
Abstract
Lanthanide-doped upconversion nanoparticles are promising for applications ranging from biosensing, bioimaging to solid-state lasing. However, their brightness remains limited by the concentration quenching effect of lanthanide activator ions, which greatly restricts their utility. Here, we develop a heterogeneous core–shell–shell nanostructure based on hexagonal NaYF4, in which Tm3+ activator and Yb3+ sensitizer are separated into the core and inner shell, while the outmost shell is used to suppress surface quenching effects. We show that this design can alleviate the activator concentration quenching effect, resulting in optimal Tm3+ concentration increasing from 1% to 8% at sub-100 W/cm2 irradiance, compared with the canonical core-only NaYF4:Yb3+/Tm3+. Moreover, under high excitation irradiance (20 MW/cm2), the optimal Tm3+ concentration could be further increased to 50%. Mechanistic investigations reveal that the spatial separation of sensitizer and activator effectively suppresses the back energy transfer from Tm3+ to Yb3+, driving the increase of optimal activator concentration. These findings enhance our understanding of lanthanide concentration quenching effect, unleashing opportunities for developing bright upconverting materials.
Similar content being viewed by others

Introduction
Lanthanide-doped upconversion nanoparticles (UCNPs) have extensive ramifications for applications in areas like biosensing1,2,3,4,5,6, bioimaging7,8,9,10,11,12,13,14 and solid-state lasing15,16,17,18. UCNPs typically consist of a low-phonon-energy material as host lattice, such as hexagonal sodium yttrium fluoride (β-NaYF4, ~350 cm−1 phonon energy) and are doped with ytterbium (Yb3+) ions as sensitizers. These sensitizer ions can harvest near-infrared (NIR) light and then non-radiatively transfer excitation energy to activator ions, such as thulium (Tm3+), erbium (Er3+) or holmium (Ho3+), producing high-energy upconversion luminescence (UCL). Recent progress in chemical synthesis has enabled precise manipulation over the size, morphology, crystalline phase and multilayer core/shell structure, offering unprecedented opportunities for modulation of luminescence color and lifetime19,20.
Nevertheless, it is still challenging to obtain bright upconversion due to the limited absorption/emission cross sections of lanthanide ions and most importantly, the presence of lanthanide concentration quenching effects. The concentration quenching phenomenon causes a decrease in brightness when the lanthanide dopant concentration exceeds a certain threshold. This is primarily due to the concentration-dependent nonradiative depopulation of the emitting states or the involved intermediary excited states. Endeavors to mitigate this quenching effect have typically involved strategies, such as coating an inert/active shell, introducing damping ions to minimize energy migration loss, or reducing surface defects to minimize surface quenching. Furthermore, improving the homogeneous distribution of dopants, using a host with an expanded unit cell dimension or increasing excitation laser irradiance has been shown to relieve cross relaxation processes and thereby alleviate the concentration quenching effects21,22,23. Despite these progress, the optimal activator concentrations remain rather low, for example, the optimal Tm3+ concentration in the NaYF4 host matrix was shown to be within the range of 0.2–1 mol% under 980 nm irradiance of sub-100 W/cm2 24,25,26. Such nanoparticles contain a limited quantity of activator ions, which significantly confines the upconversion brightness.
In this study, we reveal the critical factor of back energy transfer (BET) process from the activator (Tm3+) to the sensitizer (Yb3+) ions for underlying the lanthanide concentration quenching in the Yb3+-Tm3+ codoped upconversion systems. We demonstrate that a spatial isolation of Tm3+ and Yb3+ into the distinct domains of a heterogeneous nanostructure can efficiently suppress the BET process and elevate the optimal Tm3+ concentration from 1% to ~8% at sub-100 W/cm2, compared to the typically investigated β-NaYF4:Yb3+/Tm3+ UCNPs.
Results
Heterogeneous core–shell–shell nanostructure design
A heterogeneous core–shell–shell nanostructure was adopted to inhibit both the BET process (from the activator to the sensitizer) and the detrimental surface quenching effect simultaneously (Fig. 1a). In this design, the activator ions (e.g., Tm3+) and the sensitizer ions (e.g., Yb3+) were incorporated into the core and the first shell domain, respectively. The spatial isolation of the activator and sensitizer ions results in the inhibition of BET processes from the activator ions to the sensitizer ions, while the close contact between the sensitizer and activator ions at the core/shell interface allows efficient energy transfer (ET) across the core–shell boundary. The outermost inert layer is used to alleviate the surface quenching triggered by migration of excited energy within the first or inner shell layer domain. Specifically, a core–shell–shell nanostructure of NaYF4:x% Tm3+/NaYbF4/NaYF4 was devised, in which the inner NaYbF4 shell enriched with Yb3+ sensitizer ions was adopted to efficiently capture excitation photons at 980 nm. Subsequently, energy migration among the Yb3+ ion network takes places7, entailing efficient interfacial energy transfers to doped Tm3+ ions in the core27. Meanwhile, the NaYF4:x% Tm3+/NaYbF4 interface could effectively inhibit the BET process (3H4(Tm3+) + 2F2/7(Yb3+) → 3H6(Tm3+) + 2F2/5(Yb3+)). The outermost inert NaYF4 shell can prevent the energy dissipation in the NaYbF4 shell from the quenching effects induced by surface imperfections or external environments27,28,29. This specific nanostructure design facilitates upconversion luminescence (from NIR to VIS/UV) through a ladder-like array of the intermediate metastable states of Tm3+ ions, while minimizing nonradiative depopulations of the UCL emitting states (Fig. 2a).

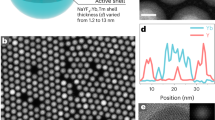
a Structural illustration of the synthesized core–shell–shell NaYF4:x% Tm3+@NaYbF4@NaYF4 nanoparticle (x = 0.5, 1, 2, 4, 8, 16, 32, 50, 70 and 100). b High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and high-resolution transmission electron microscopy (HRTEM) image (inset) of the representative core–shell–shell nanoparticles. The interfaces of core–shell–shell are marked by fuchsia dashed lines. c HAADF-STEM image and energy-dispersive X-ray mapping analysis of a single NaYF4:8% Tm3+@NaYbF4@NaYF4 nanoparticle, indicating the spatial distribution of the Yb3+, Tm3+ and Y3+ elements in the core–shell–shell structure. d Photographic images displaying upconversion luminescence of UCNPs dispersed in n-hexane under 980 nm continuous-wave laser irradiation. CSS, core–shell–shell. e Upconversion luminescence spectra acquired under 980 nm excitation at 99.3 W/cm2 for UCNPs in n-hexane. Absorbance of all samples is normalized at 975 nm, corresponding to the 2F7/2 → 2F5/2 transition of Yb3+ ions. The intensity of 800 nm luminescence peak is multiplied by ×0.1 for better data presentation. Source data are provided as a Source Data file.

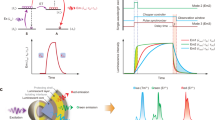
a Energy level scheme of Yb3+ and Tm3+ with proposed energy transfer processes under 980 nm laser excitation. Solid line with an arrow, absorption or emission; curve with an arrow, multiphonon relaxation; dashed line with an arrow linked by dotted line, non-radiative energy transfer; BET, back energy transfer. b The dependence of 365 and 475 nm upconversion luminescence from NaYF4:1% Tm3+@NaYbF4@NaYF4 nanoparticles on excitation laser power in a logarithmic scale. c Variation of integrated luminescence intensity of each energy level in Tm3+ ions with increasing Tm3+ doping concentration (NaYF4:x% Tm3+@NaYbF4@NaYF4, x = 0.5, 1, 2, 4, 8, 16, 32, 50, 70, 100) at irradiance of 99.3 W/cm2. The luminescence intensity from 3H4, 3H5 energy levels is multiplied by ×0.1 and ×10 for better data presentation, respectively. Source data are provided as a Source Data file.
A layer-by-layer epitaxial growth method was used to fabricate a set of the designated core–shell–shell nanoparticles of NaYF4:x% Tm3+@NaYbF4@NaYF4 (x = 0.5, 1, 2, 4, 8, 16, 32, 50, 70 and 100) (Fig. 1a). Transmission electron microscopy (TEM) images of the NaYF4:x% Tm3+ core, NaYF4:x% Tm3+@NaYbF4 core–shell and NaYF4:x% Tm3+@NaYbF4@NaYF4 core–shell–shell nanoparticles indicate an average overall size of ~24 nm, with a thickness of ~5.5 nm for the inner layer, and ~3 nm for the outermost shell layer (Supplementary Fig. 1). High resolution TEM (HRTEM, Fig. 1b inset) and selected area electron diffraction patterns (Supplementary Fig. 2b) confirm the single crystalline nature of the designated multilayered nanoparticles. Powder X-ray diffraction (XRD) results confirm that all the resulting core, core–shell, and core–shell–shell UCNPs adopt a hexagonal crystal phase (Supplementary Fig. 2a)30,31. Atomic-number-contrasted high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) imaging confirms the successful preparation of the heterogeneous core–shell–shell structure (Fig. 1b), with a clear discern of yttrium (Y) atoms (dark) from Yb atoms (bright) that are spatially distributed in the defined way27,32. Energy-dispersive X-ray analysis (EDX) verifies the presence of Yb, Tm, Y, Na, F (Supplementary Fig. 3) and the formation of the designated heterogeneous core–shell–shell structure27,31,32,33 (Fig. 1c). Inductively-coupled plasma optical emission spectroscopy (ICP-OES) indicates that the actual content of Tm3+ (mol%) within the NaYF4:x% Tm3+ core domain is close to the stoichiometric content (Supplementary Table 1).
Optical characterizations of core–shell–shell upconversion nanoparticles
Representative UCL photographic images of the core–shell–shell UCNPs suspended in hexane and their corresponding UCL spectra are depicted in Fig. 1d, e. The photographic images show both color and luminance change when varying Tm3+ concentration in the core of the core–shell–shell nanostructure. At 99.3 W/cm2 irradiance of 980 nm laser, we observed that the 8 mol% Tm3+ core–shell–shell nanoparticles emitted an unprecedentedly vibrant upconversion luminescence which markedly surpasses that of 1 mol% Tm3+ nanoparticles (the 800 nm luminescence is enhanced approximately 50-fold, Fig. 1e). In contrast, at the same excitation condition, the UCL intensity of the canonical core-only structure NaYF4:20% Yb3+, x% Tm3+ (x = 0.2, 0.5, 1, 2, 4, 8, 16) UCNPs initially climbs up and then declines when exceeding 1 mol% Tm3+, consistent with previous reports24,25,26 (Supplementary Fig. 4). These discoveries suggest that high-efficiency UCL is feasible at considerably high activator concentrations with ~100 W/cm2 of 980 nm laser radiation through spatially separating sensitizers and activators into the core and the shell domains. The spatial separation increases the average distance between them and therefore inhibits the BET from activator to sensitizer (Supplementary Fig. 5). This conclusion is supported by the result that co-doping of Tm3+ and Yb3+ ions into the intermediary shell of NaYF4@NaYF4:92% Yb3+, 8% Tm3+@NaYF4 nanoparticles will dramatically decreases UCL as compared to the one NaYF4@NaYF4:99% Yb3+, 1% Tm3+@NaYF4 nanoparticles. Furthermore, the enrichment of Yb3+ ions in the intermediary shell of NaYF4:x% Tm3+@NaYbF4@NaYF4 nanoparticles favors the intense absorption of the excitation photon energy, thereby enhancing the overall upconversion brightness (Supplementary Fig. 6). Note that the UCL brightness of core–shell–shell NaYF4:8% Tm3+@NaYbF4@NaYF4 nanoparticles (~25 nm) is about 4.8-fold higher than that of the state-of-the-art NaYF4:20% Yb3+, 1% Tm3+@NaYF4 core–shell nanoparticles with an overall size of ~ 34 nm (the inert shell thickness, 3.5 nm) (Supplementary Fig. 7). These results validate the rational design of the core–shell–shell nanostructure with spatially separated Tm3+ and Yb3+ ions. Moreover, the designed core–shell–shell architecture can also be extended to the commonly used Yb3+-Er3+ and Yb3+-Ho3+ upconversion systems to accommodate high aviator concentrations for bright upconversion (Supplementary Fig. 8).
Qualitative understanding of upconversion processes in the Yb3+-Tm3+ system
We further investigated the dependence of UCL intensity from the high-lying 1D2 state of Tm3+ on the excitation laser power (Fig. 2b). The number of photons (n) needed to fill up a certain energy level is estimated according to the relationship, \(I\propto {P}^{n}\), where \(P\) is the excitation power and \(I\) means the integrated luminescence intensity of interested energy level34. For the 475 nm luminescence, n = 2.10 was acquired, indicating that three photons are required to excite the 1G4 state. While n = 3.99 was obtained for the 365 nm UCL, suggesting that populating 1D2 state involves a three-photon energy transfer upconversion (ETU) as well as the cross relaxation 1 (CR1) process, as the n value cannot be met purely through ETU processes35,36.
Moreover, after spectral calibration, the luminescence intensity from each energy level of Tm3+ can be compared at different Tm3+ concentrations in the designated core–shell–shell nanostructure (Fig. 2c). The highest luminescence intensity derives from the 3H4 excited state, while the lowest one stems from the 3H5 state. The luminescence intensities from both energy levels were mathematically scaled appropriately, facilitating the comparison between the different energy levels. As shown in Fig. 2c and Supplementary Table 2, concentration quenching first occurred in the high-lying excited states, including 1I6, 1D2 and 1G4, and then 3H4, which can be reasonably explained by the existence of the CR2 and CR3 processes between the Tm3+ ions36,37. Compared with the result of canonical core-only NaYF4:20% Yb3+, x% Tm3+ (x = 0.2, 0.5, 1, 2, 4, 8, 16) UCNPs (Supplementary Fig. 9 and Supplementary Table 3), it was confirmed that BET from Tm3+ (3H4) to Yb3+ (2F2/5) is a dominant factor for the concentration quenching. In stark contrast, a concentration quenching effect was not observed for the 1220 nm down-shifting luminescence originating from the 3H5 energy level, revealing that 3H5 does not take part in any pronounced BET or CR processes. Instead, the down-shifting luminescence from the state 3F4 of Tm3+, populated by nonradiative relaxation from the 3H5 and radiative relaxations from the 3H4 or other upper excited states, exhibits a complex behavior.
Based on the above outlined steady-state analysis at laser irradiance of 99.3 W/cm2, we established a simplified energy level model of the Tm3+ ion to simulate the upconversion process in the Yb-Tm system26,38,39 (Supplementary Fig. 10). Luminescence intensity was calculated as a function of Tm3+ doping concentration (range from 0.1 mol% to the max value allowed in each structure). The normalized numerical computation results are in good agreement with the experimental observations for the evolution trend of concentration quenching. Especially, the optimal Tm3+ dopant concentration was improved from 1% to 8% (Fig. 3), which further verifies that the designed heterogeneous core–shell–shell nanostructure (NaYF4:x% Tm3+@NaYbF4@NaYF4) indeed can weaken the BET from Tm3+ (3H4) to Yb3+ (2F2/5), thereby inhibiting the concentration quenching effect.

a Simulated (gray) and experimental (black) UCL versus Tm3+ doping concentration for NaYF4:20% Yb3+, x% Tm3+ nanoparticles (x = 0.2, 0.5, 1, 2, 4, 8, 16). b Simulated (green) and experimental (orange) UCL versus Tm3+ doping concentration for NaYF4:x% Tm3+@NaYbF4@NaYF4 nanoparticles (x = 0.5, 1, 2, 4, 8, 16, 32, 50, 70, 100). For all simulations, the 980 nm excitation laser irradiance was set to 99.3 W/cm2. Source data are provided as a Source Data file.
Quantitative study of the heterogeneous core–shell–shell nanostructure for inhibiting BET
For quantifying the effect of heterogeneous core–shell–shell nanostructure to inhibit BET, we compared the down-shifting luminescence spectra of NaYF4:1% Tm3+@NaYbF4@NaYF4 (6.5@2@3 nm, the number indicates the radius of the core or the thickness of the shell layer) and NaYF4@NaYbF4:1% Tm3+@NaYF4 (3.5@5@3 nm) core–shell–shell UCNPs under the same 808 nm excitation condition (Fig. 4a and Supplementary Fig. 11). These two nanostructures have the same overall size, outermost inert protecting shell thickness, as well as the almost equal content ratio and doping concentrations of Yb3+/Tm3+. The NaYF4@NaYbF4:1% Tm3+@NaYF4 nanostructure shows ~2.7-fold peak intensity of 980 nm luminescence (from Yb3+ ions) relative to the NaYF4:1% Tm3+@NaYbF4@NaYF4 nanostructure, while the intensity of the 1470 nm luminescence stemming from the 3H4 energy level of Tm3+ exhibits a contrary tendency. This result gives evidence that the spatial separation of Tm3+ and Yb3+ in the designated NaYF4:1% Tm3+@NaYbF4@NaYF4 heterogenous core–shell–shell structure can efficiently weaken the BET process.

a Down-shifting luminescence spectra under 808 nm excitation at 120 W/cm2 for NaYF4:1% Tm3+@NaYbF4@NaYF4 and NaYF4@NaYbF4:1% Tm3+@NaYF4 upconversion nanoparticles dispersed in n-hexane. Absorbance of the two samples was normalized at 975 nm. The green dashed line indicates the position for comparison of peak intensity. b, c Luminescence decay profiles recorded at 1470 nm, corresponding to the 3H4 → 3H6 transition of Tm3+ ions, under excitation of 808 nm pulsed laser. The calculated average lifetime for each sample is indicated in the figures using color code consistent with the decay curve. IRF denotes the instrument response function. Source data are provided as a Source Data file.
To further support this conclusion and quantify the efficiency of BET process, we produced four pairs of samples, including the first pair NaYF4@NaYF4:1% Tm3+@NaYF4 (3.5@5@3 nm) vs. NaYF4@NaYbF4:1% Tm3+@NaYF4 (3.5@5@3 nm), the second pair NaYF4:1% Tm3+@NaYF4 (3.5@8 nm) vs. NaYF4:1% Tm3+@NaYbF4@NaYF4 (3.5@5@3 nm), the third pair NaYF4@NaYF4:8% Tm3+@NaYF4 (3.5@5.5@3.5 nm) vs. NaYF4@NaYbF4:8% Tm3+@NaYF4 (3.5@5.5@3.5 nm), and the fourth pair NaYF4:8% Tm3+@NaYF4 (3.5@9 nm) vs. NaYF4:8% Tm3+@NaYbF4@NaYF4 (3.5@5.5@3.5 nm). We measured the lifetimes of Tm3+ at 1470 nm in all the samples under 808 nm pulsed excitation, and then used Eqs. (1) and (2) to quantify the BET efficiencies. The measured lifetimes for each pair are presented in the insets of Fig. 4b, c. The efficiency (rate) of BET process from Tm3+ to Yb3+ was determined to be 66% (1722 s−1) for the NaYF4@NaYbF4:1% Tm3+@NaYF4, and to be 25% (268 s−1) for the NaYF4:1% Tm3+@NaYbF4@NaYF4 nanostructure at low Tm3+ concentration (Fig. 4b). In analogy, the BET efficiency (rate) was evaluated to be 50% (7077 s−1) for the NaYF4@NaYbF4:8% Tm3+@NaYF4, and to be 39% (2095 s−1) for the NaYF4:8% Tm3+@NaYbF4@NaYF4 nanostructure at high Tm3+ concentration (Fig. 4c). These results clearly demonstrate that the spatial separation of Tm3+ activator and Yb3+ sensitizer into different domains of the designated core–shell–shell nanostructure can effectively inhibit the BET process. Note that the magnitude of reduction of BET efficiency is smaller at high Tm3+ (8%) concentration than at low Tm3+ (1%) concentration. This is probably because the cross-relaxation process among Tm3+ ions become more severe at high doping concentrations and the reduced mean distance between Tm3+ and Yb3+ ions aggravates the BET rate. However, the spatial separation of Tm3+ and Yb3+ ion results in a substantial reduction of BET rate by 4982 s−1 at high Tm3+ (8%) concentration, compared to the one of 1454 s−1 at low Tm3+ (1%) concentration. Taken together, the designated core–shell–shell nanostructure with spatially separated sensitizer and activator can inhibit the BET process that substantially depopulate the 3H4 state (emitting the most intense 800 nm UCL), thereby alleviating the Tm3+ concentration effect. We would like to note that separating Yb3+ and Tm3+ into different domains simultaneously reduces both ET efficiency and BET efficiency. While the decreased ET efficiency lowers upconversion brightness, the reduced BET efficiency mitigates concentration quenching effects, thereby enhancing brightness. This trade-off in the designed core–shell–shell nanostructure ultimately results in brighter upconversion nanoparticles, as the brightness is more significantly influenced by inhibiting the concentration quenching effect (Supplementary Note).
Inhibition of activator concentration quenching at high excitation laser irradiance
We then assessed the power-dependent upconversion intensities for the designed core–shell–shell NaYF4:x% Tm3+@NaYbF4@NaYF4 (x = 4, 8, 16, 32, 50, 70, 100) nanoparticle at high excitation power densities (>100 W/cm2) (Fig. 5a) utilizing a home-built confocal microscope (Supplementary Fig. 12). Increasing 980 nm laser irradiance from 100 W/cm2 to 20 MW/cm2 enhances the overall (400–850 nm) UCL intensity, accompanied with a gradually decreasing slope that finally reaches a plateau for all samples. The relative magnitude of UCL intensity at 100 W/cm2 for the samples of 4% and 8% Tm3+ is consistent with the ensemble measurement results in Fig. 2c.

a Consolidated UCL (400–850 nm) log-log plotted with 980 nm laser irradiance for the core–shell–shell NaYF4:x% Tm3+@NaYbF4@NaYF4 (x = 4, 8, 16, 32, 50, 70 and 100) UCNPs. b Average single particle UCL brightness for all the core–shell–shell samples under laser irradiance of 20 MW/cm2; note that UCL from the core–shell–shell samples with 0.5, 1, and 2% Tm3+ is marked by dotted box, as these samples were not bright enough to be measured. The 800 nm luminescence (760-850 nm) intensity is marked with a gray stripe pattern. Luminescence intensity at 20 MW/cm2 in (b) was determined by single particle measurements, while the variation of each saturation curve in (a) was given by multi-particle investigations31. Source data are provided as a Source Data file.
We further quantified and compared the average upconversion brightness of single UCNPs for all the heterogenous core–shell–shell samples at 20 MW/cm2 (Fig. 5b). The detailed histograms of single particle brightness for each sample are shown in Supplementary Fig. 13. For 0.5, 1 and 2% Tm3+ doped samples, which were not bright enough to surpass the detection limit, we estimated their average brightness according to the inhibiting effect of concentration quenching for other samples. No noticeable UCL photo-bleaching or photo-blinking was observed over 60 mins single nanoparticle measurements, affirming the stable structure of the designated core–shell–shell UCNPs at high laser irradiances (Supplementary Fig. 14). The overall upconversion brightness (UCL, 400–850 nm) shows a nearly linear growth with the increment of Tm3+ concentration up to 50%, which is about six-fold higher than the optimal Tm3+ concentration of 8% at laser irradiance of 99.3 W/cm2. The brightness of the UCL band at 800 nm exhibits a similar trend as the overall upconversion brightness with an optimal Tm3+ doping concentration of 50%. Note that the intensities of shorter wavelength UCL (400–760 nm) from higher-lying energy levels (1G4, 1D2, 1I6) (three-, four- and five-photon processes) are on par with that of UCL at 800 nm (two-photon process) under single UCNP measurement. This can be attributed to the involvement of higher-order multiphoton processes and the utilization of high laser irradiances. Importantly, the high laser irradiance induced increase of the optimized Tm3+ concentration affirms the existence of the contribution of the cross-relaxation processes to the activator concentration quenching, alongside the revealed BET process from Tm3+ (3H4) to Yb3+ (2F5/2) at low laser irradiances. In other words, both the BET processes and the cross-relaxation processes collectively define the activator Tm3+ concentration quenching effect in UCNPs.
Discussion
In this work, we highlight the crucial role of back energy transfer (BET) from activator Tm3+ to sensitizer Yb3+ in the lanthanide concentration quenching of Yb3+/Tm3+-codoped UCNPs. Through the strategic design and synthesis of the heterogenous core–shell–shell structure, we demonstrated that segregating sensitizer and activator ions into adjacent layers effectively suppresses BET while entailing efficient interfacial energy transfer from Yb3+ to Tm3+, resulting in bright upconversion at high Tm3+ doping concentrations. As a consequence, the optimal Tm3+ concentration increased to ~8%, compared with the typically investigated β-NaYF4:Yb3+/Tm3+ UCNPs, under excitation power densities below 100 W/cm², which is in good agreement with rate equation modeling. At higher excitation power densities of about 20 MW/cm², the optimal Tm3+ concentration reached an unprecedented ~50% at the single particle level. Moreover, minimizing lattice defects to reduce excited energy losses may further enhance the optimal Tm3+ doping concentration32. We believe this segregated doping strategy will advance the design of bright UCNPs for a variety of applications.
Methods
Nanoparticle synthesis
NaYF4:x% Tm3+@NaYbF4@NaYF4 and other core-multishell nanoparticles were synthesized following a seed-mediated hot-injection procedure. The NaYF4:x% Tm3+ core is first synthesized to serve as a template seed for layer-by-layer epitaxial growth of the two shells in sequence. While the core-only structure NaYF4:20% Yb3+, x% Tm3+ was prepared using a coprecipitation protocol. Experimental details are described in Supplementary Methods.
Ensemble characterization
The size, shape and spatial distribution of elements were examined using a transmission electron microscope (FEI Tecnai G2 F30 S-TWIN) hyphenated energy dispersive X-ray spectroscopy operating at 300 kV accelerating voltage, while the phase and crystalline structure of samples were analyzed by a powder X-ray diffractometer (BRUKER D8 ADVANCE). Tm3+ molar content was determined with an inductively-coupled plasma optical emission spectrometer (Thermo Fisher Scientific ICAP 6000). Steady-state photoluminescence and transient decay behavior were measured by a spectrofluorometer (FLS1000, Edinburgh) integrated with 808 nm (MDL-III-808-2W, CNI) and 980 nm (MDL-III-980-2W, CNI) diode lasers at room temperature.
Calculation of BET efficiency
The back energy transfer (from Tm3+ to Yb3+) efficiency η can be quantitatively estimated on the basis of Eqs. (1) and (2)40:
where τm is the mean lifetime of energy donor (Tm3+) when in the company of energy acceptor (Yb3+), τin is the intrinsic lifetime of energy donor (Tm3+) determined by replacing energy acceptor (Yb3+) with inert Y3+, and α is the amplitude.
Single particle measurements
The luminescence intensity of a single nanoparticle was measured by a self-built confocal microscope. Supplementary Fig. 12 draws the schematic plot of experimental apparatus, wherein UCNPs are excited by a single-mode fibre coupled 980 nm diode laser (VLSS-980-B, Connet). The purpose of the first polarizer (DGL10, Thorlabs) is to produce a polarized laser beam. Based on this, a second polarizer (GTH10M, Thorlabs) is employed to manager the excitation power, facilitated with rotation of a half-wave plate (H, WPH10E-980, Thorlabs). The whole excitation and detection process: the collimated excitation beam is reflected by a short pass dichroic mirror (DM, ZT1064rdc-sp-UF1, Chroma), and focused through an air objective lens (UPlanSApo, 40×/0.95, Olympus) onto the sample slide. Photoluminescence is collected via the same objective and then split from the excitation beam by the DM. After filtered by a short pass filter (SPF, FF01-950SP-25, Semrock), the luminescence signal is coupled into a 4f-system containing a 100 μm diameter pinhole (P100K, Thorlabs), and detected by a photomultiplier tube (PMT, HPM-100-42, Becker & Hickl GmbH).
Theoretical modeling and numerical simulation
The simplified energy level diagram of the Tm3+ ion is illustrated in Supplementary Fig. 10. Upconversion process in a Yb-Tm system can be depicted by the following rate equations:
Here \({n}_{{A}_{i}}\) (i = 0–4) and \({\tau }_{{A}_{i}}\) (i = 1–4; \({\tau }_{{A}_{3}}\) = \(1/{\Gamma }_{{{\rm{r}}}}^{{{\rm{A}}}}\)) represent the population density and radiative lifetime of 3H6, 3F4, 3H5, 3H4 and 3F2,3 energy levels for Tm3+, respectively. \({w}_{i}\) (i = 0, 1) is the energy transfer rate from the 2F5/2 state of Yb3+ to the 3H5 and 3F2,3 states of Tm3+, respectively. \({w}_{b}\) is the back energy transfer rate from the 3H4 state of Tm3+ to the 2F5/2 state of Yb3+. \({c}_{1}\) denotes the rate of cross relaxation for Tm3+-Tm3+ (3H4 + 3H6 → 3F4 + 3F4). \({\beta }_{i}\) (i = 2, 4) is the nonradiative decay rate for 3H5 and 3F2,3 energy levels of Tm3+, respectively. \({n}_{{S}_{0}}\) and \({n}_{{S}_{1}}\) represent the population density of ground state and excited state for Yb3+, respectively. \({\tau }_{{S}_{1}}\)(\(1/{\Gamma }_{{{\rm{r}}}}^{{{\rm{S}}}}\)) is the lifetime of excited state for Yb3+. \({\sigma }_{S}\) is the absorption cross section of Yb3+ at 980 nm. \(h\) is Planck constant. \(\nu\) is the frequency of 980 nm laser light. \(I\) is the excitation irradiance of 980 nm laser beam.
For NaYF4:x% Tm@NaYbF4@NaYF4 core-multishell system, Yb3+ and Tm3+ are equivalently regarded to uniformly distribute in the active region (NaYF4:x% Tm3+@NaYbF4) as a co-doping condition, while holding the amount of ions constant. Nonetheless, cross relaxation of Tm3+-Tm3+ was excluded from this assumption. In addition, the influence of varying Tm concentration upon the parameters \({w}_{0},{w}_{1},{c}_{1}\) related to energy transfer between Yb3+ and Tm3+ is referred to the description provided by Lee et al.39. The rate parameters for core-only (NaYF4:20% Yb3+, 1% Tm3+) and core–shell–shell (NaYF4:1% Tm3+@NaYbF4@NaYF4) samples are listed in Supplementary Table 4.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the relevant data that support the findings of this work are available from the corresponding author upon request. Source data are provided with this paper.
References
Galvão, R. et al. Menezes, L. Single Er3+/Yb3+-codoped yttria nanocrystals for temperature sensing: experimental characterization and theoretical modeling. J. Phys. Chem. C 125, 14807–14817 (2021).
Song, N. et al. Enhancing upconversion of Nd3+ through Yb3+-mediated energy cycling towards temperature sensing. J. Rare Earths 39, 1506–1511 (2021).
Bai, Y., Li, Y., Wang, R. & Li, Y. NIR laser-treatment, anti-oxidation upconversion nanoparticles for optical temperature sensing. ChemistrySelect 6, 10263–10273 (2021).
Yu, Y. et al. Near infrared triggered dual-functional NaYF4:Yb3+/Tm3+@ NaYF4:Yb3+/Sm3+ core-shell UCNPs for temperature sensing and photothermal conversion. Phys. B Condensed Matter. 622, 413340 (2021).
Zhang, Y. et al. An upconversion nanosensor for rapid and sensitive detection of tetracycline in food based on magnetic-field-assisted separation. Food Chem 373, 131497 (2022).
Wang, X. et al. High-sensitivity sensing of divalent copper ions at the single upconversion nanoparticle level. Anal. Chem. 93, 11686–11691 (2021).
Li, H. et al. Temporal multiplexed in vivo upconversion imaging. J. Am. Chem. Soc. 142, 2023–2030 (2020).
Liao, J. et al. NIR-II emissive properties of 808 nm-excited lanthanide-doped nanoparticles for multiplexed in vivo imaging. J. Lumin. 242, 118597 (2022).
Jiang, X. et al. Noninvasive and early diagnosis of acquired brain injury using fluorescence imaging in the NIR-II window. Biomed. Opt. Express 12, 6984–6994 (2021).
Wei, Z. et al. Rapidly liver-clearable rare-earth core–shell nanoprobe for dual-modal breast cancer imaging in the second near-infrared window. J. Nanobiotechnol. 19, 369 (2021).
Xiang, Y. et al. Near-infrared mediated orthogonal bioimaging and intracellular tracking of upconversion nanophotosensitizers. Microchim. Acta 189, 120 (2022).
Xu, H. et al. Orthogonal multiplexed NIR-II imaging with excitation-selective lanthanide-based nanoparticles. Anal. Chem. 94, 3661–3668 (2022).
Jin, D. et al. Nanoparticles for super-resolution microscopy and single-molecule tracking. Nat. Methods 15, 415–423 (2018).
Liu, Y. et al. Super-resolution mapping of single nanoparticles inside tumor spheroids. Small 16, 1905572 (2020).
Chen, X. et al. Confining energy migration in upconversion nanoparticles towards deep ultraviolet lasing. Nat. Commun. 7, 10304 (2016).
Moon, B.-S. et al. Continuous-wave upconversion lasing with a sub-10 W cm−2 threshold enabled by atomic disorder in the host matrix. Nat. Commun. 12, 4437 (2021).
Shang, Y. et al. Low threshold lasing emissions from a single upconversion nanocrystal. Nat. Commun. 11, 6156 (2020).
Sun, T. et al. Ultralarge anti-Stokes lasing through tandem upconversion. Nat. Commun. 13, 1032 (2022).
Liao, J. et al. Optical fingerprint classification of single upconversion nanoparticles by deep learning. J. Phys. Chem. Lett. 12, 10242–10248 (2021).
Liu, S., Yan, L., Huang, J., Zhang, Q. & Zhou, B. Controlling upconversion in emerging multilayer core–shell nanostructures: from fundamentals to frontier applications. Chem. Soc. Rev. 51, 1729–1765 (2022).
Wen, S. et al. Advances in highly doped upconversion nanoparticles. Nat. Commun. 9, 2415 (2018).
Min, Q. et al. Atomic-level passivation of individual upconversion nanocrystal for single particle microscopic imaging. Adv. Funct. Mater. 30, 1906137 (2020).
Huang, F. et al. Transient energy trapping as a size-conserving surface passivation strategy for producing bright ultrasmall upconversion nanoprobes. Nano Energy 105, 108015 (2023).
Yin, A., Zhang, Y., Sun, L. & Yan, C. Colloidal synthesis and blue based multicolor upconversion emissions of size and composition controlled monodisperse hexagonal NaYF4: Yb,Tm nanocrystals. Nanoscale 2, 953–959 (2010).
Zhang, H., Li, Y., Lin, Y., Huang, Y. & Duan, X. Composition tuning the upconversion emission in NaYF4:Yb/Tm hexaplate nanocrystals. Nanoscale 3, 963–966 (2011).
Zhao, J. et al. Single-nanocrystal sensitivity achieved by enhanced upconversion luminescence. Nat. Nanotechnol. 8, 729–734 (2013).
Zhou, B. et al. Enhancing multiphoton upconversion through interfacial energy transfer in multilayered nanoparticles. Nat. Commun. 11, 1174 (2020).
Würth, C., Fischer, S., Grauel, B., Alivisatos, A. P. & Resch-Genger, U. Quantum yields, surface quenching, and passivation efficiency for ultrasmall core/shell upconverting nanoparticles. J. Am. Chem. Soc. 140, 4922–4928 (2018).
Rabouw, F. T. et al. Quenching pathways in NaYF4:Er3+,Yb3+ upconversion nanocrystals. ACS Nano 12, 4812–4823 (2018).
Siefe, C. et al. Sub-20 nm core–shell–shell nanoparticles for bright upconversion and enhanced Förster resonant energy transfer. J. Am. Chem. Soc. 141, 16997–17005 (2019).
Liu, Q. et al. Single upconversion nanoparticle imaging at sub-10 W cm−2 irradiance. Nat. Photonics 12, 548–553 (2018).
Su, Q. et al. Six-photon upconverted excitation energy lock-in for ultraviolet-C enhancement. Nat. Commun. 12, 4367 (2021).
Huang, K. et al. Room-temperature upconverted superfluorescence. Nat. Photonics 16, 737–742 (2022).
Pollnau, M., Gamelin, D. R., Lüthi, S. R., Güdel, H. U. & Hehlen, M. P. Power dependence of upconversion luminescence in lanthanide and transition-metal-ion systems. Phys. Rev. B 61, 3337–3346 (2000).
Maurizio, S. L., Tessitore, G., Krämer, K. W. & Capobianco, J. A. BaYF5:Yb3+,Tm3+ upconverting nanoparticles with improved population of the visible and near-infrared emitting states: implications for bioimaging. ACS Appl. Nano Mater 4, 5301–5308 (2021).
Zhan, Q. et al. Achieving high-efficiency emission depletion nanoscopy by employing cross relaxation in upconversion nanoparticles. Nat. Commun. 8, 1058 (2017).
Wei, W. et al. Cross relaxation induced pure red upconversion in activator- and sensitizer-rich lanthanide nanoparticles. Chem. Mater. 26, 5183–5186 (2014).
Liu, Y. et al. Amplified stimulated emission in upconversion nanoparticles for super-resolution nanoscopy. Nature 543, 229–233 (2017).
Lee, C. et al. Giant nonlinear optical responses from photon-avalanching nanoparticles. Nature 589, 230–235 (2021).
Weber, M. J. Optical properties of Yb3+ and Nd3+-Yb3+ energy transfer in YAlO3. Phys. Rev. B 4, 3153–3159 (1971).
Acknowledgements
This work was supported by the Fundamental Research Fund for Distinguished Scholars (Harbin Institute of Technology) (XWQQ5710001615 to G.C.), the Fundamental Research Funds for the Central Universities, China (AUGA5710052614 and AUGA8880100415 to G.C.), and the National Natural Science Foundation of China (52272270 and 51972084 to G.C.).
Funding
Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
G.C. conceived the research; D.H. prepared nanoparticles under the guidance of F.L.; D.H. built the optical system; D.H. acquired, processed and analyzed data; D.H. developed the theoretical model and performed numerical simulations; The paper was written by D.H. and G.C., with comments from H.Å.; All authors discussed the results; The project was supervised by G.C.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yung Doug Suh and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, D., Li, F., Ågren, H. et al. Inhibiting concentration quenching in Yb3+-Tm3+ upconversion nanoparticles by suppressing back energy transfer. Nat Commun 16, 4218 (2025). https://doi.org/10.1038/s41467-025-59452-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59452-4
