
Abstract
Blood-based extracellular RNA (cell-free RNA; exRNA) biomarkers require validated sample collection, processing, and quantification procedures. No study to date has systematically tested pre-analytical variables affecting transcriptome-wide exRNA analysis. By evaluating their impact on deep transcriptome profiling of microRNAs and mRNAs in blood plasma or serum, we compared ten blood collection tubes, three blood processing time intervals, and eight RNA purification methods. In addition, we assessed interactions among a selected pre-analytical variable set, resulting in 456 extracellular transcriptomes. Blood preservation tubes failed to stabilize exRNA and RNA purification methods differed significantly in performance, causing variations in concentration, detected gene numbers, replicability and observed transcriptome complexity. Critical interactions between tubes, purification methods and time intervals were identified. We provide 11 analytical performance metrics for exRNA quantification methods and put forward recommendations for both users and manufacturers of RNA purification methods and blood collection tubes, collectively, essential groundwork for exRNA-based precision medicine applications.
Similar content being viewed by others
Introduction
Biomarker studies increasingly utilize biofluids as an attractive source of molecules reflecting human health and disease states. Referred to as liquid biopsies, they have the advantage over tissue biopsies by being minimally invasive and compatible with longitudinal sample collection, enabling monitoring of the impact of treatments or other interventions over time. Most liquid biopsy biomarker studies focus on cell-free nucleic acids as candidate biomarkers. Cell-free DNA has been intensively studied and found its way into daily clinical practice for non-invasive prenatal testing1 and cancer-relevant mutation and methylation detection2, while extracellular RNA (exRNA) is relatively new to the biomarker field.
Particular molecules from various RNA classes, including microRNA (miRNA), messenger RNA (mRNA), long-noncoding RNA, and circular RNA (circRNA), have been put forward as potential biomarkers for cancers, autoimmune diseases, diabetes, and cardiovascular diseases3,4,5,6,7. However, few RNA-based biomarkers have been validated across multiple studies, due in part to differences in pre-analytical variables among studies. Furthermore, the absence of statements for adherence to best-practice standards or full reporting of pre-analytical variables in publications prevents biomarker study comparisons and replication of findings. Given the labile nature of RNA and the release of exRNA and cellular RNA by cells under stress8,9, standardized quantification is a necessity. Growing interest in exRNA as a biomarker resource requires strict implementation of standardized methods for sample collection, processing, and molecular profiling, to ensure that the biological signal of interest is not obscured by methodological variation. Blood serum and plasma are among the most studied liquid biopsies, and several pre-analytical variables, including blood collection tube type, needle type, and blood centrifugation speed and duration, have been reported to influence exRNA abundance patterns (Supplementary Data 1)10,11,12. However, studies investigating the impact of pre-analytical variables have either focused only on miRNAs or were restricted to targeted mRNA analysis by PCR-based measurement of a small number of genes (Supplementary Data 1). The impact of pre-analytical variables on the wide extracellular transcriptome remains largely underappreciated. Cases where one pre-analytical variable may affect exRNA levels depending on other variables (interactions among pre-analytical variables, Supplementary Data 1) has often been overlooked in studies or not been investigated in detail. Consortia founded to standardize pre-analytical variables, including the NIH Extracellular RNA Communication Consortium (ERCC)13,14, the Blood Profiling Atlas in Cancer (BloodPAC) Consortium15,16, SPIDIA/SPIDIA4P and CANCER-ID, have focused recommendations on cell-free DNA to date. The exRNA research community needs a more profound understanding of how pre-analytical variables impact results and specific recommendations for exRNA analysis.
Here we extensively assessed the impact of pre-analytical variables on both extracellular miRNA and mRNA profiles in a massively parallel sequencing-based study. We systematically evaluated ten blood collection tubes, three time intervals between blood draw and downstream processing, and eight RNA purification methods using the supplier-specified minimal and maximal input volumes. Impacts were assessed on deep transcriptome profiling of all miRNAs and mRNAs in healthy donor plasma and serum. More than 1.6 liters of blood was collected from 20 healthy donors to conduct experiments in triplicate or quintuplicate, resulting in 456 complete transcriptomes. To control RNA purification and library preparation workflows, 189 synthetic spike-in RNA molecules were used17,18. Two evaluation phases firmly established (1) the profound impact of each pre-analytical variable and (2) unknown interactions between pre-analytical variables. A wide variety of performance metrics, some of which are novel, were evaluated (Fig. 1), providing a comprehensive analysis of pre-analytical variables on exRNA from blood serum and plasma.

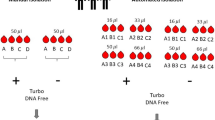
To evaluate the 8 exRNA purification methods (upper left panel), 2 blood draws from a single individual were performed to separately apply mRNA capture or miRNA sequencing. To compare RNA purification performance, 9 performance metrics were calculated. Blood was drawn from 9 individuals to evaluate 10 blood collection tube types, including 5 classic and 5 preservation tube types (upper right panel), at 3 time intervals between blood draw and processing. Preservation tubes were processed immediately (T0) and after 24 (T24) and 72 (T72) hours and classic tubes were processed immediately (T0) and after 4 (T04) and 16 (T16) hours. Both mRNA capture and miRNA sequencing were performed, and the data was analyzed using 5 performance metrics. Based on the number of miRNAs and mRNAs detected and replicate variability metrics, a dedicated selection of precise and sensitive exRNA purification methods and blood collection tubes was further evaluated in exRNAQC phase 2. For both mRNA capture and miRNA sequencing in phase 2, 5 individuals were sampled to test 3 blood collection tubes and 4 RNA purification methods. Interactions between RNA purification methods, blood collection tubes and processing time intervals were assessed by 6 performance metrics. MAP=MagNA Pure method, MAX=Maxwell method, MIR=miRNeasy method, MIRA=miRNeasy Advanced method, MIRV=mirVana method, MIRVE=mirVana method with purification protocol for small RNA enrichment, NOR=Norgen method, NUC=NucleoSpin method, QIA=QIAamp method. Designed with Freepik (free license) and Servier Medical Art (CC BY 4.0).
Results
RNA purification method influences extracellular miRNA and mRNA abundance profiles
To assess the impact of the RNA purification method on extracellular miRNA and mRNA profiles, 8 total RNA purification methods marketed for RNA purification from serum or plasma (Fig. 1) were selected for evaluation in phase 1 of the Extracellular RNA Quality Control (exRNAQC) study. Since most methods support a range of blood plasma input volumes, we tested the minimal and maximal input volumes recommended by each supplier. Blood was collected in EDTA tubes from a healthy donor, since this tube type is widely used in exRNA literature. Three technical replicates were used per condition, resulting in 45 samples processed for mRNA capture sequencing and 51 samples processed for miRNA sequencing. Residual DNA contamination was detected in the MagNA Pure method eluates (Supplementary information Fig. 1c). Hence, these data were excluded from further analyzes to ensure accurate exRNA quantification.
We calculated 9 purposely developed metrics for sequencing data to compare RNA purification method performance (Table 1, metrics described in Methods). The absolute number of mRNAs and miRNAs detected (also referred to as sensitivity) markedly differed among RNA purification methods and plasma input volumes (Fig. 2a, b). For a given RNA purification method, a higher number of mRNAs was consistently detected from the higher plasma input volume. This was not always true when different methods were compared, such as the miRNeasy Advanced method using 0.6 mL plasma versus the NucleoSpin method using 0.9 mL plasma (Fig. 2a). This finding also applied to miRNAs, excepting QIAamp, Norgen and NucleoSpin methods, which detected fewer miRNAs from the maximal plasma input volumes (Fig. 2b).

Performance metrics are shown for both mRNA capture (left panels) and miRNA (right panels) sequencing. For each unique RNA purification-plasma input volume combination, 3 technical replicates were analyzed (n = 39 for mRNA capture & n = 45 for miRNA sequencing). Absolute numbers of detected mRNAs (a) and miRNAs (b) that reached the count threshold (see “Methods”) are shown. High numbers indicate good performance. Endogenous mRNA (c) and miRNA (d) concentration. Values are log rescaled to the lowest mean of all methods and transformed back to linear scale. The mean and 95% confidence interval are shown. High concentrations indicate good performance. Replicate variability based on ALC at mRNA (e) and miRNA (f) level, respectively. Small ALC indicates good performance. Overview of all performance metrics at mRNA capture (g) and miRNA (h) sequencing level, respectively, after transforming the values to robust z-scores. High z-scores indicate good performance. Rows and columns of the heatmaps are clustered according to complete hierarchical clustering based on Euclidean distance. Average z refers to the mean of robust z-scores for a specific RNA purification method. The number that follows the name of the purification method is the plasma input volume (in ml). MAX=Maxwell method, MIR=miRNeasy method, MIRA=miRNeasy Advanced method, MIRV=mirVana method, MIRVE=mirVana method with purification protocol for small RNA enrichment, NOR=Norgen method, NUC=NucleoSpin method, QIA=QIAamp method.
Eluate RNA concentrations derived from the sequencing data correlated significantly with RNA concentrations determined by Femto Pulse electropherogram analysis (p-value < 0.001, Supplementary Figs. 2a, b). Femto Pulse analyzes also demonstrated that blood-derived exRNA was highly fragmented (Supplementary Fig. 3). RNA concentrations varied greatly among different purification methods, with a more pronounced impact on mRNA than miRNA (Fig. 2c, d). RNA concentration and yield (Supplementary Figs. 4e, f) depended on plasma input volume, with a higher input volume resulting in higher mRNA concentration and yield for a given RNA purification method. Purification methods, excepting QIAamp and Norgen, maintained this association for miRNAs (Fig. 2c, d). Although RNA purification methods with large eluate volumes (Norgen, mirVana and Maxwell) and methods with small eluate volumes (QIAamp, miRNeasy and miRNeasy Advanced) produced similar RNA yields for a given plasma input volume, methods with large eluate volumes typically resulted in lower RNA concentrations. Condensing the eluate volume prior to library preparation could potentially increase Norgen, mirVana and Maxwell method overall performance.
RNA purification efficiency (Supplementary Fig. 4g-h) is a relative measure of how well a method purifies RNA from a given plasma input volume. As expected, purification efficiency did not vary between the maximal and minimal input volumes for a given method. However, methods yielding high purification efficiency (for example mirVana method) did not always produce better RNA quantification results because of limited biofluid input volumes. If some methods would accommodate a larger biofluid input volume and/or enable a smaller eluate volume (Supplementary Information) while maintaining a high purification efficiency, manufacturers could dramatically increase the eluate RNA concentrations of their methods.
We determined a count threshold for each purification method to filter noisy data (Supplementary Figs. 4a, b & Data 2), and calculated the percentage of counts remaining (data retention, Supplementary Figs. 4c, d). Count thresholds for miRNA data were lower than those for mRNA data, resulting in higher data retention levels for miRNA analysis. This indicated higher variability in mRNA quantification compared to miRNA quantification, which was unsurprising since most exRNA purification methods were specifically developed for miRNA detection. Variability between RNA purification replicates was quantified to determine method reproducibility. Most methods performed equally well with respect to variability in miRNA count replicates (Fig. 2f), except the mirVana alternative protocol using 0.1 mL plasma input that showed higher replicate variability. For mRNA, the Norgen method using 0.25 mL plasma input and mirVana method using 0.1 mL plasma input displayed higher variability in replicates than the other methods tested (Fig. 2e). The maximal plasma input volume for any given method consistently produced less replicate variability for mRNA than the minimal plasma input volume.
We determined the average read duplication rate (Supplementary Fig. 1a) and transcriptome coverage in the mRNA capture sequencing data (Supplementary Fig. 1a). Biofluid-derived mRNA capture sequencing libraries typically have a high fraction of PCR duplicates, due to low RNA input amounts, but even small differences in duplication rate can strongly impact the total number of non-duplicated reads. For example, the purification method with the lowest duplication rate (82.2% for QIAamp purification with 4 mL plasma input) generated on average 6-fold more non-duplicated reads than the method with the highest duplication rate (97.3% for NucleoSpin purification using 0.3 mL plasma input; Supplementary Data 3).
Transcriptome coverage was determined to assess diversity in mRNA capture sequencing reads, and demonstrated substantial differences among RNA purification methods and among plasma input volumes. Transcriptome coverage was higher for any given method when maximal plasma input volumes were used (compared with minimal plasma input volumes, Supplementary Fig. 1b). Overall performance of all methods in purifying mRNA or miRNAs was calculated using robust z-score transformation of all performance metrics, providing summary plots that compare all methods tested for these 2 analysis levels (Fig. 2g, h). In general, higher plasma input volumes and lower eluate volumes usually resulted in better performance. A narrower z-score range was also observed across all methods for quantifying miRNAs, indicating less pronounced differences among RNA purification methods for miRNA analysis.
Classic blood collection tubes outperform preservation tubes for extracellular mRNA and miRNA analysis
We also evaluated various blood collection tubes and processing time intervals in exRNAQC phase 1 as possible pre-analytical variables impacting extracellular mRNA and miRNA profiles. Ten blood collection tubes were selected, including 5 classic tubes, not specifically designed to stabilize cell-free nucleic acids, and 5 manufacturer-designated preservation tubes that were purposely developed to conveniently allow more time between the blood draw and further processing steps (Fig. 1). We recruited 3 healthy volunteers and selected 3 time intervals between blood draw and processing to assess whether blood storage at room temperature produces changes in sample exRNA content (Fig. 1). For each tube type, a baseline value was established by processing the blood tube immediately after collection. To mimic same-day and next-day processing in routine lab situations, we set processing time intervals to 4 and 16 hours for classic tube types. For preservation tubes specifically marketed to stabilize extracellular nucleic acids for 7 (and up to 14) days, extended time intervals (24 and 72 hours) for plasma preparation were selected. This testing design resulted in 180 biofluid samples subsequently processed for RNA purification (using the widely used miRNeasy method with 0.2 mL biofluid input) and both mRNA capture and miRNA sequencing.
To evaluate exRNA profiles for different blood collection tubes and processing times, we calculated 5 different performance metrics (Table 1, metrics described in Methods). The stability of each performance metric over time was evaluated as fold-change between immediate processing and the 2 selected processing time intervals (illustrated in Van Paemel et al.19). Processing time intervals having no impact on the performance metric would have fold-changes close to one. Hemolysis was quantified based on absorbance units at 414 nm and evaluated by visual inspection during liquid biopsy preparation. Hemolysis in classic tube types was below the generally accepted absorbance threshold of 0.220,21 across all donors and time intervals (Supplementary Figs. 5a, 6a & 7). Contrastingly, plasma was hemolytic for at least one donor at one or multiple time points for all preservation tube types. While absolute absorbance units were generally low, longer processing time intervals did produce up to 2-fold differences in both classic and preservation tube types (Supplementary Figs. 8a, 9a). To assess RNA concentration differences in plasma/serum prepared from different blood collection tubes, performance metrics based on spike-in RNA read counts were calculated. RNA concentration remained stable over time in classic tube types (Supplementary Figs. 8b, 9b). Unexpectedly, RNA concentration was much less stable in preservation tubes, measuring the lowest levels in the RNA Streck tube (Supplementary Figs. 5b & 6b). The absolute mRNA and miRNA numbers in classic tubes remained relatively constant over time, but mean fold-changes in preservation tubes ranged from 1.86 to 4.01 (mRNA) and from 1.08 to 1.67 (miRNA, Supplementary Figs. 8c, 9c). The number of mRNAs and miRNAs detected in DNA Streck and RNA Streck tubes was substantially lower than in all other tubes (Supplementary Figs. 5c,6c). In contrast to preservation tubes, the fraction of total counts mapping to mRNAs and miRNAs (Supplementary Figs. 5d, 6d) in classic tubes remained fairly constant over time (Supplementary Figs. 8e, 9e). Replicate variability for preservation and classic tubes remained stable over time (Supplementary Figs. 8d, 9d). Clearly, preservation tubes did not show robust performance over time (summary plot for all performance metrics in Fig. 3). We conclude that the tested preservation tubes do not robustly preserve the total miRNA or mRNA quantities in plasma, and are not suited for exRNA analysis.

Per blood collection tube and per performance metric, a summary of mean fold-changes (FC) between immediate processing and the 2 selected processing time intervals is given for both mRNA (a) and miRNA (b) profiling. Ideally, the mean FC of the performance metrics approaches 1, indicating that there is little change over time. Per tube type (preservation and classic) tubes are ranked by mean value across all metrics from low (top) to high (bottom). Note that different donors were sampled and that tubes were processed at different time intervals for preservation and classic blood tubes.
RNA changes during standing time are selective for certain genes and dependent on blood collection tube type
To further characterize the observed changes in exRNA over time, we evaluated the stability of the circRNA and linear RNA fractions in each tube type after standing 4 or 16 hours (for the classic tubes) or 24 or 72 hours (for the preservation tubes). We hypothesized that the higher stability conferred by circularization would translate to a difference in abundance between these fractions over time, but this was not confirmed. Linear RNA and circRNA fractions did not significantly differ across time intervals (all adjusted p-values > 0.05; Supplementary Fig. 10). To assess how the mRNA transcript population changed over time, distributions of log2 fold-change differences between 4 or 16 hours and T0 (for classic tubes) or between 24 or 72 hours and T0 (for preservation tubes) were compared and gene set enrichment analysis (GSEA) performed. Log2 fold-change differences were higher at more prolonged standing times for both preservation and classic tubes, indicating that the abundance of a considerable number of genes changed over time, even for the high-performing classic tubes (Supplementary Fig. 12a). Nevertheless, log2 fold-change differences were higher for preservation than classic tubes. GSEA demonstrated that only EDTA and citrate tubes showed no significantly enriched gene sets after standing 4 hours (Supplementary Data 4). We also evaluated mRNA repertoire differences across blood collection tube types at the baseline time point, confirming the transcriptome differences detected by our performance metrics (Supplementary Fig. 11). Computational deconvolution revealed tube-dependent changes in RNA proportions from several immune cell types over time (Fig. 4, Supplementary Fig. 13). The estimated immune cell composition remained relatively stable in serum, citrate and ACD-A tubes that stood either 4 or 16 hours before processing. Remarkably, only citrate and ACD-A tubes showed no significant changes after standing 4 hours. Combining the findings of the GSEA and deconvolution analysis, we conclude that citrate tubes processed within 4 hours are best suited for exRNA analysis.

Colored cells represent adjusted p-values < 0.05 from beta regression models with random effects for all cell types. Tukey's method was used for pairwise comparisons (two-sided testing) while correcting for multiple testing. P-values smaller than the minimum representable value in R (1e-16) are annotated as < 1e-16. Non-significant p-values point towards blood collection tube stability over time. T0=immediate blood processing, T04, T16, T24, T72=plasma prepared 4, 16, 24 and 72 hours after blood draw, respectively. Note that different donors were sampled and that tubes were processed at different time intervals for preservation and classic tubes.
Interactions among pre-analytical variables impact exRNA quantification performance
In the second exRNAQC study phase, we evaluated whether the impact of a certain pre-analytical variable on exRNA sequencing outcome depends on other pre-analytical variables. Three classic blood collection tubes and 2 RNA purification methods were selected for this evaluation. Tube selection was based on superior performance in phase 1 and widespread clinical use (Fig. 1). RNA purification method selection was based on the number of detected m(i)RNAs (Fig. 2a, b) and replicate variability (Fig. 2e, f) from phase 1 (Fig. 5). Plasma input volume was used as an additional selection criterion, as we aimed to include at least one method requiring < 1 mL biofluid. Because purification methods performed differently for mRNAs and miRNAs (Fig. 5), different methods were selected to probe the mRNA and miRNA transcriptomes (Fig. 1). Blood was drawn from 5 healthy volunteers and processed immediately or after 4 or 16 hours, resulting in 180 samples processed for RNA purification and both mRNA capture and miRNA sequencing. Interactions were analyzed using six relevant performance metrics (Table 1, metrics described in Methods). For both mRNA capture and miRNA sequencing, several significant two-way interactions between the blood collection tube and RNA purification method or time interval were observed (Figs. 6a, b, Supplementary Figs. 14, 15). As expected, no significant interactions between RNA purification method and time interval were observed. Interactions between the blood collection tube type and RNA purification method impacted the duplication rate and number of detected mRNAs (Figs. 6a, Supplementary Fig. 14). RNA concentration and purification efficiency for mRNA was influenced by interactions between blood collection tubes and time intervals (Fig. 6a, Supplementary Fig. 14). We also analyzed mRNA abundance and GSEA across time intervals for phase 2 testing, and confirmed results from phase 1. Higher log2 fold-changes were observed for more prolonged standing times, for the selected classic tube types (Supplementary Fig. 12b & Data 4) except for the serum tube combination with the QIAamp method. Individual differential genes were only detected for the 16-hour interval compared to immediate processing, with significant abundance differences detected for 0.11–4,19% of the mRNA transcripts, while differential gene sets were observed for both the 4-hour and 16-hour interval compared to immediate processing (Supplementary Data 4). These analyses confirm that mRNA composition changes over time, even in high-performance blood collection tubes. More significant interactions affected performance metrics for miRNA sequencing than mRNA capture sequencing. The choice of RNA purification method altered performance of citrate, EDTA or serum tubes by significantly impacting purification efficiency, the number of detected miRNAs, reproducibility and miRNA/mRNA fraction (Figs. 6b, Supplementary Fig. 15). The time intervals differentially affected the RNA concentration, purification efficiency, number of detected miRNAs and miRNA/mRNA fraction performance metrics in citrate, EDTA or serum tubes (Figs. 6b, Supplementary Fig. 15). These interactions demonstrate that the impact of a certain pre-analytical variable on exRNA profiling depends on other pre-analytical variables.

Median robust z-score (see “Methods”) per method-input volume combination (13 for mRNA, 15 for miRNA) shown for the number of detected mRNAs (a) or miRNAs (b) and replicate variability metrics. The number in the labels is the plasma input volume (in ml). MAX=Maxwell method, MIR=miRNeasy method, MIRA=miRNeasy Advanced method, MIRV=mirVana method, MIRVE=mirVana method with purification protocol for small RNA enrichment, NOR=Norgen method, NUC=NucleoSpin method, QIA=QIAamp method.

For both mRNA capture and miRNA sequencing, 5 biological replicates were used for each of the 18 unique tube (n=3), purification method (n = 2) and time interval (n=3) combinations (total n=90). Shown are the interactions between pre-analytical variables for mRNA capture (a) and miRNA (b) sequencing. P-values correspond to the Wald test for the terms in the linear mixed-effects model (two-sided testing).
Recommendations for users and manufacturers
We distilled recommendations from our exRNAQC study findings that should help users to select appropriate pre-analytical variables for their specific research question (Fig. 7). Sample collection and processing should be standardized within a single study. Combining data derived from different blood collection tube types, RNA purification methods and/or processing time intervals will introduce unacceptable biases in the results. The pre-analytical variables used should also be carefully annotated and reported (according to published guidelines16,22,23,24,25) to enable study comparison and interpretation across different studies. The use of synthetic spike-in RNA allows sequencing-based quality control and optional exRNA normalization17,18. We used spike-in RNAs to assess RNA concentration and yield and to determine purification efficiency from different methods. The available biofluid volume largely determines which RNA purification method to use. When sufficient biofluid is available, we recommend maximizing the biofluid input volume to increase quantification performance. To maximize the number of detected mRNAs and minimize replicate variability, we recommend the QIAamp method for large biofluid input volumes (up to 4 mL) and the miRNeasy or miRNeasy Advanced method for small volumes (0.2-0.6 mL). The Maxwell or miRNeasy Advanced method should be used to obtain the maximal number of detected miRNAs and to minimize replicate variability. We advise using citrate blood collection tubes for exRNA sequencing, based on our performance metrics results and the gene set enrichment and deconvolution analyses. Blood processing should be completed within 4 hours after collection. The use of preservation tubes (RNA Streck, DNA Streck, Biomatrica, Roche, and PAXgene tubes) for blood collection should be avoided based on our phase 1 results (mRNA and miRNA unstable over time).

The left panel summarizes the exRNAQC recommendations for users. The right panel describes the exRNAQC recommendations for manufacturers of RNA purification methods and blood collection tubes. gDNA=genomic DNA, MAX=Maxwell method, miRNA=microRNA, MIR=miRNeasy method, MIRA=miRNeasy Advanced method, mRNA=messenger RNA, QIA=QIAamp method. Designed with Freepik (free license).
Our collected recommendations for manufacturers of RNA purification methods and blood collection tubes, based on our exRNAQC study findings, support how to evaluate newly developed products (Fig. 7). For newly developed RNA purification methods, developers or manufacturers should first evaluate their compatibility with different genomic DNA removal methods (Fig. 7)26, then assess potential performance differences using different biofluid input volumes, ideally using all 9 proposed exRNAQC performance metrics. Given that performance for miRNA quantification cannot be extrapolated to mRNA quantification, we recommend evaluating both independently. Importantly, RNA purification replicates should be included and different blood collection tubes tested to evaluate potential interactions and specify which collection tubes are recommended or should be avoided in combination with the new RNA purification method. It is crucially important for blood collection tube manufacturers to evaluate different processing time intervals to assess performance stability over time in comparison to immediate processing using the 5 exRNAQC performance metrics. Blood collection tube replicates should also be included and interaction analyses should be evaluated. Collectively, our recommendations help to set up comprehensive user guidelines for extracellular RNA evaluation and marker studies in patient samples.
Discussion
We present our comprehensive evaluation of extracellular RNA quality control in the exRNAQC study, which examined eight RNA purification methods, ten blood collection tubes, and three time intervals for blood processing as important pre-analytical variables dramatically affecting extracellular RNA quantification and analysis. Using purposely defined performance metrics, we found marked differences among RNA purification methods and demonstrated that classic blood collection tubes outperform manufacturer-designated preservation tubes. Selective changes in gene transcript abundance and the immune cell-derived RNA subpopulation during longer blood processing times indicate the need for timely blood processing into cell-free plasma, optimally in ≤ 4 hours. Importantly, interactions between pre-analytical variables further impact performance of RNA purification methods and blood collection tubes. We provide user and manufacturer recommendations to improve quality control in extracellular RNA studies from patient blood samples based on our findings (Fig. 7).
The exRNAQC study is the largest and most comprehensive sequencing-based evaluation of pre-analytical factors affecting extracellular transcriptomes to date. However, all experiments were performed in a single laboratory, and ideally, the present findings should be confirmed in a multicenter study. The exRNAQC study stands out in many aspects (Fig. 8). While most previous studies have only focused on the miRNA biotype12,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43, the exRNAQC study characterized the entire human miRNA and mRNA transcriptome. For comparisons to previous studies, the miRNA biotype was our focus from the small RNA sequencing data in exRNAQC, although other types of small RNAs are present in these raw data. Two miRNA sequencing studies compared different RNA purification methods27,30, but the impact of different blood collection tubes on the circulating miRNome was previously unexplored. Liquid biopsy collection and processing procedures were tightly controlled in exRNAQC, however, we cannot exclude that other biological and methodological pre-analytical variables had a potential impact. Wong et al. have demonstrated that the method selected for library preparation greatly affects the plasma miRNA profile30. Srinivasan et al. showed that different exRNA carrier subclasses are associated with distinct sets of miRNAs from plasma and serum, and are differentially purified by different exRNA purification methods27. It should be noted, however, that most RNA purification methods evaluated in their study specifically enrich for extracellular vesicles. From that perspective, it is not surprising to observe differences in RNA purification performance, as these differences may largely be caused by well-known differences in the purity of extracellular vesicle isolation methods44. We selected for exRNAQC only purification methods for exRNA from total plasma or serum, which in principle, should capture all circulating RNAs (including those from various carriers such as vesicles and lipoprotein and ribonucleoprotein complexes). Nevertheless, exRNAQC revealed marked performance differences for these methods, suggesting that different methods may differentially purify different exRNA carrier subclasses (e.g., extracellular vesicles, etc.). Previous studies examined the impact of pre-analytical variables on only a handful of extracellular mRNAs10,45,46, while full transcriptome mRNA characterization was applied in exRNAQC to reveal the full scope of potential exRNA population variability created by the pre-analytical variables (Fig. 8). Importantly, we demonstrate higher variability in mRNA sequencing data compared to miRNA sequencing data, resulting in RNA biotype-specific recommendations of RNA purification methods (Fig. 7). This underscores the need to include sequencing-based methods for mRNA analysis in the evaluation of pre-analytical variables affecting exRNA.

The heatmap shows studies (raw data and references in Supplementary Data 1) evaluating pre-analytical variables based on both mRNA and miRNA (top part), miRNA only (middle part) and mRNA only (bottom part). The numbers in brackets after the column names indicate the scale range. The darker the coloring, the more items were studied. Studies marked with a solid black circle evaluate interactions between pre-analytical variables. The exRNAQC study outperforms previous studies analyzing pre-analytical variables impacting exRNA analyzes in terms of the combination of evaluated metrics and shows uniqueness by studying the impact on both miRNA and mRNA.
Inefficient genomic DNA removal has previously been shown to bias sequencing-based exRNA studies47,48. Residual genomic DNA contaminated the MagNA Pure RNA eluates in exRNAQC phase 1, despite applying a genomic DNA removal strategy that worked well for all other RNA purification methods. Contamination was likely due to incompatibility between the RNA elution buffer and genomic DNA removal reagents. We excluded data from the MagNA Pure method from mRNA analysis, and emphasize here the importance of assessing successful genomic DNA removal prior to exRNA quantification.
Ten different blood collection tube types were extensively evaluated in exRNAQC. Previous reports were limited to maximally six tube types and did not assess26,27,32,34,35,36,38,45, or used only targeted PCR to asses12,31,37,42,46, performance stability over different processing times (Fig. 8). We confirmed the previous report that preservation tubes result in varying volumes of plasma that can be prepared49. Surprisingly, we also demonstrate that manufacturer-designated preservation tubes were less able to preserve extracellular mRNA and miRNA profiles than classic (including serum) tubes. This finding is particularly worrying in light of the Technical Specification (CEN/TS17742:2022) from the European Committee for Standardization recommending the use of stabilizing blood collection tubes, especially for long-term biobanking sample collection, to prevent post-collection profile changes50. The theory being that upon storage and transport of venous whole blood collected in classic tubes, blood cells may shed (vesicle-encapsulated) RNA or undergo apoptosis or mechanical lysis, releasing cell-free RNA, leading to unreliable or wrong results. In support of this theory, a previous study demonstrated a clear increase in ACTB RNA copies measured by RT-qPCR in plasma prepared after 24 or 72 hours from blood collected in EDTA tubes compared with preservation tubes (T24: 5-fold versus 2-fold increase, T72: 45-fold versus 3-fold increase, respectively)50. Similarly, a second study showed copy numbers of ACTB and several other gene transcripts significantly increased in plasma processed three days after blood collection in EDTA tubes, compared to RNA Streck tubes51, suggesting that EDTA tubes prevent clotting but not changes in the exRNA profile. CEN/TS17742:2022, however, does not specify which preservation tube was tested, and their use of RT-qPCR to assess only one or a few gene transcripts after 24- or 72-hour processing times does not comprehensively test tube performance. The global view provided by exRNAQC results includes differential abundance of all mRNAs and miRNAs for a complete picture of how profiles in classic tubes change between direct processing and 4- or 16-hour delays in processing. CEN/TS17742:2022 favors EDTA above other classic blood collection tubes, based on the study by Glinge et al. measuring abundance changes of three miRNAs over time to compare EDTA, citrate, lithium-heparin and serum tubes37. However, Glinge et al. concluded only that lithium-heparin tubes should be avoided for plasma collection and otherwise detected no significant differences in miRNA abundance in plasma from EDTA, citrate or serum tubes. Glinge et al. also claimed that variability is larger in the results from serum compared to EDTA tubes, but no data are shown to support this37. Based on the hypothesis that RNA may be released from blood cells during the clotting process in serum tubes, CEN/TS17742:2022 recommends avoiding serum tubes unless verified and validated for the intended examination. Our GSEA results from serum and EDTA tubes point towards platelet activation in blood samples before processing, providing limited support for this theory. However, platelet activation-mediated exRNA release could not be compared among blood collection tubes since the EPIC deconvolution tool has no signatures for this blood fraction52. No major differences in the exRNAQC performance metrics were identified between serum tubes and EDTA, EDTA separator, citrate and ACD-A tubes, not supporting the CEN/TS17742:2022 recommendation. Only exRNAQC pathway enrichment and cellular deconvolution analyses detected differences among classic tube types, favoring the citrate tube processed within 4 hours for stable exRNA analysis. Some caution is warranted comparing classic tube results from our exRNAQC study and CEN/TS17742:202250, since different processing times were examined. Nevertheless, based on exRNAQC results, we strongly advise to critically re-evaluate CEN/TS17742:2022. Additional preservation tube types have also been developed and marketed since initiating exRNAQC, including the cfDNA/cfRNA Preservation Blood Tube (Zymo Research, R1075), the RNA Complete BCT (Streck, 230579), and cf-DNA/cf-RNA Preservative Tube (Norgen Biotek Corp., 63980). Our posthoc evaluation using miRNA sequencing (Streck RNA Complete BCT) and mRNA capture sequencing (Norgen cf-DNA/cf-RNA Preservative Tube) demonstrated poor performance in all evaluated performance metrics. Delays in manufacturing prevented delivery (email communication) and testing of the Zymo cfDNA/cfRNA Preservation Blood Tube. We invite blood collection tube manufacturers to increase their efforts to develop a plasma or serum tube that preserves the extracellular transcriptome for at least three days.
Our study revealed multiple interactions between pre-analytical variables, impacting both mRNA and miRNA profiling. Interactions between blood collection tubes and RNA purification methods are not unexpected, given that coatings, preservatives or anticoagulants on blood collection tubes may induce changes in RNA purification conditions (specific monovalent or divalent ions, salt concentrations, pH values) that impact performance53. Importantly, the presence of these interactions demonstrates that one should not simply combine the best-performing blood collection tube and RNA purification method (from single-factor studies), but test compatibility for these pre-analytical variables when optimizing a specific sample processing workflow. This is in concordance with CEN/TS17742:2022 recommendations to use RNA purification methods recommended by the tube manufacturer, if mentioned (after verification for the intended use)50. Our comprehensive study provides guidance to select the optimal blood collection tube type, maximal processing time and RNA purification method combination for sequencing projects targeting extracellular miRNA and/or mRNAs (Fig. 8). While previous studies demonstrated that longer blood processing times altered abundance of specific mRNA or miRNAs12,37,46, transcriptome-wide interactions between blood collection tubes and time intervals were not previously addressed. Our findings confirm that standardizing the blood processing time remains crucially important even for the best-performing tubes.
The exRNAQC study represents the most comprehensive performance assessment of RNA purification methods and blood collection tubes and processing times for exRNA profiling to date. By evaluating 11 performance metrics, we show that choice of RNA purification method and blood collection tube dramatically impacts quantification of both mRNAs and miRNAs. We also demonstrate that classic blood collection tubes outperform preservation tubes and that the transcript population varies at times equivalent to overnight blood processing, potentially creating artifacts that could be mistaken as biomarkers. Interactions between RNA purification methods, blood collection tubes and processing times further indicate that compatibility of these pre-analytical variables need to be evaluated when biobanking samples.The exRNAQC study applies a comprehensive framework and metrics that can be used to evaluate performance of more recently developed commercial components for exRNA studies, and we provide recommendations to guide users planning exRNA studies and manufacturers evaluating newly developed products. Our results will enhance the reproducibility, interpretation and comparison of future exRNA studies to support exRNA research as a starting point for robust biofluid-based biomarker discovery and use.
Methods
Donor material and liquid biopsy preparation
The study was approved by the ethics committee of Ghent University Hospital (Belgian Registration number B670201733701) and written informed consent was obtained from 20 healthy donors, including 5 males and 15 females (age ranges from 27 to 54 years old; Supplementary Data 5). Sex and gender were not considered in the study design. Incapacitated or pregnant individuals, as well as individuals younger than 20 years old were excluded from the study. Donors did not receive compensation for study participation. Venous blood was collected from an elbow vein after disinfection with 2% chlorhexidine in 70% alcohol. In total, ten different blood collection tubes were used: the BD Vacutainer SST II Advance Tube (referred to as serum tube in this study; Becton Dickinson and Company, 366444), BD Vacutainer Plastic K2EDTA tube (EDTA tube; Becton Dickinson and Company, 367525), Vacuette Tube 8 mL K2E K2EDTA Separator (EDTA separator tube; Greiner Bio-One, 455040), BD Vacutainer Glass ACD Solution A tube (ACD-A tube; Becton Dickinson and Company, 366645), Vacuette Tube 9 mL 9NC Coagulation sodium citrate 3.2% (citrate tube; Greiner Bio-One, 455322), Cell-Free RNA BCT (RNA Streck tube; Streck, 230248), Cell-Free DNA BCT (DNA Streck tube; Streck, 218996), PAXgene Blood ccfDNA Tube (PAXgene tube; Qiagen, 768115), Cell-Free DNA Collection Tube (Roche tube; Roche, 07785666001), and LBgard Blood Tube (Biomatrica tube; Biomatrica, M68021-001). Immediately after blood draw, blood collection tubes were inverted five times and all tubes were transported to the laboratory for plasma or serum preparation. Tubes were immediately processed or at 4h, 16h, 24h or 72h upon blood collection. Details on the different blood draws and plasma/serum preparations are available in the Supplementary Information.
RNA isolation and gDNA removal
In total, eight different exRNA purification methods, including six spin column-based methods and two automated purification procedures, were used according to the manufacturer’s manual: the miRNeasy Serum/Plasma Kit (referred to as the miRNeasy method in this study; Qiagen, 217184), miRNeasy Serum/Plasma Advanced Kit (miRNeasy Advanced method; Qiagen, 217204), mirVana PARIS Kit (mirVana method; Life Technologies, AM1556), NucleoSpin miRNA Plasma Kit (NucleoSpin method; Macherey-Nagel, 740981.50), QIAamp ccfDNA/RNA Kit (QIAamp method; Qiagen, 55184), Plasma/Serum Circulating and Exosomal RNA Purification Kit/Slurry Format (Norgen method; Norgen Biotek Corp., 42800), Maxwell RSC miRNA Plasma and Serum Kit (Promega, AX5740 and AS1680) in combination with the Maxwell RSC Instrument (Maxwell method; Promega, AS4500), and MagNA Pure 24 Total NA Isolation Kit (Roche, 07658036001) in combination with the MagNA Pure 24 instrument (MagNA Pure method; Roche, 07290519001). Per 100 µL liquid biopsy input volume, 1 µL Sequin spike-in controls (Garvan Institute of Medical Research54) and/or 1 µL RNA purification Control (RC) spike-ins55 (IDT) were added to the lysate for TruSeq RNA Exome Library Prep sequencing and/or TruSeq Small RNA Library Prep sequencing, respectively (see Supplementary Information for concentrations). To maximally concentrate the RNA eluate, minimal eluate volumes were used, unless otherwise recommended by the manufacturer. For evaluation of the different purification methods in exRNAQC phase 1, both the minimal and maximal recommended plasma input volumes were tested in triplicate. Details on the exRNA purification methods, and Sequin and RC spike-in controls are available in the Supplementary Information.
gDNA removal of RNA samples for TruSeq RNA Exome Library Prep sequencing was performed using HL-dsDNase (ArcticZymes, 70800-202) and Heat & Run 10X Reaction Buffer (ArcticZymes, 66001). Briefly, 2 µL External RNA Control Consortium (ERCC) spike-in controls (ThermoFisher Scientific, 4456740), 1 µL HL-dsDNase and 1.4 µL reaction buffer were added to 12 µL RNA eluate, and incubated for 10 min at 37 °C, followed by 5 min at 55 °C. To RNA samples used for both TruSeq RNA Exome Library Prep sequencing and TruSeq Small RNA Library Prep sequencing, also 2 µL Library Preparation Control (LP) spike-ins56 (IDT) were added to the RNA eluate before starting gDNA removal and 1.6 µL reaction buffer was used. RNA samples solely used for TruSeq Small RNA Library Prep sequencing were not DNase treated. Here, 2 µL LP spike-ins were added to 12 µL RNA eluate before starting library preparation. Details on ERCC and LP spike-in control concentrations are available in the Supplementary Information.
mRNA capture sequencing
mRNA libraries were prepared starting from 8.5 µL RNA eluate using the TruSeq RNA Exome Kit (Illumina, 20020189, 20020490, 20020492, 20020493, 20020183), according to the manufacturer’s protocol with following adaptations: fragmentation of RNA for 2 min at 94 °C, second strand cDNA synthesis for 30 minutes at 16 °C (with the thermal cycler lid pre-heated at 40 °C), and second PCR amplification using 14 PCR cycles. Upon the first and second PCR amplification, libraries were validated on a Fragment Analyzer (Advanced Analytical Technologies), using 1 µL of library. Library concentrations were determined using Fragment Analyzer software for smear analysis in the 160 to 700 base pair (bp) range. Library quantification was qPCR-based, using the KAPA Library Quantification Kit (Roche, 07960140001), and/or based on NanoDrop 1000 measurements. Further details on the library preparation and quantification protocol are described in Hulstaert et al.18 For evaluation of the different RNA purification methods, 45 libraries were pooled on replicate level at 4 nM, yielding three pools of 15 samples, quality controlled using the KAPA Library Quantification Kit, and sequenced on a NextSeq 500 instrument (NextSeq 500/550 High Output Kit v2.5 (Illumina, 20024907, PE 2 x 75 cycles)). Loading concentrations of the three pools ranged from 2.1 pM to 2.3 pM. Percentage PhiX was 3%. For evaluation of the different blood collection tubes, all 90 libraries were pooled at 1.5 nM or the highest possible concentration, quality controlled using the KAPA Library Quantification Kit, and sequenced on a NovaSeq 6000 instrument (NovaSeq 6000 S2 Reagent Kit (Illumina, 20012861, PE 2 x 75 cycles)). Loading concentration of the pool was 324 pM. Percentage PhiX was 1 %. For exRNAQC phase 2, 90 libraries were pooled at 5.50 nM, quality controlled using the KAPA Library Quantification Kit, and sequenced on a SP100 flow cell (Illumina, NovaSeq 6000, 20027464). Loading concentration of the pool is 340 pM. Differences in read distribution across samples were subsequently used to re-pool individual libraries in order to obtain an equimolar pool. Subsequently, samples were sequenced on a S2 flow cell, at a loading concentration of 360 pM.
miRNA sequencing
Small RNA libraries were prepared starting from 5 µL RNA eluate using the TruSeq Small RNA Library Prep Kit (Illumina, RS-200-0012, RS-200-0024, RS-200-0036, RS-200-0048), according to the manufacturer’s protocol with following adaptations: the RNA 3’ adapter (RA3) and the RNA 5’ adapter (RA5) were 4-fold diluted with RNase-free water, and the number of PCR cycles was increased to 1617,57. For phase 1, samples were divided across library prep batches according to index availability. For each batch, 3 µL of small RNA library from each sample was pooled prior to automated size selection using the Pippin prep (Sage Sciences, CDH3050). Size selected libraries were quantified using qPCR, and sequenced on a MO flow cell (Illumina, NextSeq 500, 20024904) using loading concentrations ranging from 1.2 to 2.4 pM. Differences in read distribution across samples were subsequently used to re-pool individual libraries in order to obtain an equimolar pool. After size selection on a Pippin prep and qPCR quantification, these pools were sequenced on a HO flow cell (Illumina, NextSeq 500, NextSeq 500/550 High Output Kit v2.5, 20024907) using loading concentrations ranging from 1.2 to 3 pM. For phase 2, individual libraries were quantified using qPCR and pooled equimolarly across 2 pools. After size selection on a Pippin prep and qPCR quantification, library pools were sequenced on a NovaSeq 6000 SP100 flow cell with Xp workflow (Illumina, 20028401, 20043130) using a loading concentration of 270 nM.
Data analysis
In total, 456 transcriptomes were profiled and analyzed. The raw, processed and metadata were submitted to the European Genome-Phenome Archive (EGA), ArrayExpress and R2 Genomics Analysis and Visualization Platform (see Data Availability). A high-level summary of the sequencing statistics can be found in Supplementary Data 3 and Supplementary Data 6-10. Detailed pre-analytical variable information (for the BRISQ elements22,23) can be found in Supplementary Data 5.
Quality control and quantification of mRNA capture sequencing data
In case of adapter contamination indicated by FASTQC (v0.11.8; www.bioinformatics.babraham.ac.uk/projects/fastqc), adapters were trimmed with Cutadapt58 (v1.18; 3’ adapter R1: AGATCGGAAGAGCACACGTCTGAACTCCAGTCA; 3’ adapter R2 AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT). Only reads with ≥ 99% accuracy in at least 80% of bases of both mates were kept. Subsequently, FASTQ files were subsampled with Seqtk (v1.3; https://github.com/lh3/seqtk) to the lowest number of reads pairs obtained in the experiment. RNA sequencing data from exRNA samples is characterized by a high number of duplicates (Supplementary Data 3 and Supplementary Data 7), driven by the low amount of RNA in the library preparation. To improve RNA-sequencing data reproducibility (Supplementary Fig. 16), we removed these duplicates using Clumpify dedupe from BBMap (v38.26; www.sourceforge.net/projects/bbmap) with the following specifications: paired-end mode, 2 substitutions allowed, kmersize of 31, and 20 passes. For duplicate removal, only the first 60 bases of both reads were considered to account for the sequencing quality drop at the end of the reads. Strand-specific transcript-level quantification of the deduplicated FASTQ files was performed with Kallisto59 (v0.44.0). For coverage and strandedness analysis, mapped reads were obtained by STAR60 (v2.6.0c) using the default parameters (except for --twopassMode Basic, --outFilterMatchNmin 20 and --outSAMprimaryFlag AllBestScore). For all exons coverage information was retrieved by the genomeCoverageBed and intersectBed functions of BEDTools61 (v2.27.1). Strandedness information was obtained with RSeQC62 (v2.6.4). The reference files for all analyses were based on genome build hg38 (www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26) and transcriptome build Ensembl v9163. Spike annotations were added to both genome and transcriptome files.
Quality control and quantification of miRNA sequencing data
First, adapter trimming (3’ adapter: TGGAATTCTCGGGTGCCAAGG) was performed using Cutadapt (v1.16) with a maximum error rate of 0.15 and discarding reads shorter than 15 bp and those in which no adapter was found. Subsequently, low quality reads were filtered out (reads with ≥ Q20 in at least 80% of bases were kept) by FASTX-Toolkit (v0.0.14; http://hannonlab.cshl.edu/fastx_toolkit/index.html). Filtered FASTQ files were subsampled to the minimal number of reads in the experiment (Supplementary Data 6 and Supplementary Data 8) using Seqtk (v1.3). Reads were collapsed with FASTX-Toolkit and LP and RC spike reads (including possible fragments) were annotated. The non-spike reads were mapped with Bowtie64 (v1.2.2, with additional parameters -k 10 -n 1 -l 25) considering only perfect matches. Mapped reads were annotated by matching the genomic coordinates of each read with genomic locations of miRNAs (obtained from miRBase65,66,67,68,69,70, v22) and other small RNAs (tRNAs obtained from UCSC GRCh38/hg38; snoRNA, snRNA, MT_tRNA, MT_rRNA, rRNA, and miscRNA from Ensembl, v91).
Defining performance metrics
The statistical programming language R (v4.0.3; www.r-project.org) was used throughout this section and all scripts can be found at GitHub71. In total, 11 performance metrics were developed, of which nine were used for evaluation of the different RNA purification methods (exRNAQC phase 1), five for blood collection tube evaluation and six for phase 2 interaction analyses. These performance metrics are summarized in Table 1. The count threshold and replicate variability metric, that require a more detailed description, are also discussed in the following paragraphs. Detailed performance metrics results for each part of the study are available through GitHub71.
Count threshold metric: To distinguish signal from noise, we made use of pairwise count comparisons across three technical replicates for evaluation of the different RNA purification methods. We defined a count threshold for each RNA purification method and biotype in a similar manner as defined in the miRQC study72. Specifically, a threshold that reduces the fraction of single positives in technical replicates by at least 95 % (single positives are cases where a given gene has zero counts in one replicate and a non-zero value in the other one). This threshold can be used as a reproducibility metric between technical replicates. For each method-volume combination, the median threshold of the three pairwise replicate comparisons was used (Supplementary Data 2). As the blood collection tube experiment in exRNAQC phase 1 did not have technical replicates and RNA purification for all tubes was performed using MIR0.2, the median thresholds of MIR0.2 (3 counts for miRNAs; 6 counts for mRNAs) were applied here as well.
Replicate variability metric: As described in the miRQC study72, the area left of the cumulative distribution curve (ALC) was calculated by comparing the actual cumulative distribution curve of log2 fold-changes in gene or miRNA abundance between pairs of replicates to the theoretical cumulative distribution (optimal curve). Less reproducibility between samples results in more deviations from this optimal curve and therefore larger ALC-values.
Accounting for size selection bias
For the small RNA library preparation of the RNA purified using the different methods in exRNAQC phase 1, the three technical replicates of each purification method were divided over three different pools. Next, pippin prep size selection for miRNAs occurred on each pool individually. To account for size selection bias (which resulted in consistently lower sequencing counts in the second pool), we each time downsampled the miRNA counts of the other two replicates to the sum of miRNA counts of the replicate in the second pool. Down-sampling was based on reservoir sampling - random sampling without replacement (subsample_miRs.py script on GitHub71).
Transforming performance metrics into robust z-scores
For evaluation of the different RNA purification methods in exRNAQC phase 1, individual scores for performance metrics were transformed to z-scores. As the standard z-score is sensitive to outliers, we used a robust z-score transformation instead (https://asq.org/quality-press/display-item?item=E0801, https://www.ibm.com/docs/en/cognos-analytics/11.1.0?topic=terms-modified-z-score): \({{{\rm{robust\; z}}}}=\frac{x-{{{\rm{median}}}}(x)}{s}\),where s is a scaling factor that depends on the median absolute deviation (MAD): \({{{\rm{MAD}}}}={{{\rm{median}}}}\left(\left|{x}_{i}-{{{\rm{median}}}}\left(x\right)\right|\right)\). If the MAD is not zero: \(s={{{\rm{MAD}}}}*1.4826\). If the MAD equals zero: \(s={{{\rm{MeanAD}}}} \,*\, 1.2533\), where \({{{\rm{MeanAD}}}}={{{\rm{mean}}}}\left(\left|{x}_{i}-{{{\rm{mean}}}}\left(x\right)\right|\right)\).
Fold-change analyses for stability over time assessment
To evaluate tube stability across time intervals in exRNAQC phase 1 and 2, we determined several performance metrics per blood collection tube at different time intervals. We then calculated, for every tube and donor, the fold-change across different time intervals (relative to the base interval at T0, so excluding T24-72 and T04-16). A theoretical example is shown in Van Paemel et al.19.
circRNA and linear RNA fraction determination
For the assessment of blood collection tube stability over time in exRNAQC phase 1, an in-house pipeline was used to investigate the differences in fractions of circRNAs between tubes and time intervals. Starting from the raw FASTQ files from the mRNA capture sequencing, Cutadapt58 (v1.18) was used to remove the adapter sequences and reads that end up shorter than 20 bp. Next, the reads of which less than 80% of the bases had a Q-score higher than 19 were removed. Subsequently, Clumpify dedupe from BBMap (v38.26; (https://sourceforge.net/projects/bbmap) with default parameters was used to remove PCR duplicate reads. The deduplicated reads were mapped using TopHat73 (v2.1.0) with Bowtie (v1.1.2) and fusion mapping turned on. Next, the CIRCexplorer274 (v2.3.3) functions parse, annotate, assemble and denovo were used to identify and annotate known circRNAs and to identify novel circRNAs or alternative back-splicing events. Last, the circRNA ratios on back-splice junction and gene level were calculated using CiLiQuant75 (v1.0).
Differential abundance analyzes
Differential abundance analyzes were performed on the data of the blood collection tube experiment of exRNAQC phase 1. In a matrix selected for T0 samples, genes were filtered out when not present with a minimum of 10 counts in all three replicates of one tube type. For the comparison of the time intervals, only the samples for the respective tube were selected and genes were filtered out when not present with a minimum of 10 counts in all three replicates of one time interval. The filtered data was normalized with Limma voom (v3.52.4) and contrasts, comparing subsequent time intervals to T0, were fit. Genes with an absolute log2 fold-change larger than 1 and an Benjamini-Hochberg adjusted p-value < 0.05 were retained as significant. On the log2 fold-change ranked gene list, gene set enrichment analysis (GSEA) with fgsea (v1.22.0) on the MSigDB C2 pathways was performed. Pathways with an Benjamini-Hochberg adjusted p-value of less than 0.05 were retained as significantly up or down regulated.
Differences in immune cell composition over time
To further evaluate blood collection tube stability over time in exRNAQC phase 1, we first we used computational deconvolution (on subsampled data) to infer the cell type composition (proportions) of different immune cell types present in blood76. Since the origin (niche) of the expression profiles has a tremendous impact on the deconvolution results77 and it is possible that RNA coming from other cell type(s) is also present in circulation, we used EPIC52 (v1.1), a method that has a built-in reference matrix from circulating immune cells (known as BRef signature) and includes the presence of an unknown component (otherCells). Specifically, we used TPM normalized count matrices as input, as recommended by the authors52 and shown as the optimal choice for this method in a recent benchmarking study78.
Next, to evaluate differences in cell type composition of several blood immune cell types, we performed a repeated-measures analysis by means of beta regression models with random effects. For each cell type a separate model was fitted with tube and time interval as factor variables (main and interaction effects included), with donor as random effect and with tube-specific variance components (allowing for variance heterogeneity that was observed in the data exploration phase). All models were fit with the glmmTMB R package (v1.1.2.3)79. Based on the model fits, all pairwise comparisons between the three time intervals were tested for each type of tube: T0 vs T1, T0 vs T2 and T1 vs T2. For each combination of cell type and tube, the p-values were adjusted for multiple testing with Tukey's method as implemented in the emmeans.glmmTMB R function (R packages glmmTMB and emmeans (v1.7.0; https://CRAN.R-project.org/package=emmeans)). All analyzes were done with the statistical software R (v4.1.0; www.r-project.org). See GitHub71 (exRNAQC005, deconvolution) for a detailed report with the corresponding R code.
Repeated measures analyzes
For data analysis of exRNAQC phase 2, linear mixed-effects models were built with the nlme package (v3.1-157) in R. Blood collection tube, RNA purification method and time interval were included as fixed effects and donor ID as random effect. The heteroscedasticity introduced by different RNA purification methods was considered. Next, an ANOVA test was performed on the model to estimate the significance of the interactions. The normality of the residuals was checked with the qqnorm function (see GitHub71).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequencing data (FASTQ files) have been deposited in the European Genome-Phenome Archive (EGA) under study accession codes EGAS00001005263 (https://ega-archive.org/studies/EGAS00001005263; exRNAQC phase 1) and EGAS00001006499 (https://ega-archive.org/studies/EGAS00001006499; exRNAQC phase 2). These data are available under restricted access to comply with data privacy regulations. Access can be requested through EGA and requests will be evaluated by the ethics committee of Ghent University Hospital. Processed and pseudonymised count files are openly available through ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress/studies using accession ID E-MTAB-10504 - E-MTAB-10507 for exRNAQC phase 1 and E-MTAB-12541 - E-MTAB-12542 for exRNAQC phase 2). mRNA capture and miRNA sequencing for each RNA purification method (exRNAQC phase 1) are identified with exRNAQC004 and exRNAQC011 study codes, respectively. mRNA capture and miRNA sequencing for each blood collection tube type (exRNAQC phase 1) are identified with exRNAQC005 and exRNAQC013 study codes, respectively. For exRNAQC phase 2, mRNA capture and miRNA sequencing are identified with the exRNAQC017 study code. Browsable data access is provided through the R2 Genomics Analysis and Visualization Platform (http://r2platform.com/exRNAQC/). Note that in R2, data on MagNA Pure RNA eluates were excluded (because of residual DNA contamination; see Results), and that data can be analyzed using two different normalization strategies (i.e., counts normalized by variance-stabilizing transformation, DESeq2 or spike-normalized counts). We also provide consistent and standardized pre-analytical variable information to better interpret, compare, and reproduce our results. To this purpose, the transcriptomes are well annotated according to Biospecimen Reporting for Improved Study Quality (BRISQ) guidelines22,23 (Supplementary Data 5).
Code availability
All code and instructions for processing the data, and code for reproducing the manuscript figures are available on GitHub71.
References
Taylor-Phillips, S. et al. Open accuracy of non-invasive prenatal testing using cell-free DNA for detection of Down, Edwards and Patau syndromes: A systematic review and meta-analysis. BMJ Open 6, e010002 (2016).
Heitzer, E., Haque, I. S., Roberts, C. E. S. & Speicher, M. R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 20, 71–88 (2019).
Stępień, E. et al. The circulating non-coding RNA landscape for biomarker research: lessons and prospects from cardiovascular diseases. Acta Pharmacol. Sin. 39, 1085–1099 (2018).
Guay, C. & Regazzi, R. Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat. Rev. Endocrinol. 9, 513–521 (2013).
Zeng, L., Cui, J., Wu, H. & Lu, Q. The emerging role of circulating microRNAs as biomarkers in autoimmune diseases. Autoimmunity 47, 419–429 (2014).
Sole, C., Arnaiz, E., Manterola, L., Otaegui, D. & Lawrie, C. H. The circulating transcriptome as a source of cancer liquid biopsy biomarkers. Semin. Cancer Biol. 58, 100–108 (2019).
Zaporozhchenko, I. A., Ponomaryova, A. A., Rykova, E. Y. & Laktionov, P. P. The potential of circulating cell-free RNA as a cancer biomarker: challenges and opportunities. Expert Rev. Mol. Diagn. 18, 133–145 (2018).
Houseley, J. & Tollervey, D. The many pathways of RNA degradation. Cell 136, 763–776 (2009).
de Jong, O. G. et al. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J. Extracell. Vesicles 1, eCollection (2012).
Sorber, L. et al. Circulating cell-free DNA and RNA analysis as liquid biopsy: optimal centrifugation protocol. Cancers (Basel) 11, 458 (2019).
Söderström, A. C., Nybo, M., Nielsen, C. & Vinholt, P. J. The effect of centrifugation speed and time on pre-analytical platelet activation. Clin. Chem. Lab. Med. 54, 1913–1920 (2016).
Gahlawat, A. W. et al. Evaluation of storage tubes for combined analysis of circulating nucleic acids in liquid biopsies. Int. J. Mol. Sci. 20, 704 (2019).
Das, S. et al. The Extracellular RNA communication consortium: establishing foundational knowledge and technologies for extracellular RNA research. Cell 177, 231–242 (2019).
Ainsztein, A. M. et al. The NIH extracellular RNA communication consortium. J. Extracell. Vesicles 4, 27493 (2015).
Grossman, R. L. et al. BloodPAC data commons for liquid biopsy data. JCO Clin. Cancer Inform. 5, 479–486 (2021).
Febbo, P. G. et al. Minimum technical data elements for liquid biopsy data submitted to public databases. Clin. Pharmacol. Ther. 107, 730–734 (2020).
Hulstaert, E. et al. Charting extracellular transcriptomes in the human biofluid RNA Atlas. Cell Rep. 33, 108552 (2020).
Hulstaert, E. et al. Messenger RNA capture sequencing of extracellular RNA from human biofluids using a comprehensive set of spike-in controls. STAR Protoc. 2, 100475 (2021).
Van Paemel, R. et al. Genome-wide study of the effect of blood collection tubes on the cell-free DNA methylome. Epigenetics 16, 797–807 (2021).
Shah, J. S., Soon, P. S. & Marsh, D. J. Comparison of methodologies to detect low levels of hemolysis in serum for accurate assessment of serum microRNAs. PLoS One 11, 1–12 (2016).
Kirschner, M. B. et al. The impact of hemolysis on cell-free microRNA biomarkers. Front. Genet. 4, 94 (2013).
Moore, H. M. et al. Biospecimen reporting for improved study quality (BRISQ). J. Proteome Res. 10, 3429–3438 (2011).
Moore, H. M. et al. Biospecimen reporting for improved study quality (BRISQ). Cancer Cytopathol. 119, 92–102 (2011).
Compton, C. C. et al. Preanalytics and precision pathology: pathology practices to ensure molecular integrity of cancer patient biospecimens for precision medicine. Arch. Pathol. Lab. Med. 143, 1346–1363 (2019).
Merker, J. D. et al. Circulating tumor DNA analysis in patients with cancer: American society of clinical oncology and college of American pathologists joint review. J. Clin. Oncol. 36, 1631–1641 (2018).
Deleu, J. et al. Digital PCR-based evaluation of nucleic acid extraction kit performance for the co-purification of cell-free DNA and RNA. Hum. Genomics 16, 1–14 (2022).
Srinivasan, S. et al. Small RNA Sequencing across diverse biofluids identifies optimal methods for exRNA isolation. Cell 177, 446–462.e16 (2019).
Kloten, V. et al. Multicenter evaluation of circulating plasma microRNA extraction technologies for the development of clinically feasible reverse transcription quantitative PCR and next-generation sequencing analytical work flows. Clin. Chem. 65, 1132–1140 (2019).
El-Khoury, V., Pierson, S., Kaoma, T., Bernardin, F. & Berchem, G. Assessing cellular and circulating miRNA recovery: the impact of the RNA isolation method and the quantity of input material. Sci. Rep. 6, 19529 (2016).
Wong, R. K. Y., MacMahon, M., Woodside, J. V. & Simpson, D. A. A comparison of RNA extraction and sequencing protocols for detection of small RNAs in plasma. BMC Genomics 20, 1–12 (2019).
Cheng, H. H. et al. Plasma processing conditions substantially influence circulating microRNA biomarker levels. PLoS One 8, e64795 (2013).
Mitchell, P. S. et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl Acad. Sci. Usa. 105, 10513–10518 (2008).
Moret, I. et al. Assessing an improved protocol for plasma microRNA extraction. PLoS One 8, e82753 (2013).
Tan, G. W., Khoo, A. S. B. & Tan, L. P. Evaluation of extraction kits and RT-qPCR systems adapted to high-throughput platform for circulating miRNAs. Sci. Rep. 5, 9430 (2015).
Kim, D. J. et al. Plasma components affect accuracy of circulating cancer-related microRNA quantitation. J. Mol. Diagn. 14, 71–80 (2012).
McDonald, J. S., Milosevic, D., Reddi, H. V., Grebe, S. K. & Algeciras-Schimnich, A. Analysis of circulating microRNA: preanalytical and analytical challenges. Clin. Chem. 57, 833–840 (2011).
Glinge, C. et al. Stability of circulating blood-based microRNAs – pre-analytic methodological considerations. PLoS One 12, e0167969 (2017).
Androvic, P. et al. Two-tailed RT-qPCR panel for quality control of circulating microRNA studies. Sci. Rep. 9, 4255 (2019).
Feng, X., Liu, Y. & Wan, N. Plasma microRNA detection standardization test. J. Clin. Lab. Anal. 34, e23058 (2020).
Sourvinou, I. S., Markou, A. & Lianidou, E. S. Quantification of circulating miRNAs in plasma: effect of preanalytical and analytical parameters on their isolation and stability. J. Mol. Diagn. 15, 827–834 (2013).
Page, K. et al. Influence of plasma processing on recovery and analysis of circulating nucleic acids. PLoS One 8, e77963 (2013).
Murray, M. J. et al. ‘Future-proofing’ blood processing for measurement of circulating miRNAs in samples from biobanks and prospective clinical trials. Cancer Epidemiol. Biomark. Prev. 27, 208–218 (2018).
Meerson, A. & Ploug, T. Assessment of six commercial plasma small RNA isolation kits using qRT-PCR and electrophoretic separation: higher recovery of microRNA following ultracentrifugation. Biol. Methods Protoc. 1, bpw003 (2016).
Van Deun, J. et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat. Methods 14, 228–232 (2017).
Tsui, N. B. Y., Ng, E. K. O. & Lo, Y. M. D. Stability of endogenous and added RNA in blood specimens, serum, and plasma. Clin. Chem. 48, 1647–1653 (2002).
Fernando, M. R., Norton, S. E., Luna, K. K., Lechner, J. M. & Qin, J. Stabilization of cell-free RNA in blood samples using a new collection device. Clin. Biochem. 45, 1497–1502 (2012).
Zhou, Z. et al. Extracellular RNA in a single droplet of human serum reflects physiologic and disease states. Proc. Natl Acad. Sci. USA. 116, 19200–19208 (2019).
Verwilt, J. et al. When DNA gets in the way: a cautionary note for DNA contamination in extracellular RNA-seq studies. Proc. Natl Acad. Sci. USA. 117, 18934–18936 (2020).
Sorber, L. et al. A multicenter study to assess EGFR mutational status in plasma: focus on an optimized workflow for liquid biopsy in a clinical setting. Cancers (Basel) 10, 1–14 (2018).
European Committee for Standardization Technical Committee (CEN/TC) 140 “In vitro diagnostic medical devices” published standard CEN/TS 17742:2022. Molecular in vitro diagnostic examinations - Specifications for pre-examination processes for venous whole blood - Isolated circulating cell free RNA from plasma.
Qin, J., Williams, T. L. & Fernando, M. R. A novel blood collection device stabilizes cell-free RNA in blood during sample shipping and storage. BMC Res Notes 6, 380 (2013).
Racle, J., de Jonge, K., Baumgaertner, P., Speiser, D. E. & Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 6, 1–25 (2017).
Hu, W. P., Chen, Y. C. & Chen, W. Y. Improve sample preparation process for miRNA isolation from the culture cells by using silica fiber membrane. Sci. Rep. 10, 1–9 (2020).
Deveson, I. W. et al. Representing genetic variation with synthetic DNA standards. Nat. Methods 13, 784–791 (2016).
Locati, M. D. et al. Improving small RNA-seq by using a synthetic spike-in set for size-range quality control together with a set for data normalization. Nucleic Acids Res. 43, e89 (2015).
Hafner, M. et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA 17, 1697–1712 (2011).
Van Goethem, A. et al. Depletion of tRNA-halves enables effective small RNA sequencing of low-input murine serum samples. Sci. Rep. 6, 1–11 (2016).
Martin, M. et al. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17,10–12 (2022).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Wang, L., Wang, S. & Li, W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012).
Yates, A. D. et al. Ensembl 2020. Nucleic Acids Res. 48, D682–D688 (2020).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Griffiths-Jones, S. The microRNA registry. Nucleic Acids Res. 32, D109–D111 (2004).
Griffiths-Jones, S., Grocock, R. J., van Dongen, S., Bateman, A. & Enright, A. J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 34, D140–D144 (2006).
Griffiths-Jones, S., Saini, H. K., Van Dongen, S. & Enright, A. J. miRBase: tools for microRNA genomics. Nucleic Acids Res. 36, D154–D158 (2008).
Kozomara, A. & Griffiths-Jones, S. MiRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39, D152–D157 (2011).
Kozomara, A. & Griffiths-Jones, S. MiRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73 (2014).
Kozomara, A., Birgaoanu, M. & Griffiths-Jones, S. MiRBase: from microRNA sequences to function. Nucleic Acids Res. 47, D155–D162 (2019).
exRNAQCConsortium et al. Blood collection tube and RNA purification method recommendations for extracellular RNA transcriptome profiling. Zenodo https://doi.org/10.5281/zenodo.14665878 (2025).
Mestdagh, P. et al. Evaluation of quantitative miRNA expression platforms in the microRNA quality control (miRQC) study. Nat. Methods 11, 809–815 (2014).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Zhang, X. O. et al. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 26, 1277–1287 (2016).
Morlion, A. et al. CiLiQuant: Quantification of RNA junction reads based on their circular or linear transcript origin. Front. Bioinforma. 2, 834034 (2022).
Avila Cobos, F., Vandesompele, J., Mestdagh, P. & De Preter, K. Computational deconvolution of transcriptomics data from mixed cell populations. Bioinformatics 34, 1969–1979 (2018).
Schelker, M. et al. Estimation of immune cell content in tumour tissue using single-cell RNA-seq data. Nat. Commun. 8, 2032 (2017).
Avila Cobos, F., Alquicira-Hernandez, J., Powell, J. E., Mestdagh, P. & De Preter, K. Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat. Commun. 11, 5650 (2020).
Brooks, M. E. et al. glmmTMB balances speed and flexibility among packages for zero-inflated generalized linear mixed modeling. R. J. 9, 378–400 (2017).
Brand, A., Allen, L., Altman, M., Hlava, M. & Scott, J. Beyond authorship: attribution, contribution, collaboration, and credit. Learn Publ. 28, 151–155 (2015).
Vermeirssen, V. et al. Whole transcriptome profiling of liquid biopsies from tumour xenografted mouse models enables specific monitoring of tumour-derived extracellular RNA. NAR Cancer 4, 1–14 (2022).
Acknowledgements
This study was in part funded by Ghent University (BOF-GOA), Stand up to Cancer (Kom op tegen Kanker, the Flemish cancer society), the Foundation against Cancer, Research Foundation Flanders (FWO; G0B2820N), the Liquidhope Transcan-2 project, the International Waldenstroem Macroglobulinemia Foundation/Leukemia Lymphoma Society, and the European Union’s Horizon 2020 research and innovation program (grant agreement 826121). H.H.H., C.E., J.D.W., E.H., P.D., and R.V.P. were funded by a predoctoral fellowship grant from the FWO (12Q6217N, 1S07416N, 1S90621N, 1133120N,11H7523N, and 11B3718N). F.A.C was supported by a Special Research Fund postdoctoral scholarship from Ghent University (BOF21/PDO/007). A.M. was supported by a Special Research Fund scholarship from Ghent University (BOF19/DOC/228), Stand up to Cancer and a predoctoral fellowship grant from the FWO (11C1621N). A.D. was supported by a postdoctoral fellowship grant from the Special Research Fund of Ghent University (BOF17/PDO/037) and the FWO (1224021N, 1266325N). K.S. was supported by a grant from Stand up to Cancer. J.D. was supported by the Special Research Fund of Ghent University (BOF22/CDV/077). We thank Illumina for sponsoring library preparation and sequencing reagents, and Qiagen, Promega and Roche for sponsoring blood collection tubes and/or RNA purification methods. The authors would like to thank Kathy Astrahantseff for the help in manuscript writing.
Author information
Authors and Affiliations
Consortia
Contributions
Author contributions are reported according to the CRediT taxonomy80. Within the exRNAQC Consortium member list and the CRediT groups, authors are in alphabetical order and # indicates lead contributions. All authors approved the manuscript. Conceptualization: A.D., O.D.W., C.E., H.H.H., A.H., P.M., A.M., J.Va. & R.V.P. Data curation: F.A.C., A.D., C.E., H.H.H., J.K., A.M., F.P.-S., R.V.P. & J.Ve. Formal analysis: J.A., F.A.C., J.D.W.#, C.E., C.F., P.M., A.M.#, F.P.-S., J.Va., R.V.P.# & J.Ve. Funding acquisition: A.D., P.M. & J.Va. Investigation: A.D., P.D., J.D., J.D.W., B.D., H.H.H., E.H., N.N., J.N., A.P., K.S., E.V.E., R.V.P., K.V., J.Ve. & N.Y. Methodology: F.A.C., A.D., B.D., T.D., C.E., C.F., H.H.H., P.M., A.M., N.N., A.P., T.P., O.T., J.Va. & R.V.P. Project administration: A.D. Resources: P.D., J.D.W., B.D., E.H., J.K., S.K., T.R.M., A.P., G.P.S. & R.V.P. Software: J.A., F.A.C., C.E., A.M., O.T, R.V.P. & J.Ve. Supervision: K.D.P., P.M., J.Va. & T.V.M. Visualization: F.A.C., A.D., J.D.W., C.E., J.K., A.M., F.P.-S., K.S., R.V.P. & J.Ve. Writing - original draft: F.A.C., A.D., P.D., J.D., J.D.W., C.E., A.M., K.S., R.V.P. & J.Ve. Writing - review & editing: A.D., C.E., P.M., A.M. & J.Va.
Corresponding authors
Ethics declarations
Competing interests
C.F. is an employee, N.N. and T.P. are former employees, P.M. is a consultant, and J.Va is a co-founder of Biogazelle, a clinical CRO providing human biofluid extracellular RNA sequencing, now a CellCarta company. G.P.S. is a former employee and S.K. is an employee of Illumina, providing library preparation and sequencing reagents. The remaining authors declare no competing interests. Promega, Qiagen and Roche sponsored blood collection tubes and/or RNA purification methods. Funders did not influence data analysis, interpretation, and manuscript writing.
Peer review
Peer review information
Nature Communications thanks Raffaele Calogero, David John and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
The exRNAQC Consortium. Blood collection tube and RNA purification method recommendations for extracellular RNA transcriptome profiling. Nat Commun 16, 4513 (2025). https://doi.org/10.1038/s41467-025-58607-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58607-7
