal systems. For example, a 20 mg doxorubicin/kg dose of Doxil® corresponds to a lipid dose of 160 mg/kg. Second, high doses of liposomes consisting of bilayer lipids such as DSPC/cholesterol or ESM/cholesterol are essentially non-toxic. Mice have been dosed with liposomes with these lipid compositions at doses as high as 1 g/kg with no ill effects31,32. Thus liposomal LNPs would not be expected to exhibit increased toxicity.
With regard to the stability of the liposomal LNP systems, it is useful to reference the Onpattro-like formulations that have the lipid composition ionizable lipid/DSPC/cholesterol/PEG-lipid 50/10/38.5/1.5 mol/mol. If we assumed that all the cholesterol can participate in bilayer or monolayer formation, then LNP with the Onpattro lipid composition have an RB/I of ~1. However, the solubility of cholesterol in PC bilayers is limited above equimolar ratios of cholesterol to PC16 leading potential formation of cholesterol crystals33. In any event the stability data presented here suggest that the liposomal LNP with RB/I = 4 is considerably more stable than LNP with Onpattro-like compositions.
Finally, the liposomal LNP system exhibits a much different transfection profile (Fig. 2) as compared to LNP with the Onpattro-like lipid composition. This likely reflects a broader tissue distribution consistent with the longer circulation lifetimes and different organ uptake observed by SPECT/CT analysis. In turn, the longer circulation lifetimes can be ascribed to reduced adsorption of serum proteins21,34,35 as well as a higher lipid dose36,37,38. The reduced levels of serum proteins are likely correlated with the much different exterior of the liposomal LNP as compared to LNPs with the Onpattro-like composition, which exhibit, at least in part, a monolayer of lipid as compared to a bilayer. Notably, liposomal systems that exhibit lower levels of serum protein adsorption have longer circulation lifetimes10. The intriguing improvement of protein expression in the inguinal lymph node and pancreatic tissue illustrates the potential benefits of liposomal LNP systems, warranting further investigation with larger animal models and increased sample size.
In summary, LNP mRNA systems prepared at high RB/I values (9-2.3) can adopt a bilayer liposomal structure surrounding an electron dense solid core structure. These liposomal LNP exhibit efficient mRNA encapsulation and excellent in vitro transfection properties. The liposomal LNP system (RB/I = 4) exhibits enhanced circulation lifetimes and potent extrahepatic transfection in organs such as the lymph node and pancreas. The longer circulation lifetimes correspond to reduced levels of serum protein adsorption for the liposomal LNPs. The transfection potency of these liposomal LNPs appears to depend on external presentation of structures consisting of mRNA complexed with ionizable cationic lipid that are generated as the pH in the endosome decreases.
Methods
All animal protocols were approved by the Canadian Animal Care Committee and conducted in accordance with relevant University of British Columbia guidelines and regulations (A22-0044, A20-0132).
Materials
All ionizable lipid used in this study (nor-MC3/MC3/ALC-0315/SM-102) was purchased through Dr. Marco Ciufolini. While all other lipids (ESM Cat.#860061, PEG-DMG Cat.#880151, DTPA-PE Cat.#791275) were purchased from Avanti Polar Lipids. Cholesterol was purchased from Sigma Aldrich. Dulbecco’s modified Eagle medium (DMEM Cat.#12430054) and fetal bovine serum (FBS Cat.#MT35016CV) were purchased from Thermo Fisher Scientific. All other chemicals were purchased from Sigma Aldrich unless otherwise stated. mRNA encoded for the reporter protein Firefly Luciferase (FLuc) [NTx-164] and mCherry [NTx-163] were provided by NanoVation Therapeutics Inc.
LNP formulation and lipid concentration measurement
All LNP formulations were prepared by first dissolving lipids (nor-MC3/MC3/ALC-0315/SM-102, ESM/DSPC, cholesterol and PEG-DMG) into the ethanol phase at a final concentration of 10 mM with varying lipid ratio as indicated in Table S1. The lipid mixture was then rapidly mixed with an aqueous solution of mRNA of 25 mM sodium acetate (pH 4) with an amine-to-phosphate ratio (N/P) of 6. The mixing was performed with a T-junction mixer at a 1:3 ratio (ethanol: aqueous) and at final flow rate of 20 mL min−1. The resulting mixture was dialyzed overnight against >500-fold volume of pH 7.4 PBS unless stated otherwise. The LNP were then sterile filtered with a 0.2 μm Supor membrane syringe filter (Pall Corporation) and concentrated in 10 K Amicon ultracentrifugation units (EMD Millipore Corporation). The total lipid concentration was then measured using the Cholesterol E Kit (Wako Diagnostics Cat.#999-02601).
Synthesis of the NanoLuc mRNA
Plasmid DNA Preparation and Linearization
The mRNA plasmid, incorporating β-globin 5′ and 3′ UTRs and encoding nano-luciferase (sequence found in SI), was transformed into E. coli, cultured in LB broth with 100 μg/mL ampicillin, and purified using the QIAGEN Midi Prep Kit following manufacturer’s protocol. Plasmid linearization was performed using XbaI (5 μg pDNA per 20 μL; NEB Biolabs Cat# R0145T) at 37 °C for 4 hours, followed by heat inactivation at 65 °C for 20 minutes. DNA integrity was confirmed via agarose gel electrophoresis (1%, 100 V, 1 hour, SYBR Safe staining).
In vitro transcription (IVT) and purification
IVT was performed using a T7 polymerase-based reaction containing dNTPs, incorporating N1- methylpseudouridine (m1Ψ) in place of uridine (Hongene Biotech Cat#R5-027), CleanCap® AG capping reagent, and linearized pDNA (5 μg). The reaction was incubated at 37 °C for 3 hours, followed by DNase-I treatment at 37 °C for 1 hour to remove template DNA. Purification was carried out using the Monarch RNA Cleanup Kit ( > 500 μg binding capacity, NEB Biolabs Cat#T2030L).
RNA yield, quality, and dsRNA quantification
RNA yield was determined using a NanoDrop spectrophotometer by measuring 260/280 nm absorbance in triplicate. Integrity was assessed by 1% denaturing agarose gel electrophoresis (300 ng RNA per well). Full gels can be found in Source Data file. dsRNA contamination was quantified using a J2 antibody-based ELISA (Exalpha Biologicals Inc. Cat# 10613002) following manufacturer’s protocol, with absorbance measured at 450 nm.
Quantification of encapsulated RNA concentration
To measure the encapsulation of mRNA in the LNPs, RNA quantification using the Quant-it™ RiboGreen RNA assay kit (Invitrogen Cat#R11490) was performed. Particles were diluted in 1x Tris-EDTA (TE) buffer (Thermo Fisher Scientific Cat#BP2476100) to ~3 μg/mL. A standard curve using serial-diluted NanoLuc-mRNA in 1% Triton X-100 (Sigma Cat#T8787) + TE was added to the plate (0-5 μg/mL). Each diluted sample was added in two triplicate sets onto a black 96-well plate. To one triplicate set, an equal volume of TE was added, and to the other, an equal volume of 1% Triton X-100 + TE was added. Quant-iT RiboGreen RNA reagent (Invitrogen Cat#R11490) was added in 1:1 volume to the samples and fluorescence intensity was measured (Ex/Em. 500 nm/ 525 nm) on a plate reader. Measurements of samples treated with Triton X-100 represented total mRNA, while samples that only received TE represented un-encapsulated (free) mRNA. To calculate the percentage encapsulation, the difference of RFUs of total mRNA minus the free mRNA is divided by the total mRNA in the sample, multiplied by 100. Calculations of total encapsulated mRNA concentrations were performed by dividing the percent encapsulation by the total mRNA concentrations calculated from the standard curve.
LNP size analysis
Dynamic light scatter quantification of LNP diameters and polydispersity indexes (PDI) was performed using the Zetasizer Nano (Malvern Instruments Inc.). The PDI and number mean (nm) was recorded for each formulation.
Cryo-TEM imaging
LNP suspensions were concentrated to a final concentration of ~20 mg/mL of total lipid in Amicon Ultra-4 centrifugal filter units (Sigma Aldrich). LNP samples were prepared with a total lipid concentration between 20-25 mg/mL on a formvar/carbon grid (Ted Pella, Inc.) and plunge-frozen using an FEI Mark IV Vitrobot (FEI). Grids were moved into a Gatan 70° cryo tilt transfer system pre-equilibrated to at least −180 °C and then inserted into the microscope. An FEI LaB6 G2 TEM (FEI) operating at 200 kV under low-dose conditions was used to image all samples with an FEI Eagle 4 K CCD camera. All samples were imaged at 55 000× magnification with a nominal under-focus of 1–2 μm. All sample preparation and imaging were performed by the UBC High Resolution Macromolecular Cryo-Electron Microscopy Facility. Manual LNP Annotation of Cyro-TEM Images were conducted through CVAT.ai online platform (Palo Alto, CA). Each LNP was analyzed and counted for its features related to the oil core and liposomal LNP structure. LNPs were then annotated with a segmentation mask and categorized into different types of morphologies. The %Morphology is calculated using the following equation:
All pixels associated to the LNP, and its relevant internal structure were converted to cross-sectional area (CSA) in nm2. Oil droplets within a liposomal LNP were segmented as shown below, the % oil droplet to LNP is calculated using the following equations to give the interior segmented CSA of the corresponding oil droplet:
Cell culture and treatments
The human hepatocarcinoma cell line (HuH7) was cultured and maintained in DMEM media supplemented with 10% FBS, at 37 °C under 5% CO2. For assessing the transfection potency of LNPs, cells were seeded into 96-well microplates (13,000 cells/well) for 24 hours. Subsequently, cells were treated with the LNP encapsulating NanoLuc-mRNA (0.1-1 μg mRNA/mL). The treated cells were incubated for 24 hours, the media was removed prior to cells lysis using Glo Lysis buffer (100 μL; Promega Cat#E2661). The lysates were mixed at a 1:1 ratio with Nano-Glo luciferase substrate (Promega Cat#N1110) and luminescence was quantified using a Spark® Multimode Microplate reader (Tecan). The total protein content per well was determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific Cat#23225). Protein expression was calculated by normalizing to the luminescence values of treated cells against untreated cells and blank. Finally, luminescence readings were then normalized to the total protein.
BCA assay
To normalize luminescence values by total protein concentration, a Pierce™ bicinchoninic acid (BCA) Protein Assay was performed (Thermo Fisher Scientific Cat#23225). To prepare the standard curve, a serial dilution of bovine serum albumin, the protein standard (0-2 mg/mL), was prepared in PBS. In triplicates, 25 μL of each dilution was added to a clear microplate in addition to 25 μL of each lysed cell suspension. 200 μL of BCA reagent was added to each well and the plate was incubated for 30 minutes at 37 °C. Absorbance values were measured at 562 nm on a plate reader. Protein concentrations (μg/mL) in each well were determined using the standard curve.
RNA gel and wheat germ assay
mRNA encoding Nano-luciferase was extracted from LNPs and quantified using the PureLink™ RNA Mini Kit (Invitrogen Cat#12183020). The concentration of the extracted RNA was measured, and its integrity was verified using a 1% RNA agarose gel, with a loading range of 250-300 ng per well. After confirmation of RNA quality, a wheat germ extract assay was performed (Promega Cat#L4380) following the standard protocol. Following the wheat germ extract assay, the samples were diluted by a 100-fold, and luminescence was measured using an NanoLuc assay with the Nano-Glo® Luciferase Assay System (Promega Cat#N1110) following the standard protocol.
Bioluminescence imaging of LNP mRNA nano-luciferase transfection
Mice were maintained on a regular 12-hour light/12-hour dark cycle in a modified barrier animal facility at UBC following the animal protocol of A22-0044. They were maintained at a temperature range of 20‐26 °C and a 40 to 60% relative humidity that kept consistent ( ± 5%). CD-1 female mice aged between 6 to 8 weeks (Charles River Laboratories) were used throughout.
Mice were individually weighed and imaged prior to intravenous (i.v.) injection of LNP NanoLuc mRNA (0.5 mg/kg mRNA). Prior to IVIS bioluminescence imaging (10 minutes), mice were administered Nano-Glo® Fluorofurimazine In Vivo Substrate (FFz) (8.8 mM, 0.44μmol) (Promega Cat#N4100) intraperitoneally injected. Mice belonging to the same dosing groups (n = 3) were anesthetized with 3% isoflurane and placed on the imaging platform in a supine position while being maintained on 3% isoflurane via a nose cone. Mice were imaged on the Xenogen IVIS IVIS Lumina II In Vivo Imaging System (PerkinElmer), with an exposure time of 30 s at 3-, 6-, and 24-hours post i.v. injection. Bioluminescence values were quantified by measuring the radiance (photons/second/cm2/steradian) using the Living Image® software program (Caliper Life Sciences).
Immediately following the last imaging time point (24 h), mice were euthanized, and the heart, lungs, liver, spleen, kidneys, fat, muscle, skin, inguinal lymph node and pancreas were collected and imaged ex vivo at an exposure time of 30 s. Similarly, bioluminescence values were quantified by the radiance (photons/second/cm2/steradian) for each excised organ by creating a ROI for each organ using the Living Image® software program (Caliper Life Sciences).
Organ homogenization for ex vivo biodistribution
Organs of interest were excised from the 4 groups of Female CD-1 mice (n = 3) 24 hours post i.v. injection (Heart, Lungs, Liver, Spleen, Kidney, Muscle, Skin, Fat, Inguinal Lymph node and Pancreas), weighted, and reporter lysis buffer (1 mL) (Promega Cat#E4030) was added to Fastprep tubes and beads prior to homogenization. The samples were homogenized under the FastPrep-24™ Classic bead beating grinder and lysis system (MP) at 4 m/s for 2 rounds of 20 s. The homogenates were kept in -80 °C until further processing. The frozen samples were thaw at 37 °C in a water bath and centrifuged at 13523 g for 10 min and the lysate is collected for further analysis.
For NanoLuc assay measurements, the homogenate (10 μL) was diluted in reporter lysis buffer (40 μL) and sample (50 μL) was further diluted in NanoLuc substrate (50 μL) [Nano-Glo luciferase assay, 5000x dilution from stock, Promega Cat#N1110] and the luminescence were measured through an open filter.
In Vivo SPECT/CT imaging and image analysis
The current study was performed in accordance with the Canadian Council on Animal Care (CCAC) and protocol approved by the Animal Care Committee (ACC) of the University of British Columbia (A20-0132) and was conducted by UBC in vivo imaging preclinical research. Healthy CD-1 female mice (6-8 weeks old; Charles River Laboratories) were anaesthetized using 5% isoflurane for induction, between 1.5 and 2.5% in oxygen for maintenance. Mice also received a subcutaneous injection of Lactated Ringer’s solution (0.5 mL) for hydration prior to each imaging scan. Imaging was conducted using a multimodal SPECT/CT scanner (VECTor/CT, MILabs) equipped with an XUHS-2 mm mouse pinhole collimator to acquire a whole-body SPECT and CT scan. An i.v. dose containing ~120 µL (~ 450 µCi) of [111In] liposomal LNP (either nor-MC3/ALC-0315/SM-102; n = 3/group) or [111In] Onpattro-like® (n = 3) or in PBS was injected post anaesthesia. Static whole-body images were acquired at 0, 2, 5, 8, and 24 hours with acquisition durations of 12 minutes initially which were gradually increased to compensate for decay unless otherwise stated. During the imaging sessions, the mice maintained consistent body temperature by using a heating pad. The 111In photopeak window was 171 keV, with a 25% energy window width and the data reconstructed with the pixel-based ordered subset expectation maximisation (POSEM) algorithm, with a voxel size of 0.4 mm3, 16 subsets, and 6 iterations. The images were decay and attenuation corrected. CT scans were acquired with a tube setting of 55 kV and 615 µA. In total 2 frames of 180 projections over 360 degrees were acquired in step and shoot rotation mode. The acquired projection data was reconstructed using SkyScan NRecon, the reconstructed SPECT scans were post-filtered with a 3D Gaussian filter and 3D CT image were generated on 0.169 mm3 voxel size. Volumes of interest (VOIs) were manually defined using AMIDE (v.1.0.6) to determine the time activity pattern per target organ. Thus, the delineated regions were heart, lungs, liver, spleen, kidney and bone. The average organ activity per volume was obtained from the SPECT images and the Standardized Uptake Values (SUVs) were extracted from each organ. Each organ was identified using the sagittal, transverse and coronal planes of the CT images with SPECT acting as guidance. To validate the SUV value, we cross validated the imaging data with ex vivo gamma counting at the 24 h endpoint and observed comparable %ID/g using the following equation:
In order to relate the scanner units (counts/pixel) to radioactivity concentration (MBq/mL), a calibration factor was determined scanning a source with a known concentration of 111In.
Ex vivo tissue assessment of biodistribution
After the last in vivo imaging at 24 hpi, mice were sacrificed via CO2 asphyxiation under isoflurane anesthesia. Subsequently, cardiac puncture was promptly performed to collect blood, followed by the complete extraction of 10 organs of interest: n = 3/group (heart, lungs, liver, spleen, kidney, muscle, fat, inguinal lymph node, pancreas) and biological samples (blood, urine and feces). The excised organs were cleaned from blood, weighed, and measured using a calibrated gamma counter (Packard Cobra II autogamma counter, PerkinElmer) to determine postmortem residual activity and biodistribution. Radiation background was subtracted, and the net counts obtained were decay-corrected to the time of sacrifice. Total organ weights were used for the calculation of injected dose per gram of tissue (% ID/g).
Fluorescence imaging and immunofluorescence imaging of tissues
CD-1 female mice (n = 3) were intravenously administrated with LNP encapsulating mRNA encoded for mCherry (0.5 mg mRNA/kg). At 24 hpi, mice were anaesthesia (5% isoflurane) and underwent full body prefusion, 4% paraformaldehydes (PFA) (20 mL) were used to perfuse the mice through the left ventricle with an incision at the right atrium. After prefusion, the spleen and inguinal lymph node were extracted and stored in 4% PFA at 4 °C for 6 h. Fixed organs were then transferred to a 30% sucrose solution overnight and then embedded in optimal cutting temperature (OCT) compound (Tissue-Tek Cat#4583), followed by subsequent cryosection into 5-10 μm sections prior to confocal imaging and antibody staining. Tissues were stained with α-mF4/80 Alexa Fluor 488 Rat IgG2a clone:BM8 (AbLab Ref# AB0000699). SYTO™ 13 and Hoechst 33342 were used as nuclear stains. Slides were visualized using either a Leica SP8 confocal microscope using a 10x and 60x objective lens or an Olympus IXplore SR inverted fluorescence spinning disc microscope using a 10× oil objective lens. For the images taken using the Leica SP8 confocal microscope the following parameters were used: Sequential Scan, SYTO 13 (ex/em = 488/490-560 nm, gain 400, 1% laser power), mCherry (ex/em = 561/573-726 nm, gain 500, 40% laser power) and DiD-C18 (ex/em = 633/650-750 nm, gain 50, 1% laser power). For the images taken using the Olympus IXplore SR inverted fluorescence spinning disc microscope the following parameters were used: Sequential Scan, Hoechst 33342 (DAPI SD, 405 nm, 800 ms, 20% laser power), F4/80 (Green SD,488 nm, 900 ms, 60% laser power) and mCherry (Red SD, 561 nm, 900 ms, 50% laser power).
Plasma incubation and bimolecular corona analysis
LNPs formulated with DiD-C18 at 0.8 mol% were standardized to 4 mg/mL total lipid for incubation with pre-warmed CD-1 mouse plasma, gender pooled, 0.2 µm sterile filtered (BioIVT, Cat#MSE00SRM-0001284). LNP-mouse plasma Incubation was performed at 37.0 °C and 0.687 g for 1 h on a Innova 4300 (New Brunswick Scientific), at 94 µg LNP total lipid in 1 mL plasma.
Following incubation, LNPs were separated from LNP-Plasma mixture through a 15 × 1.5 cm size exclusion column, containing 20 mL Sepharose CL-4B (Sigma-Aldrich, Cat#GE17-0150-01) and PBS as the mobile phase. The elutions were collected in 0.7 mL fractions with subsequent measurements for DiD-C18 fluorescence (Ex/Em. 646 nm/ 663 nm) to track LNPs and measured using a NanoDrop™ Lite (Abs. 280 nm, Thermo Scientific) to track protein flow through the column. The earliest six fractions containing significant DiD-C18 fluorescent signal were combined and concentrated in a 30 kDa Airmcon Ultra Centrifugal filter for protein characterization by BCA, SDS-PAGE, and MS Proteomics, in addition the UV-1600PC, UV/Visible Spectrophotometer (VWR) were used to quantify LNP total lipid content by DiD-C18 absorbance (λMax 649 nm) and molar extinction coefficient (260 000 cm-1 M-1). Eluted fractions were concentrated to ~50 μL.
Protein quantification
BCA assay was performed by Pierce™ BCA Protein Assay as described by manufacturer’s instructions (Thermo Fisher Scientific Cat#23225).
Protein characterization
SDS-PAGE was performed on the LNP-Plasma elutions by standardizing protein concentration to 800 µg/mL, denaturing at 95 °C for 5 min in Laemmli Buffer, and running in a 4–15% Mini-PROTEAN® TGX™ Precast Protein Gel (Bio-Rad Cat# 4561083) at 120 V for 45 min. The resulting gel was stained for 1 h in Instant Blue Coomassie Protein Stain (Abcam Cat# ab119211) and imaged with a lightbox. Full gel can be found in Source Data file. LNP containing protein corona underwent sample preparation using the PreOmics iST-BCT kit, performed in accordance with the manufacturer’s instruction (PreOmics Cat# P.O.00001).
LC method
The following procedures were conducted by the UBC Proteomics Core Facility. Digested and purified peptides (75 ng) from the LNP protein corona were administrated and separated on-line using NanoElute 2 UHPLC system (Bruker Daltonics) with Aurora Series Gen3 (CSI) analytical column, (25 cm × 75μm 1.7μm C18 120 Å, with CSI fitting; Ion Opticks). The column was heated to 50 °C using a column toaster M (Bruker Daltonics). The NanoElute thermostat temperature was set at 7 °C. Buffer condition was as follow: buffer A consisted of 0.1% aqueous formic acid and 0.5% acetonitrile in water, and buffer B consisted of 0.1% aqueous formic acid and 0.5% water in acetonitrile. The analytical column was conditioned with 4 column volumes of buffer A before each run. The flow rate was 0.30 μL/min.
DIA-PASEF Methods
All Mass-Spec Proteomics analysis were performed at UBC’s Proteomics Core Facility. A TimsTOF Pro2 (Bruker Daltonics) coupled UHPLC was used with a captive spray ionization source operating at a capillary voltage of 1700 V, 3 L/min drying gas, and 200 °C drying temperature. Parallel Accumulation-Serial Fragmentation (PASEF) scanning was employed for DIA of the TOF measurements. Acquisition parameters were set to include the MS spectra collected from m/z range 100-1700, and from ion mobility range (1/ K0) at 0.7-1.3 V·s/cm2. For each TIMS cycle, 25 DIA-PASEF windows were utilized from m/z range 299.5-1200.5. Each window was composed of seven scans, with 3-4 steps, utilizing an equal ramp and accumulation time of 100 ms (100% duty cycle). Neighboring windows were maintained at a variable isolation width of m/z 36-61 with an overlap of m/z 1. Collision Energy was increased linearly in relation to mobility values ranging from 20 eV at 1/K0 = 0.6 V·s/cm2 to 65 eV at 1/K0 = 1.6 V·s/cm2.
DIA-NN search
All acquired data was searched using DIA-NN (version 1.8.1; PMID:31768060) against a database consisting of the Mus musculus fasta (Uniprot) and common contaminants (211 entries). Other search parameters include N-terminal M excision and Cysteine carbamidomethylation. Peptide length ranged 7-30, precursor charge ranged 2-4, precursor m/z ranged 300-1200, and fragment ion m/z ranged 200-1800. Precursor FDR was set to 1%, with 0 for settings ‘mass accuracy’, ‘MS1 accuracy’ and ‘scan window’. Settings ‘heuristic protein inference’, ‘use isotopologues’, ‘match between run (MBR)’, and ‘no shared spectra’ were all enabled. ‘Gene’ was chosen for protein inference parameter along with ‘double-pass mode’ for neural network classifier. Robust LC (high precision) was used for quantification strategy, RT-dependent mode for cross-run normalization, and smart profiling mode for library generation. The iBAQ values were calculated from a protein’s total non-normalised intensity (all peptides) divided by the number of measurable tryptic peptides.
Statistics & reproducibility
All statistical analysis was performed in GraphPad Prism Version 10. Experimental data were analyzed through a two-tailed unpaired t-test. ns = non-significant, *p < 0.05 and **p < 0.01. The exact p=value can be found in the source data file. All data are presented as mean ± S.D or mean ± S.E.M. No statistical method was used to predetermine sample size. Results from this study were replicated across multiple experiments with all replicates generating similar results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All relevant data from this study are available within the article, source data and supplementary documents. Source data are provided with this paper.
References
Kulkarni, J. A. et al. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 16, 630–643 (2021).
Cullis, P. R. & Felgner, P. L. The 60-year evolution of lipid nanoparticles for nucleic acid delivery. Nat. Rev. Drug Discov. 23, 709–722 (2024).
Leung, A. K. et al. Lipid nanoparticles containing siRNA synthesized by microfluidic mixing exhibit an electron-dense nanostructured core. J. Phys. Chem. C. Nanomater Interfaces 116, 18440–18450 (2012).
Mui, B. L. et al. Influence of polyethylene glycol lipid desorption rates on pharmacokinetics and pharmacodynamics of siRNA lipid nanoparticles. Mol. Ther. Nucleic Acids 2, e139 (2013).
Wheeler, J. J. et al. Stabilized plasmid-lipid particles: construction and characterization. Gene Ther. 6, 271–281 (1999).
Gregoriadis, G. & Davis, C. Stability of liposomes in vivo and in vitro is promoted by their cholesterol content and the presence of blood cells. Biochemical biophysical Res. Commun. 89, 1287–1293 (1979).
Papahadjopoulos, D., Jacobson, K., Nir, S. & Isac, T. Phase transitions in phospholipid vesicles. Fluorescence polarization and permeability measurements concerning the effect of temperature and cholesterol. Biochimica et. Biophysica Acta 311, 330–334 (1973).
Allen, T. M. & Chonn, A. Large unilamellar liposomes with low uptake into the reticuloendothelial system. FEBS Lett. 223, 42–46 (1987).
Klibanov, A. L., Maruyama, K., Torchilin, V. P. & Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 268, 235–237 (1990).
Johnston, M. J. et al. Characterization of the drug retention and pharmacokinetic properties of liposomal nanoparticles containing dihydrosphingomyelin. Biochim Biophys. Acta 1768, 1121–1127 (2007).
Gabizon, A. et al. Prolonged Circulation Time and Enhanced Accumulation in Malignant Exudates of Doxorubicin Encapsulated in Polyethylene-glycol Coated Liposomes. Cancer Res. 54, 987–992 (1994).
Gabizon, A. & Papahadjopoulos, D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc. Natl Acad. Sci. USA 85, 6949–6953 (1988).
Woodle, M. C. & Lasic, D. D. Sterically stabilized liposomes. Biochim Biophys. Acta 1113, 171–199 (1992).
Chander, N., Basha, G., Yan Cheng, M. H., Witzigmann, D. & Cullis, P. R. Lipid nanoparticle mRNA systems containing high levels of sphingomyelin engender higher protein expression in hepatic and extra-hepatic tissues. Mol. Ther. Methods Clin. Dev. 30, 235–245 (2023).
Smondyrev, A. M. & Berkowitz, M. L. Structure of dipalmitoylphosphatidylcholine/cholesterol bilayer at low and high cholesterol concentrations: molecular dynamics simulation. Biophys. J. 77, 2075–2089 (1999).
Huang, J., Buboltz, J. T. & Feigenson, G. W. Maximum solubility of cholesterol in phosphatidylcholine and phosphatidylethanolamine bilayers. Biochim Biophys. Acta 1417, 89–100 (1999).
Kulkarni, J. A. et al. On the formation and morphology of lipid nanoparticles containing ionizable cationic lipids and siRNA. ACS Nano 12, 4787–4795 (2018).
Kulkarni, J. A., Witzigmann, D., Leung, J., Tam, Y. Y. C. & Cullis, P. R. On the role of helper lipids in lipid nanoparticle formulations of siRNA. Nanoscale 11, 21733–21739 (2019).
Zhigaltsev, I. V. & Cullis, P. R. Morphological behavior of liposomes and lipid nanoparticles. Langmuir 39, 3185–3193 (2023).
Ciufolini, M. A. et al. MC3-type lipids and use thereof in the preparation of lipid nanoparticles. WO2022246571A1 (2023).
Chonn, A., Semple, S. C. & Cullis, P. R. Association of blood proteins with large unilamellar liposomes in vivo. Relation to circulation lifetimes. J. Biol. Chem. 267, 18759–18765 (1992).
Semple, S. C., Chonn A., Cullis, P. R. Interactions of liposomes and lipid-based carrier systems with blood proteins: Relation to clearance behaviour in vivo. Adv. Drug Deliv. Revi. 32, 3–17 (1998).
Papini, E., Tavano, R. & Mancin, F. Opsonins and dysopsonins of nanoparticles: facts, concepts, and methodological guidelines. Front Immunol. 11, 567365 (2020).
Brader, M. L. et al. Encapsulation state of messenger RNA inside lipid nanoparticles. Biophys. J. 120, 2766–2770 (2021).
Kulkarni, J. A. et al. Fusion-dependent formation of lipid nanoparticles containing macromolecular payloads. Nanoscale 11, 9023–9031 (2019).
Käs, J. & Sackmann, E. Shape transitions and shape stability of giant phospholipid vesicles in pure water induced by area to volume changes. Biophys. J. 60, 825–844 (1991).
Svetina, S. & Zeks, B. Membrane bending energy and shape determination of phospholipid vesicles and red blood cells. Eur. Biophys. J. 17, 101–111 (1989).
Mui, B. L., Dobereiner, H. G., Madden, T. D. & Cullis, P. R. Influence of transbilayer area asymmetry on the morphology of large unilamellar vesicles. Biophys. J. 69, 930–941 (1995).
Jarsch, I. K., Daste, F. & Gallop, J. L. Membrane curvature in cell biology: An integration of molecular mechanisms. J. Cell Biol. 214, 375–387 (2016).
Semple, S. C. et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 28, 172–176 (2010).
Oja, C. D., Semple, S. C., Arcadio, C. & Cullis, P. R. Influence of dose on liposome clearance: critical role of blood proteins. Biochimica et. Biophysica Acta 1281, 31–37 (1996).
Lovell, J. F. et al. Porphysome nanovesicles generated by porphyrin bilayers for use as multimodal biophotonic contrast agents. Nat. Mater. 10, 324–332 (2011).
Anindita, J. et al. The Effect of Cholesterol Content on the Adjuvant Activity of Nucleic-Acid-Free Lipid Nanoparticles. Pharmaceutics 16. https://doi.org/10.3390/pharmaceutics16020181. (2024)
Dos Santos, N. et al. Influence of poly(ethylene glycol) grafting density and polymer length on liposomes: relating plasma circulation lifetimes to protein binding. Biochim Biophys. Acta 1768, 1367–1377 (2007).
Walkey, C. D., Olsen, J. B., Guo, H., Emili, A. & Chan, W. C. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J. Am. Chem. Soc. 134, 2139–2147 (2012).
Gabizon, A., Tzemach, D., Mak, L., Bronstein, M. & Horowitz, A. T. Dose dependency of pharmacokinetics and therapeutic efficacy of pegylated liposomal doxorubicin (DOXIL) in murine models. J. Drug Target 10, 539–548 (2002).
Utkhede, D. R. & Tilcock, C. P. Effect of lipid dose on the redistribution and blood pool clearance kinetics of peg-modified technetium-labeled lipid vesicles. J. Liposome Res. 8, 381–390 (2008).
Allen, T. M. & Hansen, C. Pharmacokinetics of stealth versus conventional liposomes: effectof dose. Biochimica et. Biophysica Acta 1068, 133–141 (1991).
Acknowledgements
The authors thank Maryam Osooly for her assistance with the SPECT/CT imaging animal experiments. Cryo-TEM data collected at the High-Resolution Macromolecular Electron Microscopy (HRMEM) facility at the University of British Columbia (https://cryoem.med.ubc.ca). We thank Claire Atkinson, Joeseph Felt, Liam Worrall and Natalie Strynadka. HRMEM is funded by the Canadian Foundation of Innovation and the British Columbia Knowledge Development Fund. Mass spectrometry analysis was performed in the UBC mass spectrometry facility. We would like to thank Jason Rogalski for his technical support and suggestions for the MS proteomic experiment. This work was funded by the Canadian Institutes for Health Research (FDN 148469). M.H.Y.C was supported by the NanoMedicines Innovation Network postdoctoral fellowship in gene therapy and the CIHR Research Excellence, Diversity, and Independence (REDI) Early Career Transition Award. Y.Z. (FBD 193487), C.S. and J.L. was supported by a Frederick Banting and Charles Best Canada Graduate Scholarships Doctoral Award. Y.Z was supported by the NanoMedicines Innovation Network graduate award.
Author information
Authors and Affiliations
Contributions
M.H.Y.C. and P.R.C. contributed to the conceptualization, experimental design, project management, and writing of the manuscript. M.H.Y.C., Y.Z., K.F., J.L., C.S., E.K., and P.R.C. contributed to data collection for formulation, in vitro, animal, and protein corona experiments, as well as subsequent data analysis and manuscript editing. Y.C., M.T., H.B., and E.J. contributed to the design and synthesis of NanoLuc mRNA and data collection for mRNA analysis studies. O.Y.L.I. contributed to LNP Cryo-TEM analysis and quantification studies. C.R.R., K.S., and U.O.H. contributed to radiolabeling, SPECT/CT image and biodistribution study. A.A. contributed to mCherry-mRNA synthesis. M.H.Y.C., Y.Z., P.R.C., and D.W. contributed to the rational design of liposomal LNP systems and physicochemical characterization (Cryo-TEM studies) to elucidate LNP morphology, the mechanism of formation, and mRNA release.
Corresponding authors
Ethics declarations
Competing interests
P.R.C. has a financial interest in Acuitas Therapeutics and NanoVation Therapeutics as well as being Chair of NanoVation Therapeutics. D.W. is an employee of NanoVation Therapeutics. M.H.Y.C hold equity in NanoVation Therapeutics. Inc. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Bowen Li, Vahid Raeesi, Jens Simonsen, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, M.H.Y., Zhang, Y., Fox, K. et al. Liposomal lipid nanoparticles for extrahepatic delivery of mRNA. Nat Commun 16, 4135 (2025). https://doi.org/10.1038/s41467-025-58523-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58523-w
This article is cited by
-
Reprogramming the tumor immune microenvironment in cervical cancer: synergy between radiotherapy and immunotherapy
Molecular Cancer (2026)
-
Lipid Nanoparticle Development in Practice: Challenges and Collective Insights
Pharmaceutical Research (2026)
