
Abstract
Electrocatalytic oxygen evolution reaction (OER) is key to several energy technologies but suffers from low activity. Leveraging the lattice oxygen activation mechanism (LOM) is a strategy for boosting its activity. However, this approach faces significant thermodynamic challenges, requiring high-valent oxidation of metal ions without compromising their stability. We reveal that high-entropy alloys (HEAs) can efficiently activate the LOM through synergistic multi-path electron transfer. Specifically, the oxidation of nickel is enhanced by this electron transfer, aided by the integration of weaker Co-O bonds, enabling effective LOM at the Ni-Co dual-site. These insights allow the design of a NiFeCoCrW0.2 HEA that exhibits improved activity, achieving an overpotential of 220 mV at a current density of 10 mA cm−2. It also demonstrates good stability, maintaining the potential with less than 5% variation over 90 days at 100 mA cm−2 current density. This study sheds light on the synergistic effects that confer high activity in HEAs and contribute to the advancement of high-performance OER electrocatalysts.
Similar content being viewed by others
Introduction
Electrocatalytic oxygen evolution reaction (OER) is a critical step governing the overall water-splitting reaction and serves as an essential anodic process in several energy conversion systems1,2,3. Advancements in material innovation and a deeper grasp of reaction mechanisms will be key to addressing the challenges of high overpotentials and slow kinetics in electrocatalytic OER4,5,6,7.
Two mechanisms for OER have typically been identified: the adsorbate evolution mechanism (AEM)8,9, which is characterized by the involvement of metal bands in proximity to the Fermi level, and the lattice oxygen oxidation mechanism (LOM), which involves oxygen bands around the Fermi level10,11,12,13. While the AEM has a theoretical limit for overpotential (~370 mV)14,15,16, most advanced electrocatalysts operate through the LOM17,18,19,20. However, the challenge in the LOM pathway lies in overcoming significant thermodynamic barriers to achieve deep oxidation (i.e., with high valence) of metal ions, as indicated by the Pourbaix diagram21,22,23. Moreover, maintaining the stability of high-valent metal ions at the complex catalyst-electrolyte interface is crucial for ensuring sustainable LOM participation.
For 3 d transition metal ion centers, deep oxidation states can be achieved through high-valent metal doping or other chemical methods, thereby modulating the activation of the oxygen p-band induced by Mott-Hubbard splitting. This is particularly important in designing materials with specific catalytic and electronic properties23,24,25,26. Notable examples include the modulation of Ni or Co site, such as NiCo nanosheets27, NiCoFeP oxyhydroxides24, Fe/V co-doped Ni (oxy)hydroxides28, and Zn0.2Co0.8OOH10. The core of these strategies is introducing one or two magic hetero-elements that connect to the active metal ion center through ligand oxygen, creating synergistic effects via electronic structure modulation. One might envision that deep oxidation and thus electrocatalysis activity might be further promoted if a multi-stage process through multi-path electron synergy could be constructed, which helps bridge the significant energy gap needed to achieve high oxidation states. This aligns with the observation that multi-metal oxyhydroxides exhibit enhanced activity24,29,30.
High-entropy alloys (HEAs) with multi-compositions and uniform distribution are such candidates for multi-stage electronic synergy21,31,32,33. The abundant electronic interactions in HEAs are key to achieving deep oxidation, which is vital for activating the LOM pathway. HEAs increase configurational entropy by incorporating multiple elements, i.e., chemical disordering. This redistributes electronic energy levels, potentially enhancing electron transfer and catalytic activity. The chemical disorder allows for variable surface adsorption energy, enabling flexible adsorption and desorption, and optimizing reaction pathways. Their dynamic nature also offers self-healing capabilities, adapting to environmental changes and maintaining stable catalytic performance34,35,36. However, the guidelines on designing and constructing thermodynamically favorable LOM pathways in a targeted manner are still lacking. Although high-entropy materials, including HEAs, have attracted significant attention for their potential in OER, with several materials already reported, pinpointing the effective synergistic reaction sites is still an unresolved challenge. For instance, the specific role of each constituent element in the catalytic activity remains unclear.
In this work, we demonstrate in detail how multi-path electron synergism facilitates lattice oxygen activation, leading to enhanced oxygen evolution activity. We began our multi-stage strategy with NiOOH, a model material known for its high OER activity, obtained through electrochemical surface reconstruction. Subsequently, we systematically introduced 3d transition metals Fe, Co, and Cr, along with the high-valence element W, into pure Ni, thereby creating a series of catalysts: single (Ni), dual (NiFe), triple (NiFeCo), quadruple (NiFeCoCr), and quintuple (NiFeCoCrWx) compositions, as illustrated in Fig. 1. The introduction strategy of the elements needs to be considered for activity, surface restructuring, and electron-withdrawing capability. This progressive approach offers an opportunity to clarify the evolution of multi-path electronic synergies and influence OER kinetics.

OER mechanism shifts from AEM to LOM by increasing the number of group elements. EF: Fermi level.
Results and discussion
Principles of activating lattice-oxygen design
For the transition metal oxides, the orbital hybridization of the metal d-bands with ligand oxygen 2p bands results in a distinctive interplay of valence, electronegativity, and geometrical structure12,33. This interaction gives rise to both bonding and antibonding orbitals, specifically the (M–O) and (M–O)* orbitals37. The antibonding orbitals, denoted as (M–O)*, reflect the properties of the metal’s d-orbitals, while the bonding orbitals, (M–O), display the characteristics of oxygen (Fig. 1).
Typical to post-transition metal oxides, pronounced d-d Coulomb interactions might induce the Mott-Hubbard splitting of the d orbitals (Supplementary Fig. 1). Increasing the oxidation state or valence of the metal ions intensifies such d-d Coulomb interaction, causing the lower Hubbard band (LHB) to overlap with the oxygen 2p band, which is crucial for activating the LOM in the OER11,38. Consequently, the oxidation reaction extends beyond the antibonding orbitals (M–O)*, necessitating consideration of the oxidation-reduction processes within the oxygen p-band as well. This means that O2− can be oxidized, and O2 can be formed. Therefore, the deep oxidation of metal active sites will provide the basis for LOM to participate in OER20,39.
In our structure model, we have incorporated oxygen atoms onto the surface. These atoms undergo a relaxation process, leading to an amorphous structure (Fig. 2a and Supplementary Fig. 2). This is consistent with the experimental observations of surface reconstruction40,41,42. Figure 2a shows the typical structure for the NiFeCoCrW0.2. All subsequent calculations and statistical analyses are based on this post-relaxation amorphous structure.

a Views of the NiFeCoCrW0.2 oxide model with top amorphous layers for DFT calculations. b Difference between Ni–O 3d and 2p band center relative to the Fermi level in different models. c DOS analysis of different system models. The inset corresponds to the magnification in the range of −7 to −4 eV. d Bond order of Ni–O, Fe–O, and Co–O band of different models. e Proposed OER mechanism for NiFeCoCrW0.2 catalysts follows the LOM pathway. f Free energies of OER steps via both AEM and LOM mechanisms in different models, respectively.
In our previous work, we used DFT calculations to study three elements with OER activity: Ni, Fe, and Co. The results showed that Ni, as the active site, has a significantly lower theoretical overpotential than the other elements, highlighting its potential as the catalytic active center in this study43. Then, we examined how different elements affect the structure by analyzing the energy difference (ED) between the 3d and 2p band centers in Ni–O bonds in the amorphous layers, as depicted in Fig. 2b. Adding Fe atoms to Ni decreases the ED between the metal’s d-bands and the oxygen’s p-bands, whereas including Co atoms slightly widens this gap. Further introduction of Cr atoms yields an energy separation between the 3d and 2p orbitals similar to that of NiFe. Furthermore, in the NiFeCoCrW0.2 model, the ED in the Ni–O bonds drops substantially when we introduce W atoms. This reduction indicates that W atoms lower the d-band center, contributing to a finely-tuned local electronic structure that is advantageous for the activation of LOM11,44,45,46.
To assess the metal-oxygen interaction strength, we compared the degree of density of states (DOS) overlap between the metal and oxygen10,47,48, revealing a hierarchy of interaction strength: Fe > Ni > Co for states below 5.5 eV from the Fermi level (E–EF), as shown in Fig. 2c. Likewise, the density-derived electrostatic and chemical (DDEC) bond order analysis quantitatively captures the strength of the M–O bond (Supplementary Fig. 3)49,50. Statistical analysis of median values of bond order reveals that the Fe–O bond order surpasses that of Ni–O, with Co–O exhibiting the weakest bond order (Fig. 2d). Given the negligible OER activity of Cr and W, our study concentrates on Ni, Fe, and Co sites31,51. The distribution of DDEC bond orders reveals that Co–O bonds are weaker compared to Fe–O bonds. This is crucial for understanding the dual-site (Ni–O–Co) coupling mechanism of LOM, as the desorption of oxygen requires consideration of the breaking of two M–O bonds.
We also study the impact of incorporating Fe, Co, Cr, and W atoms on the energetics of the electrochemical processes within the AEM and LOM reaction pathways (Supplementary Fig. 4)52,53. Both reaction pathways utilize identical slabs with amorphous configurations (Supplementary Figs. 5 and 6). For the LOM, Fig. 2e outlines the quintessential five-step sequence for the detailed mechanisms, based on the distribution of d-p band centers and the strength of the M–O bonds (Supplementary Fig. 7). The energy barriers of the rate-determining steps (RDS) in both reaction pathways are keys for connecting the OER kinetics to distinct local chemical environments. The gradual incorporation of Fe, Co, Cr, and W atoms progressively reduces the energy barrier for the RDS from O* to OOH* in the AEM pathway54,55, shifting from 0.59 eV in pure Ni to a reduced 0.33 eV in the NiFeCoCrW0.2 system, as shown in Fig. 2f.
For the LOM pathway, the RDS for pure Ni is the adsorption of OH− as the first step of the OER, which demands a higher energy barrier compared to the AEM path. In the NiFe system, the RDS in the LOM pathway shifts to the desorption of oxygen, with an energy barrier of 0.58 eV that remains higher than in AEM. This is due to the need to break both Ni–O and Fe–O bonds to release O–O from the catalyst surface. The stronger Fe–O bonds, as indicated by our bond order analysis, impede the desorption of oxygen. For NiFeCo, the incorporation of Co–O bonds reduces the energy barrier for the desorption of oxygen to 0.45 eV, suggesting that the weaker Co–O bond reduces the energy barrier required for the cleavage of M–O bonds during the oxygen desorption step. Thus, the dual sites consisting of Ni and Co are more advantageous for the oxygen desorption process.
In the LOM pathway of NiFeCoCr and NiFeCoCrW0.2 systems, the RDS is the formation of the intermediate O–O bond during the OER process, with an energy barrier lower than that of the AEM pathway, indicating a shift in the preferred OER pathway from AEM to LOM10,56. This comparison highlights that the deprotonation process in the Ni–OH*–Co structure is the primary factor that impedes the reaction process. These computation results underscore that the incorporation of diverse metal-oxygen bonds not only reduces the energy barrier of the RDS but also facilitates a transition in the kinetic mechanism from AEM to LOM.
On the other hand, during the OER process, the activation of lattice oxygen leads to the desorption of oxygen from the surface and the formation of oxygen vacancies. This process requires a balance between catalytic activity and structural stability to prevent structural collapse and degradation. DFT calculations show that the vacancies formed after oxygen desorption have a strong adsorption capability for OH−, with an adsorption-free energy of −0.226 eV (Supplementary Fig. 8). This rapid and strong adsorption capability effectively mitigates the structural instability caused by vacancy formation.
Therefore, the shift in the OER mechanism from the AEM (Ni, NiFe) to the LOM (NiFeCo, NiFeCroCr, and NiFeCoCrW0.2) pathway is mainly due to two factors. First, the fine adjustment of the Ni site’s electronic structure, with closer d-p band centers, enables electron activation in the oxygen p-band for OER. Second, the desorption of O–O in the LOM pathway involves breaking metal-oxygen bonds, highlighting the importance of M–O bond strength. Introducing weaker Co–O bonds lowers the energy barrier for O–O desorption, creating an efficient Ni–Co dual-site LOM pathway.
Structural characterization
We prepared a series of metals, which are Ni, NiFe, NiFeCo, NiFeCoCr, NiFeCoW0.2, and NiFeCoCrW0.2. All these metals exhibit a face-centered cubic (f.c.c.) structure and are in the form of solid solutions (Supplementary Fig. 9)57. To maintain structural homogeneity, the atomic occupancy of W was set to x = 0.2, above which the alloys are phase-separated (Supplementary Figs. 10 and 11)58. We employed cyclic voltammetry (CV) to energize the catalyst surfaces within a voltage window of −1.5–0.8 V. This process resulted in noticeable alterations to the surfaces of the metals (Supplementary Fig. 12)59. The most significant surface reconstruction was observed in NiFeCoCrW0.2, followed by NiFeCoCr and NiFeCoW0.2, then Ni, NiFe, and the least in NiFeCo. It is worth highlighting that NiFeCoCrW0.2 developed a porous, coral-like structure, as depicted in Fig. 3a, which is beneficial for increasing the accessibility of active sites on the surface.

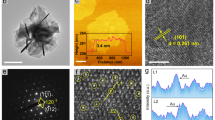
a–e Structural characteristics of NiFeCoCrW0.2 after electrochemical activation. a SEM image. b TEM images. The inset shows the SAED pattern. c HRTEM image of metal oxide on the surface. The inset is the corresponding FFT pattern. d HAADF-TEM image and the corresponding EDX elemental mappings of Ni (yellow), Fe (red), Co (pink), W (green), O (blue), and Cr (cyan). Scale bar, 0.3 μm. e The variation in pixel intensity along the area marked (yellow rectangular box) in (c). f ICP-MS analysis of metal ion concentration in electrolyte after electrochemical activation. In 1 M KOH, 500 CV scans are performed within a voltage window of −1.5 to 0.8 V, with a scan rate of 0.1 V s−1. g–j Characterization of the electronic structural features of different catalysts. g High-resolution XPS spectra of Ni 2p for different catalysts. h High-resolution XPS spectra of W 4f for different catalysts. i High-resolution XPS spectra of O 1s for different catalysts. j Statistics of relative lattice oxygen content in O 1s XPS spectra. Error bar represents the standard deviation.
The selected area electron diffraction (SAED) and high-resolution transmission electron microscopy (HRTEM) images show a crystalline-amorphous structure of the metal oxide (Fig. 3b, c and Supplementary Fig. 13), where the labeled spacings 1.5 Å, 2.1 Å and 2.4 Å correspond to the (220), (200), and (111) crystal planes (Fig. 3c) of metal oxide (NiO, PDF: 47-1049; CoO, PDF: 48-1719; FeO, PDF: 46-1312), respectively60,61. The energy-dispersive X-ray spectra (EDX) of NiFeCoCrW0.2 in Fig. 3d confirm the uniform distribution of Fe, Co, Ni, W, and O, and a negligible content of Cr elements (also reflected in the XPS results, Supplementary Fig. 18). In addition, the comparison shows that the oxidation degree of catalysts is consistent with the surface reconstruction (Supplementary Fig. 14). We also provide local intensity profiles of HRTEM images to visualize the distribution of W species at the atomic level, as shown in Fig. 3e. The presence of heavier W atoms is characterized by more pronounced intensities when juxtaposed with the lighter Ni, Fe, and Co atoms62,63. This indicates that the electronic synergy present in metal oxides is primarily related to Ni, Fe, Co, and W.
Inductively coupled plasma mass spectrometry (ICP-MS) was utilized to analyze the electrolyte solution under different CV cycles and final conditions during the electrochemical activation process, measuring the concentration of metal ions to determine the influence of the components on the extent of surface reconstruction. Figure 3f shows that Fe, Cr, and W are more leachable from the surface thus promoting reconstruction, whereas NiFeCoCrW0.2 has the highest leaching of metal atoms. In addition, under different CV cycles, the NiFeCoCrW0.2 catalyst shows a continuous increase in the leaching of Cr, W, and Fe, while Ni and Co remain stable (Supplementary Fig. 19). This is consistent with SEM morphology analysis and EDS elemental analysis (Supplementary Table 1).
Despite the near-complete loss of Cr on the reconstructed surface, its critical role in the catalyst is evident during the initial leaching process, where it enhances catalytic performance by promoting surface reconstruction. During the electrochemical activation phase, Cr is extensively leached from the catalyst surface. Although this reduces the direct involvement in electronic structure regulation, it significantly increases the active surface area of the catalyst and exposes more active sites64,65,66. Furthermore, the leaching of Cr triggers the rearrangement of other metal elements such as Ni, Fe, Co, and W, accompanied by the introduction of oxygen atoms. These changes not only maintain the structural stability of the catalyst but also allow its electronic structure to adapt to new reaction environments67.
X-ray photoelectron spectroscopy (XPS) analysis confirms full metal ion oxidation on all activated surfaces, without noticeable 0-valence metals (Supplementary Figs. 15–17). Among these, nickel in NiFeCoCrW0.2 alloy is found to have the highest oxidation state (Fig. 3g). Notably, W6+ and W5+ are detected on the W-containing catalyst’s surface68, as shown in Fig. 3h. The XPS results for Cr suggest that it has been nearly entirely leached away from the surface, which agree with the TEM-EDS analysis (Supplementary Fig. 18). In the O 1s spectrum, the peaks located at 529.7 eV and 531.3 eV correspond to lattice oxygen (M–O or OL) and metal hydroxide (M-OH), respectively (Fig. 3i and Supplementary Fig. 20)69,70. The peak at 532.4 eV indicates hydroxyl groups or water molecules adsorbed on surface oxygen vacancies (OSA)65. By comparing the oxygen species of each system, the NiFeCoCrW0.2 HEA demonstrated the highest surface concentration of OL, indicative of O–H bond decoupling in the oxide network, which is favorable for the formation of reactive lattice oxygen, as shown in Fig. 3j. Meanwhile, the peak of hydroxyl or water adsorbed by oxygen vacancies (OSA) is also elevated, which is consistent with the formation of oxygen vacancies in the LOM processes.
We performed a comprehensive X-ray absorption fine structure (XAFS) examination, encompassing both X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), to scrutinize the coordination configurations of alloys ranging from monatomic to quinary compositions. As three typical examples, NiFe, NiFeCoCr, and NiFeCoCrW0.2 were selected as representative cases to illustrate the progression from binary to ternary and quaternary electron synergies. The normalized XANES spectra of Ni and Fe/Co K-edges are shown in Fig. 4a and Supplementary Fig. 21, respectively. The pre-edge peak in XANES spectra of Ni (8330.5 eV), Fe (7111.6 eV), and Co (7708.5 eV) is correlated with the symmetry coordination of metal-oxygen71,72.

a–d XAFS analysis of different catalysts. a The XANES spectra of Ni K-edge of different catalysts and reference samples. b The FT curves of EXAFS k2χ(k) spectra of Ni K-edge. c The WT-EXAFS analysis of NiFeCoCrW0.2 catalyst. d The near-edge absorption energy at χμ = 0.5 of Ni K-edge, Co K-edge, and Fe K-edge. e The PET pathway proposed in NiFeCoCrW0.2 catalyst.
In addition, the inset is an amplification of the near-edge absorption spectrum around half-height (χμ = 0.5), which defines the correlation between absorbed energy and valence states. Distinct shifts in near-edge absorption energies are observed among the three samples, indicating that the order of valence states for Ni is as follows: NiFeCoCr0.2 exhibits the highest valence state, followed by NiFe, then NiFeCoCr, and finally NiO with the lowest valence state. In the case of Fe, the valence states decrease from NiFeCoCrW0.2 through NiFeCoCr and NiFe down to Fe2O3. For Co, the highest valence state is also found in NiFeCoCrW0.2, followed by NiFeCoCr, with CoO having the lowest valence state. Thus, the analysis suggests that in the quinary alloy NiFeCoCrW0.2, the 3d metal ions (Ni, Fe, and Co) have higher valence states compared to those in the other systems, as visually summarized in Fig. 4d.
We study the changes in the coordination environment from the k3-weighted Fourier transform (FT) EXAFS spectra (Fig. 4b and Supplementary Fig. 21). The distinct peaks observed within the 1–2 Å region of R-space correspond to the coordinated shells of Ni–O (~1.5 Å), Fe–O (~1.4 Å), and Co–O (~1.48 Å), indicating the short-range interactions between these metal ions and oxygen73. As the peak intensity correlates with M–O coordination, the coordination numbers obtained from the least-squares fitting analysis reveal a trend where the NiFeCoCrW0.2 alloy exhibits the lowest intensity and coordination number (Supplementary Table 2). This is followed by the NiFeCoCr alloy, with the NiFe binary alloy showing the highest values, suggesting a direct relationship between surface reconstruction and coordination number.
Therefore, the surface reconstruction of NiFeCoCrW0.2 forms more low-coordination centers and oxygen vacancies, which is consistent with the XPS, ICP-MS, and TEM results described above. The corresponding Ni K-edge EXAFS wavelet transforms (WT) further underscore the lowest coordination structure of Ni in NiFeCoCrW0.2, as shown in Fig. 4c. This is evidenced by the less intense peak at approximately 1.5 Å−1 in Fig. 4b. The FT peak of the Ni K-edge, located at 5.3 Å and related to a linear arrangement of three metal atoms (Supplementary Fig. 22)74,75. Considering the influence of the noise level in amplitude at high radial distance distribution, this cannot be taken as a direct reference for structural order. Furthermore, combined with the WT of Fe and Co, it demonstrates that the NiFeCoCrW0.2 alloy has stronger structural disorder compared to other systems (Supplementary Figs. 23–25)32.
These results based on electronic structures also reveal a partial electron transfer (PET) process, which is enabled by the π-donation from bridging oxygen atoms within distinct chemical environments28,76. XPS analyses evidence that the predominant valence states of Ni and Fe in NiFeCoCrW0.2, NiFeCoCr, and NiFe samples are Ni2+ and Fe3+, respectively. In the case of both NiFeCoCrW0.2 and NiFeCoCr, Co primarily exhibits a valence state of Co3+. Notably, within NiFeCoCrW0.2, W is predominantly found in the W6+ oxidation state (Fig. 3h). For binary NiFe oxide, Ni2+ has an electronic configuration of 3d8, and the π-symmetric (t2g) d orbital is fully the electrons occupied, with an electron-electron repulsion interaction with the bridging oxygen (Supplementary Fig. 26a). In contrast, the π-symmetric (t2g) d-orbitals of Fe3+ exhibit a half-filled electron configuration, which engages in π-donation interactions with the bridging oxygen77.
In the case of ternary synergistic NiFeCoCr electrocatalyst, the d-orbital electron configuration of Co3+ is characterized as 3d6 (corresponding to the traditional low-spin state) and possesses a greater electron occupancy in the π-symmetric (t2g) d-orbitals compared to Fe3+. Upon introducing Co, Fe can also capture electrons from Co through bridging oxygen, leading to competition between Co and Ni in electron transfer. This also implies that the π-donation of bridging oxygen to Fe3+ is weakened. Hence, the oxidation state of Ni in NiFeCoCr is reduced compared to that in NiFe, whereas the oxidation state of Fe in NiFeCoCr is elevated relative to NiFe, attributable to the divergent propensity of Ni and Co to engage in electron transfer with Fe through bridging oxygen.
We note that d-orbitals of W6+ are entirely vacant, endowing them with a heightened capacity for electron trapping (Supplementary Fig. 26b). To elucidate the PET effect within the tetrametallic co-lattice framework of NiFeCoCrW0.2, Fig. 4e presents a diagrammatic representation of the electronic interactions among Ni, Fe, Co, W, and O. Owing to the strong electronic modulation exerted by W6+, Ni, Fe, and Co are all facilitated to achieve a concerted transfer of electrons via the π-donation of bridging oxygen mechanism. Therefore, Ni, Fe, and Co exhibit the highest K-edge energies in the NiFeCoCrW0.2 system (Fig. 3d), which implies the highest oxidation states compared to the other systems in this work. Moreover, this multi-path electron transfer increases the delocalization of the π-symmetric (t2g) d-orbital electrons and promotes efficient electron transfer. These findings indicate that the NiFeCoCrW0.2 system establishes a more conducive electronic structural setting, which is advantageous for high-valence metal ions. This environment facilitates the integration of the LHB into the O p-band, thereby triggering the LOM process, as shown in Supplementary Fig. 27.
Electrocatalytic activity and stability
Figure 5a presents the linear sweep voltammogram (LSV) polarization curves acquired for a series of electrocatalysts within a three-electrode system in a 1 M KOH solution, where the current- and resistance- (iR) correction is 85%78. The NiFeCoCrW0.2 catalyst exhibits superior OER performance, achieving a current density of 10 mA cm−2 with a low overpotential of just 0.22 V, whereas monometallic Ni requires a higher overpotential of 0.37 V79. The binary and ternary catalysts, NiFe and NiFeCo, show improved performance with overpotentials reduced to 0.32 V and 0.3 V, respectively. Furthermore, the performance of the catalysts NiFeCoCr and NiFeCoW0.2 is also good, with overpotentials of 0.27 V and 0.25 V, respectively (Supplementary Fig. 28). In the non-Faradaic region, CV tests were conducted to estimate the double-layer capacitance (CDL), where an allometric fitting method was used instead of linear regression to fit the data points (Supplementary Fig. 29)80. The results confirm that the multi-path design of the NiFeCoCrW0.2 catalyst achieves a higher CDL value and demonstrates superior normalized specific activity compared to other catalysts (Supplementary Fig. 30). These results highlight the importance of the electronic interactions within the multi-metal oxide structure. It appears that these interactions significantly boost the catalysts’ ability to facilitate the OER process.

a LSV curves of various catalysts in O2-saturated 1 M KOH solution at a scan rate of 5 mV s−1. b Tafel plots of different catalysts corresponding to Fig. 4a. c Comparison of overpotential at a current density of 10 mA cm-2 and Tafel slope for NiFeCoCrW0.2 and previously reported OER catalysts. d Chronopotentiometry curve obtained for NiCoFeCrW0.2 catalyst at a current density of 100 mA cm-2. The inset is an SEM image after the long-term test. e LSV curves measured in KOH solutions at different pH values (13.9, 13.4, 12.9), with a scan rate of 5 mV s−1. f Specific current density at 1.45 V versus RHE as a function of pH.
The OER kinetics were further evaluated by Tafel plots (Supplementary Table 3)81,82. The NiFeCoCrW0.2 catalyst demonstrates a Tafel slope of 36.8 mV/dec, which is lower than that of NiFeCoW0.2 (38.9 mV/dec), NiFeCoCr (45.5 mV/dec), NiFeCo (43.1 mV/dec), NiFe (57.6 mV/dec), and notably lower than the monometallic Ni at 71.8 mV/dec, as depicted in Fig. 5b. This is intimately associated with the tuning of the reaction pathway and the RDS that the electrocatalyst undergoes. Furthermore, electrochemical impedance spectroscopy (EIS) was employed to examine the charge transfer behavior at the electrode-electrolyte interface, revealing that the NiFeCoCrW0.2 catalyst demonstrates rapid OER kinetics and reduced charge transfer resistance (Supplementary Fig. 31). Compared with previously reported OER electrocatalysts based on high entropy or amorphous structures, high valence metal doping, and LOM reaction pathways (Fig. 5c and Supplementary Table 4), NiFeCoCrW0.2 exhibits low overpotentials and Tafel slopes, which is one of the state-of-the-art catalysts for alkaline OER.
The electrocatalytic stability of NiFeCoCrW0.2 was confirmed by CV and chronoamperometric tests. After 3000 CV cycles, the OER polarization curves show negligible deviation from their initial state, thereby highlighting the durability and retention of catalytic performance (Supplementary Fig. 32). Furthermore, the NiFeCoCrW0.2 electrode demonstrated strong stability, maintaining performance over a continuous 90-day electrolysis period at a constant current density of 100 mA cm−2, as shown in Fig. 5d, which signifies its commendable endurance and reliability. In addition, ICP-MS analysis of the electrolyte solution at different time points under a current density of 100 mA cm−2 indicates that the ion leaching from the catalyst surface remains balanced during long-term stability tests, which also demonstrates that the restructured structure has good stability (Supplementary Fig. 33). The inset in Fig. 5d shows the SEM images of the NiFeCoCrW0.2 surface morphology after long-term testing, which preserves the oxide and porous structure after initial activation and ensures the overall activity of the catalyst (Supplementary Figs. 34 and 35). Moreover, the NiFeCoCrW0.2 catalyst shows a decrease in performance in fresh KOH solution, while displaying an improvement in performance in metal ion-rich electrolytes, indicating that the NiFeCoCrW0.2 HEA has good self-healing properties (Supplementary Figs. 36 and 37). Electron paramagnetic resonance (EPR) spectroscopy was used to verify the stability of oxygen vacancies in the NiFeCoCrW0.2 catalyst. The results confirmed significant oxygen vacancies with a strong EPR signal at g = 2.003, indicating electron capture of oxygen vacancies (Supplementary Fig. 38). The consistent EPR signal intensity during three stages of long-term stability testing further highlights the stability of the oxygen vacancies. Time-dependent XPS analysis of O 1s reveals stable oxygen states are in the multi-metal oxide structure (Supplementary Fig. 39). These factors are crucial for the stability of the catalyst83,84,85. By combining DFT analysis with entropy-driven thermodynamic and kinetic stability characteristics, it has been demonstrated that the NiFeCoCrW0.2 catalyst exhibits strong stability and durability during long-term operation67.
For the OER mechanism, the AEM pathway is dominated by four concerted proton-electron coupling transfers of metal ions at the reaction interface. On the other hand, when the chemical potential of electrons within the O p-band, which lies above the Fermi level, surpasses the redox potential of the O2/H2O couple, the energy barrier impeding electron transfer diminishes, thereby enhancing the kinetics of electron transfer (Supplementary Fig. 40)11. Consequently, the discrepancy between electron transfer dynamics and hydroxide (OH-) affinity at the oxide/electrolyte boundary leads to a non-concerted proton-electron transfer process during the RDS in the LOM mechanism, which displays a pH-dependent characteristic on the reversible hydrogen electrode (RHE) scale86.
Figure 5e shows the LSV polarization curves of these electrocatalysts measured at different pH, illustrating regular current density response with potential (Supplementary Fig. 41)87. Notably, the NiFeCoCrW0.2 exhibits a strong pH dependence on OER activity from the linear fit between the logarithm of current density and pH, which suggests a significant non-concerted proton-electron transfer in the OER process34, as shown in Fig. 5f. On the contrary, monometallic Ni exhibits intrinsic OER activity almost independent of pH. This suggests that the high valence of metal ions increases the covalency of M–O bonds and triggers the LOM path. Faster electron transfer kinetics leads to non-concerted proton-electron transfer, which manifests in a stronger dependence of OER activity on pH.
Intermediates detection
We employed in situ Raman spectroscopy to track the changes in the active intermediates during the OER process (Fig. 6a and Supplementary Fig. 42). As shown in Fig. 6b, only the Raman peaks concerning the Ni–O bond were observed in the pure nickel system. As the applied voltage increases, it transitions from Ni-OH (525 cm−1) to γ-NiOOH (480 cm−1) and β-NiOOH (562 cm−1). The Raman peak at 690 cm−1 in NiFe corresponds to Fe–O bond vibrations at low potentials (Fig. 6c)88,89. However, once the voltage surpasses 1.3 V, the signal from the Fe–O bonds diminishes, and the Raman signals are primarily attributed to the vibrations of NiOOH, indicating a high coverage of NiOOH on the catalyst surface. This illustrates the transformation of the active phase at the oxide/electrolyte interface from Ni-OH to the higher active structure Ni-OOH, which is consistent with the surface evolution in previous studies that Ni(OH)2/NiOOH redox transition takes place at ~1.4 V (vs. RHE)90.

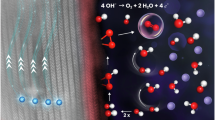
a–f In situ Raman analysis. a Schematic of cell setup for in situ Raman testing. b–f Raman spectra of Ni (b), NiFe (c), NiFeCo (d), NiFeCoCr (e), and NiFeCoCrW0.2 (f) were collected at different applied voltages (1.1–1.6 V versus RHE) in 1 M KOH solution. g LSV polarization curves of NiFeCoCrW0.2 in 1 M KOH and TMAOH with H2O and D2O as solvent, respectively. The top diagram is a schematic of TAM+ hindering O–O coupling. h Time-resolved DEMS signals of 32O2, 34O2, and 36O2 from the NiFeCoCrW0.2 catalyst during the CV process in 1 M KOH solution, with a voltage scan range of 0.3–0.8 V and a scan rate of 5 mV s−1. i Raman spectra of various catalyst surfaces after chronopotentiometry testing in 1 M KOH and TMAOH, respectively.
For NiFeCo, we observed that the Raman peaks exhibit extremely asymmetric and broad peaks at low voltage range, which is related to the co-existence of the various metal oxides (Fig. 6d and Supplementary Fig. 43). The peak at 590 cm−1 corresponds to the Fe–O bond, while the broad peak between 480 and 570 cm−1 is due to the overlap of the Ni–O and Co–O peaks. As the voltage increased, the Raman peak of Fe–O still disappears and a large amount of γ-NiOOH is formed on the surface, which is highly active for OER, followed by β-NiOOH and CoOOH. The Raman signals from β-NiOOH and CoOOH overlap, leading to an asymmetric peak between 500 and 560 cm−1, which can be attributed to both compounds (Supplementary Fig. 44). Similarly, in the NiFeCoCr and NiFeCoW0.2 systems, the intermediates eventually evolve to NiOOH and CoOOH at high potential, in agreement with the discussion in NiFeCo (Fig. 6e, f).
Most notably, a distinct Raman peak at 1069 cm−1 is observed in the spectra of the NiFeCoCrW0.2, NiFeCoW0.2, and NiFeCoCr catalysts throughout the entire voltage range. This peak is absent in the spectra of the simpler systems, such as Ni, NiFe, and NiFeCo, suggesting a unique structural feature or intermediate associated with the more complex, multi-metallic catalysts. The Raman shift of the region of 900–1100 cm−1, where the peak of oxygen species is observed, is indicative of intermediates resulting from the deprotonation of hydroxides or oxyhydroxides91,92. This observation has enabled us to precisely identify the M-OO species (here, specifically refers to Ni and Co elements), which are generated through O–O coupling and serve as crucial intermediates in the LOM pathway during the OER. In addition, NiFeCoCrW0.2 exhibits the strongest Raman signal of Ni-OO-Co species (see Supplementary Fig. 45 for more details). Here, the possible deprotonation process is OH − M1(O) − OH − M2 + OH− → OH − M1 − OO − M2 + H2O; and the subsequent OH − M1 − OO − M2 → OH − M1 − □ − M2 + O2 ↑ + e− (where □ represents the oxygen vacancy, Ov), which also explains the O–O coupling in the LOM pathway leading to the pH dependence of the OER activity. These results indicate that the dual-site Ni–O–Co is the actual active site of OER that follows the LOM pathway (Supplementary Fig. 46).
On the other hand, the negative species of superoxide (O2−) formed by deprotonation and subsequent O–O coupling of oxygen-containing intermediates has specific interactions with the tetramethylammonium cation (TMA+). We conducted a comparative analysis of LSV polarization curves in both 1 M KOH and tetramethylammonium hydroxide (TMAOH) solutions (Supplementary Fig. 47)19,93. The observed variance in OER activity among the catalysts is indicative of the degree to which lattice oxygen is activated. As shown in Fig. 6g, an increase in composition correlates with a reduction in OER activity, with the overpotential difference between the two tests increasing from 30 mV for Ni to 120 mV for NiFeCoCrW0.2 (Supplementary Fig. 48). This decrease in activity is attributed to the attack of TMA+ on O2− species, which impedes the O–O coupling essential for the LOM process. In addition, the Tafel slopes of NiFeCoCrW0.2, NiFeCoW0.2, and NiFeCoCr in TMAOH electrolytes were reduced to similar levels, illustrating the consistency of the Ni–Fe–Co trimetallic oxide activity after the exclusion of the efficient LOM pathway.
To directly verify the participation of lattice oxygen in the OER process with the NiFeCoCrW0.2 catalyst, we performed 18O isotope labeling and differential electrochemical mass spectrometry (DEMS) measurements (Supplementary Fig. 49). We conducted 5 cycles of CV testing on the 18O-labeled catalyst in the DEMS electrochemical cell. As shown in Fig. 6h, the DEMS results for the 18O-labeled NiFeCoCrW0.2 catalyst exhibit significant 18O16O (m/z = 34) peaks corresponding to the 5 CV cycles, which directly confirms the participation of lattice oxygen during the electrocatalytic OER in the NiFeCoCrW0.2 catalyst (Supplementary Fig. 50)21. The 36O is formed by the combination of a small amount of 18OH groups present on the catalyst with lattice oxygen (18O).
To further elucidate the OER mechanisms of these electrocatalysts, we investigated their OER activities in KOH and TMAOH electrolytes, utilizing H2O and D2O as solvents, respectively, to serve as buffers (Fig. 6g and Supplementary Fig. 51)94. The OER activity of the electrocatalysts, primarily governed by the LOM pathway, was notably reduced in KOH + D2O and TMAOH + D2O electrolytes, accompanied by a significant increase in the Tafel slope for catalysts such as NiFeCoCrW0.2, NiFeCoW0.2, and NiFeCoCr. This suggests that the O–O bond formation (RDS) of the LOM pathway is inhibited, owing to the introduction of D2O modifying the interaction between the intermediate and OH-, and thus ultimately changing the enthalpy of formation of the intermediate that is RDS.
We analyzed the catalyst surface using Raman spectroscopy following the i-t tests in both KOH and TMAOH solutions, with the results presented in Fig. 6i. In the case of Ni, a peak observed at 506 cm−1 in the Raman spectrum is indicative of the A1g stretching mode of the Ni–O bond within Ni(OH)2. This peak suggests a transformation from hydrated nickel hydroxide to a structure resembling nickel oxide due to dehydration21. Furthermore, the Raman spectrum identified a pair of peaks at 560 and 680 cm−1, which correspond to the Fe–O bond, and a single peak at 527 cm−1, attributed to the Ni/Co–O bond. The appearance of three distinct peaks at 458, 755, and 952 cm−1 is characteristic of the presence of TMA+ cations10. These peaks were exclusively observed on the surfaces of the NiFeCoCrW0.2, NiFeCoW0.2, and NiFeCoCr electrodes following the i-t tests conducted in the TMAOH electrolyte.
Notably, the Raman spectrum for the NiFeCoCrW0.2 catalyst displays three prominent peaks associated with TMA+ ions, which are more clearly defined than in other electrocatalytic systems. This enhanced clarity suggests a robust interaction between TMA+ ions and the negatively charged oxygen-containing intermediates (O2−), and implies a higher availability of binding sites for TMA+. These observations reinforce the idea that the OER kinetic mechanism is related to the compositional species and that the high-entropy structure promotes the initiation of an efficient electrocatalytic LOM pathway.
In summary, we have shown that a multi-path electron synergistic synergy, fine-tuned design in HEAs, facilitates the transformation of the electrocatalytic OER mechanism from the traditional AEM to a more efficient LOM. The OER activity of NiFeCoCrW0.2 is attributed to the structural and chemical optimizations, including the deep oxidation of the active Ni sites and the incorporation of weak Co–O bonds. Furthermore, the evaluation of the key intermediate μ-OO and the electrochemical OER behavior under specific test conditions provided direct evidence for distinguishing the reaction mechanisms of multi-pathway regulation systems formed by multiple components. The present work demonstrates the superiority of multi-path electronic modulation in manipulating electrocatalytic mechanistic shifts.
Methods
Materials
High-purity metal monomers (>99.9 at.%) are customized by Hebei Qinbang New Material Technology Co., Ltd. which include iron, cobalt, nickel, chromium, and tungsten. Potassium hydroxide solid (KOH solid), tetramethylammonium hydroxide solid (TMAOH solid), and tetramethylammonium hydroxide solution (TMAOH 25 wt% in H2O) were obtained from Shanghai Aladdin Biochemical Technology Co., Ltd. Ethanol and acetone sourced from Sinopharm Chemical Reagent Co., Ltd. Deuterium Oxide (D2O, ~99.9 at.% D) was obtained from Energy Chemicals Co., Ltd. The platinum electrode clips, counter electrode (Pt sheet), and reference electrode (Hg/HgO) were purchased from Shanghai LEDONLAB Co., Ltd. The high-purity water used for testing and analysis was prepared in the laboratory. All electrolytes were prepared and used immediately.
Preparation of alloys
The alloy is prepared in a high vacuum arc melting furnace. The crucible in the chamber is polished to remove the influence of impurities. Before melting, the chamber is purged with a high vacuum (5 × 10−4 Pa) and then filled with high-purity argon gas to a pressure of 0.05 MPa. Then, the titanium ingot in the crucible is melted for 3 min to further remove the oxygen in the chamber. Finally, the melting of the raw materials was carried out at a current of about 450 A and the melting time was kept at about 3 min each time. The alloy ingots underwent five cycles of re-melting, accompanied by magnetic stirring during each cycle to guarantee a homogeneous chemical composition. It should be noted here that due to the high melting point of tungsten, the incomplete melting of tungsten can be easily caused by direct mixing, which will result in the presence of an unhomogenized pure tungsten phase in the alloy. Therefore, in this work, tungsten and nickel were first binary alloyed to lower the overall melting point, and then the binary alloy was melted at high temperature with the rest of the metal raw material to obtain the alloy ingot. Except for the alloys with tungsten in the composition, which require a binary alloying step, all alloys are obtained under the same parameters in all other steps.
Preparation of OER electrodes
Preparation of the bulk material into electrodes requires electrochemical activation under alkaline conditions. The bulk alloy was utilized as the working electrode in a three-electrode setup, with a platinum sheet serving as the counter electrode and Hg/HgO functioning as the reference electrode. The CV scans in this work were performed using a scheme with a greater degree of reconstruction of the operating voltage across the positive-negative region. Each sample was performed with 500 CV scans from −1.5 V to 0.8 V with a scan rate of 100 mV s−1 in 1 M KOH, which allowed for a complete activation of the surface.
Structure characterizations
X-ray diffraction (XRD) was measured by an X-ray diffraction spectrometer (D2 Phaser, Bruker) equipped with Cu Kα (λ = 1.541 Å) radiation at 40 kV and 100 mA. Scanning electron microscopy (SEM) and energy dispersive spectroscopy (EDS) were operated by a Thermo Fisher Scios 2 dual beam focused ion beam scanning electron microscopy (FIB-SEM) at an operating voltage of 10 kV. A series of characterizations of the structure, composition, and morphology of the samples were collected using a Talos F200X (TEM), including transmission electron microscope (TEM) images, high-resolution TEM (HRTEM) images, selected area electron diffraction (SAED) and energy dispersive X-ray spectroscopy (EDX). For pre-catalysts, the preparation of TEM samples is different from that of nanomaterials. The samples were first thinned to 30 μm and further thinned by an ion milling system (Gatan 695 PIPS II). Structural analysis was achieved by TEM of the thin area near the center hole of the sample. For the analysis of the catalyst surface, the active material was stripped and dispersed into anhydrous ethanol, and then the suspension was dropped onto a microgrid copper mesh and after drying it was allowed to undergo TEM testing. Elemental dissolution from alkaline electrolytes post-electrochemical activation was identified and evaluated using an inductively coupled plasma optical emission spectrometer (ICP-OES) on the Varian Vista Pro platform. The surface oxidation state of the activated catalyst was scrutinized via X-ray photoelectron spectroscopy (XPS) on the VG Microteach ESCA 2000, employing a monochromatic aluminum X-ray source. The binding energy (BE) values for each element were calibrated against the 284.8 eV standard for the C 1s peak. The XAS measurements were conducted at the SPring-8 X-ray absorption beamline (Harima Science Garden City, Hyogo). Data analysis was performed using Athena, while phase information was calculated using Artemis, with fitting based on an atomic model that includes NiO (Coll. Code: 24014), CoO (Coll. Code: 245320), and FeO (Coll. Code: 180974). The k-space range for the Fourier transform was from 2.0 to 13.0 Å−1, and the wavelet transform was processed using HAMA Fortran version software, with a specific R-space analysis range of 0–6 Å. The EPR spectra were obtained using a BRUKER, EMXPlus spectrometer. The catalyst dimensions were 3 × 3 × 0.2 mm. The measurement parameters were as follows: magnetic field range of 3250–3450 G, microwave frequency of 9.84 GHz, modulation amplitude of 1 G, microwave power of 20 mW, and a temperature of 77 K.
Electrochemical measurements
The electrochemical performance assessments for the catalysts were conducted on an electrochemical workstation, specifically the CHI 760E, utilizing a three-electrode setup. In this arrangement, a platinum sheet with an area of 1 cm2 functioned as the counter electrode, the reference electrode was an Hg/HgO electrode, and the alloy materials themselves served as the working electrode. Here, electrochemical tests were performed on activated samples using a three-electrode system and in an O2-saturated 1 M KOH solution (Volume: 30 mL). To reduce or eliminate the influence of Fe, all electrochemical cells, Pt counter electrode, and Pt electrode clip were washed with sulfuric acid and then thoroughly rinsed with deionized water using ultrasonic cleaning before electrochemical activation and performance testing. All performance tests were performed using 1 cm2 samples as the working electrode. The potentials measured in this work (versus Hg/HgO) were calibrated relative to RHE and were processed using the following calculation:
For linear scanning voltammetry (LSV) measurements, a scan rate of 5 mV s−1 was used and the correction potential was compensated with 85%-iR to eliminate the effect of solution resistance. The Tafel slope was calculated by fitting the Tafel equation (η = blog j + a, where b is the Tafel slope and j is the current density) to the LSV data. The stability evaluation, encompassing 5000 cyclic voltammetry cycles, was carried out at a scanning velocity of 100 mV s−1.
DEMS measurements
For the 18O isotope-labeled DEMS test, the QAS 100 mass spectrometer from Linglu Instruments in Shanghai is used. In the specialized DEMS electrochemical cell, an 18O-labeled catalyst is used as the working electrode, with Hg/HgO as the reference electrode and a Pt wire as the counter electrode. The catalyst is adhered to and completely covers the glassy carbon electrode with silver glue. The membrane used in this work for DEMS testing is a Polytetrafluoroethylene (PTFE) membrane from Linglu, Shanghai, with a thickness of 40 micrometers and a pore size of approximately 20 nanometers. This PTFE membrane has a porosity greater than 50% and exhibits good hydrophobic properties, effectively preventing liquid penetration while ensuring smooth gas flow. First, a 10-cycle CV test is conducted in 1 M KOH using H218O as the solvent to achieve isotope labeling, with a voltage range of 0.2–0.8 V and a scan rate of 5 mV s−1. After the labeling process, the catalyst is thoroughly rinsed with deionized water and dried to remove H218O adsorbed on its surface. Then, a 5-cycle CV test is performed in a customized electrochemical cell with a voltage range of 0.3–0.8 V and a scan rate of 5 mV s−1. The KOH solution in the electrolyte cell is 100 mL. The gases generated during the OER process are detected in situ by mass spectrometry. In the DEMS tests, no correction or deconvolution of the ion currents was performed, and the analysis of oxygen isotopes or ion currents was conducted automatically by the DEMS without calibration or quantification of molar quantities.
In situ Raman measurements
In situ Raman spectroscopy measurements were conducted with a confocal Raman microscope (Renishaw inVia Reflex spectrometer, from the UK). A laser wavelength of 532 nm was utilized for the analysis, with a spectral range spanning from 100 to 2000 cm−1, effectively distinguishing the various phase compositions. The in situ electrochemical testing cell was composed of activated samples, a platinum wire acting as the counter electrode, and an Ag/AgCl electrode serving as the reference electrode. The working electrode was also made from the activated samples. An O2-saturated 1 M KOH solution was used for testing in the in situ Raman electrochemical cell (Volume: 10 mL). The cell underwent i-t testing with the CHI 760E electrochemical workstation, while Raman signals were simultaneously captured by a detector positioned above the sample area.
Theoretical calculation
In this research, the HEA systems were modeled using special quasi-random structures (SQSs), which were created with the aid of the Alloy Theoretic Automated Toolkit (ATAT)95,96,97, followed by surface relaxation calculations using the Vienna Ab initio Simulation Package (VASP)98. Based on the generated SQS model, first-principles calculations were performed using VASP. Spin-polarized density functional theory with the Hubbard model (DFT + U) was employed99,100,101. The interactions between the core and valence electrons were simulated by employing the Projector Augmented Wave (PAW) method in conjunction with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation function. The correlation energy values (U) for the 3d orbitals of Fe, Co, Ni, and Cr were set to 3.3 eV, 3.4 eV, 3.4 eV, and 3.4 eV, respectively102. For bulk geometry optimization, a 3 × 3 × 1 Monkhorst-Pack k-point configuration was utilized, and a 15 Å vacuum layer was set to avoid interactions under periodic boundary conditions103. The convergence criteria for forces and total energies were set to 0.05 eV/Å and 1 × 10−5 eV, respectively. In the simulation of the catalytic process, considering the experimental conditions, the catalyst surface needs to be converted into an oxide or hydroxide form, with the metal surface serving only as a precursor for the simulation setup. Since the bottom two layers of the model consist of pure metal, oxygen atoms are introduced to the catalytic surface. These oxygen atoms relax together with the metal atoms in the surface layer, forming a surface oxide layer, thereby more closely replicating the actual catalytic environment.
Data availability
The main data supporting the results of this study can be found in the article and its supplemental materials. Source data are provided with this paper or obtained from figshare repository at https://doi.org/10.6084/m9.figshare.27650352. Source data are provided with this paper.
References
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, eaad4998 (2017).
Ding, H. et al. Structural transformation of heterogeneous materials for electrocatalytic oxygen evolution reaction. Chem. Rev. 121, 13174–13212 (2021).
Roger, I., Shipman, M. A. & Symes, M. D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 1, 0003 (2017).
Chen, Z.-J. et al. Acidic enol electrooxidation-coupled hydrogen production with ampere-level current density. Nat. Commun. 14, 4210 (2023).
Song, J. et al. A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 49, 2196–2214 (2020).
Suen, N. T. et al. Electrocatalysis for the oxygen evolution reaction: recent development and future perspectives. Chem. Soc. Rev. 46, 337–365 (2017).
Subbaraman, R. et al. Trends in activity for the water electrolyser reactions on 3d M(Ni, Co, Fe, Mn) hydr(oxy)oxide catalysts. Nat. Mater. 11, 550–557 (2012).
Antipin, D. & Risch, M. Trends of epitaxial perovskite oxide films catalyzing the oxygen evolution reaction in alkaline media. JPhys. Energy 2, 032003 (2020).
Zhang, L. et al. Selenic acid etching assisted vacancy engineering for designing highly active electrocatalysts toward the oxygen evolution reaction. Adv. Mater. 33, e2007523 (2021).
Huang, Z.-F. et al. Chemical and structural origin of lattice oxygen oxidation in Co-Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329–338 (2019).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457–465 (2017).
Grimaud, A. et al. Anionic redox processes for electrochemical devices. Nat. Mater. 15, 121–126 (2016).
Lu, M. et al. Artificially steering electrocatalytic oxygen evolution reaction mechanism by regulating oxygen defect contents in perovskites. Sci. Adv. 8, eabq3563 (2022).
Koper, M. T. M. Thermodynamic theory of multi-electron transfer reactions: Implications for electrocatalysis. J. Electroanal. Chem. 660, 254–260 (2011).
Man, I. C. et al. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3, 1159–1165 (2011).
Dau, H. et al. The mechanism of water oxidation: from electrolysis via homogeneous to biological catalysis. ChemCatChem 2, 724–761 (2010).
Wang, X. et al. Understanding of oxygen redox in the oxygen evolution reaction. Adv. Mater. 34, 2107956 (2022).
Zhang, N. & Xiong, Y. Lattice oxygen activation for enhanced electrochemical oxygen evolution. J. Phys. Chem. C. 127, 2147–−2159 (2023).
Yang, C. et al. Chemical recognition of active oxygen species on the surface of oxygen evolution reaction electrocatalysts. Angew. Chem. Int. Ed. 56, 8652–8656 (2017).
Zhou, Y. et al. Enlarged Co-O covalency in octahedral sites leading to highly efficient spinel oxides for oxygen evolution reaction. Adv. Mater. 30, 1802912 (2018).
Zhang, N. et al. Lattice oxygen activation enabled by high-valence metal sites for enhanced water oxidation. Nat. Commun. 11, 4066 (2020).
Bergmann, A. et al. Reversible amorphization and the catalytically active state of crystalline Co3O4 during oxygen evolution. Nat. Commun. 6, 8625 (2015).
Zheng, X. et al. Theory-driven design of high-valence metal sites for water oxidation confirmed using in situ soft X-ray absorption. Nat. Chem. 10, 149–154 (2018).
Zhang, B. et al. High-valence metals improve oxygen evolution reaction performance by modulating 3d metal oxidation cycle energetics. Nat. Catal. 3, 985–992 (2020).
Kanan, M. W., Surendranath, Y. & Nocera, D. G. Cobalt-phosphate oxygen-evolving compound. Chem. Soc. Rev. 38, 109–114 (2009).
Wei, R. et al. Opposite electron transfer induced high valence Mo sites for boosting the water splitting performance of Ir atoms. ACS Appl. Mater. Interfaces 16, 7141–7151 (2024).
Zhao, S. et al. Ultrathin metal-organic framework nanosheets for electrocatalytic oxygen evolution. Nat. Energy 1, 16184 (2016).
Jiang, J. et al. Atomic-level insight into super-efficient electrocatalytic oxygen evolution on iron and vanadium co-doped nickel (oxy)hydroxide. Nat. Commun. 9, 2885 (2018).
Zhang, B. et al. Homogeneously dispersed multimetal oxygen-evolving catalysts. Science 352, 333–337 (2016).
Kang, Y. et al. Mesoporous multimetallic nanospheres with exposed highly entropic alloy sites. Nat. Commun. 14, 4182 (2023).
Chen, Z. J. et al. Engineering microdomains of oxides in high-entropy alloy electrodes toward efficient oxygen evolution. Adv. Mater. 33, 2101845 (2021).
Zhang, L., Cai, W. & Bao, N. Top-level design strategy to construct an advanced high-entropy Co-Cu-Fe-Mo (oxy)hydroxide electrocatalyst for the oxygen evolution reaction. Adv. Mater. 33, 2100745 (2021).
Shi, H. et al. Multicomponent intermetallic nanoparticles on hierarchical metal network as versatile electrocatalysts for highly efficient water splitting. Adv. Funct. Mater. 33, 2214412 (2023).
Tang, L. et al. High configuration entropy activated lattice oxygen for O2 formation on perovskite electrocatalyst. Adv. Funct. Mater. 32, 2112157 (2022).
Loffler, T. et al. What makes high-entropy alloys exceptional electrocatalysts? Angew. Chem. Int. Ed. 60, 26894–26903 (2021).
Zhang, N. & Chai, Y. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647–4671 (2021).
Assat, G. & Tarascon, J.-M. Fundamental understanding and practical challenges of anionic redox activity in Li-ion batteries. Nat. Energy 3, 373–386 (2018).
Sun, Y. et al. Covalency competition dominates the water oxidation structure-activity relationship on spinel oxides. Nat. Catal. 3, 554–563 (2020).
Wang, X. et al. Pivotal role of reversible NiO6 geometric conversion in oxygen evolution. Nature 611, 702–708 (2022).
Fabbri, E. et al. Dynamic surface self-reconstruction is the key of highly active perovskite nano-electrocatalysts for water splitting. Nat. Mater. 16, 925–931 (2017).
Guan, D. et al. Utilizing ion leaching effects for achieving high oxygen-evolving performance on hybrid nanocomposite with self-optimized behaviors. Nat. Commun. 11, 3376 (2020).
Huang, J. et al. Identification of key reversible intermediates in self-reconstructed nickel-based hybrid electrocatalysts for oxygen evolution. Angew. Chem. Int. Ed. 58, 17458–17464 (2019).
Zhao, H. F. et al. Crystal facets-activity correlation for oxygen evolution reaction in compositional complex alloys. Adv. Sci. 11, e2404095 (2024).
Grimaud, A. et al. Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat. Commun. 4, 2439 (2013).
Lee, Y.-L. et al. Prediction of solid oxide fuel cell cathode activity with first-principles descriptors. Energy Environ. Sci. 4, 3966 (2011).
Suntivich, J. et al. Estimating hybridization of transition metal and oxygen states in perovskites from O K-edge X-ray absorption spectroscopy. J. Phys. Chem. C. 118, 1856–1863 (2014).
Seo, D.-H. et al. The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials. Nat. Chem. 8, 692–697 (2016).
Maitra, U. et al. Oxygen redox chemistry without excess alkali-metal ions in Na2/3[Mg0.28Mn0.72]O2. Nat. Chem. 10, 288–295 (2018).
He, Z. et al. Activating lattice oxygen in NiFe-based (oxy)hydroxide for water electrolysis. Nat. Commun. 13, 2191 (2022).
Senanayake, H. S. et al. Ab initio-derived force field for amorphous silica interfaces for use in molecular dynamics simulations. J. Phys. Chem. C. 127, 16567–16578 (2023).
Cai, J. et al. Constructing Co@WC1-x heterostructure on N-doped carbon nanotubes as an efficient bifunctional electrocatalyst for zinc-air batteries. J. Power Sources 485, 229251 (2021).
Ren, X. et al. Constructing an adaptive heterojunction as a highly active catalyst for the oxygen evolution reaction. Adv. Mater. 32, e2001292 (2020).
Ding, P. et al. Circumventing the theoretical scaling relation limit for the oxygen evolution reaction. J. Phys. Chem. Lett. 15, 2859–2866 (2024).
Li, P. et al. Tuning electronic structure of NiFe layered double hydroxides with vanadium doping toward high efficient electrocatalytic water oxidation. Adv. Energy Mater. 8, 1703341 (2018).
Zuo, S. et al. Correlating structural disorder in metal (oxy)hydroxides and catalytic activity in electrocatalytic oxygen evolution. Angew. Chem. Int. Ed. 63, e202316762 (2024).
Zhao, M. et al. Lattice oxygen activation in disordered rocksalts for boosting oxygen evolution. Phys. Chem. Chem. Phys. 25, 4113–4120 (2023).
Liu, W. H. et al. Ductile CoCrFeNiMOx high entropy alloys strengthened by hard intermetallic phases. Acta Mater. 116, 332–342 (2016).
Zhang, T. et al. Three factors make bulk high-entropy alloys as effective electrocatalysts for oxygen evolution. Mater. Futures 2, 045101 (2023).
Chen, Z.-J. et al. Optimizing spin arrangement by permeability modulation of high-entropy alloys to promote O-O formation for efficient water oxidation. Sci. China Mater. 67, 598–607 (2024).
Bergmann, A. et al. Unified structural motifs of the catalytically active state of Co(oxyhydr)oxides during the electrochemical oxygen evolution reaction. Nat. Catal. 1, 711–719 (2018).
Sivanantham, A. et al. Complementary functions of vanadium in boosting electrocatalytic activity of CuCoNiFeMn high‐entropy alloy for water splitting. Adv. Funct. Mater. 33, 2301153 (2023).
Qu, J. et al. A low-temperature synthetic route toward a high-entropy 2D hexernary transition metal dichalcogenide for hydrogen evolution electrocatalysis. Adv. Sci. 10, 2204488 (2023).
Wu, Y. et al. Triggering lattice oxygen activation of single‐atomic Mo sites anchored on Ni-Fe oxyhydroxides nanoarrays for electrochemical water oxidation. Adv. Mater. 34, 2202523 (2022).
Duan, Y. et al. Anodic oxidation enabled cation leaching for promoting surface reconstruction in water oxidation. Angew. Chem. Int. Ed. 60, 7418–7425 (2021).
Malek, A., Xue, Y. & Lu, X. Dynamically restructuring NixCryO electrocatalyst for stable oxygen evolution reaction in real seawater. Angew. Chem. Int. Ed. 62, e202309854 (2023).
Luan, C. et al. Revealing dynamic surface and subsurface reconstruction of high-entropy alloy electrocatalysts during the oxygen evolution reaction at the atomic scale. ACS Catal. 14, 12704–12716 (2024).
Li, Z. et al. Empowering multicomponent alloys with unique nanostructure for exceptional oxygen evolution performance through self-replenishment. Joule 8, 2920–2937 (2024).
Zhang, R. et al. Urea-assisted synthesis of nickel sulfide arrays with enriching high-valence W species to boost Ni3+ active sites for water oxidation. Appl. Surf. Sci. 639, 158176 (2023).
Frankcombe, T. J. & Liu, Y. Interpretation of oxygen 1s X-ray photoelectron spectroscopy of ZnO. Chem. Mater. 35, 5468–5474 (2023).
Li, S. Y. et al. Sputter‐deposited high entropy alloy thin film electrocatalyst for enhanced oxygen evolution reaction performance. Small 18, 2106127 (2022).
Peng, Y. et al. Filling octahedral interstices by building geometrical defects to construct active sites for boosting the oxygen evolution reaction on NiFe2O4. Adv. Funct. Mater. 32, 2201011 (2022).
Jiang, H. et al. Tracking structural self-reconstruction and identifying true active sites toward cobalt oxychloride precatalyst of oxygen evolution reaction. Adv. Mater. 31, e1805127 (2019).
Zubair, M. et al. Vacancy promotion in layered double hydroxide electrocatalysts for improved oxygen evolution reaction performance. ACS Catal. 13, 4799–4810 (2023).
Risch, M. et al. Water oxidation by electrodeposited cobalt oxides-role of anions and redox-inert cations in structure and function of the amorphous catalyst. ChemSusChem 5, 542–549 (2012).
Risch, M. et al. Structural changes of cobalt-based perovskites upon water oxidation investigated by EXAFS. J. Phys. Chem. C. 117, 8628–8635 (2013).
Ma, J. et al. Interfacial engineering of bimetallic Ni/Co-MOFs with H-substituted graphdiyne for ammonia electrosynthesis from nitrate. ACS Nano 17, 6687–6697 (2023).
Chen, J. Y. C. et al. Operando analysis of nife and Fe oxyhydroxide electrocatalysts for water oxidation: Detection of Fe4+ by mössbauer spectroscopy. J. Am. Chem. Soc. 137, 15090–15093 (2015).
Lei, X. et al. High-entropy single-atom activated carbon catalysts for sustainable oxygen electrocatalysis. Nat. Sustain. 6, 816–826 (2023).
Risch, M. Reporting activities for the oxygen evolution reaction. Commun. Chem. 6, 221 (2023).
Morales, D. M. & Risch, M. Seven steps to reliable cyclic voltammetry measurements for the determination of double layer capacitance. JPhys. Energy 3, 034013 (2021).
Anantharaj, S. et al. The pitfalls of using potentiodynamic polarization curves for Tafel analysis in electrocatalytic water splitting. ACS Energy Lett. 6, 1607–1611 (2021).
Antipin, D. & Risch, M. Calculation of the tafel slope and reaction order of the oxygen evolution reaction between pH 12 and pH 14 for the adsorbate mechanism. Electrochem. Sci. Adv. 3, e2100213 (2023).
Thorarinsdottir, A. E., Veroneau, S. S. & Nocera, D. G. Self-healing oxygen evolution catalysts. Nat. Commun. 13, 1243 (2022).
Feng, C. et al. A self-healing catalyst for electrocatalytic and photoelectrochemical oxygen evolution in highly alkaline conditions. Nat. Commun. 12, 5980 (2021).
Gao, D. et al. High-entropy effect promoting self-healing behavior of two-dimensional metal oxide electrocatalysts for oxygen evolution reaction. ACS Catal. 14, 3700–3711 (2024).
Giordano, L. et al. pH dependence of OER activity of oxides: current and future perspectives. Catal. Today 262, 2–10 (2016).
Hausmann, J. N. et al. The pH of aqueous NaOH/KOH solutions: a critical and non-trivial parameter for electrocatalysis. ACS Energy Lett. 6, 3567–3571 (2021).
Duan, Y. et al. Scaled-up synthesis of amorphous NiFeMo oxides and their rapid surface reconstruction for superior oxygen evolution catalysis. Angew. Chem. Int. Ed. 58, 15772–15777 (2019).
Lee, W. H. et al. Electrode reconstruction strategy for oxygen evolution reaction: maintaining Fe-CoOOH phase with intermediate-spin state during electrolysis. Nat. Commun. 13, 605 (2022).
Hu, C. et al. Surface-enhanced Raman spectroscopic evidence of key intermediate species and role of NiFe dual-catalytic center in water oxidation. Angew. Chem. Int. Ed. 60, 19774–19778 (2021).
Zhu, H. et al. High-entropy alloy stabilized active Ir for highly efficient acidic oxygen evolution. Chem. Eng. J. 431, 133251 (2022).
Wang, H. Y. et al. In situ spectroscopic identification of μ-OO bridging on spinel Co3O4 water oxidation electrocatalyst. J. Phys. Chem. Lett. 7, 4847–4853 (2016).
Jia, H. et al. Stabilizing atomic Ru species in conjugated sp2 carbon-linked covalent organic framework for acidic water oxidation. Nat. Commun. 15, 5419 (2024).
Ren, X. et al. Lattice oxygen redox mechanisms in the alkaline oxygen evolution reaction. Adv. Funct. Mater. 34, 2401610 (2024).
Zunger, A. et al. Special quasirandom structures. Phys. Rev. Lett. 65, 353–356 (1990).
van de Walle, A. Multicomponent multisublattice alloys, nonconfigurational entropy and other additions to the Alloy Theoretic Automated Toolkit. Calphad 33, 266–278 (2009).
Lumley, S. C. et al. The thermodynamics of hydride precipitation: the importance of entropy, enthalpy and disorder. Acta Mater. 79, 351–362 (2014).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Ren, C. et al. A universal descriptor for complicated interfacial effects on electrochemical reduction reactions. J. Am. Chem. Soc. 144, 12874–12883 (2022).
Wang, V. et al. Vaspkit: a user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033 (2021).
Sun, P. et al. Designing 3d transition metal cation-doped MRuOx as durable acidic oxygen evolution electrocatalysts for PEM water electrolyzers. J. Am. Chem. Soc. 146, 15515–15524 (2024).
Choudhary, K. & Tavazza, F. Convergence and machine learning predictions of Monkhorst-Pack k-points and plane-wave cut-off in high-throughput DFT calculations. Comp. Mater. Sci. 161, 300–308 (2019).
Acknowledgements
We thank Prof. Gang Xu for the discussions and assistance. This work was supported by the National Thousand a Young Talents Program of China, the Fundamental Research Funds for the Central Universities (Grant No. 2018KFYXKJC009, H.-B.Y.), Wuhan Knowledge Innovation Special Project (Grant No. 2023020201020422, X.P.). The computational work was carried out on the public computing service platform provided by the Network and Computing Center of HUST.
Author information
Authors and Affiliations
Contributions
H.-B.Y. and X.P. initiated and designed this project. T.Z. synthesized and characterized the samples, and tested the performance. T.Z. and H.-F.Z. performed the DFT calculations and Z.-J.C. offered the DFT assistance. T.Z., X.P., and H.-B.Y. co-wrote the manuscript. Q.Y., N.G., L.L., N.L., J.Z., and S.-D.B. participated in the analysis of data. J.P. and X.-W.L. provided mechanistic discussions and revisions to the manuscript. All authors contributed to the discussion of the results and comments on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Omeshwari Bisen, Marcel Risch, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, T., Zhao, HF., Chen, ZJ. et al. High-entropy alloy enables multi-path electron synergism and lattice oxygen activation for enhanced oxygen evolution activity. Nat Commun 16, 3327 (2025). https://doi.org/10.1038/s41467-025-58648-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58648-y
This article is cited by
-
The promises and reality of metal–CO2 batteries
Nature Reviews Clean Technology (2025)
-
Spatial engineering and d-orbital coupling in axial dual-atom sites for bifunctional oxygen catalysis
Nature Communications (2025)
-
Electrodeposited high-entropy alloys as electrocatalysts in water electrolysis for hydrogen production: a review on impacts of composition and synthesis parameters
Frontiers of Chemical Science and Engineering (2025)
-
Unveiling the impact of Al and Cr alloying on structure and HER performance of CoCuZnMnNiFe high-entropy alloy nanoparticles
Journal of Materials Science (2025)
