
Abstract
Here, we present a multi-omics study of type 2 diabetes and quantitative blood lipid and lipoprotein traits conducted to date in Hispanic/Latino populations (nmax = 63,184). We conduct a meta-analysis of 16 type 2 diabetes and 19 lipid trait GWAS, identifying 20 genome-wide significant loci for type 2 diabetes, including one novel locus and novel signals at two known loci, based on fine-mapping. We also identify sixty-one genome-wide significant loci across the lipid/lipoprotein traits, including nine novel loci, and novel signals at 19 known loci through fine-mapping. Next, we analyze genetically regulated expression, perform Mendelian randomization, and analyze association with transcriptomic and proteomic measure using multi-omics data from a Hispanic/Latino population. Using this approach, we identify genes linked to type 2 diabetes and lipid/lipoprotein traits, including TMEM205 and NEDD9 for HDL cholesterol, TREH for triglycerides, and ANXA4 for type 2 diabetes.
Similar content being viewed by others

Introduction
Although genome-wide association studies (GWAS) have revolutionized our understanding of the genetic underpinnings of cardiometabolic disease, systemic underrepresentation of Hispanic and Latino individuals has limited the potential for improving public health and precision medicine in a population with significant cardiometabolic health disparities. Many cardiometabolic diseases are more prevalent in Hispanic/Latino populations compared to Non-Hispanic White populations; for example, recent prevalence estimates of type 2 diabetes (T2D) and low high-density lipoprotein cholesterol (HDL-C) in Hispanic/Latino populations are 15.5% and 21.9%, respectively, compared to 13.6% and 16.6% in Non-Hispanic White populations1,2. These traits are of major clinical and public health importance. Elevated blood glucose and abnormal lipid levels comprise three components of the metabolic syndrome, which has a prevalence of 36.3% in US Hispanics and is known to underlie high rates of cardiometabolic diseases, including cardiovascular, liver and kidney diseases3. With increased recognition of the importance of racially, ethnically, and ancestrally diverse participants in genetic studies4, efforts have been made to expand diversity in GWAS of complex traits5,6. However, Hispanic/Latino sample sizes remain limited. For example, recent genome-wide meta-analyses of lipid and lipoprotein (abbreviated as lipid throughout) concentrations, key biomarkers for development of cardiometabolic disease, conducted in European-ancestry populations, comprise data from ~1.6 M participants, while the largest study performed to date in Hispanics/Latinos comprised about 48,0007,8. Further, no GWAS of cardiometabolic traits in Hispanics/Latinos systematically functionally annotated gene-based findings in differential abundance analyses of transcriptomic and proteomic data directly measured in a Hispanic population.
Furthermore, despite these large studies in trans- and European-ancestry samples, there remains a gap between estimated heritability (h2) and the variance explained by known variants. For T2D, a recent SNP-based h2 explains only 19% of T2D risk9, whereas family-based h2 estimates range from 20 to 80%10,11. For lipid phenotypes, family-based h2 estimates are as high as 83%7,12,13,14,15,16; however, even in studies of over 180,000 individuals, known variants only explain 30-33% of genetic variance17,18. Previous studies have demonstrated the benefits of studying diverse populations, both for further discovery and for deeper interrogation of established loci5,6,19,20. Extending genetic research of currently underrepresented populations is an opportunity to simultaneously work to reduce a major health disparity and to improve our understanding of genetic variation underlying these traits.
Hispanic/Latino individuals represent a complex group of populations with diverse cultural traditions, foodways, religions, lifestyles, languages, cultural norms, histories, and social environments, as well as multiple sources of genetic ancestry resulting in widely varying patterns of admixture. Some genetic loci contributing to cardiometabolic traits may be more identifiable under certain environmental conditions and allele frequency differences between ancestries can affect the power to detect associations. As a result, while much of the genetic architecture of cardiometabolic disease risk is shared across populations and environmental contexts6,21, Hispanic/Latino populations likely harbor genetic effects at loci only detectable in population-specific analysis as well as population-specific variants at previously known loci22.
To address this significant research gap, we performed GWAS meta-analyses in Hispanic/Latino populations for T2D and quantitative lipid traits, including high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), total cholesterol (TC), and triglycerides (TG). We then performed a summary statistic-based transcriptome-wide association analysis, including our islet expression prediction models using data from the InsPIRE (Integrated Network for Systematic analysis of Pancreatic Islet RNA Expression) consortium23, and systematically followed up identified genes in differential abundance analyses using directly measured blood transcriptomic and proteomic data from an independent Hispanic population. These downstream analyses highlight the importance of measuring omics in underrepresented populations for functional characterization of large-scale genomic analyses.
Results
Study demographics
Characteristics of the study participants can be found in Supplementary Data 1. Of the 19 participating studies, 15 contributed to both T2D and lipids analyses, three contributed to the lipids analyses only, and one contributed only to the T2D analyses. Our meta-analyses included more females than males (~63% female). T2D cases had a 1.7 kg/m2 higher BMI and were 3.2 years older on average than controls.
Single variant T2D meta-analysis results
Twenty genome-wide significant loci were associated with T2D (Supplementary Data 3; Supplementary Fig 1a); six of the signals remained significant after adjusting for BMI (Supplementary Data 4). We identified one novel genome-wide significant T2D-associated locus, the intronic variant rs12344703 in MOB3B (odds ratio (OR) = 1.07, 95% confidence interval (CI): (1.03, 1.11), p = 2.26 × 10−8, effect allele frequency (EAF) = 0.79; Supplementary Data 3). Fine-mapping at the MOB3B locus resulted in a 95% credible set of 10 additional variants (Supplementary Data 5). Fine-mapping analyses for all T2D single variant results indicated that two of our T2D single variant results are novel signals in known loci (Supplementary Data 5). SNP-based heritability in our T2D meta-analysis was 0.106 (SE: 0.011).
Replication of novel T2D single variant signals in DIAMANTE
We queried results from the DIAMANTE European, East Asian, and South Asian analyses to assess replication of the novel variants. We did not observe replication or a consistent direction of effect for the MOB3B variant. The signal at rs12344703 is primarily driven by effects observed in SIGMA1, however a later release of data from this project did not identify significant effects, suggesting this finding may be type 1 error24.
S-PrediXcan results for T2D
We performed an S-PrediXcan analysis to identify associations between T2D and the heritable component of gene expression (GReX) in all available tissues from GTEx and islets. Across all tissues, 65 genes were identified as having GReX significantly associated with a phenotype (Supplementary Data 6). This included 19 genes that have been previously reported through single variant GWAS, 36 additional genes in close proximity to (within 1 Mb of) a previously reported GWAS variant, and 10 potentially novel genes that have neither been previously mapped to GWAS signals, nor are within 1 Mb of previously reported variants (Supplementary Data 6).
Single variant lipids meta-analysis results
Across all lipid traits, we detected 52 known loci and nine novel loci associated with at least one lipid trait (Supplementary Data 7; Supplementary Figs 1b–e, 2c–k). One novel HDL-C-associated locus was identified with sentinel variant rs11653998 (p = 1.34 × 10−9) intronic to ERBB2 (Supplementary Data 7A). For LDL-C, two novel associations were detected, with sentinel variants rs75594955 (p = 3.41 × 10−8) located in exon 10 of TUB (p = 3.41 × 10−8), and rs186143467 (p = 1.82 × 10−13) located in the intergenic region between LOC387810 and LOC101928847 (Supplementary Data 7B). One novel locus was identified for TC; the sentinel variant rs564036749 (p = 1.02 × 10−8) is located in the intergenic region between MBIP and SFTA3 (Supplementary Data 7C). Finally, six novel loci were identified for TG: rs143891608 (p = 4.42 × 10−8), between LINC01132 and LOC101927851; rs186560848 (p = 3.84 × 10−10), between LINC02106 and LOC642366; rs181676594 (p = 3.73 × 10−10), between GFOD1 and SIRT5; rs552736307 (p = 1.94 × 10−8), between TAB2 and ZC3H12D; rs8178824 intronic to APOH; and rs557199842 (p = 1.66 × 10−15), between ZNF536 and LINC01791 (Supplementary Data 7D). Fine-mapping for our novel lipid signals identified 95% credible sets for each locus (Supplementary Data 5), and revealed novel signals at 12 known lipid loci (Supplementary Data 4). SNP-based heritability for HDL-C was estimated to be 0.101 (SE: 0.017), for LDL-C was estimated to be 0.068 (SE: 0.014), for TC was estimated to be 0.087 (SE: 0.017), and for TG was estimated to be 0.126 (SE: 0.038).
Replication of novel lipids single variant signals in MVP
We queried results from the MVP European- and African-ancestry subgroup analyses to assess for replication at the sentinel variants for our novel single variant lipid results (Supplementary Data 7). Results were only available for three of our novel results; one variant, rs11653998 in the ERBB2 locus for HDL cholesterol, replicated (p = 1.97 × 10−17 in MVP European-ancestry group, p = 1.03 × 10−2 in MVP African ancestry).
S-PrediXcan results for lipids
As with T2D, we used S-PrediXcan to identify GReX-trait associations for each of the lipid traits (Supplementary Data 8). For HDL-C, 193 genes were implicated in S-PrediXcan analyses (Supplementary Data 8A), including 91 genes previously reported through GWAS, 86 additional genes in close proximity to (within 1 Mb of) a previously reported GWAS variant, and 16 potentially novel genes. One hundred sixty-eight genes were implicated through S-PrediXcan for LDL-C (Supplementary Data 8B), including 80 known GWAS genes, 67 unreported genes in known GWAS loci, and 21 potentially novel genes. For total cholesterol, 217 total genes were implicated in our S-PrediXcan analysis (Supplementary Data 8C), including 100 genes reported in GWAS, 103 genes near a GWAS variant, and 14 potentially novel genes. Finally, 220 genes were implicated for triglycerides, including 92 known GWAS loci, 99 genes in close proximity to GWAS variants, and 29 potentially novel genes (Supplementary Data 8D).
Functional annotations of study-wide significant genes
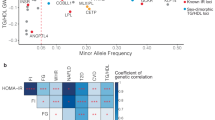
To prioritize genes for future functional studies, we annotated study-wide significant S-PrediXcan genes with tissue-specific MR results. We identified nominal evidence of causal effects on T2D for 27 genes, 89 for HDL-C, 65 for LDL-C, 99 for TC, and 97 for TG (Fig. 1; Supplementary Data 6, 8). We conducted MR for all tissues (up to 49) that a gene was study-wide significant for as denoted in the additional tissues column in Supplementary Data 6, 8.

UpSet plot (an extension of Venn diagrams to visualize overlap of more than three datasets) for type 2 diabetes (A) HDL cholesterol (B), LDL cholesterol (C) total cholesterol (D) triglycerides (E) and showing all nominally significant (p < 0.05) annotation results across gene/protein-based analyses for novel genes identified in S-PrediXcan discovery genetically regulated expression association analyses: Mendelian randomization of S-PrediXcan findings, transcriptomic association analyses, and proteomic association analyses. Intersections empty for all traits are excluded (GReX, MR, RNASeq, and Proteomics; and GReX, RNASeq, and Proteomics). GReX represents genetically regulated expression analysis (S-PrediXcan) results, MR represents Mendelian randomization results, RNASeq represents transcriptomic differential expression results, and Proteomics represents proteomic differential abundance results. Blue represents novel loci, purple represents novel genes/proteins in known loci, and pink represents known genes/proteins in known loci.
To further prioritize results by directly measured differential abundance of transcript or protein, we tested study-wide significant S-PrediXcan genes for association of measured gene expression and protein level in blood with each trait in an independent Hispanic cohort to annotate our S-PrediXcan findings. All available study-wide significant S-PrediXcan genes were tested (ntranscriptomics = 53; 127; 113; 152; 138 and nproteomics = 9; 34; 31; 39; 37 tests performed for T2D, HDL-C, LDL-C, TC, and TG respectively; the number of tests performed stratified by novelty are provided in data legends).
In the Discussion, we highlight genes identified by S-PrediXcan analyses with two or more additional supportive analyses (Mendelian randomization, differential transcriptomics, and differential proteomics analyses), including TMEM205 and NEDD9 for HDL-C, TREH for triglycerides, and ANXA4 for T2D.
Ancestry effects in single variant findings
Due to heterogeneity of admixture patterns between the studies included in this meta-analysis, we expected to observe heterogeneity of effect by study-level differences in ancestry. However, MR-MEGA detects variants with heterogeneity that are correlated with ancestry, and, for most of our single variant results, we did not observe significant ancestry-associated heterogeneity (Supplementary Data 3, 4,7). We did identify variants that are specific to (i.e., observed only in or only at any appreciable frequency in) Hispanic/Latino and, sometimes, African-ancestry populations. One example of such a variant is HDL-C-associated variant rs188287950, which is located in intron one of SIK3. This is a known HDL-C locus including APOA4 and many variants have previously been reported within one Mb of the sentinel variant (Supplementary Data 7). We observed this variant at an MAF of 0.05 in our data, but it is only observed in Admixed American populations in 1000 Genomes Project reference data (MAF = 0.05, MAF = 0 in all other 1000 Genomes populations). This variant is also observed at an MAF of 0.10 in Latino/Admixed American populations in gnomAD, and at an MAF < 0.005 in all other populations.
We also identified loci where the sentinel variant is present in other ancestry groups, but fine-mapping indicates that a haplotype distinct from previously reported associations underlies our observed signal at a known locus. An example of this is T2D-associated variant, rs1574285, at the GLIS3 locus. Nearby variants have been previously identified in other T2D GWAS, including in both the DIAMANTE European and East Asian ancestry groups25,26. However, none of the variants previously observed are contained within our 95% credible sets for this locus. These and other similar loci exhibit the importance of examining the genetic architecture in Hispanic/Latino populations for improving predictive modeling (e.g., polygenic risk scores), as has been previously noted27,28,29.
Discussion
Thousands of loci for T2D and lipid traits have been identified in GWAS and yet populations most impacted by metabolic diseases have been largely overlooked and functional interrogation of identified loci has been limited. To address these long-standing gaps, we performed meta-analyses and fine-mapping of T2D and lipid traits, functionally oriented gene-based tests, and independent functional annotation of findings in large resources of whole blood RNA sequence data and proteomics in Hispanic/Latino populations. Across all traits studied, we discovered 11 novel loci and 21 novel signals in known loci, which may constitute distinct signals from the primarily European-ancestry-derived established variants in these loci (Fig. 2), demonstrating the importance of diverse populations in genomic studies.

A novel variant in a known locus was defined as a locus that had known variants within one Mb, but no known variants contained in its 95% credible set(s). A known locus was defined as a locus with known variants contained in its 95% credible set(s). A novel locus was defined as a variant with no known variants within one Mb or contained in its 95% credible set(s). Pink represents known variants in known loci, purple represents novel variants in known loci, and blue represents novel loci. Traits included are type 2 diabetes (T2D), HDL cholesterol (HDL-C), LDL cholesterol (LDL-C), total cholesterol (TC), and triglycerides (TG).
Aggregation of single variant meta-analysis results using S-PrediXcan both across 49 GTEx tissues and models developed here using in pancreatic islet cells from the InsPIRE consortium23 identified further loci and genes of interest. We then functionally prioritized genes associated with T2D and lipid traits using Mendelian randomization to assess evidence for causality. Finally, using a multi-omics approach, we functionally annotated these prioritized genes with differential abundance analysis results from whole blood transcriptomic and proteomic data collected from participants of an independent Hispanic cohort. Our comprehensive, multi-tiered analysis identified novel genes and novel loci for T2D and lipid traits in Hispanic/Latinos, providing evidence for future functional studies.
One novel gene implicated in our single variant results for LDL-C has compelling functional evidence of impact on cardiometabolic disease. An exonic variant in TUB, rs75594955, was associated with LDL-C in our meta-regression. Mutations in TUB’s murine analog Tub produce a well-known murine model of T2D and obesity and has been shown to impact plasma lipid levels30,31. Similarly, a homozygous loss-of-function mutation in a consanguineous human family induced syndromic obesity as well as retinal dystrophy32. Expression of TUB in adipose tissue in obese individuals has also been shown to be reduced relative to non-obese controls33.
In our novel single variant results for triglycerides, we observed a variant near SIRT5, which encodes a lysine deacylase. A study of the impact of SIRT5 on bovine preadipocyte differentiation and an obese mouse model showed that SIRT5 inhibits preadipocyte differentiation, as well as lipid synthesis and lipid deposition in adipocytes34. In a separate study of an obese mouse model with hepatic SIRT5 overexpression, the authors found a reduction in triglyceride levels in the liver and increased serum triglycerides, suggesting that SIRT5 may play a role in exporting triglycerides from the liver to blood35.
Most of our novel single variant results did not replicate in other population groups, though this result is not unexpected. Differences in allele frequency (and therefore, power) may explain lack of replication in some cases, including for variants that are appreciably more frequent in Hispanic/Latino populations and in some cases entirely unobserved in other populations. Indeed, seven of our ten novel lipid results have a MAF < 0.01 in the 1000 Genomes Project European population, and eight of the ten novel lipid results have a MAF < 0.01 in the 1000 Genomes Project African populations. Other variants are common across many ancestry groups, e.g., rs28712821 for T2D; for these loci, lack of replication may be due to other causes. For example, difference in linkage disequilibrium at the locus may result in different tag variants for a causal variant common between ancestry groups, allelic heterogeneity at a locus may result in causal variants that differ by ancestry group, or differences in environmental risk or protective factors between populations may impact our power to observe effects.
Two genes implicated for HDL-C in our S-PrediXcan discovery analysis showed both nominal evidence of causality via Mendelian randomization and association with HDL-C in our independent transcriptomic association analysis, TMEM205, and NEDD9. TMEM205 has primarily been previously linked to multiple cancer phenotypes, including resistance to chemotherapeutic agents36,37. More recently, a study of a mouse model of nonalcoholic steatohepatitis revealed a role for TMEM205 in lipid metabolism38. Our results lend further support to this proposed pathway and broaden our understanding of its impact to lipoprotein levels, outside of the context of liver disease. NEDD9 has previously been found to be near genome-wide significant in a GWAS of coronary artery disease39, and in a study of gene-gene interactions that impact lipoprotein concentrations, NEDD9 was found to interact with SMAD3 to influence HDL-C, however NEDD9 has not been previously implicated for HDL-C alone40. Our findings suggest a functional role for genetic variation at this locus, through impact on regulation of NEDD9 expression, and indicate that this dysregulation may be causally impacting HDL-C concentration.
For triglycerides, we highlight two genes that were study-wide significant in S-PrediXcan and nominally significant in Mendelian randomization and either transcriptome or proteome analyses. ZNF513 was nominally associated in our transcriptomic analysis and has been previously implicated in an autosomal recessive form of retinitis pigmentosa41; neither genetic variation attributed to ZNF513 nor expression of ZNF513 has been previously linked to lipid traits, however nearby genes, including SNX1742,43, NRBP144, and GCKR8,45 have been previously identified for TG via GWAS, suggesting that this region will need further study to elucidate the role of ZNF513 and its interaction with nearby genes. ANKK1, which was nominally associated in our transcriptomic analysis, is physically near DRD2, which encodes Dopamine receptor D2. This locus has been previously linked to neuropsychiatric disorders46,47,48, as well as recently being implicated in a GWAS of TG7; an intronic variant in DRD2 was associated with TG in the GWAS.
For T2D, we identified one gene, ANXA4, that was significantly associated with T2D status in S-PrediXcan and nominally significant in Mendelian randomization and our proteomics analysis. This gene has not been previously reported in GWAS of T2D. ANXA4 expression has been shown to be impacted by knockout variants in HNF1A and PDX149, two monogenic diabetes genes; ANXA4 was also shown to be a target gene of HNF4A50. Further, the mouse gene Anxa4 is downregulated in Ipf1/Pdx1−/− pancreatic progenitor cells23. ANXA4 is also part of the GSK3β-Ikaros-ANXA4 signaling pathway, which has been demonstrated to inhibit migration of fibroblasts due to high glucose levels51. Our finding, that expression of ANXA4 may exert causal effects on T2D risk and that ANXA4 protein abundance is dysregulated in T2D, provides compelling support of a role of ANXA4 in non-monogenic diabetes risk as well.
Pancreatic islet cells are not a specific tissue type included in GTEx, yet are a central tissue in T2D pathophysiology that exhibits tissue-specific expression at key T2D genes, including INS23,26,52. Therefore, we integrated pancreatic islet cell data from the InsPIRE consortium to generate S-PrediXcan models and applied those models to our meta-analysis results. We compared the results of the pancreatic islet cell models to those using bulk pancreas tissue to identify organelle-specific genetic regulation impacting islet cells.
All four S-PrediXcan associations in islets fall within an ~2 Mb region surrounding Insulin. The S-PrediXcan model for INS itself was not significantly associated with T2D (p = 0.6). Nonetheless, these genes have molecular assays supporting their role in glucose homeostasis. For example, in vitro work shows BRSK2 phosphorylates PCTAIRE1, which in turn decreases insulin secretion in response to glucose53. TRPM5 is a receptor that has been shown through knock out mice, and subsequent in vitro work to be essential for glucose-stimulated insulin secretion54. Further, chr11p15.5-p15.4 is a region known for complex imprinting, resulting in parent-of-origin-specific expression of various genes55,56,57. OSBPL5 has methylation disruptions in insulinomas58. This region of the genome shows evidence of complex regulation, particularly in pancreatic islets. There may be mechanisms of co-regulation involved, as it contains many genes that influence glucose homeostasis; indeed, we see evidence of potential co-regulation of the associated genes based on correlation of expression in InsPIRE gene expression data (Supplementary Fig 3), however additional work in pancreatic beta cells is required to clarify.
Our S-PrediXcan analyses provide an opportunity to functionally annotate known loci, adding to our understanding of the biology of these signals by narrowing the likely causal gene(s) at the locus. In the known T2D locus on chromosome 22, we identified significant signals in both our GWAS and S-PrediXcan analyses. In our GWAS, rs16989540, in intron 27 of DEPDC5, was significantly associated with T2D status; Open Targets Genetics predicted YWHAH to be the causal gene for this signal. This locus has been previously reported in three prior studies, two of which had sample overlap with the present study and in a third study in a Maya population9,20,59. In all three studies, the signal was mapped to either DEPDC5 or YWHAH. However, S-PrediXcan functionally implicates a different gene in this locus, SLC5A1, in small intestine. The role of this gene, also known as SGLT1, in type 2 diabetes is supported by an abundance of non-GWAS evidence. Consistent with our observed direction of effect in small intestine, measured expression of this gene in the small intestine has been shown to be increased in people with type 2 diabetes60,61. Indeed, the FDA recently approved a medication, called sotagliflozin, that targets SGLT-1 and SGLT-2, reducing blood glucose and treating heart failure in T2D patients62,63. It is notable that all three GWAS that reported a signal at this locus included individuals with a large proportion of AMR ancestry, suggesting this might be a key population in which to explore drug efficacy. This is just one example of how functionally oriented analyses, using expression data from multiple tissues, can inform our interpretation of GWAS results and identify clinically actionable targets.
Our study had several limitations. Most notably, we were limited by GWAS data currently available for Hispanic/Latino populations, and even aggregating many of the extant GWAS of T2D and lipid traits in Hispanic/Latino populations in this meta-analysis resulted in a much lower sample size than analogous European-ancestry studies. This limitation must be addressed by prioritization of studies of non-European ancestry populations in biomedical research. This can only be accomplished through increased funding to engage and recruit members of diverse populations as partners in and beneficiaries of biomedical research efforts. This Euro-centric bias is pervasive, ranging from genotype array design to models of gene expression. Indeed, our S-PrediXcan analyses utilized publicly available models developed in GTEx Project data, which primarily comprises European ancestry individuals, similar to most publicly available gene expression datasets. Several studies have demonstrated that prediction performance of these models is maximized by matching genetic ancestry of the model training dataset to the testing dataset64,65,66, however it is most likely that reduced predictive performance in cross-ancestry applications will result in a reduction in power rather than increased type 1 error67, and prior application of models derived from primarily European-ancestry data to non-European-ancestry populations has indeed resulted in robust findings confirmed through replication67,68. To address this lower power, future studies must prioritize inclusion of non-European-ancestry populations in transcriptome and other omics projects, as we have done here by generating a large resource of whole blood transcriptome data in a Hispanic/Latino population, to ensure that resources like ancestry-matched GReX prediction models, and ultimately the medical advances that discoveries from omics studies will drive, are available and accessible for all populations. Finally, this study relied on meta-analysis of available imputed data, and we did not have universal access to individual-level data. Thus, we could not explore the impact of local ancestry, specific haplotypes in a locus, or leverage more diverse imputation reference panels. Further work will be needed to follow up on loci that have evidence of being population specific.
Our study reiterates the importance of large-scale studies of non-European populations for genetic discovery, presenting insight into trait biology through multiple lines of transcriptomic and proteomic data, in spite of significantly smaller sample sizes relative to current studies in European-ancestry populations. Our study also demonstrates how precision medicine advances can be made when we integrate GWAS with functional characterization through omics data, revealing candidate genes and functional variants. It is our hope that current and future efforts that prioritize inclusion of Hispanic/Latino populations, such as the All of Us research program69 and the Mexico City Prospective Study70, can substantially increase sample sizes in genetic and other omic studies to help ensure that advances in precision medicine are realized in all populations.
Methods
Ethics
Ethics statements for all studies contributing to the meta-analysis are given in Supplementary Table 1. The CCHC portion of this study was approved by the Committee for the Protection of Human Subjects of the University of Texas Health Science Center, Houston. Our study met all relevant regulations regarding the use of human study participants and was conducted in accordance with the criteria set by the Declaration of Helsinki. All study participants gave informed consent.
Genome-wide association study data
The T2D meta-analysis comprised 23,541 T2D cases and 37,434 controls from 16 contributing studies7,9,27,28,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86 and the lipid (HDL-C, LDL-C, TC, and TG) meta-analyses included up to 63,184 samples from 19 contributing studies87,88,89 (Supplementary Data 1). All participants in the contributing studies self-identify as Hispanic and/or Latino. Sex was self-reported and confirmed to match gender through genetic data. Study-specific T2D and lipid phenotype measures, definitions, and exclusions are provided in Supplementary Data 1. Individuals that reported use of lipid-lowering medication were either excluded or lipid concentrations were adjusted to account for medication use (Supplementary Data 1).
Contributing study quality control and imputation
For each study, genome-wide array data were cleaned and imputed to 1000 Genomes Project (1KG) phase 1 or 3 reference data using Minimac3 or IMPUTE290,91,92,93,94. Study-specific array, quality control, and imputation details are given in Supplementary Data 2. For the Million Veteran Program data, GWAS results were obtained from dbGaP, accession number phs001672 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001672.v12.p1], and additional details of quality control and analysis methods can be found in the original manuscripts7,9. Additional details are provided in Supplementary Data 2.
Single variant analysis and study-level quality control
T2D association analyses for each contributing study were performed via logistic regression or linear mixed model in SNPTEST95, EPACTS/EMMAX96, SOLAR97, GENESIS98, PLINK99, or R100. All models were adjusted for age, sex, and any necessary study-specific covariates (e.g., study location or batch). All studies either adjusted for principal components and/or used a mixed model to control for population substructure. All studies also either excluded closely related individuals or used a mixed model to control for close relatedness.
Association testing for the four lipid phenotypes (HDL-C, LDL-C, TC, and log-transformed TG) were conducted via linear regression in SNPTEST95, EPACTS/EMMAX96, SOLAR97, GENESIS98, PLINK99, or R100. Residual lipid values were calculated, adjusting for age, sex, and any necessary study-specific covariates (e.g., study location or batch). The residual values were inverse rank normal transformed Relatedness and population substructure were controlled for as above. Study-level quality control included removal of low-quality variants (Minimac3 r2 < 0.3 or IMPUTE2 info <0.4), low-frequency variants (minor allele count (MAC) < 14 for T2D, MAC < 6 for lipids), duplicates, variants with large MAF discrepancy with AMR (absolute value of difference > 0.2), and variants where alleles did not match the AMR reference alleles.
Single variant meta-analysis
Meta-analyses were performed using MR-MEGA, which uses a meta-regression approach to model the effect of axes of genetic variation, representing study-level differences in genetic ancestry, allowing for partitioning of heterogeneity into effects correlated with ancestry differences and any remaining heterogeneity101. We included two measures of study-level mean allele frequency differences in our meta-regression, allowing for two axes of genetic variation. Two axes of variation were selected to capture major source of admixture within Hispanic/Latino populations (AMR, EUR, and AFR). For T2D, we performed an additional meta-analysis adjusting for BMI to explore if effects at our top findings are modified by BMI. Variants with MAC < 100 in the total meta-analysis sample were excluded. Single variant tests used a genome-wide significance threshold of p < 5 × 10−8. Sentinel variants, or variants with the lowest p-value, were identified for each region (1 Mb in either direction) with p < 5 × 10−8 and annotated using ANNOVAR102 and Open Targets Genetics103,104.
Fine-mapping
Fine-mapping was performed using the method described in Magi et al., including all variants within one Mb of our sentinel variant for each region101. Briefly, we calculated a posterior probability of driving the association at a locus for each variant, then selected variants by Bayes’ factor, in descending order, summing the cumulative posterior probability until it met or exceeded 95%. The resulting 95% credible sets were queried for previously reported variants, or any variants in the NHGRI-EBI GWAS catalog within one Mb of our sentinel variant associated with the same trait105. A known signal was defined as a locus with known variants contained in its 95% credible set(s). A novel signal was a variant with no known variants within one Mb or contained in its 95% credible set(s). We defined a novel signal in a known locus as a locus that had known variants within one Mb, but no known variants contained in its 95% credible set(s).
Replication of single variant novel signals
The T2D meta-analyses were a part of a larger multi-ancestry meta-analysis effort by the DIAMANTE Consortium20. Five groups were assembled, including our Hispanic/Latino group, an African ancestry group (MEDIA), an East Asian ancestry group (AGEN-T2D)25, a European ancestry group (DIAGRAM)26, a South Asian ancestry group (SA-T2D)106, and the multi-ancestry meta-analysis of these five groups24. We queried our top novel T2D results for replication using summary statistics from the European, East Asian, and South Asian ancestry groups. For lipid traits, we queried publicly available results from the European- and African-ancestry subgroups of the Million Veteran Program (MVP) GWAS7 for each trait to assess for replication (the MVP Hispanic subgroup was included in our meta-regression).
Development of pancreatic islet cell prediction models
Because pancreatic islet cells are a central tissue in T2D pathophysiology and exhibit a tissue-specific expression profile (e.g., 40–73% of islet eQTLs replicate in GTEx), especially at T2D-relevant genes such as INS23, in addition to leveraging extant GTEx prediction models107,108 we constructed models from extant pancreatic islet RNA sequence and associated genomic data23. The InsPIRE consortium was formed to aggregate human islet RNA-Seq data and genetic data to identify eQTLs and characterize genetic regulation of gene expression in a tissue central to T2D pathogenesis23. Here, we leveraged a subset 254 participants of the InsPIRE dataset that were made available to us upon request. Given that samples collected in the US were all described as Caucasian and the remainder of sampling occurred in Europe (Geneva, Edmonton, and Oxford) we expect the proportion of the islet sample that is of Hispanic/Latino ethnicity is minimal.
For each gene, we trained in silico models of gene expression in pancreatic islet cells using genetic variants as features. Consider n samples with covariate-adjusted gene expression levels y1, y2, …, yn. For covariates, we used sex, the first four principal components (PCs) derived from the genotype data, and the first 30 PCs for expression. Each gene model used elastic net regularization, solving the following optimization problem:
The regularization term includes an L1 penalty on the effect-size vector β (enforcing sparsity) and an L2 penalty (promoting grouping effect). The parameter α = 0.5 determines the relative weights of the two penalties. This approach closely follows the conventional PrediXcan implementation107.
Functionally oriented meta-analysis
To examine the final meta-regression results in a functionally oriented context, S-PrediXcan was used to determine association of each phenotype with genetically regulated gene expression (GReX) levels109. Using publicly available Joint-Tissue Imputation models for 49 tissues developed in the Genotype-Tissue Expression (GTEx) project v8 data108 and our pancreatic islet cell models, we inferred tissue-specific GReX association with phenotype from our meta-GWAS summary statistics from MR-MEGA. As recommended by the S-PrediXcan authors, we applied this approach across all tissues, agnostic of currently known trait biology, as they found that generally accepted disease-relevant tissues are not typically enriched for GReX associations and, thus, a tissue-agnostic approach improves discovery109. We used a Benjamini-Hochberg adjustment across all tissues, genes, and phenotypes to account for multiple testing, as a Bonferroni correction for all tissues and traits would be too conservative due to correlation of both the expression models between tissues and the set of phenotypes tested. We considered an adjusted p < 0.01 to be significant.
Mendelian randomization
We assessed evidence of causality for genes identified in S-PrediXcan analyses and found to be significant after study-wide multiple test correction. The GTEx v8 tissue-specific eQTLs of target genes were used as instrumental variables for MR. We performed LD clumping to select eligible instrumental variables in each tissue separately with the LD panels from the 1000 Genome Admixed Americans (AMR), and the bigsnpr R package110,111. We used the median weighted MR method from the MendelianRandomization R package112, which offers unbiased and reliable estimation even when half of instrumental variables violate the assumptions of MR and in the presence of genetic pleiotropy112,113.
LD scores
LD scores for variants in meta-analysis results were created using HCHS/SOL genotype data and GCTA v1.93.2, with an LD window size of 1 Mb and an LD r2 cut-off of 0.01114. eQTL effects were obtained from GTEx v8 eQTL Tissue-Specific All SNP Gene Associations data (https://www.gtexportal.org/home/datasets) and filtered to retain variants within 1 Mb of the gene.
Measured transcriptomic expression analyses
To better understand molecular signatures associated with T2D and lipids for study-wide significant functionally oriented S-PrediXcan results, we directly measured whole blood gene expression in 884 (for T2D) and 696 (for lipid measures) Mexican-American individuals from the Cameron County Hispanic Cohort (CCHC) and performed association analyses between gene expression level and phenotype for each novel S-PrediXcan hit115. RNA sequencing was conducted using 150 bp paired-end reads on the Illumina NovaSeq 6000 by Vanderbilt Technologies for Advanced Genomics. Initial sequencing quality was checked by FastQC116. STAR-2.7.8a was applied to align sequencing reads to the human genome reference (UCSC, hg38)117, and the aligned reads were assigned to genes using featureCounts in the Rsubread package118. We excluded samples with less than 15 M total aligned reads or a rate of successful alignment of less than 20%. The sequencing library size was normalized using DESeq2119. Individuals taking lipid-lowering medication were excluded, and triglyceride concentrations were log-transformed. We then tested for association of gene expression of the novel genes implicated in our S-PrediXcan analyses with trait, using linear regression, with RNA expression as the dependent variable and trait as the independent variable, and adjusting for age, sex, estimated cell type proportions (including granulocytes, CD19 + B cells, CD4 T lymphocytes, CD8 T lymphocytes, and CD14+ monocytes), three genetic PCs to capture population substructure, and ten probabilistic estimation of expression residual (PEER) factors to capture hidden factors that explain variation in the expression data.
Proteomic analyses
To further explore significant S-PrediXcan findings in measured proteomic data, we measured the Olink Explore 3072 panel on 528 stored plasma samples from 271 individuals in CCHC. Normalized Protein eXpression (NPX) was generated in the full dataset, which included multiple measures for many individuals. Our proteomic analyses restricted to one time point per individual and adjusted for age, sex, and five genetic principal components in logistic (T2D) or linear regression (lipids), using the phenotypes measured at the time of specimen collection. Triglycerides were log-transformed.
Heritability analyses
Linkage disequilibrium score regression (as implemented in LDSC120) was applied to estimate SNP-based heritability. Heritability was estimated based on the relationship between GWAS summary statistics and linkage disequilibrium. To account for admixture in our Hispanic/Latino populations, cov-LDSC121 was applied to calculate the linkage disequilibrium score and was adjusted for the top ten genetic PCs from PC-AiR98,122. In this study, the linkage disequilibrium score was calculated with all 1,274,124 genotyped SNPs in 10,050 unrelated individuals from HCHS/SOL; the maximum unrelated set was calculated in PRIMUS and defined as unrelated at ≥third degree123.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Complete summary statistics for the primary meta-analyses are available in the NHGRI-EBI Catalog of human genome-wide association studies under accession numbers: GCST90528074, GCST90528075, GCST90528076, GCST90528077, GCST90528078, and GCST90528079. The Cameron County Hispanic Cohort transcriptomic and proteomic data generated in this study are available in dbGaP under accession number phs003894.v1.p1 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003894.v1.p1]. The islet gene expression imputation models generated in this study are available at https://github.com/gamazonlab/IsletCellsGReXModels. The Million Veterans Program data used in this study are available in dbGaP under the study accession number phs001672 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001672.v12.p1], analysis accession numbers pha004829.1 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/analysis.cgi?study_id=phs001672.v12.p1&pha=4829], pha004832.1 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/analysis.cgi?study_id=phs001672.v12.p1&pha=4832], pha004835.1 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/analysis.cgi?study_id=phs001672.v12.p1&pha=4835], pha004838.1 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/analysis.cgi?study_id=phs001672.v12.p1&pha=4838], and pha004946.1 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/analysis.cgi?study_id=phs001672.v12.p1&pha=4946]. Source data is provided with this paper. Source data are provided with this paper.
References
U.S. Centers for Disease Control and Prevention (ed U.S Department of Health and Human Services). https://www.cdc.gov/diabetes/php/data-research/index.html (2024).
Carroll, M. D. & Fryar, C. D. (ed National Center for Health Statistics). https://www.cdc.gov/nchs/products/databriefs/db363.htm (Hyattsville, MD, 2020).
Hirode, G. & Wong, R. J. Trends in the prevalence of metabolic syndrome in the United States, 2011-2016. JAMA 323, 2526–2528 (2020).
Popejoy, A. B. & Fullerton, S. M. Genomics is failing on diversity. Nature 538, 161–164 (2016).
Hu, Y. et al. Minority-centric meta-analyses of blood lipid levels identify novel loci in the Population Architecture using Genomics and Epidemiology (PAGE) study. PLoS Genet. 16, e1008684 (2020).
Wojcik, G. L. et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 570, 514–518 (2019).
Klarin, D. et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet. 50, 1514–1523 (2018).
Graham, S. E. et al. The power of genetic diversity in genome-wide association studies of lipids. Nature 600, 675–679 (2021).
Vujkovic, M. et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 52, 680–691 (2020).
Willemsen, G. et al. The concordance and heritability of type 2 diabetes in 34,166 twin pairs from international twin registers: the discordant twin (DISCOTWIN) consortium. Twin Res. Hum. Genet. 18, 762–771 (2015).
Poulsen, P., Kyvik, K. O., Vaag, A. & Beck-Nielsen, H. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance–a population-based twin study. Diabetologia 42, 139–145 (1999).
Divers, J. et al. The genetic architecture of lipoprotein subclasses in Gullah-speaking African American families enriched for type 2 diabetes: the Sea Islands Genetic African American Registry (Project SuGAR). J. Lipid Res. 51, 586–597 (2010).
Rao, D. C. et al. The Cincinnati Lipid Research Clinic family study: cultural and biological determinants of lipids and lipoprotein concentrations. Am. J. Hum. Genet. 34, 888–903 (1982).
Perusse, L. et al. Familial resemblance of plasma lipids, lipoproteins and postheparin lipoprotein and hepatic lipases in the HERITAGE Family Study. Arterioscler Thromb. Vasc. Biol. 17, 3263–3269 (1997).
Hokanson, J. E. et al. Pleiotropy and heterogeneity in the expression of atherogenic lipoproteins: the IRAS Family Study. Hum. Hered. 55, 46–50 (2003).
Ali, O. Genetics of type 2 diabetes. World J. Diab. 4, 114–123 (2013).
Teslovich, T. M. et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 (2010).
Willer, C. J. et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45, 1274–1283 (2013).
DIAbetes Genetics Replication Meta-analysis Consortium et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet. 46, 234–244 (2014)
Mahajan, A. et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet. 54, 560–572 (2022).
Bien, S. A. et al. Transethnic insight into the genetics of glycaemic traits: fine-mapping results from the Population Architecture using Genomics and Epidemiology (PAGE) consortium. Diabetologia 60, 2384–2398 (2017).
Below, J. E. & Parra, E. J. Genome-wide studies of type 2 diabetes and lipid traits in hispanics. Curr. Diab Rep. 16, 41 (2016).
Vinuela, A. et al. Genetic variant effects on gene expression in human pancreatic islets and their implications for T2D. Nat. Commun. 11, 4912 (2020).
Suzuki, K. et al. Genetic drivers of heterogeneity in type 2 diabetes pathophysiology. Nature. https://doi.org/10.1038/s41586-024-07019-6 (2024).
Spracklen, C. N. et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature 582, 240–245 (2020).
Mahajan, A. et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 50, 1505–1513 (2018).
Parra, E. J. et al. Genome-wide association study of type 2 diabetes in a sample from Mexico City and a meta-analysis of a Mexican-American sample from Starr County, Texas. Diabetologia 54, 2038–2046 (2011).
Below, J. E. et al. Genome-wide association and meta-analysis in populations from Starr County, Texas, and Mexico City identify type 2 diabetes susceptibility loci and enrichment for expression quantitative trait loci in top signals. Diabetologia 54, 2047–2055 (2011).
Below, J. E. et al. Meta-analysis of lipid-traits in Hispanics identifies novel loci, population-specific effects, and tissue-specific enrichment of eQTLs. Sci. Rep. 6, 19429 (2016).
Nishina, P. M., Lowe, S., Wang, J. & Paigen, B. Characterization of plasma lipids in genetically obese mice: the mutants obese, diabetes, fat, tubby, and lethal yellow. Metabolism 43, 549–553 (1994).
Noben-Trauth, K., Naggert, J. K., North, M. A. & Nishina, P. M. A candidate gene for the mouse mutation tubby. Nature 380, 534–538 (1996).
Borman, A. D. et al. A homozygous mutation in the TUB gene associated with retinal dystrophy and obesity. Hum. Mutat. 35, 289–293 (2014).
Nies, V. J. M. et al. TUB gene expression in hypothalamus and adipose tissue and its association with obesity in humans. Int. J. Obes. (Lond.) 42, 376–383 (2018).
Hong, J. et al. SIRT5 inhibits bovine preadipocyte differentiation and lipid deposition by activating AMPK and repressing MAPK signal pathways. Genomics 112, 1065–1076 (2020).
Du, Y. et al. SIRT5 deacylates metabolism-related proteins and attenuates hepatic steatosis in ob/ob mice. EBioMedicine 36, 347–357 (2018).
Wang, Y. et al. The association of transporter genes polymorphisms and lung cancer chemotherapy response. PloS one 9, e91967 (2014).
Shen, D. W. et al. Elevated expression of TMEM205, a hypothetical membrane protein, is associated with cisplatin resistance. J. Cell Physiol. 225, 822–828 (2010).
Yang, T. et al. The macrophage STING-YAP axis controls hepatic steatosis by promoting the autophagic degradation of lipid droplets. Hepatology. https://doi.org/10.1097/HEP.0000000000000638 (2023).
van der Harst, P. & Verweij, N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ. Res. 122, 433–443 (2018).
Ma, L., Clark, A. G. & Keinan, A. Gene-based testing of interactions in association studies of quantitative traits. PLoS Genet. 9, e1003321 (2013).
Li, L. et al. A mutation in ZNF513, a putative regulator of photoreceptor development, causes autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 87, 400–409 (2010).
Galla, S. J. et al. A comparison of pedigree, genetic and genomic estimates of relatedness for informing pairing decisions in two critically endangered birds: Implications for conservation breeding programmes worldwide. Evol. Appl 13, 991–1008 (2020).
de Vries, P. S. et al. Multiancestry genome-wide association study of lipid levels incorporating gene-alcohol interactions. Am. J. Epidemiol. 188, 1033–1054 (2019).
Sinnott-Armstrong, N. et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat. Genet. 53, 185–194 (2021).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
Li, H. J. et al. Novel risk loci associated with genetic risk for bipolar disorder among han chinese individuals: a genome-wide association study and meta-analysis. JAMA Psychiatry 78, 320–330 (2021).
Thompson, A. et al. Functional validity, role, and implications of heavy alcohol consumption genetic loci. Sci. Adv. 6, eaay5034 (2020).
Kimbrel, N. A. et al. Identification of novel, replicable genetic risk loci for suicidal thoughts and behaviors among US military veterans. JAMA Psychiatry 80, 135–145 (2023).
Servitja, J. M. et al. Hnf1alpha (MODY3) controls tissue-specific transcriptional programs and exerts opposed effects on cell growth in pancreatic islets and liver. Mol. Cell Biol. 29, 2945–2959 (2009).
Bolotin, E. et al. Integrated approach for the identification of human hepatocyte nuclear factor 4alpha target genes using protein binding microarrays. Hepatology 51, 642–653 (2010).
Wang, Y., Zheng, X., Wang, Q., Zheng, M. & Pang, L. GSK3beta-Ikaros-ANXA4 signaling inhibits high-glucose-induced fibroblast migration. Biochem. Biophys. Res. Commun. 531, 543–551 (2020).
GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Chen, X. Y. et al. Brain-selective kinase 2 (BRSK2) phosphorylation on PCTAIRE1 negatively regulates glucose-stimulated insulin secretion in pancreatic beta-cells. J. Biol. Chem. 287, 30368–30375 (2012).
Brixel, L. R. et al. TRPM5 regulates glucose-stimulated insulin secretion. Pflug. Arch. 460, 69–76 (2010).
Reik, W. & Maher, E. R. Imprinting in clusters: lessons from Beckwith-Wiedemann syndrome. Trends Genet. 13, 330–334 (1997).
Smith, A. C., Choufani, S., Ferreira, J. C. & Weksberg, R. Growth regulation, imprinted genes, and chromosome 11p15.5. Pediatr. Res. 61, 43R–47R (2007).
Schrier Vergano, S. A. & Deardorff, M. A. Coffin-Siris Syndrome. GeneReviews((R)) (eds Adam, M. P. et al.) (University of Washington, Seattle, 1993).
Karakose, E. et al. Aberrant methylation underlies insulin gene expression in human insulinoma. Nat. Commun. 11, 5210 (2020).
Dominguez-Cruz, M. G. et al. Pilot genome-wide association study identifying novel risk loci for type 2 diabetes in a Maya population. Gene 677, 324–331 (2018).
Dyer, J., Wood, I. S., Palejwala, A., Ellis, A. & Shirazi-Beechey, S. P. Expression of monosaccharide transporters in intestine of diabetic humans. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G241–G248 (2002).
Fiorentino, T. V. et al. Duodenal sodium/glucose cotransporter 1 expression under fasting conditions is associated with postload hyperglycemia. J. Clin. Endocrinol. Metab. 102, 3979–3989 (2017).
Avgerinos, I. et al. Sotagliflozin for patients with type 2 diabetes: a systematic review and meta-analysis. Diab. Obes. Metab. 24, 106–114 (2022).
Lexicon Pharmaceuticals, Inc. Inpefa (sotagliflozin) [package insert]. U.S. Food and Drug Administration. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/216203s000lbl.pdf (2023). Accessed April 4, 2025.
Mikhaylova, A. V. & Thornton, T. A. Accuracy of gene expression prediction from genotype data with PrediXcan varies across and within continental populations. Front. Genet. 10, 261 (2019).
Keys, K. L. et al. On the cross-population generalizability of gene expression prediction models. PLoS Genet. 16, e1008927 (2020).
Fryett, J. J., Morris, A. P. & Cordell, H. J. Investigation of prediction accuracy and the impact of sample size, ancestry, and tissue in transcriptome-wide association studies. Genet. Epidemiol. 44, 425–441 (2020).
Geoffroy, E., Gregga, I. & Wheeler, H. E. Population-matched transcriptome prediction increases TWAS discovery and replication rate. iScience 23, 101850 (2020).
Petty, L. E. et al. Functionally oriented analysis of cardiometabolic traits in a trans-ethnic sample. Hum. Mol. Genet. 28, 1212–1224 (2019).
Mapes, B. M. et al. Diversity and inclusion for the All of Us research program: a scoping review. PloS ONE 15, e0234962 (2020).
Ziyatdinov, A. et al. Genotyping, sequencing and analysis of 140,000 adults from Mexico City. Nature 622, 784–793 (2023).
Tayo, B. O. et al. Genetic background of patients from a university medical center in Manhattan: implications for personalized medicine. PloS one 6, e19166 (2011).
Kuo, J. Z. et al. Systemic soluble tumor necrosis factor receptors 1 and 2 are associated with severity of diabetic retinopathy in Hispanics. Ophthalmology 119, 1041–1046 (2012).
Qi, Q. et al. Genetics of type 2 diabetes in U.S. Hispanic/Latino individuals: results from the Hispanic Community Health Study/Study of Latinos (HCHS/SOL). Diabetes 66, 1419–1425 (2017).
Xiang, A. H. et al. Evidence for joint genetic control of insulin sensitivity and systolic blood pressure in Hispanic families with a hypertensive proband. Circulation 103, 78–83 (2001).
Cheng, L. S. et al. Coincident linkage of fasting plasma insulin and blood pressure to chromosome 7q in hypertensive Hispanic families. Circulation 104, 1255–1260 (2001).
Pojoga, L. H. et al. Variants of the caveolin-1 gene: a translational investigation linking insulin resistance and hypertension. J. Clin. Endocrinol. Metab. 96, E1288–E1292 (2011).
Varma, R. et al. Prevalence of diabetic retinopathy in adult Latinos: the Los Angeles Latino eye study. Ophthalmology 111, 1298–1306 (2004).
Palmer, N. D. et al. Genetic variants associated with quantitative glucose homeostasis traits translate to type 2 diabetes in Mexican Americans: the GUARDIAN (Genetics Underlying Diabetes in Hispanics) Consortium. Diabetes 64, 1853–1866 (2015).
Goodarzi, M. O. et al. Determination and use of haplotypes: ethnic comparison and association of the lipoprotein lipase gene and coronary artery disease in Mexican-Americans. Genet. Med. 5, 322–327 (2003).
Bild, D. E. et al. Multi-ethnic study of atherosclerosis: objectives and design. Am. J. Epidemiol. 156, 871–881 (2002).
MacCluer, J. W. et al. Genetics of atherosclerosis risk factors in Mexican Americans. Nutr. Rev. 57, S59–S65 (1999).
Cai, G. et al. Genome-wide scans reveal quantitative trait Loci on 8p and 13q related to insulin action and glucose metabolism: the San Antonio Family Heart Study. Diabetes 53, 1369–1374 (2004).
Sigma Type 2 Diabetes Consortium et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 311, 2305–2314 (2014).
Sigma Type 2 Diabetes Consortium et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 506, 97–101 (2014).
Mercader, J. M. et al. A loss-of-function splice acceptor variant in IGF2 is protective for type 2 diabetes. Diabetes 66, 2903–2914 (2017).
Prentice, R. L. et al. Design of the Women’s Health Initiative clinical trial and observational study. The Women’s Health Initiative Study Group. Control Clin. Trials 19, 61–109 (1998).
Justice, A. E. et al. Genetic determinants of BMI from early childhood to adolescence: the Santiago Longitudinal Study. Pediatr. Obes. 14, e12479 (2019).
Gao, C. et al. Exome sequencing identifies genetic variants associated with circulating lipid levels in Mexican Americans: The Insulin Resistance Atherosclerosis Family Study (IRASFS). Sci. Rep. 8, 5603 (2018).
Wagenknecht, L. E. et al. The insulin resistance atherosclerosis study (IRAS) objectives, design, and recruitment results. Ann. Epidemiol. 5, 464–472 (1995).
Abecasis, G. R. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Howie, B., Marchini, J. & Stephens, M. Genotype imputation with thousands of genomes. G3 1, 457–470 (2011).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Marchini, J. & Howie, B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 11, 499–511 (2010).
Kang, H. M. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010).
Almasy, L. & Blangero, J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 62, 1198–1211 (1998).
Gogarten, S. M. et al. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics 35, 5346–5348 (2019).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
R: A language and environment for statistical computing (R Foundation for Statistical Computing, 2020).
Magi, R. et al. Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum. Mol. Genet. 26, 3639–3650 (2017).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Ghoussaini, M. et al. Open Targets Genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 49, D1311–D1320 (2021).
Mountjoy, E. et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat. Genet. 53, 1527–1533 (2021).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019).
Loh, M. et al. Identification of genetic effects underlying type 2 diabetes in South Asian and European populations. Commun. Biol. 5, 329 (2022).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098 (2015).
Zhou, D. et al. A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat. Genet. 52, 1239–1246 (2020).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018).
Prive, F., Aschard, H., Ziyatdinov, A. & Blum, M. G. B. Efficient analysis of large-scale genome-wide data with two R packages: bigstatsr and bigsnpr. Bioinformatics 34, 2781–2787 (2018).
Genomes Project Consortium. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Yavorska, O. O. & Burgess, S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J. Epidemiol. 46, 1734–1739 (2017).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Fisher-Hoch, S. P. et al. Socioeconomic status and prevalence of obesity and diabetes in a Mexican American community, Cameron County, Texas, 2004-2007. Prev. Chronic Dis. 7, A53 (2010).
Andrews, S. FastQC: a quality control tool for high throughput sequence data [Online]. Available online at: http://www.bioinformatics (2010).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Luo, Y. et al. Estimating heritability and its enrichment in tissue-specific gene sets in admixed populations. Hum. Mol. Genet. 30, 1521–1534 (2021).
Conomos, M. P., Miller, M. B. & Thornton, T. A. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet. Epidemiol. 39, 276–293 (2015).
Staples, J. et al. PRIMUS: rapid reconstruction of pedigrees from genome-wide estimates of identity by descent. Am. J. Hum. Genet. 95, 553–564 (2014).
Acknowledgements
Acknowledgements for each participating study are listed in Supplementary Data 1. R01HL142302 partially supported J.E.B., K.E.N., and L.E.P.; J.M.M. is supported by American Diabetes Association grant #11-22-ICTSPM-16 and by NHGRI U01HG011723; R.J.F.L. is supported by R01DK110113, R01DK107786, R01HL142302, and R56HG010297; K.E.N. is supported primarily by 299 U01HG007416, with additional support provided via R01DK101855 and 15GRNT25880008; C.N.S. was supported by American Heart Association Postdoctoral Fellowships 15POST24470131 and 17POST33650016; M.B. is supported by DK062370. A.H.-C. is supported by the American Diabetes Association grant 11-23-PDF-35. Additional funding sources supporting each participating study are listed in Supplementary Data 1.
Author information
Authors and Affiliations
Consortia
Contributions
Meta-analysis level analysis: L.E.P., H.C., E.G.F., W. Zhu, C.G.D., M.G., P.L., P.S., X.Z., and E.R.G. performed work. Result interpretation and visualization: L.E.P., X.Z., A.C.S., R.Ros., J.M.L., S.K.F., K.E.N., J.B.M., S.P.F.-H., E.R.G., A.P.M., J.M.M., H.M.H., and J.E.B. Writing: L.E.P., H.C., P.L., K.E.N., H.M.H., and J.E.B. Study level analysis: L.E.P., M.G., M.P., Y.-D.I.C., T.L., T.S., C.G., D.N., X.G., Y.H., Y.W., and A.H. Study level data collection and supervision: E.P.B., G.N.N., R.J.F.L., J.T., E.I., P.G., L.S.E., A.M.S., K.D.T., A.H.X., T.B., K.R., N.D.P., J.M.N., L.E.W., R.V., R.M.-C., W.H., K.S., E.J.P., M.C., A.V.-S., N.W.-R., J.I.R., M.O.G., S.S.R., A.B., L.J.R., J.L.N., F.R.K., R.D., J.B., D.M.L., R.A.D., F.T., S.G., E.B., R.B., J.C.F., T.T.-L., C.G.-V., L.O., C.A.H., C.L.H., R.Roh., E.A.W., A.P.R., C.K., Y.L., Q.D., M.L., P.C.-B., S.K.F., K.E.N., J.B.M., S.P.F.-H., and J.E.B. DIAMANTE analysis group: L.E.P., M.B., D.W.B., J.C.C., A.M., M.I.M., M.C.Y.N., X.S., C.N.S., W. Zha., A.P.M., J.M.M., and J.E.B. Meta-analysis supervision: S.K.F., K.E.N., J.B.M., S.P.F.-H., E.R.G., A.P.M., J.M.M., H.M.H., and J.E.B. Critical review of manuscript: L.E.P., H.C., E.G.F., W. Zhu, C.G.D., M.G., P.L., P.S., X.Z., A.C.S., R. Ros., J.M.L., M.B., D.W.B., J.C.C., A.M., M.I.M., M.C.Y.N., X.S., C.N.S., W. Zha., M.P., E.P.B., G.N.N., R.J.F.L., Y.-D.I.C., J.T., E.I., P.G., L.S.E., T.L., T.S., A.M.S., K.D.T., A.H.X., T.B., K.R., C.G., N.D.P., J.M.N., L.E.W., D.N., R.V., R.M.-C., X.G., Y.H., W.H., K.S., E.J.P., M.C., A.V.-S., N.W.-R., J.I.R., M.O.G., S.S.R., A.B., L.J.R., J.L.N., F.R.K., R.D., J.B., D.M.L., R.A.D., F.T., Y.W., S.G., E.B., R.B., A.H., J.C.F., T.T.-L., C.G.-V., L.O., C.A.H., C.L.H., R. Roh., E.A.W., A.P.R., C.K., Y.L., Q.D., M.L., P.C.-B., S.K.F., K.E.N., J.B.M., S.P.F.-H., E.R.G., A.P.M., J.M.M., H.M.H., and J.E.B.
Corresponding author
Ethics declarations
Competing interests
A.M. and M.I.M. are employees of Genentech and a holders of Roche stock. L.S.E. is now an employee of Bristol Myers Squibb (BMS) and a holder of BMS stock. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Tinashe Chikowore, Fasil Tekola-Ayele, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Petty, L.E., Chen, HH., Frankel, E.G. et al. Large-scale multi-omics analyses in Hispanic/Latino populations identify genes for cardiometabolic traits. Nat Commun 16, 3438 (2025). https://doi.org/10.1038/s41467-025-58574-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58574-z
This article is cited by
-
Elucidating the susceptibility genes between insomnia and migraine by integrating genetic data and transcriptomes
The Journal of Headache and Pain (2026)
-
Nutrigenomics meets multi-omics: integrating genetic, metabolic, and microbiome data for personalized nutrition strategies
Genes & Nutrition (2025)
