
Abstract
Inherently chiral calix[4]arenes represent a distinct class of chiral frameworks whose chirality arises from the dissymmetry of the entire molecule. Although these chiral scaffolds have been widely applied in various research fields, their catalytic enantioselective synthesis remains largely underexplored. Herein, we report the enantioselective synthesis of inherently chiral calix[4]arenes using an organocatalyzed desymmetrization method. By using chiral phosphoric acid catalysis, the asymmetric electrophilic amination reactions of the phenol-containing prochiral calix[4]arenes led to a range of inherently chiral calix[4]arenes with high yields and enantioselectivities. Significantly, the practicability of this method is underscored by its successful implementation using as little as 0.05 mol% of chiral catalyst, without compromising reaction efficiency and enantioselectivity. Moreover, the aminophenol moiety in the products could be easily modified to produce unique calix[4]arenes with diverse N,O-heterocycles, as well as the simple meta-amino-substituted chiral calix[4]arene, which have shown promising potential in the development of new chiral catalysts.
Similar content being viewed by others
Introduction
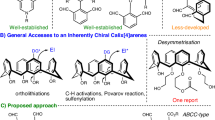
Within the macrocyclic arenes family1, calixarenes stand out as one of the most popular synthetic macrocyclic hosts due to their unique structures, easy functionalizations and wide array of applications2,3,4,5,6,7,8. The asymmetric substitutions on the rigid cone conformation of calix[4]arenes make them non-superimposable with their mirror images, a concept termed Inherent Chirality by Böhmer in 1994 to differentiate this unique chirality from traditional forms such as point, axial, planar and helical chirality9. Generally, there are two main methods to induce inherent chirality on the calix[4]arene scaffold: one involves having different substituents on the upper or lower rims, providing a specific sense of rotation to the system10, while the other entails a single meta-substitution on one phenyl ring of calix[4]arenes to break the symmetry of the calix[4]arene framework (Fig, 1A)11,12. Given the unique structure of these inherently chiral calix[4]arenes (ICCs), they show promise for potential applications in the fields such as chiral recognition13,14,15,16 and asymmetric catalysis17,18,19. However, accessing these enantioenriched ICCs continuous to be a longstanding challenge20, which is still heavily rely on resolution of racemic mixtures using chiral high-performance liquid chromatography (HPLC) separation or diastereoselective synthesis aided by chiral auxiliaries21,22,23,24,25, thereby impeding further functional studies.

A Inherently chiral calix[4]arenes and their applications. B Catalytic enantioselective synthesis of ICCs through Pd-catalyzed intramolecular C–H arylation. C Catalytic enantioselective synthesis of ICCs through CPA-catalyzed sequential Povarov reaction and aromatizations. D This work: Catalytic enantioselective synthesis of ICCs through CPA-catalyzed aromatic amination.
Catalytic enantioselective synthesis is recognized as the most efficient and direct method for asymmetric synthesis of chiral molecules; however, the exploration of this approach for producing ICCs has been limited26. In 1998, McKervey and co-workers pioneered the enantioselective synthesis of inherently chiral calix[4]arenes through lipase-catalyzed acylative desymmetrization of the lower-rim’s hydroxyl groups, although the yield was poor27. However, it was not until two decades later that the catalytic enantioselective synthesis of ICCs through the desymmetrization of the upper-rim was achieved. In 2022, Cai and co-workers developed the enantioselective synthesis of inherently chiral calix[4]arenes through the Pd-catalyzed intramolecular asymmetric C–H arylation, yielding the fluorenone-containing calix[4]arenes with high enantioselectivities28 (Fig. 1B). Intriguingly, almost simultaneously, the Tong group presented their research employing the same substrates but a different catalyst, which produced distinct chiral products via a sequence of enantioselective 1,5-palladium migration and intramolecular C–H arylation29. Recently, our group and the Liu group independently disclosed the asymmetric synthesis of quinoline-containing ICCs through sequential chiral phosphoric acid (CPA)-catalyzed Povarov reaction and oxidative aromatizations, with the cyclization step leading to the desymmetrization of the prochiral arylamine-containing calix[4]arene substrates30,31 (Fig. 1C). It is noteworthy that significant progress has also been made in the enantioselective synthesis of heterocalix[4]arenes, despite these compounds exhibiting a distinct 1,3-alternate conformation rather than a cone conformation. The Tong and Wang group developed several highly efficient enantioselective macrocyclization methods for asymmetric synthesis of inherently chiral heterocalix[4]arenes32,33,34,35, as well as the (dynamic) kinetic resolution methods36,37.
While several other efficient methods have been disclosed for the enantioselective synthesis of ICCs recently38,39,40, these approaches are primarily limited to cyclization reactions. Given the broad potential applications of ICCs, there is a significant demand for new methodologies that enable their catalytic enantioselective and structurally diverse synthesis. The catalytic asymmetric electrophilic aromatic substitutions of electron-rich arenes, including phenols and anilines, represent a potent approach for producing chiral molecules with axial41,42,43,44,45,46,47, planar48,49,50 or helical chirality51. However, its utilization in the enantioselective construction of inherently chiral calix[4]arenes has not yet been disclosed52. Herein, we introduce the asymmetric synthesis of ICCs through CPA-catalyzed53,54,55,56 aromatic aminative desymmetrization57,58 of prochiral phenol-containing calixarene substrates (Fig. 1D). Notably, through simple derivatization of the chiral products, the simple meta-amino group-substituted chiral calix[4]arene, as well as those containing diverse N,O-heterocycles, could be readily afforded.
Results
Reaction condition optimizations
We began our study by selecting the prochiral 4-OH group-containing calix[4]arene 1a as a model substrate, envisioning that an asymmetric ortho-selective substitution reaction would lead to the generation of inherently chiral calix[4]arene (Table 1). Encouragingly, treatment of 1a with dibenzyl azodicarboxylate 2a in the presence of CPA catalyst A1 (10 mol%) in toluene at 20 oC afforded the expected ortho-C–H amination product 3a in 91% yield with 85% ee. (entry 1) It is noteworthy that 3a existed as an equilibrium mixture of two diastereomers (85:15 dr after 24 h), probably due to the partially restricted rotation of the C–N bond59. Subsequently, a series of BINOL-derived CPA catalysts bearing various aryl groups at the 3,3’-positions were screened in this reaction (entries 2-9). We were delighted to find that the CPA A9 with the 9-(10-Ph-anthracenyl) substituents yielded the chiral calix[4]arene 3a in 91% yield with 97% ee (entry 9). Next, a range of solvents were also examined for this reaction (entries 10-13). Interestingly, while high enantioselectivities were generally achieved across different solvents, polar solvents led to poor yields, with almost no reaction observed in tetrahydrofuran (THF). Given the rapid reaction rate in DCM, the amount of the CPA catalyst was reduced to 1 mol%, which can still yield product 3a in 88% yield with identical 97% ee after 24 h (entry 14).
Substrates scope investigation
With the optimal conditions established, we set out to investigate the scope of this method for enantioselective synthesis of inherently chiral calix[4]arenes (Fig. 2). Initially, a range of commercially available azodicarboxylates were investigated under the standard conditions, which yielded the corresponding chiral calix[4]arene products in good yields with high enantioselectivities (up to 99% ee, 3b–3e). It was worth mentioning that all these products existed as equilibrium diastereomeric mixtures as well. Cyclic diazodicarboxamide was identified as an effective electrophilic amination reagent for achieving enantioselective desymmetrization of 1a as well, though the reaction temperature had to be reduced to −40 °C due to its higher reactivity (3f). A series of para-substituted phenyl groups on these cyclic diazodicarboxamides were compatible, yielding chiral calix[4]arene products with high enantioselectivities (3g–3k). Notably, only one pair of peaks were observed in the chiral HPLC analysis of these compounds, which may be attributed to the low epimerization barrier exhibited by these cyclic diazodicarboxamide-containing products. The absolute structure of 3g was determined using X-ray crystallography, revealing an (M) configuration for its inherent chirality and an (Sa) configuration for the C–N axial chirality. Accordingly, the configurations of other ICC products were assigned by analogy to this structure. Additionally, various cyclic diazodicarboxamides with meta-substituted (3l–3m), ortho-substituted phenyl groups (3n–3o), 1-naphthyl group (3p) and benzyl group (3q) were well tolerated with this method, resulting in the formation of chiral calix[4]arenes with high ee values. Moreover, the modification of the ether groups at the lower rim of the calix[4]arene substrates were studied, which showed good compatibility (3r–3t).

Reactions were performed with prochiral calix[4]arenes 1 (0.05 mmol), azo reagents 2 (0.06 mmol) and CPA catalyst (0.0005 mmol, 1 mol%) in DCM (1.5 mL) at 20 °C for 24 h. Isolated yields were reported. The dr and ee values were determined by HPLC analysis using a chiral stationary phase. The products not reporting dr values indicated that only one diastereomer was observed by HPCL analysis (>20:1 dr). aThese reactions were performed at −40 °C.
After evaluating the electrophilic amination reagents and the lower rim substitutions, we turned our attention to the compatibility of the upper rim modifications (Fig. 3A). The prochiral phenol-containing calix[4]arenes 1u, featuring an additional para-bromo substitution on the 3-phenyl moiety, was also amenable to this method, yielding 3u in 88% yield with 99% ee, which existed as an equilibrium diastereomeric mixture as well. Various substitutions at the para-position of the 3-phenyl ring was examined, suggesting favorable compatibility with groups like iodo (3v), phenyl (3w), alkenyl (3x), alkyl group (3y–3z) and boronic ester (3aa). In addition, the alkoxyl group was well-tolerated, which exclusively gave the C–H amination product at the phenol ring (3ab). Furthermore, the diamination reaction of the 1,3-diphenol-containing prochiral calix[4]arene 4a was explored under similar conditions expect with 2.4 equivalent of azodicarboxylate 2a and 2 mol% of the CPA catalyst (Fig. 3B). Encouragingly, this reaction efficiently produced the expected di-aminated chiral calix[4]arene 5a in 86% yield with >99% ee. Moreover, additional substitutions at the para-positions of the 2,4-diphenyl rings were studied, which indicated that dibromo and various substituted phenyl groups are all well accommodated. This led to the generation of ICCs with modifications across all upper rims of the four phenyl rings in enantiopure form (5b–5e, >99% ee), likely due to the enantioenrichment achieved through sequential asymmetric amination reactions via a kinetic resolution process. Notably, exclusive diastereoselectivity was observed for the C–N atropisomerism in these di-amination products. Moreover, the C4 symmetric tetra-hydroxy-calix[4]arene 4f was studied with this method. However, treatment of 4f with 5.0 equivalent of azodicarboxylate 2a under the standard conditions resulted in a complex mixture, without the isolation of the expected tetra-aminated product 5f’. Instead, reducing the amount of 2a to 2.0 equivalent led to the formation of the bis-aminated product 5f in 58% yield with >99% ee, which indicated that further amination on 5f might pose challenges due to steric hindrance.

A Scope for synthesis of ICCs regarding substitutions on the 3-phenyl moiety. B Scope for synthesis of ICCs through diamination reaction. aReactions were performed with prochiral calix[4]arenes 1 (0.05 mmol), azodicarboxylate 2 (0.06 mmol) and CPA catalyst (0.0005 mmol, 1 mol%) in DCM (1.5 mL) at 20 °C for 24 h. Isolated yields were reported. The ee and dr values were determined by HPLC analysis using a chiral stationary phase. The products not reporting dr values indicated that only one diastereomer was observed by HPCL analysis (>20:1 dr). bReactions were performed with prochiral calix[4]arenes 4 (0.05 mmol), azodicarboxylate 2 (0.12 mmol) and CPA catalyst (0.001 mmol, 2 mol%) in DCM (1.5 mL) at 20 °C for 24 h.
Studies of the epimerization of C–N atropisomers
Given the emerging interest in enantioselective synthesis of molecules featuring multiple stereogenic elements60,61, the C–N axial chirality present in these inherently chiral calix[4]arene products was studied (Fig. 4). The two C–N diastereomers of 3a, (M, Sa)-3a and (M, Ra)-3a, could be obtained individually through column chromatography; however, these two compounds would later equilibrate into a mixture of diastereomers over time (Fig. 4A). Detailed monitoring this process via HPLC analysis revealed an equilibrium ratio of 76:24 ((M, Sa)-3a:(M, Ra)-3a) in a mixed hexane/isopropyl alcohol solution (10:1) after approximate 20 h, which suggested that (M, Sa)-3a was 0.7 kcal/mol more stable than (M, Ra)-3a, with a C–N bond rotation barrier of 23.9 kcal/mol. This level of rotation barrier categorizes the C–N atropisomers in 3a as class 2 atropisomers according to LaPlante’s classification62, explaining the observed epimerization between these diastereomeric products. Moreover, given that only one diastereomer was observed for the amination products derived from cyclic diazodicarboxamides, the potential C–N atropisomerism in a product like 3g was investigated by DFT calculation (Fig. 4B). The investigations revealed that (M, Sa)-3g is thermodynamically 1.8 kcal/mol more stable than (M, Ra)-3g, consistent with the X-ray crystallographic results. Moreover, the calculated energy barrier for epimerization was determined to be 20.6 kcal/mol, significantly lower than that of 3a. This reduced barrier would facilitate more rapid equilibration between these conformations at room temperature, ultimately leading to the exclusive observation of a single diastereomer in the chiral HPLC analysis.

A Experimental studies of the energies of the two diastereomers of 3a and its epimerization barrier. B DFT calculation of the energies of the two diastereomers of 3g and its epimerization barrier.
Investigations of the reaction mechanism
To elucidate the mechanism and origin of stereoselectivity in this reaction, a set of control experiments were performed (Fig. 5). Replacing the OH group with an OMe group in the calix[4]arene substrate (6a) halted the formation of the desired C–H amination product 7a, which was consistent with the observation for the alkoxyl-substituted product 3ab (Fig. 5A). These results underscored the crucial role of the potential hydrogen bonding between the phenol moiety and the CPA catalyst, which was also supported by the unfavorable outcomes observed in polar solvents (see entries 11–13 in Table 1). Given that the further di-amination reaction of the mono-amination product may potentially serve a role in kinetic resolution (KR), its influence on the high enantioselectivity achieved in these reactions was studied. However, treatment of racemic 3a with the standard conditions failed to produce the di-amination product 8a, which indicated that the amination substitution significantly decreased the reactivity of the phenol ring, and that no KR process was involved in the reaction using azodicarboxylates 2a (Fig. 5B). In contrast, the KR of racemic 3f through amination with cyclic diazodicarboxamide 2f proceed smoothly to give the achiral di-amination product 8f in 51% yield and the recovered 3f in 48% yield with 41% ee, corresponding to a selectivity factor (s)63 of 3.2 (Fig. 5C). This result suggested that in the amination reaction with cyclic diazodicarboxamides, the subsequent second amination step may enhance the enantioselectivity through a KR process, aligning with our observation that the diamination byproducts could be detected in the reactions using cyclic diazodicarboxamides. Based on these findings and previous studies42,64,65, a plausible reaction mechanism was proposed (Fig. 5D). Through dual activation of the phenol moiety and the diazo reagents via hydrogen bonding with the CPA catalyst, the addition of the ortho-position of phenol to diazo reagent produced the dearomatized intermediate with both inherent chirality and central chirality. Following this enantiodetermining step, the rapid keto-enol tautomerism resulted in the ortho-C–H aminated phenol products (M)-3. We propose that the origin of inherent chirality in these reactions could be attributed to the central chirality of the aminative dearomatization reaction of phenol. Due to the unique cone-shaped structure of calix[4]arenes, the diazo reagent could exclusively access the phenol moiety from the outer face. Accordingly, the addition through route a is expected to give the (M, Rc)-INT A, while the addition through route b should lead to the formation of (P, Sc)-INT B. In the previous research of asymmetric aminative dearomatization of 2-naphthols by You and co-workers, it was found that the (R)-BINOL-derived 3,3’-di-(9-anthracenyl)-substituted CPA catalyst typically produced the product with an (R)-configuration66, which was in alignment with our observation favoring the generation of (M, Rc)-INT A. Moreover, Sunoj and co-workers conducted thorough density functional theory (DFT) calculation on the origin of enantioselectivity in the aforementioned work67, which suggested that the parallel alignment of the two reactants relative to one of the anthracenyl substituents, along with the multiple C−H···π interactions between the substrates and the anthracenyl substituents of the CPA catalyst, elucidates the preferential addition to the Re face of the phenol’s α-position.

A Control experiment of the O-substitution of calix[4]arene. B Kinetic resolution of (±)−3a using 2a. C Kinetic resolution of (±)−3f using 2f. D Proposed reaction mechanism.
Derivatizations and applications of the chiral products
To showcase the practicability of this method, a large-scale asymmetric reaction was conducted (Fig. 6A). By utilizing the standard conditions, 304 mg of 1a (0.5 mmol) was readily converted into the inherently chiral calix[4]arene 3a (430 mg) in 95% yield with 96% ee as a diastereomeric mixture. In light of the high reactivity observed in this reaction, an investigation on reducing the catalyst amount was conducted (Fig. 6B). When calix[4]arene 1a reacted with azodicarboxylates 2a employing 0.1 mol% of CPA catalyst A9, product 3a was obtained with a consistent 96% ee, albeit in a reduced yield. Encouragingly, elevating the reaction concentration from 0.05 M to 0.2 M and temperature from 20 °C to 40 °C notably enhanced the yield while preserving the ee value. Remarkably, reducing the catalyst loading further to 0.05 mol% did not lead to a decrease in either yield or enantioselectivity. Even though using a 0.02 mol% of CPA catalyst led to slight decreases in yield and enantioselectivity, these outcomes still underscore the significant practicality of this method. The derivatizations of these ICCs products were also studied to demonstrate the utilities of this method. Treatment of the calix[4]arene 3e with trifluoroacetic acid (TFA) in air yielded the aminoquinone-containing calix[4]arene 9 in 87% yield with 94% ee, with the removal of the O-propyl group (Fig. 6C). Notably, with the cleavage of the substituted hydrazine moiety in this step, the intricate C–N atropisomerism in 3e was eliminated, leading to the generation of a useful amino group-containing product 9. Subsequent reduction of the quinone moiety via catalytic hydrogenation was followed by in situ cyclization employing triphosgene, which led to the formation of benzoxazolone-containing 10 in 91% yield. Extensive studies were also conducted on the derivatizations of the chiral calix[4]arene product 3a that features the N-Cbz-substituted hydrazine moiety (Fig. 6D). The Pd/C-catalyzed hydrogenation of 3a resulted in the formation of product 11 containing 1,2-benzoquinone, likely through the oxidation of the aminophenol intermediate to ortho-iminoquinone and subsequent hydrolysis. Notably, product 11 could readily undergo condensation with cis-1,2-diaminocyclohexane, giving rise to quinoxaline-containing compound 12 after dehydrogenation using chloranil. To prevent the oxidation of the electron-rich aminophenol product 13, in situ cyclization of 13 with triphosgene afforded the benzoxazolone-containing product 14 in 90% yield with 98% ee. Moreover, the in-situ protection of the aminophenol 13 with BzCl afforded the N-benzoylated 15, which, upon acid-catalyzed dehydration, led to the formation of benzoxazole-containing calix[4]arene 16. With these distinct inherently chiral calix[4]arene structures, we aimed to develop new chiral catalysts based on these frameworks (Fig. 6E). Following the conversion of the phenol 3a into a triflate, Pd/C-catalyzed hydrogenation resulted in the formation of a simple meta-amino group-substituted calix[4]arene 17 with 84% yield and 96% ee, which served as a crucial inherently chiral intermediate in the modifications of ICCs68,69. Furthermore, the N-dimethylation of 17 followed by oxidation yielded the inherently chiral aniline N-oxide 18 in 78% yield with 94% ee, which found potential application in chiral recognition of mandelic acid through 1H NMR spectroscopy (see Supplementary Fig. 4 in SI for details). In addition, condensation of 17 with thiophosgene and subsequent coupling with either (R,R)- or (S,S)-N1,N1-dimethylcyclohexane-1,2-diamine afforded the diastereomeric tertiary amine-thiourea bifunctional catalysts 19a and 19b. These two catalysts, along with the commercially available tertiary amine-thiourea 19c, were subsequently examined in the asymmetric [4 + 2] cycloaddition reaction involving 2-benzothiazolimine 20 and homophthalic anhydride 2170 (Fig. 6F). Notably, catalyst 19a facilitated the synthesis of product 22 in 65% yield with 15:1 dr and 86% ee, outperforming the results obtained with the commercially available catalyst 19c. In contrast, the diastereomeric catalyst 19b yielded the product 22 in 52% yield with 5:1 dr and −42% ee, highlighting a significant matched–mismatched relationship between the point chirality and inherent chirality of the chiral moieties within the catalyst. Moreover, we were delighted to find that the simple chiral meta-amino-substituted calix[4]arene 17 could serve as a chiral primary amine catalyst in the asymmetric α-amination reaction of aldehyde 23 with azodicarboxylate 2e,71 which produced the α-tertiary amine derivative 24 with 88% yield and excellent enantioselectivity control (88% ee) (Fig. 6G).

A Large scale asymmetric synthesis. B Studies on reducing catalyst loading. C Derivatizations of the chiral calix[4]arene 3e. D Derivatizations of chiral calix[4]arene 3a. E Derivatizations of 3a into meta-amino-substituted calix[4]arenes. F Application of calix[4]arene derived catalyst in asymmetric [4 + 2] cycloaddition reaction. G Application of calix[4]arene derived catalyst in asymmetric α-amination reaction of aldehyde.
Discussion
In conclusion, we have developed an effective method for catalytic asymmetric synthesis of inherently chiral calix[4]arenes via an enantioselective desymmetrization strategy. By employing a chiral phosphoric acid catalyst, the asymmetric electrophilic ortho-C–H amination reaction of the phenol moiety with various azo reagents broke the symmetry of prochiral calix[4]arenes, resulting in a range of inherently chiral calix[4]arenes with modifications at both upper rim and lower rim, in high yields and enantioselectivities. The intriguing C–N atropisomerism in the products was studied, and a series of control experiments were conducted to elucidate the reaction mechanism and origin of stereoselectivity. The large-scale reaction, along with studies on low catalyst loading (reduced to as low as 0.05 mol%), showcased the high practicality of this method. Moreover, the versatile derivatizations of the chiral calix[4]arene products into diverse inherently chiral frameworks, especially their successful applications in the development of new chiral organocatalysts, underscore the significant potential of this methodology.
Methods
General procedure for asymmetric synthesis of inherently chiral calix[4]arenes 3
To solution of 1 (0.1 mmol, 61 mg, 1.0 equiv.) and CPA (R)-A9 (0.001 mmol, 1 mol%) in DCM (1 mL) was added a solution of azodicarboxylate 2 (0.12 mmol, 36 mg, 1.2 equiv.) in DCM (0.5 mL) at 20 °C or −40 °C. After stirring for 24 h at this temperature, the reaction mixture was directly purified by column chromatography (silica gel, petroleum ether/ethyl acetate = 15:1) to afford the products 3 as a white foam.
General procedure for asymmetric synthesis of inherently chiral calix[4]arene 5
To solution of 4 (0.05 mmol, 1.0 equiv.) and CPA (R)-A9 (0.0001 mmol, 2 mol%) in DCM (1 mL) was added a solution of 2a (0.12 mmol, 35.8 mg, 2.4 equiv.) in DCM (0.5 mL) at 20 oC. After stirring for 24 h, the reaction mixture was directly purified by column chromatography (silica gel, petroleum ether/ethyl acetate) to afford the products 5 as a white foam.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and Supplementary Information file, or from the corresponding author upon request. The source data are provided with this paper. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition numbers CCDC 2381033 (for 3g). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Source data are provided with this paper.
References
Han, X.-N., Han, Y. & Chen, C.-F. Recent advances in the synthesis and applications of macrocyclic arenes. Chem. Soc. Rev. 52, 3265–3298 (2023).
Neri, P., Sessler, J. L. & Wang, M.-X. Calixarenes and Beyond. 1 edn, (Springer International Publishing AG, Cham, 2016).
Homden, D. M. & Redshaw, C. The use of calixarenes in metal-based catalysis. Chem. Rev. 108, 5086–5130 (2008).
Wang, M.-X. Nitrogen and oxygen bridged calixaromatics: synthesis, structure, functionalization, and molecular recognition. Acc. Chem. Res. 45, 182–195 (2012).
Nimse, S. B. & Kim, T. Biological applications of functionalized calixarenes. Chem. Soc. Rev. 42, 366–386 (2013).
Guo, D.-S. & Liu, Y. Supramolecular chemistry of p-sulfonatocalix[n]arenes and its biological applications. Acc. Chem. Res. 47, 1925–1934 (2014).
Kumar, R. et al. Revisiting fluorescent calixarenes: from molecular sensors to smart materials. Chem. Rev. 119, 9657–9721 (2019).
Ma, Z., Li, Y., Sun, X.-Q., Yang, K. & Li, Z.-Y. Calixarene promoted transition-metal-catalyzed reactions. Chin. J. Org. Chem. 41, 2188–2201 (2021).
Böhmer, V., Kraft, D. & Tabatabai, M. Inherently chiral calixarenes. J. Incl. Phenom. Mol. Recognit. Chem. 19, 17–39 (1994).
Li, M., Ho, C. K. S., On, I. K. W., Gandon, V. & Zhu, Y. Inherently chiral resorcinarene cavitands through ionic catalyst-controlled cross-coupling. Chem. 10, 3323–3341 (2024).
Lhoták, P. Direct meta substitution of calix[4]arenes. Org. Biomol. Chem. 20, 7377–7390 (2022).
Tlustý, M., Eigner, V., Dvořáková, H. & Lhoták, P. The formation of inherently chiral calix[4]quinolines by Doebner–Miller reaction of aldehydes and aminocalixarenes. Molecules 27, 8545 (2022).
Li, S.-Y., Xu, Y.-W., Liu, J.-M. & Su, C.-Y. Inherently chiral calixarenes: synthesis, optical resolution, chiral recognition and asymmetric catalysis. Int. J. Mol. Sci. 12, 429–455 (2011).
Zheng, Y.-S. & Luo, J. Inherently chiral calixarenes: a decade’s review. J. Incl. Phenom. Macrocyclic 71, 35–56 (2011).
Luo, J., Zheng, Q.-Y., Chen, C.-F. & Huang, Z.-T. Facile synthesis and optical resolution of inherently chiral fluorescent calix[4]crowns: enantioselective recognition towards chiral leucinol. Tetrahedron 61, 8517–8528 (2005).
Shirakawa, S., Moriyama, A. & Shimizu, S. Design of a novel inherently chiral calix[4]arene for chiral molecular recognition. Org. Lett. 9, 3117–3119 (2007).
Durmaz, M., Halay, E. & Bozkurt, S. Recent applications of chiral calixarenes in asymmetric catalysis. Beilstein J. Org. Chem. 14, 1389–1412 (2018).
Shirakawa, S., Kimura, T., Murata, S.-i & Shimizu, S. Synthesis and resolution of a multifunctional inherently chiral calix[4]arene with an abcd substitution pattern at the wide rim: the effect of a multifunctional structure in the organocatalyst on enantioselectivity in asymmetric reactions. J. Org. Chem. 74, 1288–1296 (2009).
Shirakawa, S. & Shimizu, S. Synthesis of an inherently chiral calix[4]arene amino acid and its derivatives: their application to asymmetric reactions as organocatalysts. Eur. J. Org. Chem. 2009, 1916–1924 (2009).
Arnott, G. E. Inherently chiral calixarenes: synthesis and applications. Chem. Eur. J. 24, 1744–1754 (2018).
Yakovenko, A. V. et al. Diastereoselective lower rim (1S)-camphorsulfonylation as the shortest way to the inherently chiral calix[4]arene. Org. Lett. 9, 1183–1185 (2007).
Xu, Z.-X., Zhang, C., Zheng, Q.-Y., Chen, C.-F. & Huang, Z.-T. A new approach to enantiopure inherently chiral calix[4]arenes: determination of their absolute configurations. Org. Lett. 9, 4447–4450 (2007).
Hodson, L. et al. Facile synthesis of a C4-symmetrical inherently chiral calix[4]arene. Chem. Commun. 57, 11045–11048 (2021).
Iwamoto, K., Shimizu, H., Araki, K. & Shinkai, S. Syntheses and optical resolution of calix[4]arenes with molecular asymmetry. Systematic classification of all possible chiral isomers derivable from calix[4]arene. J. Am. Chem. Soc. 115, 3997–4006 (1993).
Herbert, S. A., van Laeren, L. J., Castell, D. C. & Arnott, G. E. Inherently chiral calix[4]arenes via oxazoline directed ortholithiation: synthesis and probe of chiral space. Beilstein J. Org. Chem. 10, 2751–2755 (2014).
Tang, M. & Yang, X. Catalytic enantioselective synthesis of inherently chiral molecules: recent advances. Eur. J. Org. Chem. 26, e202300738 (2023).
Browne, J. K., McKervey, M. A., Pitarch, M., Russell, J. A. & Millership, J. S. Enzymatic synthesis of nonracemic inherently chiral calix[4]arenes by lipase-catalysed transesterification. Tetrahedron Lett. 39, 1787–1790 (1998).
Zhang, Y.-Z., Xu, M.-M., Si, X.-G., Hou, J.-L. & Cai, Q. Enantioselective synthesis of inherently chiral calix[4]arenes via palladium-catalyzed asymmetric intramolecular C–H arylations. J. Am. Chem. Soc. 144, 22858–22864 (2022).
Zhang, X., Tong, S., Zhu, J. & Wang, M.-X. Inherently chiral calixarenes by a catalytic enantioselective desymmetrizing cross-dehydrogenative coupling. Chem. Sci. 14, 827–832 (2023).
Jiang, Y.-K. et al. Organocatalytic enantioselective synthesis of inherently chiral calix[4]arenes. Angew. Chem. Int. Ed. 63, e202407752 (2024).
Yu, S. et al. Catalytic enantioselective synthesis of inherently chiral calix[4]arenes via sequential Povarov reaction and aromatizations. Angew. Chem. Int. Ed. 63, e202410628 (2024).
Tong, S. et al. CatalytiC Enantioselective Synthesis and Switchable Chiroptical Property of Inherently Chiral Macrocycles. J. Am. Chem. Soc. 142, 14432–14436 (2020).
Jiang, Y.-F., Tong, S., Zhu, J. & Wang, M.-X. Inherently chiral nor-heteracalixarenes: design and synthesis via enantioselective intramolecular Suzuki–Miyaura reaction. Chem. Sci. 15, 12517–12522 (2024).
Li, X.-C., Cheng, Y., Wang, X.-D., Tong, S. & Wang, M.-X. De novo synthesis of inherently chiral heteracalix[4]aromatics from enantioselective macrocyclization enabled by chiral phosphoric acid-catalyzed intramolecular SNAr reaction. Chem. Sci. 15, 3610–3615 (2024).
Ishibashi, K., Tsue, H., Takahashi, H. & Tamura, R. Azacalix[4]arene tetramethyl ether with inherent chirality generated by substitution on the nitrogen bridges. Tetrahedron 20, 375–380 (2009).
Lu, Q.-L., Wang, X.-D., Tong, S., Zhu, J. & Wang, M.-X. Catalytic enantioselective synthesis of inherently chiral macrocycles by dynamic kinetic resolution. ACS Catal. 14, 5140–5146 (2024).
Mao, Y.-Q., Zhang, Y., Tong, S., Zhu, J. & Wang, M.-X. Asymmetric synthesis of enantioenriched zigzag-type molecular belts by kinetic resolution and subsequent transformations. Org. Lett. 25, 3936–3940 (2023).
Zhang, L. et al. Enantioselective electrosynthesis of inherently chiral calix[4]arenes via a cobalt-catalyzed aryl C–H acyloxylation. Green. Chem. 26, 10232–10239 (2024).
Li, T. et al. Simultaneous construction of inherent and axial chirality by cobalt-catalyzed enantioselective C-H activation of calix[4]arenes. Nat. Commun. 15, 7673 (2024).
Qian, P.-F. et al. Asymmetric synthesis of chiral calix[4]arenes with both inherent and axial chirality via cobalt-catalyzed enantioselective intermolecular C−H annulation. Angew. Chem. Int. Ed. 63, e202412459 (2024).
Luo, H.-Y. et al. Chiral selenide/achiral sulfonic acid cocatalyzed atroposelective sulfenylation of biaryl phenols via a desymmetrization/kinetic resolution sequence. J. Am. Chem. Soc. 144, 2943–2952 (2022).
Yang, J. et al. Chiral phosphoric acid-catalyzed remote control of axial chirality at boron–carbon bond. J. Am. Chem. Soc. 143, 12924–12929 (2021).
Miyaji, R., Asano, K. & Matsubara, S. Bifunctional organocatalysts for the enantioselective synthesis of axially chiral isoquinoline N-oxides. J. Am. Chem. Soc. 137, 6766–6769 (2015).
Mori, K. et al. Enantioselective synthesis of multisubstituted biaryl skeleton by chiral phosphoric acid catalyzed desymmetrization/kinetic resolution sequence. J. Am. Chem. Soc. 135, 3964–3970 (2013).
Gustafson, J. L., Lim, D. & Miller, S. J. Dynamic kinetic resolution of biaryl atropisomers via peptide-catalyzed asymmetric bromination. Science 328, 1251–1255 (2010).
Bao, H., Chen, Y. & Yang, X. Catalytic asymmetric synthesis of axially chiral diaryl ethers through enantioselective desymmetrization. Angew. Chem. Int. Ed. 62, e202300481 (2023).
Ye, Z., Xie, W., Wang, D., Liu, H. & Yang, X. Atroposelective synthesis of diarylamines via organocatalyzed electrophilic amination. ACS Catal. 14, 4958–4967 (2024).
Zhu, D. et al. Enantioselective synthesis of planar-chiral sulfur-containing cyclophanes by chiral sulfide catalyzed electrophilic sulfenylation of arenes. Angew. Chem. Int. Ed. 63, e202318625 (2024).
Wang, D., Shao, Y.-B., Chen, Y., Xue, X.-S. & Yang, X. Enantioselective synthesis of planar-chiral macrocycles through asymmetric electrophilic aromatic amination. Angew. Chem. Int. Ed. 61, e202201064 (2022).
Yu, S., Bao, H., Zhang, D. & Yang, X. Kinetic resolution of substituted amido[2.2]paracyclophanes via asymmetric electrophilic amination. Nat. Commun. 14, 5239 (2023).
Liu, X. et al. Enantioselective synthesis of [4]helicenes by organocatalyzed intermolecular C-H amination. Nat. Commun. 15, 732 (2024).
Zhang, X.-Y. et al. Enantioselective synthesis of inherently chiral sulfur-containing calix[4]arenes via chiral sulfide catalyzed desymmetrizing aromatic sulfenylation. Nat. Commun. 15, 9929 (2024).
Akiyama, T., Itoh, J., Yokota, K. & Fuchibe, K. Enantioselective Mannich-type reaction catalyzed by a chiral brønsted acid. Angew. Chem. Int. Ed. 43, 1566–1568 (2004).
Uraguchi, D. & Terada, M. Chiral Brønsted acid-catalyzed direct Mannich reactions via electrophilic activation. J. Am. Chem. Soc. 126, 5356–5357 (2004).
Akiyama, T. Stronger brønsted acids. Chem. Rev. 107, 5744–5758 (2007).
Parmar, D., Sugiono, E., Raja, S. & Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived brønsted acid and metal catalysis: history and classification by mode of activation; brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem. Rev. 114, 9047–9153 (2014).
Xu, Y., Zhai, T.-Y., Xu, Z. & Ye, L.-W. Recent advances towards organocatalytic enantioselective desymmetrizing reactions. Trends Chem. 4, 191–205 (2022).
Borissov, A. et al. Organocatalytic enantioselective desymmetrisation. Chem. Soc. Rev. 45, 5474–5540 (2016).
Wang, D., Jiang, Q. & Yang, X. Atroposelective synthesis of configurationally stable nonbiaryl N–C atropisomers through direct asymmetric aminations of 1,3-benzenediamines. Chem. Commun. 56, 6201–6204 (2020).
Zhang, H.-H., Li, T.-Z., Liu, S.-J. & Shi, F. Catalytic Asymmetric Synthesis of Atropisomers Bearing Multiple Chiral Elements: An Emerging Field. Angew. Chem. Int. Ed. 63, e202311053 (2024).
Gaucherand, A. et al. Enantioselective synthesis of molecules with multiple stereogenic elements. Chem. Soc. Rev. 53, 11165–11206 (2024).
LaPlante, S. R. et al. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem. 54, 7005–7022 (2011).
Liu, W. & Yang, X. Recent advances in (dynamic) kinetic resolution and desymmetrization catalyzed by chiral phosphoric acids. Asian J. Org. Chem. 10, 692–710 (2021).
Chen, Y., Zhu, C., Guo, Z., Liu, W. & Yang, X. Asymmetric synthesis of hydroquinolines with α,α-disubstitution through organocatalyzed kinetic resolution. Angew. Chem. Int. Ed. 60, 5268–5272 (2021).
Xia, Z.-L., Zheng, C., Xu, R.-Q. & You, S.-L. Chiral phosphoric acid catalyzed aminative dearomatization of α-naphthols/Michael addition sequence. Nat. Commun. 10, 3150 (2019).
Tlustý, M., Dvořáková, H., Čejka, J., Kohout, M. & Lhoták, P. Regioselective formation of the quinazoline moiety on the upper rim of calix[4]arene as a route to inherently chiral systems. N. J. Chem. 44, 6490–6500 (2020).
Tlustý, M., Slavík, P., Kohout, M., Eigner, V. & Lhoták, P. Inherently chiral upper-rim-bridged calix[4]arenes possessing a seven membered ring. Org. Lett. 19, 2933–2936 (2017).
Wang, S.-G., Yin, Q., Zhuo, C.-X. & You, S.-L. Asymmetric dearomatization of β-naphthols through an amination reaction catalyzed by a chiral phosphoric acid. Angew. Chem. Int. Ed. 54, 647–650 (2015).
Changotra, A., Das, S. & Sunoj, R. B. Reversing enantioselectivity using noncovalent interactions in asymmetric dearomatization of β-naphthols: the power of 3,3′ substituents in chiral phosphoric acid catalysts. Org. Lett. 19, 2354–2357 (2017).
Liu, S.-J. et al. Rational design of axially chiral styrene-based organocatalysts and their application in catalytic asymmetric (2+4) cyclizations. Angew. Chem. Int. Ed. 61, e202112226 (2022).
Liu, C., Zhu, Q., Huang, K.-W. & Lu, Y. Primary amine/CSA ion pair: a powerful catalytic system for the asymmetric enamine catalysis. Org. Lett. 13, 2638–2641 (2011).
Acknowledgements
The authors thank the NSFC (grant Nos. 22171186 X.Y., 22222107 X.Y.) and ShanghaiTech University start-up funding for financial support. Prof. Guangxin Liang was thanked for sharing the optical rotation polarimeter. The authors acknowledge the support from the Analytical Instrumentation Center (# SPST-AIC10112914), SPST, ShanghaiTech University, and Dr. Na Yu for the X-ray crystallographic analysis.
Author information
Authors and Affiliations
Contributions
M.Y. performed most of the experimental studies. W.X. performed the studies on the configurational stability of the C–N atropoisomers. S.Y. and T.L. prepared some of the substrates. X.Y. directed the project and wrote the paper with the feedback from other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Gareth E. Arnott, Jun-Long Niu and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yuan, M., Xie, W., Yu, S. et al. Catalytic enantioselective synthesis of inherently chiral calix[4]arenes via organocatalyzed aromatic amination enabled desymmetrization. Nat Commun 16, 3943 (2025). https://doi.org/10.1038/s41467-025-59221-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59221-3
