
Abstract
The insertion of either C−H bond or C−O bond via bond cleavage has proven to be a very attractive strategy for the construction of C−C and C−O bonds in organic synthesis. However, such divergent catalytic asymmetric reactions for the selective formation of C(sp3)−H insertion and formal C(sp3)−O insertion products from the same precursors are rarely explored. Herein, we report a ligand-controlled divergent asymmetric C(sp3)−H insertion and formal C(sp3)−O insertion reaction via vinyl cations by a non-diazo approach, leading to the practical and atom-economical assembly of a range of chiral spiro and fused polycyclic pyrroles in generally moderate to excellent yields with generally excellent chemo- and enantioselectivities. Importantly, this protocol not only represents a rare example of successful ligand-controlled asymmetric divergent insertion reaction, but also constitutes an enantioselective 1,6-C−H insertion and an asymmetric carbenoid insertion into acetals via a non-diazo approach.
Similar content being viewed by others
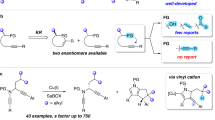
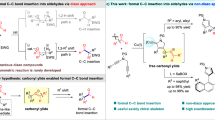
Introduction
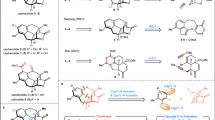
Catalytic enantioselective transformations of C−H bond and C−O bond of O-containing molecules have emerged as powerful tools in organic synthesis over the past decades1,2,3. Among these, the metal carbene-involved asymmetric C(sp3)−H bond insertion4,5,6,7,8,9,10,11 and C(sp3)−O bond insertion12,13,14,15,16,17 have attracted particular attention because this approach provides rapid, efficient, and economical ways for the assembly of chiral O-containing molecules, especially the biologically significant O-heterocycles. In this respect, compared to the well-developed asymmetric metal carbene insertion into C(sp3)−H bond by 1,5-H shift18,19,20,21,22,23,24,25,26,27,28,29,30,31,32, the related asymmetric 1,6-C−H insertions are difficult due to the kinetic favorability of 1,5-C–H insertion, and thus have been less explored (Fig. 1a)33,34,35,36,37,38,39,40. Moreover, the relevant Stevens-type rearrangement, which led to the formal C−O insertion products via oxonium ylide intermediates, would occur simultaneously in these 1,6-C−H insertion reactions, thereby resulting in low chemoseletivities of C−H insertion and C−O insertion products (Fig. 1a)33,34,35,36,37,38,39,40. To date, the controllable highly chemo- and enantioselective 1,6-C−H insertion and the related Stevens-type rearrangement from the same oxygen-containing starting materials remains unexplored. Alternatively, we suppose that the highly reactive oxygen-containing substrates such as acetals could be potentially applicable to the divergent asymmetric C(sp3)−H insertion and formal C(sp3)−O insertion41,42,43. For instance, Doyle and co-workers reported the intramolecular divergent asymmetric insertion of rhodium carbenoids into acetals via C(sp3)−H insertion and formal C(sp3)−O insertion by using diazoesters as carbene precursors, but only poor chemoselectivities (12/88–78/22) and generally moderate enantioselectivities (85–99% ees; 33–66% ees) were observed (Fig. 1b)41. In addition, Davies and co-workers also demonstrated the related intermolecular asymmetric insertion of rhodium carbenoids into acetals, affording the corresponding C(sp3)−H insertion and formal C(sp3)−O insertion products, respectively, with generally moderate enantioselectivities (68–91% ees; 0–47% ees) and with poor chemoselectivities (31/69–43/57) in some cases (Fig. 1b)42. Thus, the development of emerging approaches for divergent asymmetric insertion is highly desirable, especially those with excellent chemo- and enantioselectivities and via a non-diazo approach.

a Asymmetric C(sp3)−H and formal C(sp3)−O insertion via metal carbenes. b Divergent asymmetric carbenoid insertion into acetals. c This work: ligand-controlled divergent asymmetric insertion via vinyl cations.
In order to address the issue of asymmetric divergent insertion reactions mentioned above, we proposed that the vinyl cation chemistry could be an appropriate option due to the unique carbene-like reactivity of vinyl cations44,45. Recently, our group has developed a variety of chiral copper-catalyzed asymmetric transformations through vinyl cation intermediates via a remote control of enantioselectivity46,47,48,49,50,51,52,53,54,55,56,57, including intramolecular aromatic C(sp2)−H functionalization46, vinylic C(sp2)−H functionalization48, C(sp3)−H functionalization by kinetic resolution54, cyclopropanation46, [1,2]-Stevens-type rearrangement49,53, one-carbon ring expansion55, dearomatization56, intermolecular annulations with styrenes47 and ketones50, oxidation and X−H insertion51, as well as formal C−C bond insertion into aldehydes52. Inspired by these results and our recent study on developing ynamide chemistry for heterocycle synthesis58,59,60,61,62,63,64, we envisioned that the acetal group might trap the vinyl cation intermediates A, formed through copper-catalyzed cyclization of N-propargyl ynamides65,66,67, to afford intermediates B and C, eventually leading to the corresponding chiral spiro and fused polycyclic pyrroles via 1,6-H shift and 1,2-rearrangement, respectively (Fig. 1c). In this article, we report the results of our studies on the ligand-controlled divergent asymmetric insertion through copper-catalyzed diyne cyclization via vinyl cations, allowing the practical and atom-economical assembly of a range of chiral spiro and fused polycyclic pyrroles in generally moderate to excellent yields with generally excellent chemo- and enantioselectivities (Fig. 1c). To the best of our knowledge, this protocol not only represents a rare example of successful ligand-controlled asymmetric divergent insertion reaction, but also constitutes an enantioselective 1,6-C−H insertion and an asymmetric carbenoid insertion into acetals via a non-diazo approach.
Results
Screening of reaction conditions
At the outset, the acetal-substituted N-propargyl ynamide 1a was selected as the model substrate to probe the possible reaction pathway (Table 1). The reaction was first examined in the presence of Cu(CH3CN)4PF6 (10 mol %), various chiral ligands, NaBArF4 (12 mol %), and DCM as the solvent at 30 °C. By employing (S)-BINAP (L1) as the chiral ligand, the C(sp3)−H insertion product 2a and formal C(sp3)−O insertion product 3a were formed with poor chemo- and enantio- selectivities (entry 1). In addition, other bisphosphine ligands were found to be less effective for the reaction in terms of both chemo- and enantioselectivities (see the Supplementary Table 1). Then, we turned our attention to Tang’s sidearm-modified bisoxazoline ligands (SaBOX)68, which were successfully applied in our previous copper-catalyzed diyne cyclization46,47,48,49,50,51,52,53,54,55,56. To our delight, the C(sp3)−H insertion product 2a could be obtained as the major product with 22–71% ees when SaBOX ligands L2–L6 were used (entries 2–6). Subsequent solvent and temperature screening further enhanced the chemo- and enantioselectivities for this C−H insertion (entries 7–12). When SaBOX ligand L6 was employed as the chiral ligand in CHCl3/DCB = 1/3 at −30 °C for 80 h, the desired tricyclic pyrrole 2a could be afforded in 74% yield with 92% ee and 99/1 chemoselectivity of 2a/3a (entry 12). Interestingly, further modification of sidearms and skeletons on the SaBOX ligands L7–L10 led to the formal C(sp3)−O insertion product 3a as the main product with <5–73% ees (entries 13–16). In particular, similar screening of solvent and temperature (entries 17–19) revealed that the tetracyclic pyrrole 3a was delivered in 77% yield with 92% ee and 8/92 chemoselectivity of 2a/3a by using L10 as chiral ligand in DCE at 0 °C for 7 h (entry 19). Thus, the skeleton of chiral ligands contributed significantly to this asymmetric divergent insertion, exhibiting excellent chemoselectivity and enantioselectivity.

Reaction scope study
With the optimized reaction conditions in hand (Table 1, entry 12), we then explored the scope of this chiral copper-catalyzed C(sp3)−H insertion reaction. As depicted in Fig. 2, a variety of diynes 1 were well-tolerated to deliver the target chiral C(sp3)−H insertion products 2 in mostly good to excellent yields with excellent enantioselectivities. We first investigated the scope of ynamide moieties with different typical N-protecting groups, including Ts, SO2Mes, SO2Ph, Bs and Ms groups, and the desired chiral spiro tricyclic pyrroles 2a–2e were obtained in 59–85% yields with 82–94% ees. Subsequently, a wide range of various aryl-substituted N-propargyl ynamides bearing both electron-withdrawing groups such as F, Cl, Br, CN, NO2 and CF3 and electron-donating groups such as Me, OMe in positions 4 and 5 were studied, allowing the formation of the expected spirocyclic pyrroles 2f–2s in moderate to good yields with 85–94% ees. Of note, slight formation of C−O insertion product 3l (2l/3l = 92/8) was observed in case of MeO-substituted diyne 1l. In addition, the reaction proceeded smoothly for other electron-rich aniline-substituted N-propargyl ynamides 1t and 1u, delivering the corresponding products 2t (70%, 90% ee) and 2u (83%, 77% ee), respectively. However, the use of PMP-substituted diyne 1v (R1 = PMP) only led to the desired product 2v in 32% yield with 68% ee and 67/33 chemoselectivity of 2v/3r (see the Supplementary Fig. 1). It is notable that the reaction of the thiophene-linked diyne 1w only afforded the desired product 2w in low efficiency (17%) with 89% ee under the optimized conditions (see the Supplementary Fig. 2). Finally, it should be noted that attempts to extend this C−H insertion to the Me- and styryl-substituted diynes 1x–1y (R1 = Me, styryl) and diynes with non-electron-rich aryl groups only led to the formation of complicated mixtures under the optimal and related conditions (see the Supplementary Fig. 3). Importantly, unique chemoselectivities were achieved in all these cases except diynes 1l and 1v. Thus, this protocol not only constitutes a direct asymmetric C(sp3)−H insertion via vinyl cations, but also features an enantioselective 1,6-C−H insertion and an asymmetric insertion of metal carbenoids into acetals via a non-diazo approach.

Reaction conditions: 1 (0.15 mmol), Cu(CH3CN)4PF6 (0.015 mmol), L6 (0.018 mmol), NaBArF4 (0.018 mmol), CHCl3/DCB (0.5 mL/1.5 mL), −30 °C, N2, 61–168 h, in Schlenk tubes; yields were those for the isolated products; ees were determined by HPLC analysis. a2l/3l = 92/8. PG protecting group, Bs 4-bromobenzenesulfonyl. SO2Mes = 2,4,6-trimethylbenzenesulfonyl.
Next, we investigated the substrate scope of this enantioselective formal C(sp3)−O insertion using the same type of diynes under the optimized reaction conditions (Table 1, entry 19). As summarized in Fig. 3, this insertion reaction occurred smoothly with various substituted diynes 1, and the corresponding fused tetracyclic pyrroles 3 were formed in generally moderate to good yields with generally excellent enantioselectivities and excellent diastereoselectivities (>50:1 dr). Diynes with different sulfonyl protecting groups were first examined, and the expected polycyclic pyrroles 3a–3e were furnished in 47–80% yields with 90–92% ees. Additionally, the desired chiral products 3f–3m could be afforded in 47–70% yields with 90–98% ees by using different aryl-substituted N-propargyl ynamides bearing both electron-withdrawing and -donating substituents (R1 = aryl). In particular, ortho-substituted diynes were also tolerated, leading to the expected products 3n–3o with excellent enantioselectivities (91–95% ees) albeit in moderate yields (29–43%) probably due to the increased steric hindrance. Interestingly, other electron-rich aryl-substituted N-propargyl ynamides could be readily transformed into the corresponding products 3p–3s in 53–79% yields with 75–93% ees in this C−O insertion, and higher temperature (15 °C) was needed in the latter two cases (3r–3s). Moreover, this enantioselective insertion was also extended to the six-member-ring acetal-substituted diyne and the thiophene-linked diyne 1w, affording the desired chiral dioxepane product 3t (60%, 95% ee) and 3u (68%, 93% ee), respectively. Again, our attempts to expand this C−O insertion to the Me- and styryl-substituted diynes 1x–1y (R1 = Me, styryl) only led to complicated mixtures (see the Supplementary Fig. 3). Of note, ratios of 80/20–91/9 of 3/2 chemoselectivities were detected in some cases (3b–3c, 3f–3h, 3j–3m, 3q, 3s). Significantly, this approach represents a rare example of successful enantioselective formal C(sp3)−O insertion via ylide-type rearrangement69,70. The absolute configuration of product 3f was confirmed by X-ray diffraction (see the Supplementary Fig. S9).

Reaction conditions: 1 (0.15 mmol), Cu(CH3CN)4PF6 (0.015 mmol), L10 (0.018 mmol), NaBArF4 (0.018 mmol), DCE (2 mL), 0 °C, N2, 1–91 h, in Schlenk tubes; yields were those for the isolated products; ees were determined by HPLC analysis; ratios of 3/2 were measured by 1H NMR using 1,3,5-trimethoxybenzene as the internal standard. a30 °C. b15 °C. cBy using L11 as chiral ligand.
It is notable that attempts to develop the intermolecular asymmetric reaction of the phenyl-substituted acetals with diynes have been unsuccessful as yet (see the Supplementary Fig. 4).
Synthetic applications
To demonstrate the utility of this asymmetric divergent methodology, the preparative scale reaction was carried out under the standard reaction conditions, and the corresponding chiral C−H insertion product 2a and C−O insertion product 3a were obtained in 72% yield with 92% ee and in 70% yield with 92% ee and >50:1 dr, respectively Fig. 4). Further synthetic applications of these products were then explored (Fig. 4). For instance, the N-protecting group (Ts) of 2a could be easily removed by the treatment of KOH, affording the unprotected tricyclic pyrrole 4 in 89% yield. In addition, the Diels-Alder reaction of 2a with DMAD (dimethyl acetylene dicarboxylate) reagent led to the bridged product 5 in 73% yield with >20:1 dr. Interestingly, the 1,3-dioxolane ring of 2a could be opened to deliver product 6 in 89% yield under the acidic conditions. Moreover, the treatment of the NMe2 group of 2a with MeI, followed by the aryl Grignard reagent, could afford the corresponding Pd-catalyzed cross-coupling product 7 in 95% yield. Besides, NBS bromination of the pyrrole moiety of 7 and subsequent nucleophilic attack by MeOH led to the corresponding product 8 in 96% yield with 5:1 dr. Moreover, the bridged product 9 could be formed in 97% yield with 2.7:1 dr via [4 + 2] cycloaddition between the pyrrole ring of 7 and the in situ generated benzyne. Further selective hydrogenation of the double bond on the bridged-ring of 9 with Pd/C under H2 atmosphere (5 MPa) allowed the formation of product 10 in 67% yield with excellent dr (>20 :1). To our delight, the acetal moiety of 10 could be readily cleaved into the carbonyl group in the presence of TsOH•H2O, furnishing the desired chiral ketone 11 in 97% yield bearing five continuous stereocenters. Of note, attempts to directly cleave the acetal moiety of 2a or 4−5 or 7−9 into the carbonyl group were unsuccessful yet probably due to the aromatization-driven force. The absolute configurations of products 5 and 11 were unambiguously confirmed by X-ray diffraction (see the Supplementary Figs. S10 and S11), which also determined the absolute configuration of chiral C(sp3)−H insertion products 2. Next, several related transformations of product 3a were also investigated. It was found that the deprotection of 3a, followed by Boc protection, could afford the expected tetracyclic pyrrole 13 in excellent yield (90%, 2 steps). Meanwhile, selective bromination of the pyrrole moiety of 12 with DBDMH (1,3-dibromo-5,5-dimethylhydantoin) led to the monobrominated product 14 in 81% yield. Importantly, almost no erosion of the enantiopurity of the compounds was detected in all these transformations.

Reagents and conditions: (i) KOH (5 equiv), EtOH/THF = 1/1, 30 °C, 9 h. (ii) DMAD (10 equiv), toluene, 100 °C, 59 h. (iii) TsOH•H2O (40 mol %), acetone/H2O = 9/1, 60 °C, 12 h. (iv) MeI (10 equiv), MeCN, 80 °C, 2 h. (v) Pd(PPh3)2Cl2 (5 mol %), PhMgBr (5 equiv), THF, 30 °C, N2, 5 min. (vi) NBS (1 equiv), MeOH/MeCN = 1/4, −25 °C, 5 min. (vii) 2-(trimethylsilyl)phenyl triflate (2 equiv), CsF (3 equiv), MeCN, 40 °C, N2, 2 h. (viii) H2 (5 MPa), Pd/C (20 mol %), EtOH/DCM = 4/1, 50 °C, 72 h. (ix) TsOH•H2O (2 equiv), acetone/H2O/DCE = 9/1/1, 80 °C, 12 h. (x) Boc2O (4 equiv), 4-DMAP (20 mol %), Et3N (4 equiv), DCM, 0 °C–rt, 2 h. (xi) DBDMH (0.6 equiv), THF, 0 °C, 5 min.
Mechanistic investigations
To probe the reaction mechanism, several control experiments were also conducted. A deuterium labeling experiment with the substrate [D]-1a was first carried out, and it was found that the deuterium atom was completely retained in prodcuts [D]-2a and [D]−3a under the related standard conditions, respectively (Fig. 5a, see the Supplementary Figs. S5 and S6). Then, the kinetic isotope effect (KIE) experiment was performed with the mixture of diynes 1a and [D]−1a, and the result (kH/kD = 2.7) suggests that the cleavage of C(sp3)–H bond might be the rate-determining step in this chiral copper-catalyzed C(sp3)−H insertion reaction (Fig. 5b, see the Supplementary Figs. S7 and S8).

a The reaction of [D]−1a under the related standard conditions. b KIE experiments.

a Free energy diagram for the formation of C(sp3)−H insertion product 2a. b Free energy diagram for the formation of C(sp3)−O insertion product 3a. Relative free energies (ΔG, in kcal/mol) were computed at the B3LYP-D3(BJ)/6-311 + + G(d,p)-SDD-PCM(CHCl3/DCB = 1/3)//B3LYP-D3/6-31 G(d)-LANL2DZ level of theory and B3LYP-D3(BJ)/6-311 + + G(d,p)-SDD-PCM(DCE)//B3LYP-D3/6-31 G(d)-LANL2DZ level of theory.

The geometries and relative free energies (ΔΔG, in kcal/mol) of the transition states CuL6-(S)-TSC1/CuL6-(R)-TSC1 and CuL10-(S)-TSC2/CuL10-(R)-TSC2 with the chiral ligand L6 and L10. All hydrogen atoms are omitted for clarity except for those involved in critical interactions. Relative free energies (ΔΔG, in kcal/mol) were computed at the B3LYP-D3(BJ)/6-311 + + G(d,p)-SDD-PCM(CHCl3/DCB = 1/3)//B3LYP-D3/6-31 G(d)-LANL2DZ level of theory and B3LYP-D3(BJ)/6-311 + + G(d,p)-SDD-PCM(DCE)//B3LYP-D3/6-31 G(d)-LANL2DZ level of theory. Color code: red = O; white = H; gray = C; yellow = S; blue = N; brown = Cu.
On the basis of the aforementioned experimental observations, our previous studies46,47,48,49,50,51,52,53,54,55,56 and comprehensive computational analysis (see the Supplementary Tables 2, 3 and Supplementary Data 1), the free energy diagram of the reaction from 1a to 2a and 3a in different mechanisms and catalyzed by Cu(I) species coordinated by different ligands (L6/L10) is exhibited in Fig. 6. At the beginning, the reaction is initialized via a preferential coordination of the Cu(I) catalyst to activate the electron-richer amide-tethered C≡C bond of 1a to produce the precursor A, followed by an intramolecular cyclization to afford the vinyl cation intermediate B. Starting from the vinyl cation intermediate B, the reaction can proceed via two distinct pathways, each yielding a different product. In path a, the intermediate B undergoes a formal vinyl cation insertion into the C−H bond (i.e., a 1,6-hydride shift followed by an electrophilic cyclization) resulting in the copper carbene intermediate D1. In path b, the intermediate B undergoes a nucleophilic addition to form the carbocation intermediate C2, followed by an electrophilic cyclization leading to a structurally different copper carbene intermediate D2. In both pathways, the formation of the copper carbene intermediates D1 and D2 is followed by a base (1a)-assisted formal 1,4-hydride shift, as previously reported56, leading to the final products 2a and 3a, respectively. Analysis of the free energy diagram disclosed that the reaction mechanism and resulting products are modulated by different ligand-coordinated Cu(I) catalysts. When the reaction is catalyzed by L6-ligated Cu(I) species, path a is energetically more favorable, with the rate-determining step being the vinyl cation insertion into the C−H bond, undergoing a free energy barrier of 13.0 kcal/mol and leading to product 2a. This result is also consistent with the above KIE experiment (b). In contrast, when the reaction is catalyzed by L10-ligated Cu(I) species, path b is more favorable, with the rate-determining step being the nucleophilic cyclization step, with a barrier height of 8.3 kcal/mol, resulting in product 3a. The final products of the reaction catalyzed by L6/L10-ligated Cu(I) species exhibited by free energy diagram above are in line with the final products obtained in the aforementioned experimental works.
The enantio-determining step for the formation of chiral products 2a and 3a was then studied on the basis of theoretical calculations by using chiral ligands L6 and L10 coordinated to the Cu(I) center in this irreversible enantio-determining electrophilic cyclization process (Fig. 7). In the light of further inspection of the structures of these enantio-determining transition states, it is found that there is a significant C−H···π interaction between the substrate and the bulky group of branched phenyl group of L6 in [CuL6]-(S)-TSC1, that stabilizes the transition state, and thus accounts for the free energy difference of 3.8 kcal/mol between the two enantio-determining transition states, eventually leading to the enantioselectivity of C(sp3)−H insertion product 2a. Likewise, there is a quite strong π···π interaction between the substrate and the branched phenyl group of L10 in [CuL10]-(S)-TSC2, stabilizing the transition state, resulting in the energy difference of 2.0 kcal/mol between the two enantio-determining transition states, and giving rise to the enantioselectivity of formal C(sp3)−O insertion product 3a.
Discussion
In summary, a ligand-controlled divergent asymmetric C(sp3)−H insertion and formal C(sp3)−O insertion reaction by chiral copper-catalyzed diyne cyclization via vinyl cations has been developed. This non-diazo approach enables the practical and atom-economical assembly of a variety of chiral spiro and fused polycyclic pyrroles in generally moderate to excellent yields with generally excellent chemo- and enantioselectivities. Importantly, this protocol not only represents a rare example of successful ligand-controlled asymmetric divergent insertion reaction, but also constitutes an enantioselective 1,6-C−H insertion via a non-diazo approach. Furthermore, computational studies have been carried out to understand the asymmetric divergent reaction mechanism, and the origin of chemo- and stereoselectivity. We anticipate that this method will offer further perspectives and explorations for asymmetric divergent synthesis, insertion reaction, and vinyl cation chemistry.
Methods
General
For 1H, 13C and 19F nuclear magnetic resonance (NMR) spectra of compounds in this manuscript and details of the synthetic procedures as well as more reaction conditions screening, see Supplementary Information.
General procedure for the synthesis of chiral spirocyclic 1,3-dioxolanes 2
To a dry 10 mL Schlenk tube charged with a stir bar were added Cu(CH3CN)4PF6 (5.6 mg, 0.015 mmol), L6 (9.8 mg, 0.018 mmol), NaBArF4 (16.0 mg, 0.018 mmol) and CHCl3/DCB (0.25 mL/0.75 mL) sequentially under N2 atmosphere. The solution was stirred at room temperature for 2 h. After cooling to −30 °C, the solution of N-propargyl ynamide 1 (0.15 mmol) in CHCl3/DCB (0.25 mL/0.75 mL) was added into the reaction dropwise. The resulting mixture was stirred at −30 °C for 61–168 h and the progress of the reaction was monitored by TLC. Upon completion, the resulting mixture was concentrated under reduced pressure and purified by column chromatography on silica gel (eluent: hexanes/ethyl acetate) to afford the desired chiral spirocyclic 1,3-dioxolane 2.
General procedure for the synthesis of chiral fused polycyclic 1,4-dioxanes 3
To a dry 10 mL Schlenk tube charged with a stir bar were added Cu(CH3CN)4PF6 (5.6 mg, 0.015 mmol), L10 (16.4 mg, 0.018 mmol), NaBArF4 (16.0 mg, 0.018 mmol) and DCE (1 mL) sequentially under N2 atmosphere. The solution was stirred at room temperature for 2 h. After cooling to 0 °C, the solution of N-propargyl ynamide 1 (0.15 mmol) in DCE (1 mL) was added into the reaction dropwise. The resulting mixture was stirred at 0 °C for 1–91 h and the progress of the reaction was monitored by TLC. Upon completion, the resulting mixture was concentrated under reduced pressure and purified by column chromatography on silica gel (eluent: hexanes/ethyl acetate) to afford the desired chiral fused polycyclic 1,4-dioxane 3.
Data availability
Data for the crystal structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition numbers CCDC 2383734 (3 f), 2383735 (5), 2383736 (11). Copies of these data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study, including experimental procedures and compound characterization, are available within the paper and its Supplementary Information files, or from the corresponding authors on request.
References
Ghorai, D., Tóth, B. L., Lanzi, M. & Kleij, A. W. Vinyl and alkynyl substituted heterocycles as privileged scaffolds in transition metal promoted stereoselective synthesis. Acc. Chem. Res. 57, 726–738 (2024).
Zhang, Q., Wu, L.-S. & Shi, B.-F. Forging C–heteroatom bonds by transition-metal-catalyzed enantioselective C–H functionalization. Chem 8, 384–413 (2022).
Woźniak, L. et al. Catalytic enantioselective functionalizations of C–H bonds by chiral iridium complexes. Chem. Rev. 120, 10516–10543 (2020).
Liu, Z., Sivaguru, P., Zanoni, G. & Bi, X. N‑triftosylhydrazones: a new chapter for diazo-based carbene chemistry. Acc. Chem. Res. 55, 1763–1781 (2022).
He, Y. et al. Recent advances in transition-metal-catalyzed carbene insertion to C–H bonds. Chem. Soc. Rev. 51, 2759–2852 (2022).
Zhu, D., Chen, L., Fan, H., Yao, Q. & Zhu, S. Recent progress on donor and donor–donor carbenes. Chem. Soc. Rev. 49, 908–950 (2020).
Lombard, F. J. & Coster, M. J. Rhodium(II)-catalysed intramolecular C–H insertion α- to oxygen: reactivity, selectivity and applications to natural product synthesis. Org. Biomol. Chem. 13, 6419–6431 (2015).
Yamaguchi, J., Yamaguchi, A. D. & Itami, K. C−H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed. 51, 8960–9009 (2012).
Doyle, M. P., Duffy, R., Ratnikov, M. & Zhou, L. Catalytic carbene insertion into C–H bonds. Chem. Rev. 110, 704–724 (2010).
Slattery, C. N., Ford, A. & Maguire, A. R. Catalytic asymmetric C−H insertion reactions of α-diazocarbonyl compounds. Tetrahedron 66, 6681–705 (2010).
Davies, H. M. L. & Beckwith, R. E. J. Catalytic enantioselective C–H activation by means of metal-carbenoid-induced C–H insertion. Chem. Rev. 103, 2861–2904 (2003).
Shi, C.-Y., Zhou, B., Teng, M.-Y. & Ye, L.-W. Recent advances in asymmetric [1,2]-Stevens-type rearrangement via metal carbenes. Synthesis 55, 2118–2127 (2023).
Jana, S., Guo, Y. & Koenigs, R. M. Recent perspectives on rearrangement reactions of ylides via carbene transfer reactions. Chem. Eur. J. 27, 1270–1281 (2021).
Empel, C., Jana, S. & Koenigs, R. M. Advances in [1,2]-sigmatropic rearrangements of onium ylides via carbene transfer reactions. Synthesis 53, 4567–4587 (2021).
Ford, A. et al. Modern organic synthesis with α‑diazocarbonyl compounds. Chem. Rev. 115, 9981–10080 (2015).
Murphy, G. K., Stewart, C. & West, F. G. Intramolecular generation and rearrangement of oxonium ylides: methodology studies and their application in synthesis. Tetrahedron 69, 2667–2686 (2013).
Sweeney, J. B. Sigmatropic rearrangements of ‘onium’ ylids. Chem. Soc. Rev. 38, 1027–1038 (2009).
Dishman, S. N., Laconsay, C. J., Fettinger, J. C., Tantillo, D. J. & Shaw, J. T. Divergent stereochemical outcomes in the insertion of donor/donor carbenes into the C–H bonds of stereogenic centers. Chem. Sci. 13, 1030–1036 (2022).
Qiu, S., Gao, X. & Zhu, S. Dirhodium(II)-catalysed cycloisomerization of azaenyne: rapid assembly of centrally and axially chiral isoindazole frameworks. Chem. Sci. 12, 13730–13736 (2021).
Zhang, X. et al. Catalytic asymmetric C(sp3)–H carbene insertion approach to access enantioenriched 3‑fluoroalkyl 2,3-dihydrobenzofurans. ACS Catal. 11, 14293–14301 (2021).
Murai, T. et al. Conformational control in dirhodium(II) paddlewheel catalysts supported by chalcogen-bonding interactions for stereoselective intramolecular C–H insertion reactions. ACS Catal. 11, 568–578 (2021).
Dydio, P. & Key, H. M. et al. An artificial metalloenzyme with the kinetics of native enzymes. Science 354, 102–106 (2016).
Zhu, D. et al. Enantioselective intramolecular C–H insertion of donor and donor/donor carbenes by a nondiazo approach. Angew. Chem. Int. Ed. 55, 8452–8456 (2016).
Lu, P. & Mailyan, A. et al. Enantioselective synthesis of (–)-maoecrystal V by enantiodetermining C–H functionalization. J. Am. Chem. Soc. 136, 17738–17749 (2014).
Soldi, C. et al. Enantioselective intramolecular C–H insertion reactions of donor–donor metal carbenoids. J. Am. Chem. Soc. 136, 15142–15145 (2014).
Boultadakis-Arapinis, M., Lemoine, P., Turcaud, S., Micouin, L. & Lecourt, T. Rh(II) carbene-promoted activation of the anomeric C–H bond of carbohydrates: a stereospecific entry toward α- and β-ketopyranosides. J. Am. Chem. Soc. 132, 15477–15479 (2010).
Takeda, K., Oohara, T., Anada, M., Nambu, H. & Hashimoto, S. A polymer-supported chiral dirhodium(II) complex: highly durable and recyclable catalyst for asymmetric intramolecular C–H insertion reactions. Angew. Chem. Int. Ed. 49, 6979–6983 (2010).
Doyle, M. P., Hu, W., Wee, A. G. H., Wang, Z. & Duncan, S. C. Influences of catalyst configuration and catalyst loading on selectivities in reactions of diazoacetamides. Barrier to equilibrium between diastereomeric conformations. Org. Lett. 5, 407–410 (2003).
Saito, H. et al. 2-Aryl-3-methoxycarbonyl-2,3-dihydrobenzofurans via the Rh(II)-catalyzed C–H insertion process. Org. Lett. 4, 3887–3890 (2002).
Davies, H. M. L., Grazini, M. V. A. & Aouad, E. Asymmetric intramolecular C–H insertions of aryldiazoacetates. Org. Lett. 3, 1475–1477 (2001).
Doyle, M. P. et al. Diastereocontrol for highly enantioselective carbon-hydrogen insertion reactions of cycloalkyl diazoacetates. J. Am. Chem. Soc. 116, 4507–4508 (1994).
Doyle, M. P., Oeveren, A., Westrum, L. J., Protopopova, M. N. & Clayton, T. W. Jr. Asymmetric synthesis of lactones with high enantioselectivity by intramolecular carbon-hydrogen insertion reactions of alkyl diazoacetates catalyzed by chiral rhodium(II) carboxamides. J. Am. Chem. Soc. 113, 8982–8984 (1991).
Bergstrom, B. D., Merrill, A. T., Fettinger, J. C., Tantillo, D. J. & Shaw, J. T. Divergent asymmetric synthesis of panowamycins, TM−135, and veramycin F using C–H insertion with donor/donor carbenes. Angew. Chem. Int. Ed. 61, e202203072 (2022).
Han, F. et al. Cyclometalated chiral-at-ruthenium catalyst for enantioselective ring-closing C(sp3)–H carbene insertion to access chiral flavanones. ACS Catal. 12, 10304–10312 (2022).
Nickerson, L. A. et al. Enantioselective synthesis of isochromans and tetrahydroisoquinolines by C–H insertion of donor/donor carbenes. Chem. Sci. 11, 494–498 (2020).
Ito, M. et al. Diastereo- and enantioselective intramolecular 1,6-C–H insertion reactions of α-diazo esters catalyzed by chiral dirhodium(II) carboxylates. Tetrahedron Lett 56, 1397–1400 (2015).
Ye, T., García, F. C. & McKervey, M. A. Chemoselectivity and stereoselectivity of cyclisation of adiazocarbonyls leading to oxygen and sulfur heterocycles catalysed by chiral rhodium and copper catalysts. J. Chem. Soc. Perkin Trans 1, 1373–1379 (1995).
McKervey, M. A. & Ye, T. Asymmetric synthesis of substituted chromanones via C–H insertion reactions of α-diazoketones catalysed by homochiral rhodium(II) carboxylates. J. Chem. Soc., Chem. Commun. 11, 823–824 (1992).
Eberlein, T. H., West, F. G. & Tester, R. W. The Stevens [1,2]-shift of oxonium ylides: a route to substituted tetrahydrofuranones. J. Org. Chem. 57, 3479–3482 (1992).
McCarthy, N. et al. A new rhodium(II) phosphate catalyst for diazocarbonyl reactions including asymmetric synthesis. Tetrahedron Lett. 33, 5983–5986 (1992).
Doyle, M. P., Ene, D. G., Forbes, D. C. & Tedrow, J. S. Highly enantioselective oxonium ylide formation and Stevens rearrangement catalyzed by chiral dirhodium(II) carboxamidates. Tetrahedron Lett. 38, 4367–4370 (1997).
Davies, H. M. L., Yang, J. & Nikolai, J. Asymmetric C–H insertion of Rh(II) stabilized carbenoids into acetals: a C–H activation protocol as a Claisen condensation equivalent. J. Organomet. Chem. 690, 6111–6124 (2005).
Cambeiro, F., López, S., Varela, J. A. & Saá, C. Cyclization by catalytic ruthenium carbene insertion into Csp3–H bonds. Angew. Chem. Int. Ed. 51, 723–727 (2012).
Liu, X.-J., Xu, Y., Tang, C., Qian, P.-C. & Ye, L.-W. Unactivated C(sp3)–H functionalization via vinyl cations. Sci. China Chem. 65, 20–30 (2022).
Niggemann, M. & Gao, S. S. Are vinyl cations finally coming of age? Angew. Chem. Int. Ed. 57, 16942–16944 (2018).
Hong, F.-L. et al. Generation of donor/donor copper carbenes through copper-catalyzed diyne cyclization: enantioselective and divergent synthesis of chiral polycyclic pyrroles. J. Am. Chem. Soc. 141, 16961–16970 (2019).
Hong, F.-L. et al. Copper-catalyzed asymmetric reaction of alkenyl diynes with styrenes by formal [3+2] cycloaddition via Cu-containing all-carbon 1,3-dipoles: access to chiral pyrrole-fused bridged [2.2.1] skeletons. J. Am. Chem. Soc. 142, 7618–7626 (2020).
Zhu, X.-Q. & Hong, P. et al. Copper-catalyzed asymmetric cyclization of alkenyl diynes: method development and new mechanistic insights. Chem. Sci. 12, 9466–9474 (2021).
Hong, F.-L. & Shi, C.-Y. et al. Copper-catalyzed asymmetric diyne cyclization via [1,2]-Stevens-type rearrangement for the synthesis of chiral chromeno[3,4-c]pyrroles. Angew. Chem. Int. Ed. 61, e202115554 (2022).
Qi, L.-J. & Li, C.-T. et al. Enantioselective copper-catalyzed formal [2+1] and [4+1] annulations of diynes with ketones via carbonyl ylides. Angew. Chem. Int. Ed. 61, e202210637 (2022).
Chen, Y.-B. et al. Construction of axially chiral arylpyrroles via atroposelective diyne cyclization. Angew. Chem. Int. Ed. 62, e202303670 (2023).
Li, C.-T. et al. Asymmetric formal C–C bond insertion into aldehydes via copper-catalyzed diyne cyclization. Nat. Commun. 14, 7058 (2023).
Zhou, J.-J. & Meng, Y.-N. et al. Copper-catalyzed enantioselective diyne cyclization via C(sp2)–O bond cleavage. Chem. Sci. 14, 3493–3500 (2023).
Chen, Y.-B. et al. Enantioselective functionalization of unactivated C(sp3)–H bonds through copper-catalyzed diyne cyclization by kinetic resolution. Nat. Commun. 15, 2232 (2024).
Li, F.-S. et al. Asymmetric one-carbon ring expansion of diverse N-heterocycles via copper-catalyzed diyne cyclization. Sci. Adv. 10, eadq7767 (2024).
Zheng, Y.-X. et al. Asymmetric Büchner reaction and arene cyclopropanation via copper-catalyzed controllable cyclization of diynes. Nat. Commun. 15, 9227 (2024).
Jia, S. & Mei, G. Construction of axially chiral arylpyrroles via atroposelective diyne cyclization. Chin. J. Org. Chem. 43, 2261–2263 (2023).
Hu, L. & Zhao, J. Ynamide coupling reagents: origin and advances. Acc. Chem. Res. 57, 855–869 (2024).
Hu, Y.-C., Zhao, Y., Wan, B. & Chen, Q.-A. Reactivity of ynamides in catalytic intermolecular annulations. Chem. Soc. Rev. 50, 2582–2625 (2021).
Lynch, C. C., Sripada, A. & Wolf, C. Asymmetric synthesis with ynamides: unique reaction control, chemical diversity and applications. Chem. Soc. Rev. 49, 8543–8583 (2020).
Hong, F.-L. & Ye, L.-W. Transition metal-catalyzed tandem reactions of ynamides for divergent N-heterocycle synthesis. Acc. Chem. Res. 53, 2003–2019 (2020).
Luo, J. et al. Exploiting remarkable reactivities of ynamides: opportunities in designing catalytic enantioselective reactions. ACS Catal. 10, 13978–13992 (2020).
Evano, G., Theunissen, C. & Lecomte, M. Ynamides: powerful and versatile reagents for chemical synthesis. Aldrichimica Acta 48, 59–70 (2015).
Wang, X.-N. et al. Ynamides in ring forming transformations. Acc. Chem. Res. 47, 560–578 (2014).
Ahrens, A. et al. Synthesis of fulvene vinyl ethers by gold catalysis. Chem. Eur. J. 26, 5280–5287 (2020).
Tavakkolifard, S. et al. Gold-catalyzed regiospecific annulation of unsymmetrically substituted 1,5-diynes for the precise synthesis of bispentalenes. Chem. Eur. J. 25, 12180–12186 (2019).
Wurm, T. et al. On the gold-catalyzed generation of vinyl cations from 1,5-diynes. Angew. Chem. Int. Ed. 56, 3364–3368 (2017).
Liao, S., Sun, X.-L. & Tang, Y. Side arm strategy for catalyst design: modifying bisoxazolines for remote control of enantioselection and related. Acc. Chem. Res. 47, 2260–2272 (2014).
Li, Z., Parr, B. T. & Davies, H. M. L. Highly stereoselective C–C bond formation by rhodium-catalyzed tandem ylide formation/[2,3]-sigmatropic rearrangement between donor/acceptor carbenoids and chiral allylic alcohols. J. Am. Chem. Soc. 134, 10942–10946 (2012).
Li, Z. & Davies, H. M. L. Enantioselective C–C bond formation by rhodium-catalyzed tandem ylide formation/[2,3]-sigmatropic rearrangement between donor/acceptor carbenoids and allylic alcohols. J. Am. Chem. Soc. 132, 396–401 (2010).
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (22125108, 22121001 and 22331004 for L.-W.Y.; 22122109 and 22271253 for X.H.), National Key R&D Program of China (2022YFA1504301, X.H.), Zhejiang Provincial Natural Science Foundation of China (LDQ23B020002, X.H.), the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (SN-ZJU-SIAS-006, X.H.), CAS Youth Interdisciplinary Team (JCTD-2021-11, X.H.), Fundamental Research Funds for the Central Universities (226-2022-00140 and 226-2022-00224, 226-2023-00115 and 226-2024-00003, X.H.), the State Key Laboratory of Clean Energy Utilization (ZJUCEU2020007, X.H.), the State Key Laboratory of Physical Chemistry of Solid Surfaces (202210, X.H.), the Leading Innovation Team grant from Department of Science and Technology of Zhejiang Province (2022R01005, X.H.) and Open Research Fund of School of Chemistry and Chemical Engineering of Henan Normal University (2024Z01, X.H.). Calculations were performed on the high-performance computing system at Department of Chemistry, Zhejiang University.
Author information
Authors and Affiliations
Contributions
C.-T.L., J.-Z.L., H.-X.D., Z.-S.W. and B.Z. performed experiments. X.H. designed DFT calculations. L.-G.L. and R.-A.N. performed DFT calculations. L.-W.Y. conceived and directed the project and wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, CT., Liu, LG., Li, JZ. et al. Ligand-controlled divergent asymmetric C(sp3)−H and C(sp3)−O insertion via vinyl cations. Nat Commun 16, 4107 (2025). https://doi.org/10.1038/s41467-025-59328-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59328-7
This article is cited by
-
Enantioselective dearomative single-atom skeletal editing of benzofurans
Nature Communications (2025)
