
Abstract
Lupin crops provide nutritious seeds as an excellent source of dietary protein. However, extensive genomic resources are needed for crop improvement, focusing on key traits such as nutritional value and climate resiliency, to ensure global food security based on sustainable and healthy diets for all. Such resources can be derived either from related lupin species or crop wild relatives, which represent a large and untapped source of genetic variation for crop improvement. Here, we report genome assemblies of the cross-compatible species Lupinus cosentinii (Mediterranean) and its pan-Saharan wild relative L. digitatus, which are well adapted to drought-prone environments and partially domesticated. We show that both species are tetraploids, and their repetitive DNA content differs considerably from that of the main lupin crops L. angustifolius and L. albus. We present the complex evolutionary process within the rough-seeded lupins as a species-based model involving polyploidization and rediploidization. Our data also provide the foundation for a systematic analysis of genomic diversity among lupin species to promote their exploitation for crop improvement and sustainable agriculture.
Similar content being viewed by others

Introduction
The genus Lupinus (lupins) is part of the highly diverse legume family (Fabaceae), which has undergone spectacular evolutionary radiation1. Some species, such as Lupinus albus, L. angustifolius, L. luteus and L. mutabilis, are economically important animal feed crops that are particularly resilient to drought2. Others, such as L. albus and L. mutabilis, are considered as orphan crops dedicated to food production via the cultivation of traditional varieties and landraces in restricted geographical areas. L. cosentinii and L. digitatus are part-domesticated species associated with drought tolerance, and others have the potential for agricultural exploitation, including L. atlanticus and L. pilosus3,4. Lupins have attracted interest due to their nutritional seeds5 and potential for sustainable farming6,7. Lupin seeds can contribute to a healthy human diet8,9 due to their protein content of up to 40%10,11, and accordingly they are considered an important component of local and global food security12. The development of genomic tools could facilitate pre-breeding and breeding processes by exploiting the rich diversity of wild and domesticated lupin species13, which we describe here as crop wild relatives (CWRs), meaning wild species or weedy plants that are taxonomically related to domesticated lupins or can be used in agriculture (e.g. de novo domestication). However, to fully exploit genomics for crop improvement, the number of whole-genome and pangenome sequences available for legume crops must increase, including not only major domesticated gene pools but also wild species as well minor crops of the primary, secondary and tertiary gene pool14,15. The following whole-genome sequences are available for legume CWRs: peanut (Arachis hypogaea), A. duranensis and A. ipaensis16; soybean (Glycine max) and G. soja17; mung bean (Vigna radiata var. radiata), V. reflexo-pilosa var. glabra and V. radiata var. sublobata18; and a chickpea (Cicer arietinum) super-pangenome including wild species such as C. reticulatum, C. judaicum and C. pinnatifidium19,20.
Whole-genome sequences have been published for three lupin crops, namely L. angustifolius21,22, L. albus23,24 and most recently L. mutabilis25, providing insight into key aspects of lupin genome structure, diversity and evolution. However, information from CWRs is needed to take full advantage of lupin genetic resources. The genetic diversity of lupins has been highlighted by studies of chromosome number and genome size26,27, as well as epigenomic28 and phylogenetic analysis29, and more recently the development of pangenomes for L. albus30 and L. angustifolius22.
There are ~275 lupin species conventionally divided into New World and Old World types, reflecting two main geographical centers of species diversity4. The distribution of Old World lupins has resulted from both climate change and human activities beginning in the Pleistocene epoch, whereas evolution within New World lupins has been enhanced by processes such as ecological differentiation and intensive hybridization. The main changes in the gene pool of wild populations may reflect disruptive differentiation caused by occasional hybridization and subsequent intergradations with escaped or neglected sporadically domesticated strains31. Human activity in Europe may have influenced the distribution of L. angustifolius32. Most annual and perennial lupins belong to the New World group and are found mainly in North America and the Andes, but only one species has been domesticated (L. mutabilis, 2n = 48). The Old World lupins comprise ~15 annual species distributed around the Mediterranean basin as well as North and East Africa4, some of which are smooth-seeded and others rough-seeded species. The smooth-seeded species L. albus, L. angustifolius and L. luteus have been domesticated, along with the rough-seeded species L. cosentinii4. The domestication of lupins involved the introduction of desirable traits such as permeable seeds, non-shattering pods, early flowering and low alkaloid levels in seeds. L. mutabilis also has typical traits of the classical legume domestication syndrome, but the seed alkaloid content remains high in domesticated lines33.
The somatic chromosome number of Old Word lupins varies widely (2n = 32, 36, 38, 40, 42, 50 or 52), with a basic chromosome number of x = 5–13. However, the highest chromosome numbers (2n = 40–52) tend to be found in the heterogeneous smooth-seeded group, whereas the morphologically and genetically more homogeneous rough-seeded species have fewer chromosomes (2n = 32–42)26. In contrast, most New World lupins have a somatic chromosome number of 2n = 36 or 48, the exceptions being L. bracteolaris, L. linearis (both 2n = 32 or 34), L. cumulicola and L. villosus (both 2n = 52), but the basic chromosome number is proposed to be x = 6 in all cases34,35. Multiple chromosome rearrangements have occurred among the Old World lupins, revealing a complex evolutionary process that suggests polyploidy23,27,36. There is evidence that L. albus and L. angustifolius evolved by genome duplication and/or triplication21,23,36,37 from a diploid Lupinus ancestor24. Furthermore, processes such as aneuploidy may be unique to the Old World lupins38,39, but aneuploid reduction from ancestral species has been reported in the legume family38.
Lupins belong to the early-branching papilionoid genistoid clade1, and are placed in the core genistoid clade. The estimated age of genistoid diversification is ~56 Ma40, soon after the emergence of papilionoids ~58.6 Ma40,41. Numerous whole-genome duplication (WGD) events have been identified42,43 following legume-common tetraploidy (LCT, ~59 Ma) and ancient core eudicot-common hexaploidy (ECH, ~130 Ma)44. Evidence of these common events remains in papilionoid species such as peanut45 and soybean44, highlighting their important impact in shaping legume genome structure and evolution. In papilionoids, one WGD has been shared by an entire subfamily, in the common ancestor of all papilionoids42,43, even though several WGDs have occurred in papilionoids, as well as one whole-genome triplication (WGT) in the most recent common ancestor of Genisteae42. The genistoid clade shows the highest frequency of polyploidy but is poorly characterized and only weakly supported as a sister clade to the remaining core papilionoids (Doyle 2012). Early data suggest that the genistoid basic chromosome number was x = 9 and the most common somatic chromosome number was 2n = 1843,46. The WGT event has been identified in L. albus ~ 22 Ma24 and L. angustifolius 20–30 Ma37,43, indicating that the lupin diploid ancestor had a basic chromosome number of x = 924.
A more comprehensive view of polyploidization in legumes requires a denser sampling of taxa across the genistoid clade43,47. Old World lupin species: L. cosentinii Guss. (2n = 32) and L. digitatus Forsk. (2n = 36) are recognized to be drought tolerance, and L. digitatus serves as a source of drought-tolerance genes. L. cosentinii is native to the western Mediterranean coast but has been introduced in Austria, Romania, South Africa and, more recently in several parts of Australia4,48. Furthermore, L. cosentinii cv Erregulla was domesticated de novo in Australia from local wild germplasm in the 20th century, and has desirable traits such as soft, low-alkaloid seeds, non-shattering pods and early flowering48. In contrast, L. digitatus is native to the pan-Saharan region4 and seeds of domesticated L. digitatus have been found in the tombs of Egyptian Pharaohs, suggesting domestication began >4000 years ago49. Interspecific crosses of rough-seeded lupins yielded the highest frequency of viable F1 hybrids for L. cosentinii, L. digitatus and L. atlanticus, suggesting genomic similarity4,50.
Here, we present high-quality genome assemblies for two rough-seeded lupin species (L. cosentinii and L. digitatus), revealing their genetic architecture and the consequences of polyploidy during lupin evolution. We also propose a model that will enable further studies within the Lupinus genus and the genistoid clade. Our comparative genomics analysis with other, smooth-seeded lupins provides insights into the complex evolutionary history of Lupinus, including potential rediploidization events following polyploidization.
Results and discussion
De novo genome assemblies of rough-seeded lupins
We used a combination of methods to generate genome assemblies of the rough-seeded lupin species L. cosentinii (2n = 32) and its wild rough-seeded relative L. digitatus (2n = 36). First, we produced PacBio HiFi reads (Supplementary Data 1, Supplementary Data 2, Supplementary Fig. 1) with ~55× coverage (~32.8 Gbp) for L. cosentinii and ~43× coverage (18.9 Gbp) for L. digitatus. We then used HiCanu to generate 650 and 492 Mbp assemblies for L. cosentinii and L. digitatus, respectively (Supplementary Data 3, Supplementary Data 4). The assemblies were polished using 39 and 37 Gbp of Illumina 150PE reads (Supplementary Data 1, Supplementary Data 2). Purging reduced the assembly size to 588 Mbp for L. cosentinii and 435 Mbp for L. digitatus (Supplementary Data 3, Supplementary Data 4). We applied two sequential approaches to scaffold the contigs, first with 560 Gbp (L. cosentinii) and 722 Gbp (L. digitatus) of Bionano optical maps (based on 4.7 million and 5.4 million molecules for L. cosentinii and L. digitatus, respectively), then with 60.4 Gbp (L. cosentinii) and 53 Gbp (L. digitatus) of chromosome-level Illumina Hi-C data (Supplementary Data 1, Supplementary Data 2, Supplementary Fig. 2). The resulting L. cosentinii genome (588 Mbp) had 19 scaffolds (~426 Mbp, ~72% of the assembled genome) and 709 further contigs, whereas the L. digitatus genome (435 Mbp) consisted of 22 scaffolds (~378 Mbp, ~87% of the assembled genome) and 339 remaining contigs (Table 1, Fig. 1). Benchmarking universal single-copy orthologs (BUSCO) reported 96.3% and 95.3% completeness along with 23.6% and 22.5% duplicated genes in L. cosentinii and L. digitatus, respectively. Both genomes were assembled at the highest possible level, including both Bionano optical maps and Hi-C data, but the high level of assembly duplication and genome ploidy hindered the reconstruction process, resulting in a more fragmented and less contiguous product than expected. This highlights the difficulties encountered when reconstructing complex genomes, such as those of plants, where even two of the most powerful genome scaffolding technologies can be ineffective. Lupin genomes that are already published (L.albus and L. angustifolius; Supplementary Fig. 3) were therefore used as references for comparison and downstream analysis. The L. cosentinii genome was larger than that of L. digitatus, which was similar to the reported 451-Mbp genome size of L. albus23, but both genomes reported here were much smaller than the 653-Mbp genome of L. angustifolius22. The recently reported 620-Mbp L. mutabilis genome25 has a BUSCO completeness of 94.8% with a duplication rate of 21.4% on the “Fabales” BUSCO database. Considering the BUSCO completeness in all five lupins, they each present a completeness level of ~96% and feature a similar proportion of duplicated genes. Our lupin genomes are similar in size to those of the common bean (Phaseolus vulgaris) and the model legumes Medicago truncatula and Lotus japonicus, which are ~580 Mbp51, ~430 Mbp52 and ~470 Mbp53, respectively.

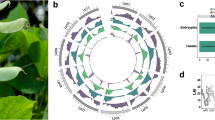
a Genome assemblies and annotations of L. cosentinii, left panel shows L. cosentinii plant and seed. b Genome assemblies and annotations of L. digitatus, left panel shows L. digitatus plant and seed. (1) Circular maps. (2) GC content of the genomes. (3) Gene density. (4) Repetitive element density. For ease of comparison, only scaffolds from L. cosentinii and L. digitatus are represented here. Source data are provided as a Source Data file.
All lupin genomes based on whole-genome sequencing (WGS) were smaller than values estimated by flow cytometry because the latter technique is based on relative genome sizes (absolute sizes are more difficult to validate). Furthermore, the variable characteristics of plant tissues (e.g., abundance of secondary metabolites) and the use of different buffers, reagents and reference standards, can influence genome size determination54. Long-read sequencing therefore provides more precise estimates and can address inconsistencies, while also improving the accuracy of flow cytometry standards. Genome sizes based on k-mer analysis are usually smaller than those estimated by flow cytometry due to collapsed repeat regions as well as polyploidy55,56.
Gene structure and composition of repetitive sequences
An ab initio prediction supported by RNA-Seq data (22–32 samples, ~30 million Illumina 150PE fragments each) was used to annotate the two genomes. We predicted the presence of 34,780 and 31,260 genes in the L. cosentinii and L. digitatus genomes, respectively. For 26,860 (77.2%) and 25,478 (81.5%) of these genes, functional annotations were also present in high-confidence databases (SwissProt, RefSeq and TAIR) (Supplementary Data 5 and 6) and in the proteomes of L. albus and L. angustifolius (Table 2). Functional annotation with Gene Ontology (GO) terms was possible for 23,544 L. cosentinii (67.7%) and 23,019 L. digitatus genes (73.6%). Repetitive DNA accounted for 352.5 Mbp (60%) and 206.3 Mbp (47.4%) of the L. cosentinii and L. digitatus genomes, respectively. The major classes of repetitive elements were simple repeats, representing 22.4% and 15.5% of the L. cosentinii and L. digitatus genomes, and long terminal repeat (LTR) retroelements, representing 21.3% and 20.1% of the L. cosentinii and L. digitatus genomes, respectively (Table 3, Fig. 2a). Both L. albus and L. angustifolius have a repetitive DNA content of 50–60%, compared to 64% for the recently characterized L. mutabilis genome, and have a much lower simple repeat content (~1%) than L. cosentinii and L. digitatus (~17%) but a higher content of LTR elements (~36% compared to ~21%).

a Sequence characteristics of repetitive DNA content in the genomes of L. albus, L. angustifolius, L. cosentinii and L. digitatus. b The distribution of indels (insertions and deletions) in the two assembled genomes, L. cosentinii and L. digitatus. The distribution was considered in the four main families of transposable elements (LTRs, DNA transposons, simple repeats and unclassified repeats). Source data are provided as a Source Data file.
Repetitive DNA and polyploidization are known to be key factors in the evolution of plant genomes57, including lupins7. The repetitive DNA content of L. cosentinii and L. digitatus was comparable to the other lupins (50–60%), irrespective of species or ploidy, showing that no extensive amplification or reduction of repeat sequences caused significant variation in the overall content after speciation or polyploidization. However, both L. cosentinii and L. digitatus had a smaller portion of LTR elements than L. albus and L. angustifolius. Although LTR elements are the most prevalent transposable elements in plant genomes, the abundance of specific superfamilies can vary greatly between species and even within varieties of the same species. Several related species share the ability to amplify a superfamily58, but LTR elements with a high copy number in one species may have a low copy number in a close relative59. Along with polyploidization, LTR elements may therefore be important for lupin genome evolution as suggested for the Fabaceae family more widely60. Indeed, retrotransposons and tandem repeats/microsatellites have influenced the evolution of L. angustifolius, and different processes such as repeat amplification, proliferation and clearance underlie the lineage-specific dynamics of repetitive sequences in lupins61. L. cosentinii and L. digitatus feature a higher number of simple repeats than L. albus and L. angustifolius, suggesting simple repeats played an important role in shaping the genome diversification in lupins. A large increase in the content of simple repeats was reported in the L. angustifolius pangenome relative to the reference genome22. Given the extensive haplotype variation for transposable elements in many species62, a large number of accessions of the same lupin species should be sequenced to investigate the dynamic evolution of polyploid plant genomes.
We aligned the Illumina WGS data on the two assembled genomes, revealing 21,645 deletions and 72 insertions in L. cosentinii as well as 441 deletions and 1831 insertions in L. digitatus (Fig. 2b). We focused our analysis of these structural variations (SVs) in the two main families of transposable elements. L. cosentinii featured more deletions in LTR elements (3420) and DNA transposons (11,160) than L. digitatus (304 and 93, respectively), whereas L. digitatus was characterized by the presence of more insertions (717 in LTR elements and 633 in DNA transposons). L. cosentinii featured more deletions in unclassified repeats (1283).
The repetitive DNA graphs for the four lupins showed that DNA transposons were concordant in all four species, especially L. cosentinii and L. digitatus (Fig. 3), suggesting low divergence between these two genomes. L. albus and L. angustifolius DNA transposons showed peaks of divergence at ~10% but nevertheless followed the curves of the other two genomes. Conversely, the LTR elements were concordant between L. albus and L. angustifolius, both of which shared an initial peak at twice the percentage of the other two lupins until 20% divergence. Notably, although L. cosentinii and L. digitatus showed mostly coherent curves, L. cosentinii displayed a peak at ~22% divergence representing 2% of the genome. The LTR elements included in this peak contained 441 deletions (2% of the total deletions).

a DNA transposons. b LTR elements. The graphs display the proportion of the genome (%) on the y-axis and the degree of divergence based on the kimura distance on the x-axis. The K values range from 1 to 40, indicating the level of evolutionary divergence from younger to older transposable elements. Source data are provided as a Source Data file.
DNA transposons showed the highest divergence peak (<10%) in L. albus, revealing the accumulation and homogenization of new DNA transposons, and that contributions to the total abundance of these elements in the L. albus genome are from recently evolved copies. In contrast, the most abundant peaks were observed at 20% for DNA transposons in the genomes of L. digitatus and L. cosentinii, suggesting that older copies are more abundant than newly evolved copies in these species. However, a discordant repeat landscape was observed in L. angustifolius, with two or more ancient peaks for DNA transposons, hinting at an abrupt change and distinct patterns of recently evolved copies of repeat elements. The divergence peak for LTR elements was <10%, suggesting active dissemination and homogenization of these new copies in the genomes of L. digitatus, L. angustifolius and L. albus. In L. cosentinii, the peak was observed at 22% divergence, indicating that older copies are more abundant in this genome.
Consequences of polyploidy during lupin evolution
We anticipated that L. cosentinii and L. digitatus would show some degree of polyploidy, like other legumes63. Accordingly, Genomescope/Smudgeplot analyses indicated that both species are tetraploid (Fig. 4b, d). A high degree of homozygosity (~99.94%) was evident in both species, shown by the single major peak in the k-mer distribution (Fig. 4a, c64;). The distribution of biallelic single nucleotide polymorphisms (SNPs) in the genome assemblies of both species also indicated tetraploidy. The delta log–likelihood scores, calculated from the difference between the free model and the diploid, triploid and tetraploid models, were 1,202,737, 896,833 and 316,770, respectively in L. cosentinii, but 746,976, 428,940 and 101,168, respectively in L. digitatus (Fig. 5a, b). The low scores in the tetraploid model therefore favor tetraploidy. The same analysis was applied to individual sequences from both species to determine whether some sequences or chromosomes have a different ploidy to the rest of the genome (a sign of aneuploidy). However, the lowest scores for all sequences again favored the tetraploid model (Fig. 5c, d). Our data therefore indicate that L. cosentinii and L. digitatus are tetraploid species.

a L. cosentinii GenomeScope profile. b L. cosentinii k-mer distribution. c L. digitatus GenomeScope profile. d L. digitatus k-mer distribution.

The lower the delta log–likelihood score, the better the fit to the corresponding model. a Prediction scores on the L. cosentinii whole genome assembly and b on the longest 21 sequences (N90). c Prediction scores on the L. digitatuswhole genome assembly and d on the longest 26 sequences (N80). Source data are provided as a Source Data file.
Given the evidence for WGT events (resulting in 2n = 6x = 54), we presumed that lupins with chromosome numbers of 2n = 32 (L. cosentinii) and 2n = 36 (L. digitatus) possesses basic chromosome numbers of x = 9 if they were subject to multiple chromosome rearrangements, leading changes of 22 and 18 chromosomes (may indicate an entire set of subgenomic chromosomes) in L. cosentinii and L. digitatus, respectively, due to rediploidization after WGT to establish tetraploidy. We hypothesized that both ECH and LCT might also affect lupin genome evolution. Assuming that the eudicot common ancestor is hexaploid with a basic chromosome number x = 7, the legume common ancestor is tetraploid. But ignoring the basic chromosome number, which is proposed to be x = 1144, x = 1623 or various43— and a diploid lupin ancestor (x = 9)—we propose the scheme shown in Supplementary Fig. 4. In addition, given that the genistoid clade and lupin diploid ancestor had x = 9 chromosomes24,43, we deduced that L. digitatus (2n = 36) possesses two diploid subgenomes, indicating a basic chromosome number of x = 9, whereas L. cosentinii (2n = 32) might have a basic chromosome number of x = 8. However, the basic chromosome number might be x = 9 for both species if a progressive rediploidization has shaped the current genomes of L. cosentinii and L. digitatus. We hypothesize that rediploidization may have occurred during the period of evolution separating L. cosentinii and L. digitatus from L. albus, involving changes affecting 18 or 14 chromosomes, respectively. This is supported by the fact that L. cosentinii and L. digitatus have the smallest chromosome number in the Lupinus genus, indicating the pressure on plants to reduce their chromosome number.
Although we have identified two diploid subgenomes, it was not possible to verify whether they have been shaped by autotetraploidization or allotetraploidization events. This includes numerous rearrangements and rediploidization events that might span tens of millions of years, exemplifying Old World lupin genome evolution, assuming a phylogenetic relationship in which L. cosentinii, L. digitatus and L. albus split ~4.5, ~0.5, and ~7.5 Ma, respectively65. Rediploidization has been reported in L. albus24, but more studies are needed to demonstrate a polyploidization–rediploidization model in lupins, as previously shown in mangrove species66.
As two rough-seeded species, L. cosentini and L. digitatus occupy a position in the Lupinus phylogenetic tree (Ainouche and Bayer 1999) that separates smooth-seeded lupins into two groups, suggesting they are genetically closer to L. albus (one of the closest smooth-seeded lupins to rough-seeded species) than L. angustifolius65 but also indicating unique evolutionary changes. Indeed, these species adapted to semi-desert and warm Mediterranean conditions, and facilitated their survival in rapidly changing environments by increasing genomic plasticity. Polyploidization may have promoted the diversification of lupins, as shown by the unique morphology of rough-seeded lupins, with scabrous-tuberculate testa48. The seed alkaloid content of rough-seeded lupins is moderate while growing naturally or though long selection, giving hope the untreated seeds of rough-seeded lupins could be used directly as food/feed4,67.
Polyploidization happened early in the evolution of the genistoid clade, indicating that WGD may have predated the divergence of Old World and New World lupins43. The chromosome number of rough-seeded species is small, like the species found in South America. Additionally, the annual American lupins are the only species with chromosome numbers of 2n = 32 or 3434. This might suggest the chromosome number 2n = 36 arose independently at least twice within the genus, an indication of convergent evolution in the context of ploidy. On the other hand, the chromosome number in Old and New World lupins may raise questions about the geographical origin of lupins, which is proposed to be in the Old World68. In the northern hemisphere, where the genistoid clade is thought to have originated during the Paleocene epoch38, evolutionary studies of these two groups of lupins will shed new light on the evolution of the entire Lupinus genus. The rough-seeded lupins described here can be used as a model to investigate the evolution of American lupins.
The complex evolution of the Lupinus genus is characterized by remarkable diversity in genome size, basic and somatic chromosome numbers, and chromosome rearrangements, in contrast to other legume genera. For example, Phaseolus and Cajanus (phaseoloids) feature mostly diploid species with the same chromosome number 2n = 22, whereas Arachis (dalbergioids) features both diploids (2n = 20) and tetraploids (2n = 40), and Dalbergia (dalbergioids) features exclusively diploid species with the chromosome number 2n = 2069. Interestingly, the Lupinus diploid ancestor with a basic chromosome number of x = 924 was confirmed across the genistoids38. However, the basic chromosome number may differ in early-diverging genistoid species, including those in the genus Sophora such as S. flavescens (2n = 18, diploid)70 and S. japonica (2n = 28, ploidy unreported)71, and also Crotalaria spp. (2n = 14, 16, or 32)72. Furthermore, the genistoid genus Ulex has a chromosome number of 2n = 32, 64 or 96 and Genista has a chromosome number of 2n = 48, 44 (described as aneuploid), 72 or 9673. L. digitatus (2n = 36) is the only known Old World lupin providing a direct example of x = 9, corresponding to the Lupinus diploid ancestor. In contrast, L. cosentinii has a different basic chromosome number (x = 8 or 9) and refutes the hypothesis that species with chromosome numbers such as 2n = 32 are aneuploids36,68 or underwent various chromosomal rearrangements. However, x = 8 is considered a primitive basic number of the genistoids.
Comparative genomics in lupin species
We compared our L. cosentinii and L. digitatus genome assemblies to the annotated and curated L. albus23 and L. angustifolius22 genome assemblies. Systematic pairwise comparisons revealed large syntenic blocks conserved in all four genomes (Fig. 6, Supplementary Figs. 5–10). The largest blocks were 24.3, 19.9, 17.3, 10.9, 9.9 and 9.8 Mbp in length, consisting of 1649, 783, 1190, 775, 593 and 508 collinear genes in the L. digitatus vs L. cosentinii (LdLc), L. digitatus vs L. albus (LdLa), L. albus vs L. cosentinii (LaLc), L. angustifolius vs L. cosentinii (LnLc), L. angustifolius vs L. digitatus (LnLd) and L. albus vs L. angustifolius (LaLn) comparisons, respectively (Supplementary Fig. 11). The degree of duplication showed a similar distribution when considering the total number of genes and genes located in smaller syntenic blocks. The average degree of duplication when considering all genes was similar in L. cosentinii (1.36) and L. digitatus (1.35) but increased to 1.43 in both species when considering the four smaller syntenic blocks (Supplementary Table 1). The rate of synonymous substitutions (Ks) calculated for duplicated BUSCO genes suggested that L. cosentinii and L. digitatus are more closely related to each other than the other genome combinations, as confirmed by the LdLc density curve (light blue) being lower than the others. In contrast, the LaLn (green) and LnLc (blue) density curves were the highest peaks in the graph, suggesting that the relationship between L. albus and L. angustifolius and that between L. angustifolius and L. cosentinii are the most distant among the pairwise combinations (Fig. 7a).

a Collinear blocks between L. digitatus (Ld) and L. cosentinii (Lc). b Collinear blocks between L. albus (La) and L. cosentinii (Lc). c Collinear blocks between L. albus (La) and L. digitatus (Ld). d Collinear blocks between L. angustifolius (Ln) and L. cosentinii (Lc). e Collinear blocks between L. angustifolius (Ln) and L. digitatus (Ld). f Collinear blocks between L. angustifolius (Ln) and L.albus (La). Source data are provided as a Source Data file.

a Distribution of Ks scores calculated from the coding sequences of orthologous genes from the six comparisons. The Ks values are shown on the x-axis, and their density is on the y-axis. b UpSet plot comparing the shared gene families (orthogroups) among the four lupin species. The histogram displays the number of gene families shared by the genomes indicated below the x-axis. The histogram is sorted by the number of shared families in each possible combination of the four genomes. c A phylogenetic tree constructed for the four lupin species using the STAG method in OrthoFinder. The values indicate the branch lengths. The scale bar represents the number of differences between the sequences. Source data are provided as a Source Data file.
To explore the evolution of the gene families among the four species, we used orthologous clustering to define 25,663 gene families (Fig. 7b). Most (19,203) were common to all the species, followed by the gene families shared by L. albus, L. cosentinii and L. digitatus (3221). The number of single-species gene families was similar in L. cosentinii and L. digitatus (94 and 88, respectively). The species tree inferred by Orthofinder indicated that the most closely related species were L. digitatus and L. cosentinii, followed by L. albus (Fig. 7c), reflecting the relationship known so far about Lupinus species (e.g. Drummond et al.65). The number of expanded gene families (2784 and 2751 in L. cosentinii and L. digitatus, respectively) and the number of contracted gene families (4071 and 4075, respectively) were similar in the two new assemblies when compared to L. albus. The same numeric similarity was observed when we used L. angustifolius as the reference species (Supplementary Table 2).
In conclusion, we have described whole-genome assemblies of the rough-seeded lupin species L. cosentinii and L. digitatus, providing insight into lupin genomics and evolution, and adding to the genetic resources available for lupin breeding and crop improvement. These two annotated assemblies provide key reference genomes for lupin and, more generally, the genistoid clade. Importantly, we provide evidence that both species are tetraploid. Our data provide insight into the role of genome duplication during lupin evolution but further evidence from other wild and domesticated species would help to complete the picture, enabling us to understand the domestication, agricultural improvement, environmental adaptability and evolution of legume crops, and facilitating the exploitation of legumes as part of a healthy and sustainable diet. The analysis of lupin gene families provided insights into their relationship with phenotypic diversification and species adaptation, which will facilitate the exploitation of underutilized legume species by identifying genes that can be used in crop breeding programs. Our work will underpin the development of improved lupin crops by exploiting the genetic diversity of CWRs and orphan crops to promote the conservation and sustainable utilization of lupins as a source of high-quality dietary protein, and to promote the domestication of a greater variety of wild lupin species.
Methods
Plant materials
The characteristics of L. cosentinii and L. digitatus are summarized in Supplementary Table 3. We selected L. cosentinii 98460 based on its seed production to secure enough seeds for further research and multiplication. Accession L. cosentinii 98460, CV population, country of collection Morocco, was obtained from the Polish Lupinus Collection (Poznan Plant Breeders Ltd, Wiatrowo branch, Poland). We used the only L. digitatus accession provided by US Department of Agriculture (ID: PI 660697, collected in Spain). For both species we developed single-seed descent (SSD) lines, and then multiplied them to conserve genetic resources. Seeds of both species were scarified, vernalized for 21 days and then sown in 7.5-L pots containing a 1:1 mix of peat and vermiculite. The plants were grown in a phytotron at 22/18 °C (day/night temperature) with a 16-h photoperiod, 60–65% relative humidity, and watering as required.
Extraction of high-molecular-weight DNA
For PacBio sequencing, high-molecular-weight DNA was extracted from 1 g frozen young leaf material that was ground to powder under liquid nitrogen. Nuclei were isolated in NIBTM buffer (10 mM Tris, 10 mM EDTA, 0.5 M sucrose, 80 mM KCl, 8% PVP (MW 10 kDa), 100 mM spermine, 100 mM spermidine, pH 9.0) supplemented with 0.5% Triton X-100 and 0.2% 2-mercaptoethanol, followed by filtration through 100-μm and 40-μm cell strainers and centrifugation (2500 g, 10 min, 4 °C)74. DNA was then extracted from nuclei using the Genomic-tip 100/G kit (Qiagen) and eluted in low-EDTA TE buffer (10 mM Tris, 0.1 mM EDTA, pH 9.0). DNA size and integrity were analyzed by pulsed-field gel electrophoresis (PFGE) using the CHEF Mapper system (Bio-Rad Laboratories) with a 5–450 kbp run program. DNA was quantified using the Qubit DNA BR Assay Kit and a Qubit fluorimeter (Thermo Fisher Scientific) and its purity was evaluated by spectrophotometry using a Nanodrop 2000 (Thermo Fisher Scientific). PacBio libraries were prepared from both species using the SMRTbell prep kit v3.0, followed by SMRT sequencing on a Sequel II device (Pacific Biosciences).
Whole-genome library preparation for Illumina sequencing
We fragmented 700 ng of high-molecular-weight DNA using an S220 sonicator (Covaris) and a WGS library was generated for both species using the KAPA Hyper Prep kit with a PCR-free protocol according to the manufacturer’s instructions (Roche). We applied final size selection by using a 0.7-fold ratio of AMPureXP beads (Beckman Coulter). The sequence length was assessed by capillary electrophoresis on a 4150 TapeStation (Agilent Technologies) and the library was quantified by qPCR against a standard curve with the KAPA Library Quantification Kit (Roche). Libraries were sequenced on a NovaSeq6000 Illumina platform in 150PE mode.
DNA extraction and Bionano optical mapping
Ultra-high-molecular-weight DNA was extracted from fresh sprouts or leaves (<2 cm in length) of L. cosentinii 98460 and L. digitatus PI 660697, which were kept in the dark for ~16 h before extraction75. DNA was isolated from ~0.4 g of sprouts using the Bionano Prep High Polysaccharides Plant Tissue DNA Isolation Protocol (Bionano Genomics, document number 30128, revision C). For each species, two agarose plugs were prepared according to Staňková et al.76. DNA extracted from one plug was assessed for length and concentration by PFGE as above. DNA from the second plug was used for Bionano optical mapping following the direct label and stain (DLS) protocol (Bionano Genomics). The labeled and stained DNA was loaded onto a Bionano Saphyr (Bionano Genomics).
Hi-C library preparation for Illumina sequencing
Hi-C libraries were prepared from 0.52 g of frozen young leaves of L. cosentinii 98460 and L. digitatus PI 660697 using the Proximo Hi-C (Plant) Kit and protocol v4.0 (Phase Genomics), incorporating three additional wash steps and 12 PCR amplification cycles. The quality of the Hi-C libraries was assessed using a D1000 ScreenTape Kit on a 2200 TapeStation (Agilent Technologies), and the quantity was determined by qPCR using primers that anneal to the adapter sequences. Libraries were sequenced on a NovaSeq 6000 Illumina platform in 150PE mode.
RNA-Seq library preparation for Illumina sequencing
We prepared 22 RNA samples from L. cosentinii 98460 (six samples of young leaves, four of fully developed leaves, four petioles, four pods and four roots) as well as 32 from L. digitatus PI 660697 (six of leaves, six petioles, six pods, five stems, four apical stems, three lateral roots and two main roots). Total RNA was isolated from 30 mg of ground plant tissue using the SV Total RNA Isolation System Kit (Promega) and its concentration and integrity were assessed using the RNA 6000 Nano Kit on a Bioanalyzer (Agilent Technologies). All samples showed an RNA integrity number (RIN) > 7. Samples were quantified using the Qubit RNA HS Assay Kit (Thermo Fisher Scientific). We pooled 2–3 RNA samples from the same tissue for library preparation to make pools of five different L. cosentinii tissues (young leaves, fully developed leaves, petioles, pods and roots) and seven different L. digitatus tissues (leaves, pods, stems, lateral roots, main roots, petioles and apical stems). RNA-Seq libraries were generated using the TruSeq stranded mRNA ligation kit (Illumina) from 1000-ng RNA samples, after poly(A) capture and according to the manufacturer’s instructions. Library quality and size were assessed by capillary electrophoresis using a 4150 TapeStation as above, and their quantity was determined by real-time PCR against a standard curve using the KAPA Library Quantification Kit as above. The libraries were pooled at equimolar concentrations and sequenced on a Novaseq6000 device in 150PE mode.
De novo genome assembly from PacBio Hi-Fi reads
PacBio Hi-Fi reads were assembled de novo using HiCanu v2.1.177 with default parameters. Completeness was evaluated using BUSCO v5.4.778 and the Fabales_obd10 database comprising 5,366 genes. Illumina WGS data were evaluated using FastqQC v0.11.9 and low-quality segments and sequencing adapters were removed using Fastp v0.21.079. Filtered reads were aligned on the assembly using bwa-mem2 v0.7.17 and residual base-level errors were corrected by three rounds of polishing using Pilon v1.23. To evaluate the effectiveness of this approach, we applied variant calling using GATK HaplotypeCaller v4.2.280 before and after polishing. We also used purge_haplotigs v1.1.281 to remove putative haplotype duplications. BLAST v2.9.0+82 was used to screen all remaining reads against the NCBI nr database to confirm that all reads belonged to the kingdom Viridiplantae, thus ensuring the absence of contamination. BLAST results were filtered considering a minimum identity coverage of 80% and minimum query coverage of 40%. BLAST was also used to screen mitochondrial (https://ftp.ncbi.nlm.nih.gov/refseq/release/mitochondrion/) and chloroplast (https://ftp.ncbi.nlm.nih.gov/refseq/release/plastid/) RefSeq databases and published L. albus organelle sequences23 to exclude organelle DNA.
Scaffolding with Bionano optical maps
Bionano sequencing outputs were filtered to remove molecules <150 kbp in length before de novo assembly and alignment on the corresponding genome maps using Bionano Solve v3.7.1 (https://bionanogenomics.com/support/).
Chromosome-level scaffolding with Hi-C data
The Hi-C raw reads were aligned on the Bionano genome assemblies using the Juicer v1.6 pipeline83 before a second round of scaffolding using 3d-dna v18.09.2284 with default parameters. Before the misjoin correction step, the Hi-C contact matrix was manually curated with juicebox v1.11.08.
Structural annotation
Repetitive elements in all four genomes were identified with HiTE v3.285. To evaluate the presence of indels in the two assembled genomes, Illumina reads from the polishing step were aligned and structural variations were called in the repeated regions of the genome by applying the TEPID pipeline v0.1586,87. Genomic divergence was computed with Parsing-RepeatMasker-Outputs v5.8.288.
RNA-Seq data were aligned on the assembled genome using Hisat2 v2.2.189 with a maximum intron length of 60 kbp. The alignments were then converted into intronic hints, retaining only those supported by at least 10 reads. RNA-Seq data were also assembled into transcripts using Trinity v2.1590. Only the primary isoform of all reconstructed genes, namely those classified as ‘main’ and ‘complete’ by Evidential Gene v2018, were retrieved and aligned on the assembled genome using gmap v2017-11-1591 for use as extrinsic evidence. Finally, proteins from the closely related species L. albus (https://phytozome-next.jgi.doe.gov/info/Lalbus_v1) were aligned on the genome assembly using Genome Threader v1.7.192. The extrinsic evidence extracted from the three different sources described above was then used for final ab initio gene prediction with Augustus v3.3.393 trained using Fabales BUSCO genes (BUSCO v5.4.7, Fabales_odb10 database). The predicted genes were filtered using InterProScan v5.52-86.094 to identify genes structurally related to known protein domains.
Functional annotation
Genes were functionally annotated based on the analysis of homology (BLAST v2.9.0+, keeping only the best hits for each gene) and protein domains (InterProScan). For homology-based analysis, we considered three levels of confidence: (1) genes with functional annotations in SwissProt (https://www.uniprot.org/uniprotkb?facets=reviewed%3Atrue&query=%2A), RefSeq plant databases (https://ftp.ncbi.nlm.nih.gov/refseq/release/plant/) and/or TAIR were labeled as high confidence; (2) genes were labeled as medium confidence if we retrieved functional annotations based on the L. albus proteome; and (3) genes that were not annotated using the first two levels were screened against the NCBI nr database to obtain a descriptive annotation. The alignments were filtered by percentage coverage and identity, both with thresholds of 80%. GO terms were derived from homology-based analysis at the first and second confidence levels (if the function was concordant) and from InterProScan analysis.
Ploidy analysis
The level of ploidy in L. cosentinii and L. digitatus was assessed using two methods, the first based on k-mer distribution and the second on biallelic SNP frequencies, applied to Illumina reads after noise reduction. For the first approach, the k-mers in WGS Illumina reads were counted using KMC v3.2.295. The k-mer distributions were analyzed using Genomescope2.0 and Smudgeplot96 with parameter –homozygous due to the high level of homozygosity. For the second approach, Gaussian mixture models were used to estimate the ploidy level with nQuire97. Reads were mapped to the genome and biallelic SNP frequencies were calculated. A delta log-likelihood score was then calculated between a free model and three fixed models (diploid, triploid and tetraploid). The lowest fixed-model score points to the most likely ploidy. This analysis was applied to the whole L. cosentinii and L. digitatus datasets and also to 26 individual sequences in L. cosentinii (corresponding to ~80% of the genome assembly) and 21 in L. digitatus (corresponding to ~90% of the genome assembly). If some sequences showed a lower score in a different ploidy model than the rest of the genome, this could be interpreted as a sign of aneuploidy. The rationale behind the use of two approaches was to validate the predicted ploidy level independently from the homozygosity of the two assembled genomes.
Comparative genomics
Orthofinder v2.5.498 was applied to all four species with default parameters (-S diamond). Genes in an orthogroup from the same species were considered paralogs and members of the same gene family. The file N0.tsv inside the “Phylogenetic Hierarchical Orthogroups” folder was used for downstream analysis, representing the different gene families. A phylogenetic tree was built based on the OrthoFinder results and converted to its ultrametric format using treePL.
Synteny was evaluated using MCScanX99 with default parameters. Specifically we used MCScanX_h, allowing the exploitation of orthologous genes from L. albus, L. angustifolius, L. cosentinii and L. digitatus predicted by Orthofinder. We tested the pairwise comparisons L. digitatus vs L. cosentinii (LdLc), L. albus vs L. digitatus (LaLd), L. albus vs L. cosentinii (LaLc), L. abus vs L. angustifolius (LaLn), L. angustifolius vs L. cosentinii (LnLc) and L. angustifolius vs L. digitatus (LnLd). The Ks distribution was evaluated considering only duplicated BUSCO genes. The coding regions of the orthologous gene pairs from the six pairwise comparisons were used to calculate Ka/Ks ratios in the MCScanX downstream analysis package “add_kaks_to_synteny”.
Variation in gene family sizes were characterized using Cafe5 v5.1.0100 with the -k 7 parameter followed by GO functional enrichment analysis of the expansion (gain) and contraction (loss) events in the gene families. Cafe5 was applied to all four species and the evaluation of gene family expansion/contraction and GO enrichment were achieved by comparing each assembled genome against one of the two published genomes, independently. GO enrichment analysis was implemented using the ‘enricher’ method of the clusterProfiler library101, considering only significant results (p < 0.05).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequence read data generated in this study have been deposited in the Sequence Read Archive (SRA) of the National Center of Biotechnology Information (NCBI) under BioProject ID PRJNA1080360 (L. cosentinii and L. digitatus) [https://www.ncbi.nlm.nih.gov/search/all/?term=PRJNA1080360], and Biosample IDs SAMN40127157 (L. cosentinii) [https://www.ncbi.nlm.nih.gov/biosample/?term=SAMN40127157] and SAMN40126867 (L. digitatus) [https://www.ncbi.nlm.nih.gov/biosample/?term=SAMN40126867]. The genome assemblies and annotations are publicly available under the BioProject ID PRJNA1080360 and can also be accessed at Figshare: L. cosentinii [https://doi.org/10.6084/m9.figshare.25367899] and L. digitatus [https://doi.org/10.6084/m9.figshare.25367935]. Seeds of L. cosentinii and L. digitatus are available upon request. Source data are provided with this paper.
Change history
20 August 2025
The following sentence was omitted from the acknowledgments section of this paper, ‘INCREASE has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 862862. This publication reflects only the author’s view and neither the Research Executive Agency (REA) nor the European Commission are responsible for any use that may be made of the information it contains’. The original article has been corrected.
References
Cardoso, D. et al. Reconstructing the deep-branching relationships of the papilionoid legumes. South Afr. J. Bot. 89, 58–75 (2013).
Kroc, M. et al. Towards development, maintenance, and standardized phenotypic characterization of single-seed-descent genetic resources for lupins. Curr. Protoc. 1, e191 (2021).
Gladstones, J. S. Lupins as crop plants. Field Crop Abstr. 23, 123–148 (1970).
Gladstones J. S. Distribution, origin, taxonomy, history and importance. in Lupins as Crop Plants: Biology, Production, and Utilization (eds Gladstones J. S., Atkins C. A. & Hamblin J.) (CAB International, 1998).
Nartea, A. et al. Legume byproducts as ingredients for food applications: preparation, nutrition, bioactivity, and techno-functional properties. Compr. Rev. Food Sci. Food Saf. 22, 1953–1985 (2023).
Shrestha, S., Lvt, Hag, Haritos, V. S. & Dhital, S. Lupin proteins: structure, isolation and application. Trends Food Sci. Technol. 116, 928–939 (2021).
Singh K. B., Kamphuis L. G. & Nelson M. N. The Lupin Genome (Springer, 2020).
Bulut, M. et al. A comprehensive metabolomics and lipidomics atlas for the legumes common bean, chickpea, lentil and lupin. Plant J. 116, 1152–1171 (2023).
Bellucci, E. et al. The INCREASE project: intelligent collections of food-legume genetic resources for European agrofood systems. Plant J. 108, 646–660 (2021).
Rawal VN, D. K. (eds) The Global Economy of Pulses (FAO, 2019).
Zhao, J. et al. Global systematic review with meta-analysis reveals yield advantage of legume-based rotations and its drivers. Nat. Commun. 13, 4926 (2022).
FAO. Tracking Progress on Food and Agriculture-related SDG Indicators 2023 (FAO, 2023).
Bohra, A. et al. Reap the crop wild relatives for breeding future crops. Trends Biotechnol. 40, 412–431 (2022).
Schreiber, M., Jayakodi, M., Stein, N. & Mascher, M. Plant pangenomes for crop improvement, biodiversity and evolution. Nat. Rev. Genet. 25, 563–577 (2024).
Cortinovis, G. et al. Adaptive gene loss in the common bean pan-genome during range expansion and domestication. Nat. Commun. 15, 6698 (2024).
Bertioli, D. J. et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 48, 438–446 (2016).
Stupar, R. M. Into the wild: the soybean genome meets its undomesticated relative. Proc. Natl. Acad. Sci. USA 107, 21947–21948 (2010).
Kang, Y. J. et al. Genome sequence of mungbean and insights into evolution within Vigna species. Nat. Commun. 5, 5443 (2014).
Varshney, R. K. et al. A chickpea genetic variation map based on the sequencing of 3,366 genomes. Nature 599, 622–627 (2021).
Khan, A. W. et al. Cicer super-pangenome provides insights into species evolution and agronomic trait loci for crop improvement in chickpea. Nat. Genet. 56, 1225–1234 (2024).
Hane, J. K. et al. A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: insights into plant-microbe interactions and legume evolution. Plant Biotechnol. J. 15, 318–330 (2017).
Garg, G. et al. A pan-genome and chromosome-length reference genome of narrow-leafed lupin (Lupinus angustifolius) reveals genomic diversity and insights into key industry and biological traits. Plant J. 111, 1252–1266 (2022).
Hufnagel, B. et al. High-quality genome sequence of white lupin provides insight into soil exploration and seed quality. Nat. Commun. 11, 492 (2020).
Xu, W. et al. The genome evolution and low-phosphorus adaptation in white lupin. Nat. Commun. 11, 1069 (2020).
Pancaldi, F. et al. The genome of Lupinus mutabilis: Evolution and genetics of an emerging bio-based crop. Plant J. 120, 881–900 (2024).
Naganowska, B., Wolko, B., Sliwinska, E. & Kaczmarek, Z. Nuclear DNA content variation and species relationships in the genus Lupinus (Fabaceae). Ann. Bot. 92, 349–355 (2003).
Susek, K., Bielski, W. K., Hasterok, R., Naganowska, B. & Wolko, B. A first glimpse of wild lupin karyotype variation as revealed by comparative cytogenetic mapping. Front Plant Sci. 7, 1152 (2016).
Susek, K. et al. Epigenomic diversification within the genus Lupinus. PLoS ONE 12, e0179821 (2017).
Aïnouche, A. & Bayer, R. Molecular phylogeny, diversification and character evolution in Lupinus (Fabaceae) with special attention to Mediterranean and African lupines. Plant Syst. Evol. 246, 211–222 (2004).
Hufnagel, B. et al. Pangenome of white lupin provides insights into the diversity of the species. Plant Biotechnol. J. 19, 2532–2543 (2021).
Plitmann, U. Evolutionary history of the old world lupines. TAXON 30, 430–437 (1981).
Mousavi-Derazmahalleh, M. et al. Exploring the genetic and adaptive diversity of a pan-Mediterranean crop wild relative: narrow-leafed lupin. Theor. Appl. Genet. 131, 887–901 (2018).
Atchison, G. W. et al. Lost crops of the Incas: Origins of domestication of the Andean pulse crop tarwi, Lupinus mutabilis. Am. J. Bot. 103, 1592–1606 (2016).
Maciel, H. S. & Schifino-Wittmann, M. T. First chromosome number determinations in south-eastern South American species of Lupinus L. (Leguminosae). Bot. J. Linn. Soc. 139, 395–400 (2002).
Conterato, I. F. & Schifino-Wittmann, M. T. New chromosome numbers, meiotic behaviour and pollen fertility in American taxa of Lupinus (Leguminosae): contributions to taxonomic and evolutionary studies. Bot. J. Linn. Soc. 150, 229–240 (2006).
Susek, K. et al. Impact of chromosomal rearrangements on the interpretation of lupin karyotype evolution. Genes 10, (2019).
Kroc, M., Koczyk, G., Święcicki, W., Kilian, A. & Nelson, M. N. New evidence of ancestral polyploidy in the genistoid legume Lupinus angustifolius L. (narrow-leafed lupin). TAG Theor. Appl. Genet. Theor. Angew. Genet. 127, 1237–1249 (2014).
Doyle J. J. Polyploidy in legumes. in Polyploidy and Genome Evolution (eds Soltis P. S. & Soltis D. E.) (Springer Berlin Heidelberg, 2012).
Drummond, C. S. Diversification of Lupinus (Leguminosae) in the western New World: derived evolution of perennial life history and colonization of montane habitats. Mol. Phylogenet. Evol. 48, 408–421 (2008).
Lavin, M., Herendeen, P. S. & Wojciechowski, M. F. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst. Biol. 54, 575–594 (2005).
Gepts, P. et al. Legumes as a model plant family. Genomics for food and feed report of the cross-legume advances through genomics conference. Plant Physiol. 137, 1228–1235 (2005).
Zhao, Y. et al. Nuclear phylotranscriptomics and phylogenomics support numerous polyploidization events and hypotheses for the evolution of rhizobial nitrogen-fixing symbiosis in Fabaceae. Mol. Plant 14, 748–773 (2021).
Cannon, S. B. et al. Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Mol. Biol. Evol. 32, 193–210 (2015).
Wang, J. et al. Hierarchically aligning 10 legume genomes establishes a family-level genomics platform. Plant Physiol. 174, 284–300 (2017).
Zhuang, W. et al. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat. Genet. 51, 865–876 (2019).
Goldblatt P. Cytology and the phylogeny of leguminosae. in Advances in Legume Systematics, Part 2. (ed Polhill R. M. RP) (Royal Botanic Gardens, 1981).
Cardoso, D. et al. Revisiting the phylogeny of papilionoid legumes: new insights from comprehensively sampled early-branching lineages. Am. J. Bot. 99, 1991–2013 (2012).
Kole C. Wild Crop Relatives: Genomic and Breeding Resources (Springer-Verlag, 2011).
Gresta, F. et al. Lupins in European cropping systems. in Legumes in Cropping Systems (eds Murphy-Bokern D., Stoddard F. & Watson C.) (CABI, 2017).
Gupta, S., Buirchell, B. J. & Cowling, W. A. Interspecific reproductive barriers and genomic similarity among the rough-seeded Lupinus species. Plant Breed. 115, 123–127 (1996).
Schmutz, J. et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet 46, 707–713 (2014).
Pecrix, Y. et al. Whole-genome landscape of Medicago truncatula symbiotic genes. Nat. Plants 4, 1017–1025 (2018).
Li H., Jiang F., Wu P., Wang K., Cao Y. A high-quality genome sequence of modellLegume Lotus japonicus (MG-20) provides insights into the evolution of root nodule symbiosis. Genes 11, 483 (2020).
Nix, J., Ranney, T. G., Lynch, N. P. & Chen, H. Flow cytometry for estimating plant genome size: revisiting assumptions, sources of variation, reference standards, and best practices. J. Am. Soc. Hort. Sci. 149, 131–141 (2024).
Wright, J. et al. Chromosome-scale genome assembly and de novo annotation of Alopecurus aequalis. Sci. Data 11, 1368 (2024).
Li, K., Xu, P., Wang, J., Yi, X. & Jiao, Y. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nat. Commun. 14, 6556 (2023).
Pulido, M. & Casacuberta, J. M. Transposable element evolution in plant genome ecosystems. Curr. Opin. Plant Biol. 75, 102418 (2023).
Estep, M. C., DeBarry, J. D. & Bennetzen, J. L. The dynamics of LTR retrotransposon accumulation across 25 million years of panicoid grass evolution. Heredity 110, 194–204 (2013).
Hawkins, J. S., Proulx, S. R., Rapp, R. A. & Wendel, J. F. Rapid DNA loss as a counterbalance to genome expansion through retrotransposon proliferation in plants. Proc. Natl. Acad. Sci. USA 106, 17811–17816 (2009).
Yang, L.-L. et al. Lineage-specific amplification and epigenetic regulation of LTR-retrotransposons contribute to the structure, evolution, and function of Fabaceae species. BMC Genom. 24, 423 (2023).
Aïnouche, A. et al. The repetitive content in lupin genomes. in The Lupin Genome (eds Singh K. B., Kamphuis L. G., & Nelson M.N.) (Springer International Publishing, 2020).
Wang, Q. & Dooner, H. K. Remarkable variation in maize genome structure inferred from haplotype diversity at the bz locus. Proc. Natl. Acad. Sci. USA 103, 17644–17649 (2006).
Zhu, H., Choi, H. K., Cook, D. R. & Shoemaker, R. C. Bridging model and crop legumes through comparative genomics. Plant Physiol. 137, 1189–1196 (2005).
Kajitani, R. et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395 (2014).
Drummond, C. S., Eastwood, R. J., Miotto, S. T. S. & Hughes, C. E. Multiple continental radiations and correlates of diversification in Lupinus (Leguminosae): testing for key innovation with incomplete taxon sampling. Syst. Biol. 61, 443–460 (2012).
Feng, X. et al. Genomic evidence for rediploidization and adaptive evolution following the whole-genome triplication. Nat. Commun. 15, 1635 (2024).
Święcicki, W. et al. Chromatographic fingerprinting of the Old World lupins seed alkaloids: a supplemental tool in species discrimination. Plants 8, 548 (2019).
Nevado, B., Atchison, G. W., Hughes, C. E. & Filatov, D. A. Widespread adaptive evolution during repeated evolutionary radiations in New World lupins. Nat. Commun. 7, 12384 (2016).
Hung, T. H. et al. Reference transcriptomes and comparative analyses of six species in the threatened rosewood genus Dalbergia. Sci. Rep. 10, 17749 (2020).
Qu, Z., Wang, W. & Adelson, D. L. Chromosomal level genome assembly of medicinal plant Sophora flavescens. Sci. Data 10, 572 (2023).
Lei, W. et al. Chromosome-level genome assembly and characterization of Sophora japonica. DNA Res. 29, dsac009 (2022).
Mondin, M. & Aguiar-Perecin, M. L. R. Heterochromatin patterns and ribosomal DNA loci distribution in diploid and polyploid Crotalaria species (Leguminosae, Papilionoideae), and inferences on karyotype evolution. Genome 54, 718–726 (2011).
Bacchetta, G., Brullo, S., Velari, T. C., Chiapella, L. F. & Kosovel, V. Analysis of the Genista ephedroides group (Fabaceae) based on karyological, molecular and morphological data. Caryologia 65, 47–61 (2012).
Zhang, M. et al. Preparation of megabase-sized DNA from a variety of organisms using the nuclei method for advanced genomics research. Nat. Protoc. 7, 467–478 (2012).
Canaguier, A. et al. Oxford Nanopore and Bionano Genomics technologies evaluation for plant structural variation detection. BMC Genom. 23, 317 (2022).
Staňková, H. et al. BioNano genome mapping of individual chromosomes supports physical mapping and sequence assembly in complex plant genomes. Plant Biotechnol. J. 14, 1523–1531 (2016).
Nurk, S. et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305 (2020).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: novel and streamlined workflows along with boader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinforma. 19, 460 (2018).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Hu, K. et al. HiTE: a fast and accurate dynamic boundary adjustment approach for full-length transposable element detection and annotation. Nat. Commun. 15, 5573 (2024).
Horvath, R., Minadakis, N., Bourgeois, Y. & Roulin, A. C. The evolution of transposable elements in Brachypodium distachyon is governed by purifying selection, while neutral and adaptive processes play a minor role. eLife 12, RP93284 (2024).
Stuart, T. et al. Population scale mapping of transposable element diversity reveals links to gene regulation and epigenomic variation. eLife 5, e20777 (2016).
Kapusta, A., Suh, A. & Feschotte, C. Dynamics of genome size evolution in birds and mammals. Proc. Natl. Acad. Sci. USA 114, E1460–E1469 (2017).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Gremme, G., Brendel, V., Sparks, M. E. & Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Inf. Softw. Technol. 47, 965–978 (2005).
Hoff, K. J. & Stanke, M. Predicting genes in single genomes with AUGUSTUS. Curr. Protoc. Bioinforma. 65, e57 (2019).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Kokot, M., Długosz, M. & Deorowicz, S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33, 2759–2761 (2017).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Weiß, C. L., Pais, M., Cano, L. M., Kamoun, S. & Burbano, H. A. nQuire: a statistical framework for ploidy estimation using next generation sequencing. BMC Bioinforma. 19, 122 (2018).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Mendes, F. K., Vanderpool, D., Fulton, B. & Hahn, M. W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518 (2020).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Acknowledgements
This study was supported by the National Science Centre, Poland (grant nos. HARMONIA 7 2015/18/M/NZ2/00422 and OPUS 18 2019/35/B/NZ8/04283 to KS). We thank Andrea Benazzo and Robert Hasterok for critical comments that improved the manuscript. We acknowledge the support provided by the Horizon 2020 Project INCREASE, grant agreement number 862862 (R.P. and K.S.; https://www.pulsesincrease.eu). INCREASE has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 862862. This publication reflects only the author’s view and neither the Research Executive Agency (REA) nor the European Commission are responsible for any use that may be made of the information it contains.
Author information
Authors and Affiliations
Contributions
K.S. conceptualized the study, designed the experiments, wrote the manuscript, interpreted data and supervised the project. L.V. carried out bioinformatic analysis, prepared the figures, and helped to write the manuscript. M.T. cultivated the plants under controlled conditions. M.K. analyzed the plants, extracted nucleic acids, assisted with data interpretation and manuscript preparation. E.F assisted with the bioinformatic analysis and drafted the corresponding part of the manuscript. E.C. and A.R.L. performed laboratory experiments. U.K.T. assisted with bioinformatic analysis and manuscript preparation. H.J. assisted with bioinformatic analysis. M.D. supervised genome sequencing and assembly. M.D., M.N.N., P.B., D.E., R.P. and S.A.J. assisted with data interpretation. R.P., M.D. and S.A.J. contributed to the substantive revision and editing of the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests
Peer review
Peer review information
Nature Communications thanks Abdelkader Ainouche, who co-reviewed with Jean Keller; Lars Kamphuis and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Susek, K., Vincenzi, L., Tomaszewska, M. et al. The unexplored diversity of rough-seeded lupins provides rich genomic resources and insights into lupin evolution. Nat Commun 16, 4358 (2025). https://doi.org/10.1038/s41467-025-58531-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58531-w
