
Abstract
Lung cancer is the leading cause of cancer-related deaths worldwide. Although immunotherapy such as anti-programmed death-1 and its ligand 1 (PD-1/L1) is a standard treatment for advanced non-small cell lung cancer (NSCLC), many patients do not derive benefit directly. Several studies have elucidated new strategies to improve the antitumor immune response through gut microbiota modulation. However, it remains largely debatable regarding how gut microbiota remotely affect lung cancer microenvironment and subsequently modulate immunotherapy response. Here we show that commensal Bifidobacterium-derived extracellular vesicles (Bif.BEVs) can modulate the therapeutic effect of anti-PD-1 therapy in NSCLC. These Bif.BEVs are up-taken by lung cancer cells predominantly via dynamin-dependent endocytosis and upregulate PD-L1 expression through TLR4-NF-κB pathway. They also efficiently penetrate murine intestinal and patient-derived lung cancer organoids. Oral gavage of these Bif.BEVs result in their accumulation in tumors in mice. Using a syngeneic mouse model, Bif.BEVs are found to synergize the anti-tumor effect of anti-PD-1 via modulation of key cytokines, immune response and oncogenic pathways, and increase in tumor-infiltrating CD8+ T cells. Our study therefore identifies a link between Bif.BEVs and the tumor microenvironment, providing an alternative mechanism to explain how gut microbiota can influence immunotherapy response, particularly in tumors located anatomically distant from the gut.
Similar content being viewed by others


Introduction
The past decade of research has made critical development in the treatment of lung cancer that has substantially improved overall survival of patients. Immunotherapy using immune checkpoint inhibitors (ICIs) targeting programmed death-1 and its ligand 1 (PD-1/L1) and cytotoxic T- lymphocyte-associated protein 4 (CTLA-4) has emerged as a treatment of choice for the routine care of variety of cancers including non-small cell lung cancer (NSCLC)1,2,3,4. These ICIs have proven their efficacy by prolonging patients’ survival1,2,3,4. However, only a small proportion of cancer patients directly derive benefit from immunotherapy, and occasional serious immune related adverse events (irAEs) could further limit its therapeutic value. Finding modifiable modulating factors is therefore urgently needed and clinically relevant.
Microorganisms, particularly gut microbiota, have shown the potential to modulate cancer immunotherapy5,6. For example, Sivan et al., in 2015 demonstrated that the commensal Bifidobacterium (Bif.B) in the gut can enhance the antitumor effects of anti-PD-L1 therapy7. Additionally, in a colitis model, Bif.B mitigated intestinal immunopathology without affecting the antitumor response to anti-CTLA-4 treatment8. These findings are supported by evidence from human studies showing that Bif.B is more abundant in NSCLC patients who respond well to ICIs9. Our previous clinical study also found that enrichment of this genus of bacteria correlated with less irAEs10. These studies collectively underscore the role of gut microbiota in modulating host immune status and response to cancer immunotherapy11, justifying our focus on Bif.B in this study. Furthermore, since Bif.B is commercially available as a probiotic, it offers a practical option for clinical application.
The tumor microenvironment (TME) or tumor immune microenvironment (TIME) dictates the response to immunotherapy12. Since gut microbiota reside inside of the intestinal tract where cellular tight junctions and a mucin layer over the intestinal epithelium prevent them from crossing the intestinal barrier, it remains elusive how gut microbiota can remotely affect the TIME of lung cancer and its subsequent response to immunotherapy. While bacterial metabolites (e.g., short chain fatty acids; SCFA), migration of locally activated immune cells, and altered cytokine/chemokines have all been speculated to address the effect of gut microbiota on cancer immunotherapy13, we propose here an alternative model involving bacteria-derived extracellular vesicles (BEVs), to connect gut microbiota remotely to the TIME of NSCLC.
All eukaryotic and prokaryotic cells constantly interact with their surrounding by means of cellular components, among which, the production, uptake and secretion of extracellular vesicles (EVs) is one primordial feature14. The communication between bacteria and with host cells happens either through direct contact or by release of various metabolites, nucleic acids-DNA-RNA, proteins, toxins, lipoglycans and membrane vesicles15,16. Both Gram-positive and negative bacteria release EVs and are comprised of cell membrane and cytoplasm with their size range between 10-400 nm17. These EVs can deliver numerous immune-stimulatory or modulatory signals from donor cells to the recipient cells that can further regulate immune responses in mammalian host cells making them potential immunotherapeutic agents17,18.
In this study, we aimed to elucidate how BEVs derived from the Gram-positive bacteria Bif.B (Bif.BEVs) can remotely modulate the TIME of NSCLC, and subsequent response to anti-PD-1 immunotherapy. We utilized 2D cell culture, 3D organoids and an in-vivo syngeneic mouse model in a stepwise approach to delineate the process.
Results
Bif.BEVs show characteristic EV features and carry lipoteichoic acid (LTA) of Bif.B origin
Several publications have shown that bacteria secrete EVs that can be collected and enriched19,20. Bif.BEVs have their characteristic morphology, size range and origin similar to mammalian-derived EVs21. To begin with, we assessed the release of Bif.BEVs and characterized them. We first used scanning electron microscopy (SEM) to visualize the surface of Bif.B. SEM showed vesicle-like structures protruding/blebbing outside on the surface of Bif.B (Fig. 1a). We noted a significant number of Bif.B that were secreting such vesicles. To further characterize those secreted vesicles, Bif.B were grown in anaerobic culture for 48 h and then supernatant was collected. Bif.BEVs were isolated by using an ultracentrifugation approach as described in Methods. The size of Bif.BEVs was determined by NanoSight nanoparticle tracking analysis (NTA). Isolated Bif.BEVs ranged from 30-500 nm with the majority of the Bif.BEVs falling in the 119 nm and 242 nm range, the size that well defines EVs (Fig. 1b). Isolated Bif.BEVs were further subjected to transmission electron microscopy (TEM) to visualize Bif.BEVs and assess their morphology. All Bif.BEVs showed characteristic cup-shaped morphology (Fig. 1b inset). It was further important to confirm that isolated BEVs originated from Bif.B. To assess, immunogold labeling against surface associated LTA was performed on the Bif.B and the sections of bacteria were visualized by TEM to identify anti-LTA-gold particles on the bacterial surface. The immunogold TEM micrograph clearly showed the presence of LTA on the surface of bacteria (Fig. 1c). Interestingly, the anti-LTA-immunogold labeling was also observed on the vesicles, likely secreted Bif.BEVs, in the close vicinity of the Bif.B (Fig. 1c inset). To further validate the presence of LTA in Bif.BEVs, western blot was performed. LTA was found present in lysates from both Gram-positive Bif.B cells and their BEVs (Bif.BEVs) but not from Gram-negative E. coli-derived EVs (Eco.BEVs). Anti-LPS western blot was used to differentiate Gram-positive Bif.B and Bif.BEVs lysates from Gram-negative Eco.BEVs lysate (Fig. 1d). Based on these, we concluded that Bif.BEVs have characteristic morphology of BEVs and contain LTA as an integral component of their cargo. To clarify, there are two primary reasons why LTA was chosen as the marker for Bif.BEVs: 1) LTA is a major component of the cell wall in Gram-positive bacteria, anchoring to the cell membrane22; 2) According to the 2023 Position Paper from the International Society for Extracellular Vesicles (ISEV) on minimal information for studies of extracellular vesicles (MISEV2023), LTA serves as a specific biomarker for BEVs derived from Gram-positive bacteria23. This biomarker is advantageous due to the availability of commercially available antibodies23.

a SEM Image showing vesicle-like bodies blebbing outside on the surface of Bif.B. b NTA showing size distribution of secreted EVs along with particle concentration of Bif.BEVs. Inset showing negative staining TEM image of isolated Bif.BEVs (scale bar = 100 nm). c Immunogold electron micrograph of anti-LTA-labeled Bif.B. Inset showing enlarged image of anti-LTA-bound Bif.BEVs. Data shown here are the best of three independent experiments. d Comparative western blot analysis of Bif.BCL, Bif.BEVs and E. coli EVs’ lysate (Eco.BEVs) probed for anti-LTA and anti-LPS. Data shown here are the representative of three independent experiments. Source data are provided as a Source Data file.
Uptake of Bif.BEVs utilizes dynamin-dependent endocytosis and modulates the expression of immune checkpoints in lung cancer cells
Although studies showed Bif.B can synergize ICIs in anti-tumor effect7,9, there is lack of data to suggest Bif.BEVs may also possess immunomodulatory capacity. To address this, we started investigation from a 2-D in vitro system. We first questioned whether Bif.BEVs can be taken up by the cells as a key step to exert biological function. As shown in Suppl. Fig. 1a, human lung cancer A549 cells were treated for 24 h with isolated Bif.BEVs fluorescently labeled with PKH67, a membrane dye. Immunofluorescent imaging of cells demonstrated uptake of green PKH67-labeled Bif.BEVs in A549 cells. Similar uptake was also observed using murine lung cancer LL/2 cells (Suppl. Fig. 1b).
To explore the possible mechanism of cellular uptake of Bif.BEVs, we used various inhibitors including amiloride (AM), cytochalasin D (CD), dynasore (DS) and nystatin (NS). AM inhibits the Na + /H+ exchanger that is crucial for macrocytosis; CD inhibits actin that is indispensable for both macrocytosis and phagocytosis; DS primarily inhibits dynamin dependent endocytosis; and NS inhibits lipid rafts that play a role in caveolin-dependent endocytosis24. Various cell lines, like murine LL/2 (Fig. 2a) and human A549 (Suppl. Fig. 2) (along with Calu-1 and H1792 cells; Suppl. Fig. 3), were pre-treated with endocytosis inhibitors for 2 h and then with PKH67 stained 5 µg/ml Bif.BEVs for 24 h. Looking at the data from the microscopic images of various cell lines as described above, it was discovered, that the uptake of Bif.BEVs in LL/2 cells was consistently and most prominently inhibited by DS (Fig. 2a), suggesting dynamin-dependent endocytosis represents one of the major mechanisms for cellular uptake of Bif.BEVs, although other processes could have also contributed. The quantitative data demonstrates a significant inhibition of Bif.BEVs uptake upon treatment with DS (Fig. 2b). Further, LL/2 cells were pre-treated with endocytosis inhibitors for 2 h and then with 5 µg/ml Bif.BEVs and western blot analysis of LTA was performed (Fig. 2c, Suppl. Fig. 4). It was observed that the expression of LTA was lowest in DS treated cells, confirming that the uptake of Bif.BEVs heavily relied on dynamin-dependent endocytosis (Fig. 2c). To further validate our findings, we generated isogenic LL/2 cells with stable dynamin I/II knockdown using an shRNA approach. After incubating these LL/2 cells with Bif.BEVs, we observed a reduced detection of LTA in dynamin-deficient cells, indicating a decreased uptake of Bif.BEVs (Suppl. Fig. 5a). Additionally, we examined changes in genes associated with dynamin-dependent endocytosis, including dynamin I/II/III, clathrin, and EPS15. Exposure to Bif.BEVs in LL/2 cells led to the upregulation of dynamin II/III, clathrin, and EPS15 (Suppl. Fig. 5b–f). These effects were inhibited by DS, with the most notable reduction seen in dynamin expression. Together, these results suggest that dynamin is crucial for the endocytosis of Bif.BEVs.

a Immunofluorescent detection of uptake of PKH67 (green) stained Bif.BEVs (5 µg/ml) by LL/2 cells after 24 h post treatment with endocytosis inhibitors. DAPI (blue) was used to visualize nuclei. The scale bar is 20 µm. b Quantification of fluorescence intensity of images from 2a by ImageJ analysis using 17 different areas. Statistical comparisons of medians were conducted using the Kruskal-Wallis test. For all box plots, the central line inside each box represents the median (50th percentile). The box bounds correspond to the first quartile (Q1, 25th percentile) and third quartile (Q3, 75th percentile). The whiskers extend to the minimum and maximum values. Exact p values are indicated in the figure. c Western blot analysis of anti-LTA in LL/2 cells after pre-treatment with endocytosis inhibitor and Bif.BEVs (5 ug/ml). GAPDH was used as internal control. d Western blot analysis of PDL-1, CD80, PVR/CD155 in lung cancer cells (A549, H460, LL/2) along with immortal noncancerous lung cells BEAS-2B after treatment with either Bif.BEVs (5 µg/ml) or cell lysates (5 µg/ml). GAPDH and actin were used as internal controls. e Western blot analysis of TLR-4, TLR-7 and NF-κB after treatment with Bif.BEVs (5 µg/ml). Actin was used as internal loading control. f Western blot analysis of NF-κB and PD-L1 after pre-treatment with NF-κB inhibitor for 2 h and Bif.BEVs (5 µg/ml) for 48 h. All Western blots except using BEAS-2B (due to slow growth) have been repeated at least twice in independent experiments. Source data are provided as a Source Data file.
Noticing that Bif.BEVs can be actively taken up by lung cancer cells, we then questioned if Bif.BEVs can subsequently modulate the expression level of immune checkpoints, e.g., PD-L1. To test this, murine LL/2 and human lung cancer cells A549, H460 and lung immortal noncancerous cells BEAS-2B were treated with Bif.BEVs at a concentration of 5 µg/ml Bif.BEVs for 48 h (Fig. 2d). Using Western blot analysis, we assessed the expression levels of immune checkpoints, including PD-L1, CD80 and PVR/CD155. This selection was based on the availability of corresponding antibodies in clinical use (either as standard-of-care such as anti-PD-1/L1 and anti-CTLA-4, or in clinical trials such as anti-TIGIT). Bif.BEVs showed significant and consistent increase in PD-L1 expression in human (A549, H460) and mouse (LL/2) lung cancer cells (Fig. 2d, Suppl. Fig. 4), as well as immortalized nontumorigenic lung BEAS-2B cells. We also noted increased expression of CD80 in all cell lines treated by Bif.BEVs. There was no obvious change noted for PVR/CD155 (Fig. 2d, not tested for LL/2 due to lack of antibody). The Bif.B bacterial cell lysates (Bif.BCL) was able to induce similar change, suggesting possible a shared component of Bif.BEVs and Bif.BCL is responsible for this effect. While the regulation of each immune checkpoint ligand’s expression could vary, in this paper, we focused on PD-L1 expression. Since LTA has been reported to regulate PD-L1 expression via the Toll-like receptor (TLR)- NF-κB pathway25,26,27, we wondered if this could be the mechanism of Bif.BEVs inducing PD-L1 expression. To test this, LL/2 cells were treated with 5 µg/ml Bif.BEVs for 48 h and western blot analysis of the levels of several TLRs (TLR-2, TLR-4, TLR-6 TLR-7) was performed. Increased expression of both TLR-4 and NF-κB was noted (Fig. 2e, Suppl. Fig. 4) when the cells were treated with Bif.BEVs as compared to control. Further, to confirm our results, we pre-treated the cells with an NF-κB inhibitor for 2 h and then treated with 5 µg/ml Bif.BEVs for 48 h as illustrated in Fig. 2f. It was noted that treatment with Bif.BEVs led to increased expression of NF-κB and PD-L1 which was abolished after pretreatment with the NF-κB inhibitor (Fig. 2f, Suppl. Fig. 4). This provides a mechanism of PD-L1 upregulation, i.e, the dynamin-dependent endocytosis of Bif.BEVs activated the TLR-4-NF-κB pathway which then induced the expression of PD-L1.
Biodistribution of BEVs in organoids and in-vivo syngeneic mouse models
Noticing Bif.BEVs could be taken up by cancer cells in a 2D culture system, we wondered if Bif.BEVs could get into and accumulate inside cells in a 3D environment, as well as in vivo due to their nanoscale in size. If so, Bif.BEVs can probably penetrate gut epithelial lining, enter the blood circulation, and eventually accumulate in the tumor microenvironment of lung cancer – this seems a plausible model to explain the remote effect of gut microbiota on lung cancer immunotherapy response. Here, we are connecting the dots to address this possibility. To begin with, we developed intestinal organoids by collecting the crypts from 15-week-old C57BL/6 mice and processing them to culture organoids. Intestinal organoids grew into typical crypt-villus architecture and had regenerative capabilities in culture (Fig. 3a). After 7 days in culture, organoids were treated with PKH26 (red) labeled Bif.BEVs (10 µg/ml) for 24 h along with no BEV control (Fig. 3b). After treatment, the organoids were stained with 1 µg/ml of Hoechst for 30 min at 37 °C. The confocal microscopy showed the presence of Bif.BEVs in both the intestinal cells and crypt lumen, therefore providing evidence of Bif.BEVs’ internalization into the intestinal cells and penetration through into the lumen of crypts (Fig. 3b).

a Bright field imaging of 7-day old intestinal organoid developed from 15-week-old C57BL/6 mice. b Confocal imaging showing uptake of PKH26 (red) stained Bif.BEVs (10 µg/ml) by intestinal organoids along with no BEVs control after 24 h. DAPI (blue) was used to visualize nuclei. c Bright field image of 14-day old organoid developed from a lung cancer patient. d Immunofluorescent detection of patient-derived lung cancer organoids treated with PKH67 (green) stained Bif.BEVs (10 µg/ml) for 24 h. DAPI (blue) was used to visualize nuclei. e Immunofluorescent detection of patient-derived lung cancer organoids treated with DS for 2 h and then with PKH67 (green) stained Bif.BEVs (10 µg/ml) for 24 h. DAPI (blue) was used to visualize nuclei. Scale bar = 10 µm. a–e are representatives of three independent experiments with all scale bars = 10 µm. f Syngeneic tumor bearing C57BL/6 mice were orally administered with either DiD labeled Bif.BEVs (2 × 1010/mouse) or unlabeled in PBS. The organs were excised 8-10 h post Bif.BEVs administration for imaging using IVIS imaging system. The epifluorescence was measured using radiant efficiency. The images shown are representative of four tumors. The graph on the right shows quantification of radiant efficiency in the tumors. The histogram bars depict mean ± SEM values, analyzed using an unpaired t-test with Welch’s correction (two-sided). Exact p-values are indicated in the figure. Source data are provided as a Source Data file.
Next, we wanted to know if Bif.BEVs can also be taken up by patient-derived lung tumor spheroids or organoids. The bright field image in Suppl. Fig. 6a shows 14-day old spheroid developed from human lung adenocarcinoma (LUAD) tissue. The floating spheroids were treated with PKH67 (green) labeled EVs (5 µg/ml) for 24 h. After treatment, the spheroids were visualized using confocal microscopy and significant uptake of BEVs (green) was noted (Suppl. Fig. 6b). We also developed 3D organoids from LUAD tissue as shown in Fig. 3c (bright field image, 14 day old). Immunofluorescence imaging of organoids treated with PKH67 labeled Bif.BEVs (10 µg/ml) for 24 h (Fig. 3d) showed significant uptake of Bif.BEVs and their presence in the luminal side and interstitium, which further provided evidence that Bif.BEVs can easily cross 3D tissue/organ (Fig. 3d) and are therefore likely capable of penetrating into tumor as well. On the contrary, when LUAD organoids were treated with DS inhibitor, reduced uptake of PKH67-labeled Bif.BEVs were noted as shown in Fig. 3e, which strongly supported our hypothesis.
Since 3D organoids can develop apical/basal polarity which could affect EV uptake, we also treated LUAD organoids with the same inhibitors shown in Fig. 2a–c. The organoids were then dissociated into a single-cell suspension to quantify the uptake of Bif.BEVs via flow cytometry. We measured the mean fluorescent intensity of cells exhibiting green fluorescence, indicating the uptake of PKH67-stained Bif.BEVs. We observed consistent inhibition with various endocytosis inhibitors, with the strongest effect again noted when DS was used. However, unlike the 2D context, we also saw significant inhibition with AM, suggesting that macropinocytosis might also play a significant role in Bif.BEVs’ endocytosis in the 3D model (Suppl. Fig. 7). Interestingly, cell polarity proteins were found to promote macropinocytosis28.
Observing that Bif.BEVs can be internalized into 3D structures ex vivo, we aim to validate our hypothesis through in vivo studies using mice. Here we established a syngeneic tumor model by injecting LL/2 cells subcutaneously into the flanks of syngeneic C57BL/6 mice. After tumor reached 1.5 cm3 in size, Bif.BEVs were labeled with far-red fluorescent dye (DID [1,1-Dioctadecyl-3,3,3,3-tetramethylindodicarbocyanine]) and orally administered to the mice19. Eight hours later, mice were sacrificed, and various tissue and organs were harvested and imaged using IVIS imaging (Fig. 3f). The organs were imaged side by side from mice treated with DID-labeled Bif.BEVs vs. PBS dissolved Bif.BEVs to have a direct comparison. The radiant efficiency was measured and a significant increase in far-red fluorescence was observed in various organs (intestine, liver, kidney, lung, and tumors) in mice treated with DID-labeled Bif.BEVs. A strong radiant efficiency was noted in tumors as shown by the quantification of tumors in a bar diagram (Fig. 3f), suggesting significant distribution of Bif.BEVs inside of tumors. In addition, a various degree of Bif.BEVs accumulation was observed in other organs such as the intestine, lungs, kidney, and liver etc. Altogether, these biodistribution data clearly demonstrated that Bif.BEVs can cross the intestinal barrier and travel to distant tumor sites to potentially modulate the TME/TIME.
Bif.BEVs enhanced therapeutic effect of anti-PD-1 in NSCLC
Since Bif.BEVs can upregulate PD-L1 expression and cross through various tissues/organs, including tumor, we wanted to evaluate whether Bif.BEVs can modulate the therapeutic effect of anti-PD-1 in NSCLC. Here, we used the LL/2 syngeneic model as it is a notoriously poorly immunogenic tumor model29,30. As a proof-of-concept experiment, we used syngeneic model by subcutaneously injected the LL/2 cells in both flanks of C57BL/6 mice as described in Methods. The treatment scheme is shown in Fig. 4a. The mice were subdivided into 5 groups: untreated control (Control), IgG Control (IgG), anti-PD-1 only (anti-PD-1), anti-PD-1+Bif.BEVs (2 × 1010), anti-PD-1+Bif.B. The tumors were allowed to grow for a week and then treatment started. When compared to the Control group, both anti-IgG and anti-PD-1 groups showed nearly similar growth without significant changes as shown in the graph in Fig. 4b, confirmed low efficacy of anti-PD-1 only in LL/2 mouse model. But anti-PD-1+Bif.BEVs (2 X 1010) and anti-PD-1+Bif.B groups showed significant decrease in tumor growth as compared to control group. Interestingly, anti-PD-1+Bif.BEVs (2 X 1010) showed nearly comparable growth with the anti-PD-1+Bif.B group (Fig. 4b). The tumor weights also recapitulated the observations from tumor volume (Fig. 4c). These observations raised a question if Bif.BEVs can have a dose dependent response towards tumor suppression. To answer, in a separate experiment, LL/2 tumors were treated with a 10-fold higher amount of Bif.BEVs to see the effect. In this experiment, mice were divided in four groups as Control, Bif.BEVs (2 × 1010), anti-PD-1+Bif.BEVs (2 × 1010) and anti-PD-1+Bif.BEVs (2 × 1011). All the groups were treated in the similar fashion (Fig. 4d). Here, we intentionally included a Bif.BEVs group to know whether the effect in the previous experiment was primarily due to Bif.BEVs vs. the combinatorial effect with anti-PD-1. It was noted that Bif.BEVs (2 × 1010) alone did not result in significant decrease in tumor growth as compared to the control group, but interestingly, the anti-PD-1+Bif.BEVs (2 × 1011) group (10x more Bif.BEVs) in combination with anti-PD-1 showed significant reduction in tumor volume and weight as compared to anti-PD-1+Bif.BEVs (2 × 1010) and control groups (Fig. 4e, f). Overall, these two experiments clearly demonstrated that BEVs derived from commensal Bif.B can enhance the therapeutic efficacy of anti-PD-1 in NSCLC.

a Schematic representation showing the timeline of tumor cell implantation, tumor formation and treatment with either Bif.BEVs followed by anti-PD-1 or isotype control. Created in BioRender83. b Tumor growth curves showing tumor volumes after administration of control IgG, anti-PD-1, anti-PD-1+Bif.BEVs, anti-PD-1+Bif.B along with control group. The total number of tumors assessed was n = 6 per group. The inset displays representative tumor sizes. Comparison of means at different time points was performed using a two-way ANOVA. c Boxplot showing the changes in tumor volumes after treatment with control, IgG control, anti-PD-1, anti-PD-1+Bif.BEVs, and anti-PD-1+Bif.B. The total number of tumors assessed was n = 6 per group. The Mann-Whitney test (two-tailed) was used to compare the medians. d Schematic representation of a separate experiment that used increasing number of Bif.BEVs. Created in BioRender84. e Tumor growth curves showing tumor volumes in the groups of Control, Bif.BEVs only (2 × 1010), anti-PD-1+Bif.BEVs (2 × 1010) and anti-PD-1+Bif.BEVs (2 × 1011) respectively. The total number of tumors assessed was n = 10 per group. Inset shows the representative image of changes in the tumor growth with increasing concentration of Bif.BEVs as described above. Comparison of means at different time points was performed using a two-way ANOVA. f Boxplot showing the changes in tumor volumes after treatment with control, Bif.BEVs only (2 × 1010), anti-PD-1+Bif.BEVs (2 × 1010), anti-PD-1+Bif.BEVs (2 × 1011). The total number of tumors assessed was n = 10 per group. The Kruskal-Wallis test was used to compare the medians with those of the control group. All tumor growth curve graphs (b & e) present data as the mean ± SEM. In all box plots (c & f), the central line within each box represents the median (50th percentile), while the box boundaries correspond to the first quartile (Q1, 25th percentile) and third quartile (Q3, 75th percentile). The whiskers extend to the minimum and maximum values. All p values are shown in the figure. Source data are provided as a Source Data file.
Bif.BEVs plus anti-PD-1 mitigated tumor growth via modulating cytokine levels
To investigate how Bif.BEVs potentiated anti-PD-1 effect, we first measured the level of Ki67, a reliable marker for tumor cell proliferation31,32. As shown in Fig. 5a, the numbers of Ki67+ cells by IHC were significantly lower in both the anti-PD-1+Bif.BEVs (2 × 1010) and anti-PD-1+Bif.BEVs (2 × 1011) groups. The lower magnification images (10x) (Suppl. Fig. 8) were confirmatory of Bif.BEVs’ capacity of potentiating the tumor suppressive effect of anti-PD-1 through reducing the proliferation of tumor cells.

a Representative images of Ki67 staining in tumor sections from the groups of control, anti-PD-1, Bif.BEVs only (2 × 1010), anti-PD-1+Bif.BEVs (2 × 1010), anti-PD-1+Bif.BEVs (2 × 1011). Images are displayed at 20x magnification. b The expression level of TNF-α, IL-2 and IFN-γ were measured by real time RT-PCR using tumor specimens harvested from different groups, with n = 5 from each group. All graphs present data as the mean ± SEM, with comparison to the untreated control (UC) group performed using an unpaired two-sided t test. Exact p values are displayed in the figure. Source data are provided as a Source Data file.
Cytokine release is a key element in determining the fate of tumor growth. Cytokines secreted by both the tumor and immune cells in the TIME may either induce or suppress tumor progression33,34. We analyzed major cytokines (TNF-α, IL-2, IFN-γ and IL-10) by quantitative PCR to understand the mechanism of Bif.BEV-induced tumor suppression in combination with anti-PD-1 therapy. The relative expression levels were measured in the tumor tissues from all the 5 groups including control, anti-PD-1, Bif.BEVs (2 × 1010), anti-PD-1+ Bif.BEVs (2 × 1010), anti-PD-1+Bif.BEVs (2 × 1011) as shown in Fig. 5b. It was noted that the level of proinflammatory cytokine TNF-α35, was significantly decreased in anti-PD-1+BEVs (2 × 1011) as compared to control group (Fig. 5b). TNF-α signaling was recently found to impair the accumulation of CD8+ tumor-infiltrating T lymphocytes, and its blockade synergized anti-PD-1 therapy36. IL-2 plays a critical role in the activation of immune cells37. We observed that the expression of IL-2 was significantly increased in anti-PD-1+Bif.BEVs (2 × 1011) group clearly indicating the activation of T cells (Fig. 5b). Lastly, we also measured the levels of IFN-γ, which plays important role in stimulation of antitumor immune response by coordinating both innate and adaptive immune responses38. The proapoptotic cytokine IFN-γ showed significant increase in both anti-PD-1+Bif.BEVs (2 × 1010) and anti-PD-1+Bif.BEVs (2 × 1011) groups as compared to control, therefore resulting in enhanced anti-tumor immunity (Fig. 5b). There was no statistical difference in IL-10 expression levels (Suppl. Fig. 9c). Overall, these results clearly indicated that Bif.BEVs along with anti-PD-1 could mitigate tumor growth at least through the modulation of major cytokines directly involved in cellular apoptosis, inflammation, and immune cell activation.
Bif.BEVs in combination with anti-PD-1 increased tumor infiltrating lymphocytes
The lymphoid components of the immune system including CD8+ T cells, CD4+ T cells, B cells and innate lymphoid cells all play important roles in antitumor immune response39. Here we sought to determine whether the differential response of LL/2 syngeneic tumor to Bif.BEVs plus anti-PD-1 was also due to differential T-cell responses. As shown in Fig. 6a, b, the numbers of CD3+ and CD8+ cells by IHC staining are significantly higher in both anti-PD-1+Bif.BEVs (2 × 1010) and anti-PD-1+Bif.BEVs (2 × 1011) groups, but no significant changes were noted in anti-PD-1 and Bif.BEVs alone groups when compared to the control group. There was no statistical significance observed for CD4+ cells as shown in Suppl. Fig. 10. These results indicated that Bif.BEVs could potentiate anti-PD-1 through increasing tumor infiltrating cytotoxic T lymphocytes. Since Bif.B could systematically alter the gut microbiota in a regulatory T-cell (Treg) dependent manner11, we also assessed the effect of Bif.BEVs on intratumoral Foxp3+ Treg population via IHC in tumor tissue sections, however no significant difference was observed among the groups (Suppl. Fig. 9a, b).

a Immunohistochemistry showing assessment of tumor infiltrating lymphocytes (CD3+, CD8+) in tumor specimen from control, anti-PD-1, Bif.BEVs (2 × 1010), anti-PD-1+Bif.BEVs (2 × 1010) and anti-PD-1+Bif.BEVs (2 × 1011) treated mice. Scale bar = 100 µm. All the imaging data presented here are representative of n = 10 tumors. b Quantification of CD3+ and CD8+ cells of tumor infiltrating lymphocytes in tumors from mice treated with anti-PD-1, Bif.BEVs (2 × 1010), anti-PD-1+Bif.BEVs (2 × 1010), anti-PD-1+Bif.BEVs (2 × 1011) along with control. Each dot represents the average number of cells counted from 5 representative images from each tumor. The graphs present data as the mean ± SEM, with comparison to the control group performed using an unpaired two-sided t test. Exact p values are displayed in the figure. UC: untreated control. Source data are provided as a Source Data file.
Bif.BEVs upregulated PD-L1 expression in tumor cells and modulated key immune and oncogenic pathways
To further investigate the impact of Bif.BEVs on TME/TIME, we combined multiplexed immunofluorescence imaging using the Nanostring GeoMx digital spatial profiling (DSP) instrument with bulk RNA sequencing (RNAseq). GeoMx DSP allows for the simultaneous measurement of multiple markers of interest in tumor tissue samples with high spatial resolution (Suppl. Fig. 11). As shown in Fig. 7a, we used pancytokeratin staining (green) to select areas with similar quantities of tumor cells for comparison, revealing that Bif.BEVs significantly increased PD-L1 expression in these cells (yellow stained). Additionally, when combined with the anti-PD-1 agent, there was a notable increase in tumor-infiltrating immune cells (stained red with an anti-CD45 antibody), which together may contribute to an enhanced anti-tumor effect (merged images).

a GeoMx DSP instrument was used to assess PD-L1 expression levels. A representative tumor section from each group is shown for illustration. Pancytokeratin (green) was used to stain cancer cells, enabling the selection of regions with similar cancer cell density for comparison. PD-L1 (yellow staining) showed an overall increase in expression in groups treated with Bif.BEVs. Additionally, there was an increase in immune cells (stained red for CD45), particularly in the group treated with higher dose of Bif.BEVs. Together, these changes may enhance the anti-tumor effect when combined with anti-PD-1 treatment. This experiment has been repeated at least twice using tissue sections. b Quantification of PD-L1 expression level using immunofluorescence imaging. Five regions of interest (ROIs) were randomly selected from each stained slide of tumors harvested from each experimental group for analysis. An additional ROI of the same size was placed outside of the tissue boundary for background correction. For each ROI, the corrected total fluorescence (CTF) was calculated by subtracting the measured background integrated density from the integrated density and then normalizing to the area of the ROI, allowing for comparison of CTF across different stained slides. x1 and x2: compared to the control (Ctrl) group, p = 0.040 and 0.0047 respectively, using an unpaired two-sided t test with Welch’s correlation. y: compared to the Anti-PD-1 group, p = 0.011 using a two-sided t test. Error bars represent standard errors. Source data are provided as a Source Data file.
The increase in PD-L1 expression was statistically significant in the Bif.BEVs (2 × 1010) and anti-PD-1+Bif.BEVs (2 × 1011) groups compared to the control group (Fig. 7b, p = 0.040 and 0.0047 respectively), but not in the anti-PD-1+Bif.BEVs (2 × 1010) group (p = 0.382). We speculate that, aside from possible selection bias in tumor sections and areas analyzed, the observed PD-L1 upregulation in this group may have been offset by the killing of PD-L1 positive cancer cells following the addition of the anti-PD-1 agent. This hypothesis is supported by the observation that the anti-PD-1 group exhibited lower PD-L1 expression than the control group, and the anti-PD-1+Bif.BEVs (2 × 1010) group showed significantly higher PD-L1 expression compared to the anti-PD-1 group (p = 0.011, Fig. 7b).
We also examined the protein levels of key cytokines, TNF-α, IL-2, and IFN-γ, whose mRNA levels were reported in Fig. 5b. Our goal was to assess whether their protein levels correlate with mRNA expression and whether they are predominantly modulated in cancer versus immune cells. As shown in Suppl. Figs. 12–14, the protein levels of these cytokines generally correspond to their mRNA levels, confirming the impact of Bif.BEVs on cytokine production. However, the correlation is not perfect. For instance, while Bif.BEVs alone increased IL-2 protein level, this effect was not significant at the mRNA level (Fig. 5b). This discrepancy could stem from differences between mRNA expression and actual protein levels, as well as potential selection biases in the tumor regions or tissue sections analyzed. Nonetheless, a general concordance in trends was observed. As expected, IL-2 and IFN-γ staining signals correlated well with the CD45 signal, which marks immune cells (Suppl. Fig. 15), suggesting these cytokines are primarily produced by immune cells. However, the correlation between TNF-α expression and the relative abundance of cancer versus immune cells was less pronounced. This suggests that the contributions of immune and cancer cells to TNF-α production are not as markedly distinct as for IL-2 and IFN-γ (Suppl. Fig. 15).
To further investigate how Bif.BEVs contribute to immune boosting, we conducted bulk RNAseq on tumors from mice treated with either anti-PD-1 alone or anti-PD-1 plus Bif.BEVs. This analysis revealed substantial changes in gene expression, leading to alterations in pathways relevant to immunotherapy response. Notable findings include the upregulation of MHC peptide assembly and presentation, increased production or response to IFN-α or γ, enhanced T cell mediated cytotoxicity and enhanced (aka reduced negative regulation of) toll-like receptor 4 (TLR4) signaling pathway (Fig. 8a). Additionally, we observed significant downregulation of angiogenesis pathways (Fig. 8b). Finally, Bif.BEVs were found to downregulate several critical cancer growth pathways that contribute to immune suppression, such as Ras-MAPK and TGF-β signaling pathways (Suppl. Fig. 16 and Suppl. Data 1).

a Bulk RNA sequencing (RNAseq) revealed that the addition of Bif.BEVs to the anti-PD-1 treatment resulted in alterations in several immune response pathways. These included the upregulation of MHC peptide assembly and presentation, increased production or response to interferon α or γ, enhanced T cell-mediated cytotoxicity, and enhanced Toll-like receptor 4 signaling (through reduced negative regulation). b Bulk RNAseq also demonstrated that the addition of Bif.BEVs significantly downregulated angiogenesis-related pathways. The p-values were calculated using the GSEA method implemented in the “clusterProfiler” function in the “gseGO” R package.
Discussion
The human gastrointestinal tract is the host for a vast variety of microbes and plays a vital role in maintaining GI homeostasis and host health40. Several studies have correlated dysbiosis in gut commensal microbiota with cancer, respiratory diseases, cardiovascular diseases, diabetes, etc.18,41,42. In addition, we and others have demonstrated the gut microbiota associates, predicts and modifies the treatment response and irAEs in cancer immunotherapy10,43,44,45. As the TIME dictates the immunotherapy response12, investigating the link between gut microbiota and the TIME (could be remote) therefore emerged as an important question in the era of cancer immunotherapy13.
In this study, we sought to use BEVs to explain the remote effect of the gut microbiota on lung cancer immunotherapy. EVs are released by all domains of life: eukaryotes, fungi, archaea, bacteria, and carry variety of proteins, lipids, DNA/RNA - therefore serve as a mode of inter- and intra- kingdom communication46,47,48. This creates an opportunity for host-microbe interaction49. Both Gram-positive and negative bacteria shed bilayered EVs outside into the environment21. The Gram-negative bacterium has inner and outer lipopolysaccharide membranes and studies revealed that EVs released from them contain LPS50. The Gram-positive bacterium has a thick cell wall rich in peptidoglycan and it is still debatable regarding how EVs are being released, although via bubbling/blebbing of cytoplasmic membrane has been proposed47,51,52. In our study, we showed that the BEVs released by Gram-positive Bif.B contain LTA (Fig. 1). This supports the idea that BEVs from Gram-positive bacteria are from the cytoplasmic membrane as this is where LTA is anchored53,54.
Our data have identified that Bif.BEVs can easily get into epithelial cells (Fig. 2, Suppl. Figs. 1–3), and modulate the expression of PD-L1, not only in human (A549, H460) and murine LL/2 lung cancer cells, but also in noncancerous immortal lung epithelial cells (BEAS-2). This process of endocytotic uptake is dynamin dependent (Fig. 2a–c, Suppl. Fig. 5), which is consistent with previous reports on other EVs55 although ours is arguably the first time to show this process using Gram-positive BEVs. We further observed that the upregulation of PD-L1 by Bif.BEVs was through a TLR-4-NF-κB signaling pathway (Fig. 2e–f), which supports the finding of Antonangeli et al. showing that NF-κB regulates PD-L1 expression56. We therefore demonstrated the uptake of BEVs by cancer cells can have functional impact.
It has been hypothesized that EVs can travel through the blood and reach to distant organs. In fact, EVs were found to be present in plasma of patients suffering from severe infections57,58. A study done by Tulkens et al., showed that higher levels of systemic LPS-positive EVs were present in patients with intestinal barrier dysfunction59. These intrigued us to understand if BEVs are capable of reaching tumors at remote sites and subsequently affect the TIME. Here observed uptake of Bif.BEVs in murine intestinal organoids (Fig. 3a & b), and in patient-derived lung cancer organoids (Fig. 3c & d). As expected, oral gavaged Bif.BEVs in mice were found to be in the liver, kidneys, intestine, lungs, and tumors (Fig. 3f). Of note, tumors exhibited quite significant intensity of fluorescence suggesting a good amount of Bif.BEVs uptake. Our experiments therefore provided evidence that EVs from the gut microbiota (including Gram-positive bacteria) can travel to reach distant organs and tumors in remote sites.
Previous studies showed potential synergy of anti-PD-1/L1 along with Bif.B for cancer control7,9. Here, we established that oral administration of Bif.BEVs could also enhance the therapeutic effect of anti-PD-1 in syngeneic LL/2 tumors in a dose dependent manner (Fig. 4), suggesting that a shared component such as LTA plays an essential role. In fact, LTA can exert differential immune modulatory effects depending on the bacterial source from which it is isolated60. It has been reported that oral administration of purified LTA from Lactobacillus rhamnosus GG can delay skin tumorigenesis induced by UV radiation through increased level of IFN-γ and cytotoxic T-cells61, and using LTA-deficient Lactobacillus acidophilus strain, an immune modulatory role of LTA was clearly demonstrated62. Further investigation of the role of LTA and other BEV components in enhancing immunotherapy response is therefore essential. In addition, given that higher levels of Bif.BEV were associated with a stronger anti-tumor effect when combined with anti-PD-1, it is reasonable to speculate that a higher dose of Bif.B might also enhance the anti-tumor effect. However, since the immunomodulatory role of Bif.B in cancer immunotherapy is well-established7,8,9, and the focus of our current project is to investigate the role of BEVs in elucidating the remote effects of gut microbiota on lung cancer immunotherapy, we intend to explore the dose-dependent effects of Bif.B in future studies.
To further connect Bif.BEVs to the TIME and immunotherapy response, we examined tumor tissue sections from each mouse group. We were able to attribute modulation of key cytokines (e.g. upregulation of IFN-γ, IL-2) and increasing tumor-infiltrating CD8+ T-cells to enhanced immunotherapy response. More importantly, we utilized multiplexed immunofluorescence imaging using the GeoMx DSP instrument and bulk RNAseq to demonstrate that Bif.BEVs upregulated PD-L1 expression and modulated several pathways critical for immunotherapy responses. These pathways include enhanced MHC peptide assembly and presentation, increased interferon production or response, boosted T cell-mediated cytotoxicity, and enhanced TLR4 signaling pathway. The upregulation of MHC peptide assembly and presentation aligns with the capability of BEVs to enter tumor cells and stimulate both class I and II MHC presentation51. In fact, the induction of MHC presentation is recognized as a key mechanism through which intratumoral bacteria may influence cancer immunotherapy63. Increased interferon activity is also crucial for tumor killing through immunotherapy64. Moreover, the activation of TLR4 signaling has shown positive immune-modulatory effect65,66,67. Notably, our in vitro studies confirmed that Bif.BEVs activated TLR4 signaling (Fig. 2e). Additionally, we observed significant downregulation of angiogenesis pathways. Reduced angiogenesis not only inhibits cancer growth but also counteracts the immunosuppressive tumor microenvironment38. Interestingly, Bif.B has been reported to inhibit angiogenesis68. Finally, Bif.BEVs were found to downregulate several critical cancer growth pathways that contribute to immune suppression, including the Ras-MAPK and TGF-β signaling pathways. However, it is important to recognize that the TIME is a highly complex ecosystem comprising diverse cell populations, many of which extend beyond the scope of this study. Additionally, the composition of the TIME can be influenced by factors such as oncogenes, tumor suppressors, and aberrant signaling pathways12. To gain a more comprehensive understanding of how BEVs impact the TIME, we recognize the need to expand our investigations. Future studies will encompass a broader range of models, including patient-derived tumoroids and genetically engineered mouse models, particularly those with specific genetic contexts, such as STK11 mutations69. In addition, new techniques such as single cell RNAseq will be employed. These efforts aim to provide a comprehensive delineation of the potential of BEVs in modulating immunotherapy for lung cancer through further analysis of their impact on TIME.
We need to emphasize that while Bif.BEVs enhance the efficacy of anti-PD-1 treatment, further studies are required to determine their impact in combination with anti-PD-L1 agents. Both anti-PD-1 and anti-PD-L1 therapies have become essential components of lung cancer treatment, yet they can have distinct biological effects, as noted in previous research70. Notably, the PD-L1:CD80 cis-interaction is believed to contribute to these differences. This interaction limits PD-1 function71, reduces CD80:CTLA-4 interactions, and promotes CD80:CD28 co-stimulation70. The anti-PD-L1 agent can disrupt PD-L1:CD80 cis-interaction, resulting in a shift from co-stimulatory CD80:CD28 to coinhibitory CD80:CTLA-4 interactions, ultimately leading to CD80 endocytosis70. Given that both PD-L1 and CD80 are upregulated by Bif.BEVs (Fig. 2d), we strongly suspect that significant cis-interaction may occur between PD-L1 and CD80, potentially leading to differing effects of anti-PD-L1 and anti-PD-1 therapies. Additionally, Bif.BEVs may influence PD-L2 expression, as gut microbiota have been shown to do so72,73, further contributing to the potential differences, since only anti-PD-1 agents can inhibit the PD-1:PD-L2 interaction. However, verifying these hypotheses will require further investigation beyond the scope of the current study, which we plan to address in future research.
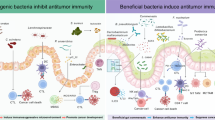
In conclusion, through a series of experiments, we have demonstrated, for the first time, that BEVs derived from the Gram-positive commensal Bifidobacteria are readily endocytosed in a dynamin-dependent manner. These vesicles possess the capability to modulate the expression of immune checkpoint ligands, and importantly, they can reach tumor tissues remotely and augment the anti-tumor effects of anti-PD-1 by orchestrating changes in the TIME, as succinctly summarized in Fig. 9 created by a free online software BioRender. Our study pioneers a novel understanding of how the gut microbiota can remotely wield influence over the TIME, thereby shaping the response to immunotherapy. It also presents an innovative strategy for harnessing the potential of the microbiota to bolster overall health.

In this model, we employed BEVs to establish a connection between gut microbiota and the TIME in lung cancer. Bif.BEVs exhibit the remarkable ability to traverse the gut lining, enter the bloodstream, and be internalized by lung cancer cells through the dynamin-dependent endocytosis pathway. Once inside, they bind to TLR4 and activate NF-κB, consequently leading to an increase in the expression of PD-L1. When combined with anti-PD-1 therapy, Bif.BEVs significantly elevate IFN-γ and IL-2 levels while reducing TNF-α levels. Furthermore, they enhance the infiltration of CD3+ and CD8+ lymphocytes into the tumor, and modulate several key immune response and oncogenic pathways. Collectively, these effects potentiate the anti-tumor efficacy of anti-PD-1 agents in lung cancer. Created in BioRender85.
Methods
This research complies with all relevant ethical regulations. We have obtained written approvals from the University of Kansas Medical Center (KUMC)’s Institutional Biosafety Committee (IBC) for various in vitro experiments, Institutional Animal Care and Use Committee (IACUC) for mice studies, and Institutional Research Committee (IRB) for using human lung cancer tissue to generate organoids and spheroids and use them for relevant experiments in this study. The source data are provided with this paper.
Cell culture
The lung cancer cell lines A549, H460 and LL/2 were purchased from American Type Culture Collection (ATCC) and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Corning-USA) supplemented with 10% FBS (Atlanta biosciences) with 100 units/ml Penicillin-Streptomycin (Sigma) at 37 °C with 5% CO2. The immortalized lung epithelial cell line BEAS-2B was grown in LHC-9 Medium (Gibco) with added components as described by vendor. Cells were maintained as per manufacturer’s instructions.
Human samples
Human fresh lung tumor tissues (∼1–4 cm3) were obtained from surgically resected lung specimens from the University of Kansas Cancer Center (KUCC) Biospecimen Repository Core Facility (BRCF), under the Institutional Research Committee (IRB) approved protocol. All the experimental protocols were conducted in compliance with the IRB guidelines of KUCC. Based on histopathological analysis, samples were confirmed as tumor or normal. The diagnosis was confirmed by pathologist (RM) at the University of Kansas Medical Center (KUMC). The samples were processed for the generation of lung cancer organoids as described in lung organoid culture section.
Bacterial culture and Isolation of BEVs
For the convenience of future clinical translation, we used a probiotic Bif.B cocktail, which was purchased from Seeking Health LLC, Bellingham, WA. The cocktail is a mixture of Bifidobacterium infantis (Bi-26), Bifidobacterium bifidum/lactis (Bb-02), Bifidobacterium longum (Bl-05), Bifidobacterium bifidum (Bb-06), Bifidobacterium lactis (Bl-04), Bifidobacterium lactis (Bl-07) and Bifidobacterium breve (Bb-03), as previously used and reported7. The lyophilized cocktail capsules contain 12 billion colony forming units. The bacteria were cultured in Bifidus Selective Medium (BSM) broth (Sigma) at 37 °C for 48 h in an anaerobic chamber (Bactron). Bifidobacterial supernatant was first centrifuged at 400x g for 10 min at 4 °C to eliminate bacterial pellet followed by 12,000 x g for 20 min at 4 °C to eliminate dead bacterial debris in a SORVALL Legend XTR centrifuge. The supernatant was then passed through a 0.22 µm vacuum filter. The resulting cell-free supernatant fractions were further spun at 200,000 x g for 2 h at 4 °C to collect BEVs in a SORVALL WX + ULTRA SERIES (Surespin 630 swinging bucket rotor) centrifuge. The BEV pellet was then washed with PBS and ultracentrifuged at 200,000 x g for 2 h. The pellet was finally resuspended in 100 µl PBS for further experimentation or stored at -80 °C until use. To quantitate, BEVs were lysed, and the amount of BEV protein recovered was assessed using BCATM Protein Assay Kit (Thermo scientific) according to the manufacturer’s instruction.
Uptake of bacterial extracellular vesicles
Cellular uptake
For the uptake of BEVs, the protocol from Xiao et al., 2022 was followed74. Briefly, A549, H460, BEAS-2B and LL/2 cells were seeded on coverslips in 6-well plate and allowed to attach for overnight. The next day, the BEVs were stained with either PKH67 (green, Cat# MINI67-KT) or PKH26 (red, Cat# MINI26-KT) fluorescent cell linker kit purchased from Sigma according to manufacturer’s protocol. The cells were treated with 5 µg/ml of stained EVs or as mentioned in the experiment for 24 h. Cells were then washed with Hank’s balanced salt solution (HBSS)/0.01% sucrose to remove the unabsorbed EVs and fixed with 2% paraformaldehyde (PFA). Finally, cells were washed with HBSS washing solution and mounted using UltraCruz® Aqueous Mounting medium with DAPI (Cat# sc-24941). Images were captured using a Nikon 80i microscope.
Organoid uptake
Organoids were seeded in Matrigel until maturation on idibi plates (Cat # 81218) and then treated with Bif.BEVs (10 µg/ml) stained with either PKH67 or PKH26 fluorescent cell linker according to manufacturer’s protocol for 24 h. Carefully, media was aspirated, and the cells were then washed with 0.25% TritonX-100/PBS to remove the unabsorbed Bif.BEVs. Further, organoids were fixed with fresh 2% PFA/PBS at room temperature (RT) for 20 min and then washed with PBS twice. Organoids were stained with 200 µl DAPI at a final concentration of 5 µg/ml for 20 min at 37 °C and then washed three times with PBS. Stained organoids were imaged using a confocal microscope.
Endosomal uptake Inhibition
LL/2 and A549 cells (2 × 105) were seeded in a 6-well plate with added coverslips into each well. The next day, media was changed from each well and cells were pre-treated with the endosomal inhibitors Dynasore (DS), Cytochalasin D (CD), Nystatin (NS), Amiloride (AM) according to Jones et al. 19 or vehicle control for 2 h. After due time, the cells were treated with 5 µg/ml Bif.BEVs stained with PKH67 for 24 h. The next day, media from each well was removed and cells were washed with PBS. Washed cells were fixed in warm 2% PFA for 15 min at RT followed by two washes with PBS for 5 min each. Further, cells were either incubated with Hoechst (1 µg/ml) for 5 min followed by a PBS wash and mounting with DABCO or mounted with vectashield with DAPI. Cells were visualized and imaged with a Nikon 80i microscope.
Nanoparticle tracking analysis (NTA)
Bif.BEVs were quantified for their particle size and concentration by NTA using a NanoSight LM10 instrument (Malvern Instruments). Bif.BEV preparations were first diluted in PBS and pre-run on NTA to obtain an optimal concentration between 105 and 108 particles/ml. For final measurement, approximately 500 µl of diluted sample was vortexed prior to injection and used for each analysis. The videos were collected in triplicate from 3 dilutions and were averaged to get the resulting counts for each sample. Each video of moving particles was 60 sec in duration, with a shutter speed of 30 ms and camera gain of 680. Software settings for analysis include detection threshold, 6; blur, auto; minimum expected particle size, 50 nm. A minimum of 200 particle tracks were completed for each video and the data were analyzed using the NTA 2.3 analytical software (Malvern). Data are presented as the average and standard deviation of the triplicate.
Intestinal organoid culture
C57BL/6 J mice were used to derive small intestinal organoid cultures. Briefly, small intestinal tissues (10 cm length cut into ~2 mm pieces) were collected in sterile PBS and washed several times with PBS supplemented with 2% penicillin-streptomycin. The crypts were isolated using Gentle Cell Dissociation Reagent (Stemcell technologies; Cat# 07174) by immersing tissue for 90 min at 4 °C with agitation. After dissociation of crypts, the supernatants were passed through a 70-micron cell strainer (Corning). Crypts in the filtrates were pelleted by centrifugation at 300 x g for 5 min at 4 °C and resuspended in 1 ml of serum deprived DMEM/F12 and counted. Around 4000 crypts were pelleted and resuspended in 1:1 ratio using 200 µl serum-free IntestiCult organoid growth medium (Stemcell technologies, Vancouver, Canada) mixed with 200 µl Matrigel (Corning). About 60-70 µl of the crypt suspension was added as drop/well on the surface of prewarmed low-adhesion 24-well plates and kept at 37 °C for 20 min for gelification after 20 min of incubation. Then, 700 µl of IntestiCult organoid growth medium was added to each well to support organoid development from seeded crypts. Organoids were grown at 37 °C in 5% CO2 incubator for 14 days. Mature organoids were used for different experiments or passaged using gentle cell dissociation reagent (Stemcell Technologies) or stored.
Patient-derived lung cancer organoid culture and relevant studies
For culturing patient-derived lung cancer organoids, the protocol by Kim et al.75 was followed with some modifications as described below75. The lung tumor tissues (2-10 mm3) were obtained from surgically resected specimens of NSCLC patients and collected in DMEM with 20% FBS and 1% Pen-Strep on ice by BRCF staff and delivered to lab within 1 h of collection. Tissues were washed thrice with cold HBSS from Sigma (Cat# H6648) with antibiotics and chopped finely with a sterile scalpel blade. Fine pieces of tissues were then incubated in Advanced DMEM/F12 (Gibco, Cat#12634-010) media containing 0.001% DNase, 1 mg/ml collagenase, 2% Pen-Strep, and 50 μg/ml amphotericin B at 37 °C for 1 h with intermittent agitation. The suspensions were repeatedly pipetted up and down and passed through 70 μm cell strainers (MIDSCITM, MO, USA). The strained cells were centrifuged at 300 x g for 3 min, and the pellets were resuspended in 500 μl of serum-free medium DMEM/F12 supplemented with 20 ng/ml of bFGF (Gibco; cat# 13256029), 50 ng/ml human EGF (Gibco; cat# PHG0311), 1X N2 (Gibco; cat# A13707-7), 1X B27 (Gibco; cat# 12587-010), 10 μM ROCK inhibitor (Stem cell; cat# 72304), and 1% Pen-Strep. The cells in media were mixed with 50% Matrigel (Corning; cat# 356231) and plated on prewarmed 24-well plate in the form of droplets. The plate was kept at 37 °C for 20 min for gelification. After gelation, 700 µl of compete media was added to the well. The medium was replaced every 4–5 days, and the organoids were passaged after 2–3 weeks. Further after 14 days, mature organoids were either used for experiments, stored, or passaged.
To study endocytosis in 3D organoid structures, we seeded organoids in Matrigel on MatTek plates (Cat. # P35GC-1.5-14-C) and allowed them to mature. After maturation, the organoids were either untreated or pretreated with various endocytosis inhibitors, including AM, CD, DS, and NS as above-mentioned, respectively. Two hours later, we added PKH67-labeled Bif.BEVs (10 µg/ml) and incubated the organoids for 24 hours. Following incubation, the organoids were washed with warm PBS, fixed with fresh 2% PFA at room temperature for 20 minutes, and then collected into a 15 ml Falcon tube. The organoids were centrifuged at 112 x g for 5 minutes at 4 °C, washed with cold PBS, and then centrifuged again at 250 x g for 5 minutes at 4°C. The organoids were resuspended in 2 ml of 1x TrypLE (Cat. # 12605010, ThermoFisher, USA) and incubated for 10 minutes at 37 °C for dissociation. After dissociation, 10 ml of cold Advanced DMEM/F12 (Cat. # 12634028, ThermoFisher, USA) was added, followed by centrifugation at 112 x g for 5 minutes at 4 °C, and washing/resuspension in 1x PBS twice. These steps resulted in efficient dissociation of the organoids into a single-cell suspension. The uptake of Bif.BEVs was then quantified by flow cytometry by measuring the mean fluorescent intensity of cells exhibiting green fluorescence, indicating the uptake of PKH67-stained Bif.BEVs.
Western blot analysis
The lysates from bacterial EVs and total cell lysates were separated on SDS/PAGE gels and transferred to PVDF membrane (Immobilon®-PSQ Transfer membrane, Millipore). The membranes were blocked at RT with 10% milk for 2 h, followed by overnight incubation with primary antibodies at 4 °C with gentle shaking. The primary antibodies used included anti-PD-L1 [E1L3N(R)], Cat # 13684S) at 1:1000, CD80 (Cat #15416), PVR/CD155 (Cat #81254), TLR-2 (Cat #13744), TLR-4 (Cat # 14358), TLR-6 (Cat # 12717), TLR-7 (Cat # 82658), NF-kB (Cat# 8242) and anti-GAPDH (14C10, Cat # 2118S) at 1:1000 were purchased from Cell Signaling Technology. Anti-ACTIN (Clone C4, Cat # 691001) at 1:5000 was purchased from MP Biomedicals. The anti-lipoteichoic acid (LTA; Cat # MA1-7402) and anti-LPS (Cat # PA1-73178) were purchased from Invitrogen and used according to manufacturer’s instructions. The dynamin II antibody (Cat # sc-166669) was purchased from Santa Cruz Biotechnology (CA). Details are summarized in Suppl. Table 1. Membranes were then washed three times for 10 min with 1x TBST and incubated with suitable secondary antibody overnight. The next day, after three washes for 10 min with 1x TBST, the signal was developed using PierceTM ECL Plus Western Blotting Substrate from Thermo Scientific and exposed to CL-XPosureTM Film from Thermo Scientific. The membranes were stripped using RestoreTM PLUS Western Blot Stripping Buffer (Cat # 46430; Thermo Scientific) and reprobed accordingly. The Western blot results were further quantified using the densitometric method with ImageJ, a free software.
mRNA analysis
For the investigation of differential gene expression related to dynamin-dependent endocytosis
LL/2 cells (2 × 105) were seeded in a 6-well plate and experiments were performed when cell confluency reached 70-80%. Cells were washed twice with PBS and pre-treated with DS, followed by treatment with 5 µg/ml Bif.BEVs, including control groups. After 24 hours of incubation, RNA was extracted using the All Prep. DNA/RNA Mini Kit (Cat. # 80204, QIAGEN). The RNA concentration was measured by NanoDrop 2000 (Thermo Fisher Scientific Inc.), and cDNA was synthesized using 1 µg of total RNA with the Applied Biosystems™ High-Capacity cDNA Reverse Transcription Kit (Cat # 43-688-535 14), which contains MultiScribe Reverse Transcriptase. The cDNA synthesis cycling conditions were as follows: 25 °C for 10 min, 37 °C for 120 min, 85 °C for 5 min, and then stored at 4 °C until removal from the thermocycler and kept at -20 °C until further use. RT-qPCR analysis was performed using a QuantStudio 7 real-time PCR system with TaqManTM Fast Advanced Master Mix (Cat. # 4444557, Applied Biosystems), along with TaqManTM Gene Expression Assays for the following targets: Dynamin I (Assay ID: Mm01342903_m1), Dynamin II (Assay ID: Mm00514582_m1), Dynamin III (Assay ID: Mm00554098_m1), Clathrin (Cltc) (Assay ID: Mm01303974_m1), Epidermal growth factor receptor pathway substrate 15 (Eps15) (Assay ID Mm01258456_m1), PD-L1 (Assay ID Mm03048248_m1), and GAPDH (Assay ID Mm99999915_g1). mRNA expression levels were quantified using the ΔΔCT method to calculate fold changes.
For mouse tumor tissue studies
The mouse tumor tissues were used to extract the total RNA using the TRIzol reagent (Invitrogen, Carlsbad, CA). The concentration was measured by NanoDrop 2000 (Thermo Fisher Scientific Inc.). cDNA synthesis was performed using 1 µg of total RNA via Applied Biosystems™ High-Capacity cDNA Reverse Transcription Kit (Cat # 43-688-14) containing MultiScribe Reverse Transcriptase. The cycling conditions were 25°C, 10 min; 37°C, 120 min; 85°C, 5 min; 4°C until removed from thermocycler and stored at -20°C until used. qPCR analysis was performed using TaqManTM Fast Advanced Master Mix (Cat # 4444557, Applied Biosystems) along with primers from TaqManTM Gene Expression Assays to target TNF-α (Assay ID; Mm00443258_m1), IFN-γ (Assay ID; Mm01168134_m1), IL-2 (Assay ID; Mm00434256_m1) and IL-10 (Assay ID; Mm 01288386_m1). GAPDH (Assay ID; Mm99999915_g1) was used as control for normalization. The fold change in mRNA expression levels was measured by using the ΔΔCT method.
Immunohistochemistry
The tumor tissues were formalin-fixed paraffin-embedded (FFPE) and were cut into 4 µM thin sections. The tumor sections were then deparaffinized by immersing them in Xylene for 10 min (with a change of Xylene at 5 min) and rehydrated in graded series of ethanol solutions from 100% (3 min), 95% (3 min), 80% (3 min), 70% (3 min), 50% (3 min) and finally deionized water (5 min). Further, deparaffinized tumor sections were heated in 0.01 M citrate buffer, pH 6.0 (with 0.05% tween-20) for antigen retrieval in a pre-warmed steamer for 30 min and then cooled to room temperature. Sections were gently washed in deionized water and then twice in PBS (5 min each). Tumor sections were blocked in serum blocking solution (10% normal goat serum in TBS-T for 1 h) and then washed twice with PBS-T. Sections were stained for 1 h at RT with primary antibody (1:100, anti-CD3ε (D4V8L), Cat # 99940); (1:100, anti-CD4 (D7D2Z), Cat # 25229); (1:200, anti-CD8α (D4W2Z) XP, Cat # 98941); (1:200, anti-Ki67 (SP6) Cat #ab16667); diluted in blocking solution. Sections were washed twice (5 min) in PBS and stained for 30 min in dark at RT with conjugated goat anti-rabbit polymer HRP secondary antibody MACH2 (Biocare medical). Finally, the sections were counterstained with hematoxylin, dehydrated in graded series of ethanol and cleared in Xylene. After staining, the sections were mounted and imaged using a Nikon 80i microscope.
Immunofluorescence imaging and analysis using the GeoMx digital spatial profiling (DSP) instrument
The tumor tissue samples mentioned above were also used to study several markers of interest using Nanostring’s GeoMx® Digital Spatial Profiler, which provides high spatial resolution. For this study, the following primary antibodies were used: anti-pancytokeratin (Novus Biologicals, cat # NBP2-33200AF532, 1 μg/ml), anti-CD45 (Novus Biologicals, cat # NBP2-34528AF594, 5 μg/ml), anti-PD-L1 (ThermoFisher, cat # 17952-1-AP, 7 μg/ml), anti-TNF-α (ThermoFisher, cat # 60291-1-Ig, 15 μg/ml), anti-IL-2 (ThermoFisher, cat # 26156-1-AP, 3.3 μg/ml), and anti-IFN-γ (ThermoFisher, cat # MM700, 5 μg/ml). Slides were stained with primary antibodies overnight at 4 °C. For unconjugated primary antibodies, a secondary antibody conjugated to a fluorescent dye (e.g., Alexa Fluor™ 532, 594, or 647) was applied for 60 minutes at room temperature. To visualize the cell nuclei, the slides were counterstained with Syto-13 for 15 minutes at room temperature. The stained tissue slides were then imaged using the GeoMx DSP instrument to capture high-resolution images of the tissue sections. To identify representative areas, we followed these steps: 1) Avoided regions that could introduce artifacts, such as tissue edges or loose sections. 2) Selected areas with uniform pancytokeratin or DNA staining to ensure comparable cellular density. As a general approach for this project, pancytokeratin was used to label cancer cells, while CD45 was used to label immune cells. For quantifications, such as for PD-L1, immunofluorescence images captured on the GeoMx instrument were analyzed using Image J software (version 1.54 d). Briefly, five regions of interest (ROIs) were randomly selected from each stained slide of tumors harvested from each experimental group for analysis. An additional ROI of the same size was placed outside of the tissue boundary for background correction. For each ROI, the corrected total fluorescence (CTF) was calculated by subtracting the measured background integrated density from the integrated density and then normalizing to the area of the ROI, allowing for comparison of CTF across different stained slides.
Bulk RNA sequencing (RNAseq) and its analysis
We used tumor samples from the following groups to study the impact of Bif.BEVs on anti-PD-1 treatment: Anti-PD-1 only vs. Anti-PD-1+Bif.BEVs (2 × 1011). Tumor sections were deparaffinized and RNA extracted from tissue using QIAGEN RNeasy FFPE Kit, followed by a quality check and RNA concentration measurement using NanoDrop. The RNAseq was then performed via an Illumina platform through the Genome Sequencing Core Facility in the University of Kansas Medical Center.
The raw sequencing dataset was first quality-trimmed using Trimmomatic76 with default parameters. The trimmed reads were then mapped to the human genome (GRCh38) using STAR77 in paired-end mode. Next, the mapped reads were summarized into gene-level read counts using “featureCounts” from the Rsubread package78, with RefSeq as the gene annotation database. DESeq279 was then used to identify genes with significant expression differences by comparing the Anti-PD-1+Bif.BEV group with the Anti-PD-1 only group. Genes without any mapped reads, labeled as “NA” in the DESeq2 output, were filtered out. The remaining genes were sorted based log2 fold change from DESeq2. Gene set enrichment analysis (GSEA)80 was performed on the sorted gene list using Gene Ontology81 as the pathway database, utilizing “gseGO” from the R package “clusterProfiler”82. The minimum and maximum gene set sizes were set to 5 and 1000, respectively. All enriched pathways with adjusted p-values less than 0.1 were retained for further manual inspection.
Electron microscopy
Scanning electron microscopy (SEM) on Bif.B
For SEM, the Bif.B cocktail was grown overnight in an anaerobic chamber and the bacterial pellet was washed with PBS three times to remove the growth media. Next, the pellet was fixed in 70% ethanol and then dehydrated in graded series of ethanol for 5 min/step. The grids were viewed in a Hitachi S-2700 SEM with Quartz PCI digital capture (Hitachi, Inc.).
Negative staining of isolated Bif.BEVs
The Bif.BEVs were negatively stained using carbon filmed copper grids (300 mesh, Electron Microscopic Sciences, Hatfield, PA); the grids were discharged through one cycle, at 15 mAmp for 20 sec in Easy-Glow (PELCO) apparatus. The isolated Bif.BEVs were mixed by vortexing and suspension of 20 µl drops was placed on wash plate well. The carbon coated grids were placed on the sample drops upside down and allowed to absorb samples for a minimum of 20 min. The coated grids were then washed six times by resting them on droplets of deionized distilled water for 10 seconds on each drop. After washing, the grids were stained on drop of 1% uranyl acetate stain (Electron Microscopic Sciences, Hatfield, PA) by placing them again upside down for nearly 10 seconds. The carbon grids were blot dried on filter paper in a dish and then air dried for 15 min. The dried grids with Bif.BEVs were photographed at 100 KV JEM-1400 under transmission electron microscope.
Immunogold labeling of Bif.BEVs with anti-LTA
The Bif.B cocktail was grown in overnight culture and the next day the pellet was washed twice with HBSS, pH 7.0. The pellet was blocked in 5% goat serum with 1% BSA (blocking buffer) pH 7.4 for 15 min at 37 °C to block unspecific antigens. The pellet was then incubated with anti-LTA primary antibody (1:50) diluted in blocking buffer for overnight at 4 °C on a shaker. The next day, the Bif.B cells were centrifuged and washed three times with HBSS to remove unbound antibody and incubated with goat anti-mouse-gold (Cat #25129, Electron Microscopic Sciences, Hatfield, PA) secondary antibody (1:20) for 1 h in blocking solution for goat gold conjugates (Cat #25596, 10 nm Electron Microscopic Sciences, Hatfield, PA). Finally, bacteria were washed twice with HBSS and centrifuged at 100 x g for 5 min to pellet. The pellet was then fixed in 1% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) for 2 h at RT. Further the pellet was osmicated, stained, dehydrated, and embedded to get the ultrathin section on nickel grid. The sections were visualized and imaged at 100 KV JEM-1400 transmission electron microscope.
Animal studies
C57BL/6 J mice were purchased from Jackson Laboratory. All animals were maintained in a specific pathogen-free animal facility. Animal studies were IACUC approved (ACUP numbers: 2019–2536) at the KUMC animal facility. All experiments were performed on 8–10 weeks old female mice. All mice had free access to food (Oval Pellet, LabDiet 5K67, Cat #0006673) and drinking water (disposable ultra-pure water, Innovive) ad libitum, ensuring adequate intake of all essential nutrients throughout the study period. The housing conditions were as follows: Light cycle – Standard (beginning the first Sunday in November): 5 AM to 7 PM; Daylight Saving Time (DST) (beginning the second Sunday in March): 6 AM to 8 PM. Temperature and humidity were maintained at 68–75 °F and 30–70%, respectively. The mouse studies were performed by developing syngeneic tumors using LL/2 cells. LL/2 cells were suspended in PBS with 1:1 ratio of Matrigel (Corning). Subcutaneous injections were done on both flanks of mice using 100 ul of LL/2 cell suspension (1 × 106) by sterilizing the inoculation site with ethanol under inhaled isoflurane general anesthesia. The tumors were measured with a digital caliper three times a week. The first pilot experiment shown in Fig. 4a was subdivided in 5 groups with 6 tumors in each group: untreated control (Control), IgG Control (anti-IgG), anti-PD-1 only (anti-PD-1), anti-PD-1+Bif.BEVs (2 × 1010), anti-PD-1+Bif.B. The tumors were allowed to grow for 7 days and then treatment was started. From day 7, mice in group 4, anti-PD-1+Bif.BEVs were orally gavaged with (2 × 1010) Bif.BEVs/mouse three times a week. The particle concentration was measured by NTA as described above. On day 7, 14, 21 of the experiment, the mice in group 5 (anti-PD-1+Bif.B), were orally gavaged with 200 µl of Bif.B (1 × 109 CFU/mouse) in PBS. These 2 groups were also injected with either anti-IgG or anti-PD-1 from day 9, thrice a week as described below. For anti-IgG and anti-PD-1 groups, mice received 150 µg of antibody, isotype control (InVivo MAB rat IgG2a isotype control, clone 2A3, BioXCell) and (InVivoPlus anti-mouse PD-1 (CD-279), clone RMP1-14, BioXCell) respectively per IP injection three times a week. After the tumors reached 1.5 cm diameter in the control group the mice were sacrificed to collect the tumors. The formula used to calculate tumor volume was V = (W2 x L)/2, where W is tumor width and L is tumor length. The maximal tumor size allowed by our IACUC protocol is 2 cm3, which was not exceeded in this study. The tumors were measured, weighed, and further stored in formalin or liquid nitrogen based on experimental requirements. In the second experiment shown in Fig. 4d, mice were divided into four groups, with five mice per group including: untreated control (Control), Bif.BEVs (2 × 1010), anti-PD-1+Bif.BEVs (2 × 1010), and anti-PD-1+Bif.BEVs (2 × 1011). Here, group 2, 3 and 4 were orally gavaged with Bif.BEVs on Day 7 and further thrice a week. The anti-PD-1 doses were also given to the respective groups as mentioned above in the similar fashion. The tumors were measured, resected, and stored as per experimental needs.
Statistics & reproducibility
Data are compared using either the mean or median. When comparing means, data are expressed as the mean ± standard error of the mean (SEM) or standard deviation, and statistical significance is assessed using Student’s t test or ANOVA. When comparing medians, nonparametric tests such as Kruskal-Wallis and Mann-Whitney tests are used, with data presented as box plots. All statistical analyses were performed using GraphPad Prism 5.0 software (San Diego, CA). A p values of <0.05 was considered statistically significant. Unless otherwise specified, three independent experiments were conducted. No statistical method was used to predetermine the sample size. Investigators were not intentionally blinded during experiments or outcome assessments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All experimental data supporting the findings of the study are available within the article and its Supplementary Information. Additional RNAseq data generated in this study have been deposited in the NCBI’s BioProject with the accession number: PRJNA1230548, which can be accessed via the following link: http://www.ncbi.nlm.nih.gov/bioproject/1230548. Source data are provided with this paper.
Code availability
The code is available in the Supplementary Information under the section named “RNAseq Analysis Workflow”.
References
Gandhi, L. et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med 378, 2078–2092 (2018).
Paz-Ares, L. et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med 379, 2040–2051 (2018).
Hellmann, M. D. et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N. Engl. J. Med 381, 2020–2031 (2019).
Sezer, A. et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet 397, 592–604 (2021).
Strouse, C., Mangalam, A. & Zhang, J. Bugs in the system: bringing the human microbiome to bear in cancer immunotherapy. Gut Microbes, 1-4 (2018).
Chau, J. & Zhang, J. Tying small changes to large outcomes: The cautious promise in incorporating the microbiome into immunotherapy. Int. J. Mol. Sci. 22 (2021).
Sivan, A. et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089 (2015).
Wang, F., Yin, Q., Chen, L. & Davis, M. M. Bifidobacterium can mitigate intestinal immunopathology in the context of CTLA-4 blockade. Proc. Natl Acad. Sci. USA 115, 157–161 (2018).
Lee, S. H. et al. Bifidobacterium bifidum strains synergize with immune checkpoint inhibitors to reduce tumour burden in mice. Nat. Microbiol 6, 277–288 (2021).
Chau, J. et al. Prospective correlation between the patient microbiome with response to and development of immune-mediated adverse effects to immunotherapy in lung cancer. BMC Cancer 21, 808 (2021).
Sun, S. et al. Bifidobacterium alters the gut microbiota and modulates the functional metabolism of T regulatory cells in the context of immune checkpoint blockade. Proc. Natl Acad. Sci. USA 117, 27509–27515 (2020).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550 (2018).
Ross, T. J. & Zhang, J. The Microbiome-TIME Axis: A Host of Possibilities. Microorganisms 11 (2023).
van Niel, G., D’Angelo, G. & Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19, 213–228 (2018).
Hughes, D. T. & Sperandio, V. Inter-kingdom signalling: communication between bacteria and their hosts. Nat. Rev. Microbiol 6, 111–120 (2008).
Tulkens, J., De Wever, O. & Hendrix, A. Analyzing bacterial extracellular vesicles in human body fluids by orthogonal biophysical separation and biochemical characterization. Nat. Protoc. 15, 40–67 (2020).
Chronopoulos, A. & Kalluri, R. Emerging role of bacterial extracellular vesicles in cancer. Oncogene 39, 6951–6960 (2020).
Kim, O. Y. et al. Bacterial outer membrane vesicles suppress tumor by interferon-gamma-mediated antitumor response. Nat. Commun. 8, 626 (2017).
Jones, E. J. et al. The uptake, trafficking, and biodistribution of bacteroides thetaiotaomicron generated outer membrane vesicles. Front Microbiol 11, 57 (2020).
Yaghoubfar, R. et al. Effect of akkermansia muciniphila, faecalibacterium prausnitzii, and their extracellular vesicles on the serotonin system in intestinal epithelial cells. Probiotics Antimicrob. Proteins 13, 1546–1556 (2021).
Nahui Palomino, R. A., Vanpouille, C., Costantini, P. E. & Margolis, L. Microbiota-host communications: Bacterial extracellular vesicles as a common language. PLoS Pathog. 17, e1009508 (2021).
Zhang, P. & Liu, Z. Structural insights into the transporting and catalyzing mechanism of DltB in LTA D-alanylation. Nat. Commun. 15, 3404 (2024).
Welsh, J. A. et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 13, e12404 (2024).
Rennick, J. J., Johnston, A. P. R. & Parton, R. G. Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat. Nanotechnol. 16, 266–276 (2021).
Cecil, J. D. et al. Differential responses of pattern recognition receptors to outer membrane vesicles of three periodontal pathogens. PLoS One 11, e0151967 (2016).
Wang, Y. et al. Adjuvant effect of the novel TLR1/TLR2 agonist diprovocim synergizes with anti-PD-L1 to eliminate melanoma in mice. Proc. Natl Acad. Sci. USA 115, E8698–E8706 (2018).
Ruiz, P. A., Hoffmann, M., Szcesny, S., Blaut, M. & Haller, D. Innate mechanisms for bifidobacterium lactis to activate transient pro-inflammatory host responses in intestinal epithelial cells after the colonization of germ-free rats. Immunology 115, 441–450 (2005).
Lambies, G. et al. Cell polarity proteins promote macropinocytosis in response to metabolic stress. bioRxiv (2024).
Lechner, M. G. et al. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J. Immunother. 36, 477–489 (2013).
Mosely, S. I. et al. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol. Res 5, 29–41 (2017).
Graefe, C. et al. Optimized Ki-67 staining in murine cells: a tool to determine cell proliferation. Mol. Biol. Rep. 46, 4631–4643 (2019).
Tan, P. H. et al. Immunohistochemical detection of Ki67 in breast cancer correlates with transcriptional regulation of genes related to apoptosis and cell death. Mod. Pathol. 18, 374–381 (2005).
Setrerrahmane, S. & Xu, H. Tumor-related interleukins: old validated targets for new anti-cancer drug development. Mol. Cancer 16, 153 (2017).
Perez-Ruiz, E. et al. Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature 569, 428–432 (2019).
Berraondo, P. et al. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 120, 6–15 (2019).
Bertrand, F. et al. TNFalpha blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 8, 2256 (2017).
Jiang, T., Zhou, C. & Ren, S. Role of IL-2 in cancer immunotherapy. Oncoimmunology 5, e1163462 (2016).
Zhao, S. et al. Low-dose apatinib optimizes tumor microenvironment and potentiates antitumor effect of PD-1/PD-L1 blockade in lung cancer. Cancer Immunol. Res 7, 630–643 (2019).
Paijens, S. T., Vledder, A., de Bruyn, M. & Nijman, H. W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol. Immunol. 18, 842–859 (2021).
Hou, K. et al. Microbiota in health and diseases. Signal Transduct. Target Ther. 7, 135 (2022).
Li, W. Z., Stirling, K., Yang, J. J. & Zhang, L. Gut microbiota and diabetes: From correlation to causality and mechanism. World J. Diab. 11, 293–308 (2020).
Tang, W. H., Kitai, T. & Hazen, S. L. Gut microbiota in cardiovascular health and disease. Circ. Res 120, 1183–1196 (2017).
Huang, C. et al. Relating gut microbiome and its modulating factors to immunotherapy in solid tumors: A systematic review. Front Oncol. 11, 642110 (2021).
Liu, B., Chau, J., Dai, Q., Zhong, C. & Zhang, J. Exploring gut microbiome in predicting the efficacy of immunotherapy in non-small cell lung cancer. Cancers (Basel) 14 (2022).
Baruch, E. N. et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 371, 602–609 (2021).
Bittel, M. et al. Visualizing transfer of microbial biomolecules by outer membrane vesicles in microbe-host-communication in vivo. J. Extracell. Vesicles 10, e12159 (2021).
Brown, L., Wolf, J. M., Prados-Rosales, R. & Casadevall, A. Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol 13, 620–630 (2015).
Johnston, E. L., Heras, B., Kufer, T. A. & Kaparakis-Liaskos, M. Detection of bacterial membrane vesicles by NOD-like receptors. Int. J. Mol. Sci. 22 (2021).
Kuehn, M. J. & Kesty, N. C. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 19, 2645–2655 (2005).
Schwechheimer, C. & Kuehn, M. J. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat. Rev. Microbiol 13, 605–619 (2015).
Buzas, E. I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol. 23, 236–250 (2023).
Toyofuku, M., Nomura, N. & Eberl, L. Types and origins of bacterial membrane vesicles. Nat. Rev. Microbiol 17, 13–24 (2019).
Zhang, B. & Perez, C. Stabilization and crystallization of a membrane protein involved in lipid transport. Methods Mol. Biol. 2127, 283–292 (2020).
Hesser, A. R., Schaefer, K., Lee, W. & Walker, S. Lipoteichoic acid polymer length is determined by competition between free starter units. Proc. Natl Acad. Sci. USA 117, 29669–29676 (2020).
Mulcahy, L. A., Pink, R. C. & Carter, D. R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell Vesicles 3 (2014).
Antonangeli, F. et al. Regulation of PD-L1 Expression by NF-kappaB in Cancer. Front Immunol. 11, 584626 (2020).
Alberro, A., Iparraguirre, L., Fernandes, A. & Otaegui, D. Extracellular vesicles in blood: sources, effects, and applications. Int. J. Mol. Sci. 22 (2021).
Namork, E. & Brandtzaeg, P. Fatal meningococcal septicaemia with “blebbing” meningococcus. Lancet 360, 1741 (2002).
Tulkens, J. et al. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 69, 191–193 (2020).
Jeong, J. H. et al. Differential immune-stimulatory effects of LTAs from different lactic acid bacteria via MAPK signaling pathway in RAW 264.7 cells. Immunobiology 220, 460–466 (2015).
Weill, F. S. et al. Lipoteichoic acid from lactobacillus rhamnosus GG as an oral photoprotective agent against UV-induced carcinogenesis. Br. J. Nutr. 109, 457–466 (2013).
Khan, M. W. et al. Modulating intestinal immune responses by lipoteichoic acid-deficient lactobacillus acidophilus. Immunotherapy 4, 151–161 (2012).
Kalaora, S. et al. Identification of bacteria-derived HLA-bound peptides in melanoma. Nature 592, 138–143 (2021).
Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012).
Seo, Y. D. et al. Toll-Like Receptor 4 agonist injection with concurrent radiotherapy in patients with metastatic soft tissue sarcoma: A Phase 1 Nonrandomized Controlled Trial. JAMA Oncol. 9, 1660–1668 (2023).
Farias, A. et al. A TLR4 agonist improves immune checkpoint blockade treatment by increasing the ratio of effector to regulatory cells within the tumor microenvironment. Sci. Rep. 11, 15406 (2021).
Nelson, M. H. et al. Toll-like receptor agonist therapy can profoundly augment the antitumor activity of adoptively transferred CD8(+) T cells without host preconditioning. J. Immunother. Cancer 4, 6 (2016).
Xu, D. et al. Feasibility between bifidobacteria targeting and changes in the acoustic environment of tumor tissue for synergistic HIFU. Sci. Rep. 10, 7772 (2020).
Skoulidis, F. et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 8, 822–835 (2018).
Zhao, Y. et al. PD-L1:CD80 Cis-heterodimer triggers the co-stimulatory receptor cd28 while repressing the inhibitory PD-1 and CTLA-4 Pathways. Immunity 51, 1059–1073.e1059 (2019).
Sugiura, D. et al. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 364, 558–566 (2019).
Park, J. S. et al. Targeting PD-L2-RGMb overcomes microbiome-related immunotherapy resistance. Nature 617, 377–385 (2023).
Park, J. S., Gazzaniga, F. S., Kasper, D. L. & Sharpe, A. H. Microbiota-dependent regulation of costimulatory and coinhibitory pathways via innate immune sensors and implications for immunotherapy. Exp. Mol. Med. 55, 1913–1921 (2023).
Xiao, H., Ye, X., Vishwakarma, V., Preet, R. & Dixon, D. A. CRC-derived exosomes containing the RNA binding protein HuR promote lung cell proliferation by stabilizing c-Myc mRNA. Cancer Biol. Ther. 23, 139–149 (2022).
Kim, M. et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 10, 3991 (2019).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Harris, M. A. et al. The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261 (2004).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. (Camb.) 2, 100141 (2021).
Vishwakarma, V. https://biorender.com/h49i850 (2025).
Vishwakarma, V. https://biorender.com/l69p944 (2025).
Vishwakarma, V. https://biorender.com/l74q234 (2025).
Acknowledgements
We are greatly thankful to the patients who consented for providing samples and KUMC’s Biorepository Core Facility (BRCF) for collecting the samples to generate patient-derived lung cancer spheroids and organoids for this study. We are truly grateful to Drs. Shahid Umar and Sufi Thomas for allowing us to use their anaerobic chamber. The Electron Microscope Research Laboratory is supported, in part, by NIH/NIGMS COBRE grant P20GM104936. The JEOL JEM-1400 transmission electron microscope was purchased with funds from NIH grant 1S10RR027564. We acknowledge the Imaging Core supported by the KIDDRC (NIH U54 HD 090216) at the University of Kansas Medical Center, Kansas City, KS 66160. The Leica STED microscope is supported by NIH 1S10OD023625. JZ has received startup funds and E3DT grant from KUMC to perform these studies. Immunofluorescence staining and imaging using the GeoMx DSP platform were supported by the Kansas Institute for Precision Medicine COBRE BBV Core (P20GM130423). C.Z. is funded by National Science Foundation CAREER award (DBI-1943291).
Author information
Authors and Affiliations
Contributions
R.P., M.A.I., J.S., and G.R. designed and performed the experiments, and generated the data. RP wrote the initial draft of the manuscript. A.M., V.V., C.K., S.C., H.P. and Q.D. aided at different stages of experiments. R.M. provided histological and pathological expertize. W.S. provided insightful thoughts to the manuscript. C.Z. analyzed RNAseq data. M.A.M. aided in immunological analyses. J.Z. conceived and designed the study, provided supervision to R.P., M.A.I., J.S. and G.R., criticized and extensively revised the manuscript. All authors provided their comments and approved the final version of this manuscript.
Corresponding author
Ethics declarations
Competing interests
JZ has served as a scientific advisor/consultant for: AstraZeneca, Bayer, Biodesix, BMS, Cardinal Health, Daiichi Sankyo, Fosun, Hengrui, Eli Lilly, Mirati, Nexus Health, Novartis, Novocure, Regeneron, Sanofi and Takeda. He is on the speakers’ bureau for AstraZeneca, Regeneron, Sanofi and MJH Life Sciences. He has also received research funding or support from Abbvie, AstraZeneca, BeiGene, Biodesix, BridgeBio, BMS, Champions Oncology, Eli Lilly, Genentech, Hengrui, InnoCare Pharma, Janssen, Kahr Medical, Merck, Mirati, Nilogen and Novartis. In addition, he is the inventor of a patent application submitted by KUMC with the Application Serial No 63/705,859, based on the findings from this project. MAM has served as a paid consultant for Johnson & Johnson and Design-Zyme and has received research funding from Targovax. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Federica Michielin, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Preet, R., Islam, M.A., Shim, J. et al. Gut commensal Bifidobacterium-derived extracellular vesicles modulate the therapeutic effects of anti-PD-1 in lung cancer. Nat Commun 16, 3500 (2025). https://doi.org/10.1038/s41467-025-58553-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58553-4
This article is cited by
-
Gut microbiota shapes cancer immunotherapy responses
npj Biofilms and Microbiomes (2025)
-
Bacterial Extracellular Vesicles: Emerging Regulators in the Gut–Organ Axis and Prospective Biomedical Applications
Current Microbiology (2025)
-
The emerging role of microbiota in lung cancer: a new perspective on lung cancer development and treatment
Cellular Oncology (2025)
