
Abstract
Microscopic colitis (MC) is a chronic inflammatory disease of the large intestine that primarily affects older adults and presents with chronic diarrhea. The etiology is unknown and there are currently no FDA approved medications or biomarkers for treatment or monitoring of the disease. Emerging evidence have implicated the gut microbiome and metabolome disturbances in MC pathogenesis. We conduct a comprehensive analysis of gut microbial and metabolic changes in a cohort of 683 participants, including 131 patients with active MC, 159 with chronic diarrhea, and 393 age- and sex-matched controls without diarrhea. Stool microbiome and metabolome are profiled using whole-genome shotgun metagenomic sequencing and ultra-high performance liquid chromatography–mass spectrometry, respectively. Compared to controls, eight microbial species including pro-inflammatory oral-typical Veillonella dispar and Haemophilus parainfluenzae, and 11 species, including anti-inflammatory Blautia glucerasea and Bacteroides stercoris are enriched and depleted in MC, respectively. Pro-inflammatory metabolites, including lactosylceramides, ceramides, lysophospholipids, and lysoplasmalogens, are enriched in active MC. Multi-omics analyses reveal robust associations between microbial species, metabolic pathways, and metabolites, suggesting concordant disruptions in MC. Here, we show distinct shifts in gut microbiome and metabolome in MC that can inform the development of non-invasive biomarkers and novel therapeutics.
Similar content being viewed by others

Introduction
Microscopic colitis (MC) is an inflammatory bowel disease (IBD) characterized by chronic relapsing watery diarrhea. MC is one of the most frequent causes of chronic diarrhea, especially in women older than 65 years, and is associated with a significant impairment in quality of life1. Yet, the pathophysiology of MC is largely unknown. One proposed mechanism is that MC occurs as a result of inappropriate immune response to environmental-induced perturbances in the gut microbiome2. This hypothesis is supported by the observation that fecal diversion can lead to endoscopic remission and reduction of pro-inflammatory cytokines, including TNF-α, while restoration of the gut continuity leads to disease recurrence and resurgence of pro-inflammatory cytokines3. Several smaller-scale studies have attempted to characterize the changes in the gut microbiome in MC and reported lower alpha diversity and a higher microbial dysbiosis index in patients with active MC compared to controls4,5. Further cross-sectional taxonomic profiling have revealed reduced abundance of Ruminococcaceae6, Coriobacteriaceae7, Clostridiales8, Collinsella9, and Akkermansia4,10, and increased abundance of pro-inflammatory Desulfovibrionales7 and Prevotella8 comparing MC to controls.
Longitudinal data are scarce, with some reported decreased abundance of Collinsella, Ruminococcaceae, Coriobacteriaceae and Clostridiales in active MC compared to remission6. A recent study from our group using metagenomic sequencing of stool samples reported depletion of Alistipes putredinis in MC5. Despite these previous endeavors, research to-date has been limited by small sample sizes (n = 10–52)4,5,6,7,8,9,10, reliance on 16S rRNA sequencing techniques, which provides lower resolution data on composition and function of the gut microbiome4,6,7,8,9, use of non-diarrhea controls that could not account for confounding by stool consistency4,6,7,8,9,10,11, lack of more direct data on function of the gut microbiome (e.g., metabolomics or transcriptomics)4,5,6,7,9,10 and limited data on differences in the gut microbiome according to disease activity6,7,8,9,10.
Besides taxonomic composition and functional potential, the gut microbiome also contributes to and is influenced by the intestinal metabolomic landscape. In fact, some small molecules, including those derived from gut microbiome, such as short-chain fatty acids (SCFA), have been found to play an important role in host immune response12. However, few studies have analyzed the microbiome-metabolome correlations in patients with MC. In this work, we establish a large cohort of 683 participants, including 131 patients with MC and two comparator groups—393 age-and sex-matched controls without diarrhea and 159 individuals with chronic diarrhea. We comprehensively characterize microbiome and metabolome alterations in MC, assess longitudinal changes associated with disease activity, and identify potential microbial and metabolic signatures that can inform biomarker development for MC.
Results
Patients with MC have a pro-inflammatory and aerotolerant microbiome
After quality control, we included 683 participants, including 131 patients with active MC, 159 patients with chronic diarrhea, and 393 age- and sex-matched controls without diarrhea for metagenome analysis (Table 1). Individuals with MC were in average older, more likely to be female, and had lower BMI as compared to chronic diarrhea controls. Our longitudinal cohort included 66 pairs of stool samples from patients with MC in both active and remission phase, among which 27 had collagenous colitis (CC) and 33 had lymphocytic colitis (LC). The median time between active to remission samples was 159 days (interquartile range, IQR: 89–238 days).
After quality control and filtering the metagenomic reads (relative abundance ≥ 0.01% and prevalence ≥ 10%), 467 microbial species were included in the downstream analyses. For the overall composition of the gut microbiome, we observed that the alpha diversity (Chao 1 index) was significantly lower in active MC as compared to controls without diarrhea (Wilcoxon p < 0.001) but not chronic diarrhea controls (Wilcoxon p = 0.446) (Fig. 1a). Similarly, in longitudinal analysis, the gut microbiome in active MC had a lower alpha diversity (Chao 1 index) as compared to remission (Wilcoxon p = 0.033) (Fig. 1b). These trends were consistent across subgroup analyses for MC subtypes and were also observed in the Shannon diversity index (Supplementary Figs. 1A–C and 2A–E). Principal coordinate analysis (PCoA) based on the Bray–Curtis dissimilarity and Aitchinson distances of microbial composition showed no significant difference between the structure of the gut microbiome according to disease status or activity (Fig. 1c, d and Supplementary Fig. 3A, B). These findings were corroborated in our PERMANOVA analysis where both disease type (MC vs chronic diarrhea vs control without diarrhea) and disease activity (active vs remission) explained 2.2% and 1.8% of the variation observed in stool microbiome composition, but neither reached statistical significance (Supplementary Fig. 3C).

a, b Alpha diversity (Chao 1 index) in the cross-sectional (a) and longitudinal cohorts (b). Cross-sectionally, the alpha diversity in the MC microbiome is lower than that of controls without diarrhea but not chronic diarrhea group. Longitudinally, the alpha diversity is higher in the remission phase compared to active phase among patients with MC. Data are shown as median with interquartile range (IQR, 25th and 75th). Whiskers indicated 1.5*IQR. P-values are two-sided without multiple testing adjustment. Source data are provided as a Source Data file. c, d Principal coordinate analysis (PCoA) based on Bray–Curtis dissimilarity show no significant differences in microbial composition comparing MC to chronic diarrhea or controls without diarrhea (c) and active MC versus MC in remission (d). Source data are provided as a Source Data file.
We compared the relative abundance of taxonomic features at species level. In our cross-sectional analysis, compared to controls without diarrhea, patients with MC had 46 enriched species and 234 depleted species (Benjamini–Hochberg FDR q < 0.25) while compared to chronic diarrhea, patients with MC had 36 enriched species and 17 depleted species (Benjamini–Hochberg FDR q < 0.25, Supplementary Fig. 4). Eight species were enriched in MC compared to both controls, while 11 species were depleted (Fig. 2a). Among the enriched species, Veillonella dispar, V. parvula, V. rogosae, and Haemophilus parainfluenzae are pathologic oral bacteria which have also been shown to be enriched in other types of IBD13,14,15 (Fig. 2b). Similarly, several depleted species, including Collinsella spp., Mediterraneibacter butyricigenes, Firmicutes spp., Blautia glucerasea, and Bacteroides stercoris have also been shown to be decreased in other types of IBD16,17,18,19,20. Notably, many of these species are known to have anti-inflammatory properties (e.g., B. stercoris20) or produce metabolites such as SCFA (e.g., B. glucerasea19) that can suppress inflammation. We then compared the top differentially abundant species identified cross-sectionally in the longitudinal cohort, and the results showed seven species were depleted in active MC compared to MC in remission, while no species were enriched (Fig. 2c). Six out of the seven depleted species (except Nitrosopumilus_SGB14899) in active disease were obligate anaerobes (such as Clostridiales_bacterium_NSJ_32 and Collinsella_SGB4121), supporting the notion that in the pro-inflammatory state there is excessive oxidative stress15,21. In exploratory analysis, we showed that most MC-enriched species were consistently enriched in both LC and CC compared to controls while MC-depleted species were only consistently depleted in CC (Supplementary Fig. 5). This finding suggests that there may be unique differences in the gut microbiome composition according to histologic subtypes of CC and LC.

a The heatmap shows the β coefficient of species with altered abundance comparing MC to both controls in the cross-sectional cohort. We use the following model to adjust for confounding factors: Log (Microbiome features) ~ intercept + Disease type (MC vs Chronic diarrhea vs Controls without diarrhea) + BSS (> 5 vs ≤ 5) + age + sex + BMI. Source data are provided as a Source Data file. *** FDR q < 0.05; ** 0.05 ≤ FDR q < 0.1; * 0.1 ≤ FDR p < 0.25. b Examples of log-transformed abundances of altered species in MC group (n = 131) in the cross-sectional cohort (Controls without diarrhea: n = 393, Chronic diarrhea: n = 159). V. dispar, V. parvula, and H. parainfluenzae are enriched in MC, while Methylobacterium_SGB15164, M. butyricigenes, and B. stercoris are depleted. Data are shown as median with interquartile range (IQR, 25th and 75th). Whiskers indicated 1.5*IQR. Source data are provided as a Source Data file. c Examples of log-transformed abundances of altered species in active MC (n = 66) versus MC in remission (n = 66) in the longitudinal cohort. All species are depleted in active MC compared to remission. Benjamini–Hochberg FDR q-values are derived from Wilcoxon signed rank tests. Data are shown as median with interquartile range (IQR, 25th and 75th). Whiskers indicated 1.5*IQR. Source data are provided as a Source Data file.
Patients with MC have a pro-inflammatory metabolome and disturbed lipid metabolism
We profiled the gut metabolome in 335 patients, including 92 patients with active MC, 138 patients with chronic diarrhea, and 105 controls without diarrhea. In the longitudinal cohort, 33 MC patients had metabolome data. The PCoA plot for metabolite composition revealed more distinct distributions of MC vs control without diarrhea group compared to the PCoA plot of microbial composition (Supplementary Fig. 6A). Moreover, we found strong positive correlation (Spearman rho = 0.579, p < 0.001) between the first axes of ordination of metabolome and microbiome datasets, suggesting strong coupling of microbial composition, metabolic profile, and disease type. Similarly in the longitudinal cohort, we observed greater distinction between active versus remission phase of MC by metabolite composition than microbiome (Supplementary Fig. 6B). Specifically, there was a strong negative correlation (Spearman rho = −0.550, p < 0.001) between the first axes of ordination of metabolome and microbiome, suggesting that the overall compositional alterations were similar between the two data types when comparing the active versus remission phases of MC. This indicates a coordinated shift in gut microbial and metabolic composition with disease activity.
Enrichment analyses were performed for the 1423 known metabolites using MaAsLin2. We first compared the abundance of metabolite classes in MC versus each control group (Supplementary Fig. 7A, B). Among the altered metabolite classes, 14 were enriched and three were depleted in MC as compared to both control groups (Fig. 3a). Top enriched metabolite classes included lactosylceramides (LCER), long chain polyunsaturated fatty acids (LC-PUFAs), lysoplasmalogens, fatty acid metabolism (acyl carnitine, long chain saturated) metabolites, ceramides, and lysophospholipids. Top depleted metabolite classes included fatty acid/dicarboxylate, sterol, and androgenic steroids. Many of these metabolite classes have also been shown to be altered in IBD12. Within the altered metabolite classes, there were 114 altered individual metabolites (Fig. 4a and Supplementary Data 1). Among the top metabolite classes, we conducted detailed enrichment analyses of individual metabolites for the four classes—LCER, ceramides, lysophospholipids, and lysoplasmalogens based on their consistent results when comparing to both controls and prior literature suggesting potential biological role in other IBD12,22,23 and immune-mediated diseases24. In our detailed enrichment analyses, the total levels of LCER (n = 3), ceramide (n = 5), lysophospholipid (n = 14), and lysoplasmalogen (n = 8) metabolites were elevated in active MC compared to the two control groups and the remission phase of MC (Fig. 3b–e). In addition, we also found elevated levels of individual metabolites from each of the four metabolite classes in active MC, including lactosyl-N-palmitoyl-sphingosine (d18:1/16:0), N-palmitoyl-sphingosine (d18:1/16:0), 1-stearoyl-GPC (18:0), and 1-stearyl-GPE (O-18:0), which have all been shown to be enriched in other immune-mediated diseases including other forms of IBD12,22,23,24 (Fig. 4b–e). Overall, the metabolomics data revealed profound disturbance of lipids (sphingolipids and phospholipids) metabolism in active MC.

a The box plots show 14 enriched metabolite classes (median t-statistics > 0) and three depleted metabolite classes (median t-statistics < 0) in MC compared to both controls after multi-variable adjustment. The dotted lines illustrated the significance threshold of an individual metabolite. T-statistics are derived from MaAsLin using the model: Log (Metabolite features) ∼ intercept + Disease type (MC vs Chronic diarrhea vs Controls without diarrhea) + BSS (> 5 vs ≤ 5) + age + sex + BMI. Data are shown as median with interquartile range (IQR, 25th and 75th). Whiskers indicated 1.5*IQR. Source data are provided as a Source Data file. b–e Enrichment analyses of metabolite class in the cross-sectional (top) (MC: n = 131, Controls without diarrhea: n = 393, Chronic diarrhea: n = 159) and longitudinal (n = 66) cohorts (bottom). We use the following model to adjust for confounding factors in the longitudinal cohort: Log (Metabolite features) ~ intercept + BSS (> 5 vs ≤ 5) + age + sex + BMI. Data are shown as median with interquartile range (IQR, 25th and 75th). Whiskers indicated 1.5*IQR. P-values are derived from the logistic regression and are two-sided without multiple testing adjustment. Source data are provided as a Source Data file.

a The volcano plots demonstrate alteration of individual metabolites’ relative abundance in MC compared to both controls. Red color indicates enriched relative abundance, turquoise color indicates depleted relative abundance, and gray color indicates no change in relative abundance. Coefficients and q-values are derived from MaAsLin using the same model. Source data are provided as a Source Data file. b–e Enrichment analyses of candidate individual metabolites in the cross-sectional (top) (MC: n = 131, Controls without diarrhea: n = 393, Chronic diarrhea: n = 159) and longitudinal (n = 66) cohorts (bottom). We use the following model to adjust for confounding factors in the longitudinal cohort: Log (Metabolite features) ~ intercept + BSS ( > 5 vs ≤ 5) + age + sex + BMI. Data are shown as median with interquartile range (IQR, 25th and 75th). Whiskers indicated 1.5*IQR. P-values are derived from the logistic regression and are two-sided without multiple testing adjustment. Source data are provided as a Source Data file.
LCER and ceramides are components of the broader sphingolipid metabolism pathway (Supplementary Fig. 8), known to be enriched in IBD25. Besides LCER and ceramides, we identified another four MC-enriched metabolites in this pathway, including palmitoyl sphingomyelin (d18:1/16:0) (a sphingomyelin), N-palmitoyl-sphinganine (d18:0/16:0) (a dihydroceramide), palmitoyl dihydrosphingomyelin (d18:0/16:0) (a dihydrosphingomyelin), and glycosyl-N-palmitoyl-sphingosine (d18:1/16:0) (a glucosylceramide) (Fig. 4a). Additionally, our metabolomic analysis revealed MC-specific alterations in several individual metabolites (Supplementary Data 1), such as enriched 1-dihomo-linolenylglycerol (20:3) (a monoacylglycerol), alanylhistidine (a dipeptide), and genistein (a food component/plant), none of which have been previously associated with UC or CD12. In exploratory analysis, we examined the differences in the metabolic profiles between MC and controls according to histologic subtypes of collagenous colitis (CC) and lymphocytic colitis (LC) and observed similar patterns (Supplementary Fig. 9).
Links between microbiome composition, microbial metabolic pathways, and metabolomic profiles
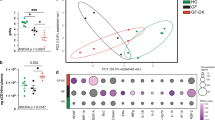
To understand how gut microbiome may alter the metabolomic landscape, we tested the correlations between altered species and altered metabolites (Fig. 5a). Overall, we saw a strong correlation between perturbed microbes and altered metabolites in MC, suggesting that our observed metabolic changes are likely linked to compositional changes in the gut microbiome. We used sphingolipids (Fig. 5b) to illustrate the strong concordance between species and metabolites. Levels of sphingolipid metabolites, which were elevated in MC, were positively correlated with relative abundance of V. parvula, which was enriched in MC (Fig. 5c). Conversely, levels of sphingolipid metabolites were negatively correlated with relative abundance of Methylobacterium_SGB15164, which was depleted in MC (Fig. 5d). We also considered the possibility that metabolomic disturbance in MC may be related to dynamic changes in the function of the gut microbiome and therefore additionally evaluated the correlations between: (1) all microbial species and altered metabolites and (2) all microbial metabolic pathways and altered metabolites.

a Metabolites and species are classified and clustered as MC-enriched (brown) or control-enriched (blue, including controls without diarrhea and chronic diarrhea). Metabolites are further grouped by metabolite class. The abundance of metabolites and microbes covariate concordantly with a clear pattern that MC-enriched species and metabolites (or control-enriched species and metabolites) are positively correlated, while MC-enriched species and control-enriched metabolites (or control-enriched species and MC-enriched metabolites) are negatively correlated. Source data are provided as a Source Data file. *** FDR q < 0.05; ** 0.05 ≤ FDR q < 0.1; * 0.1 ≤ FDR p < 0.25. b Correlations between sphingolipids (MC-enriched metabolite) and species. Source data are provided as a Source Data file. c, d Examples of individual correlations between MC-enriched sphingolipids and V. parvula (MC-enriched) or Methylobacterium SGB15164 (control-enriched). Concordantly, positive correlations are observed between MC-enriched species and MC-enriched sphingolipids, while negative correlations are found between control-enriched species and MC-enriched sphingolipids. Source data are provided as a Source Data file.
For correlations between all microbial species and altered metabolites, we reported the species highly correlated with sphingolipids, lysophospholipids, and lysoplasmalogens, all of which are pro-inflammatory (Supplementary Fig. 10A–C). From these correlations, we highlighted the key species, which have altered abundance in MC in our study or have been reported to be associated with MC in literature, and generated heatmaps (Supplementary Fig. 11A, B, E). We also demonstrated that upstream biosynthetic enzymes (EC3.1.3.27: Phosphatidylglycerophosphatase and EC4.1.1.65: Phosphatidylserine decarboxylase) in MC-enriched species (V. parvula and H. parainfluenzae) were positively correlated with abundance of lysophospholipids, suggesting a potential role of these pathologic species in synthesizing these pro-inflammatory metabolites (Supplementary Fig. 11C, D). Among species with altered abundance (Fig. 2a), pathologic oral species (such as H. parainfluenzae and Veillonella spp.) which were more abundant in MC had a positive correlation with elevated pro-inflammatory metabolites, while species (such as Methylobacterium_SGB15164, M. butyricigenes, and B. glucerasea) that were depleted in MC had a negative correlation with pro-inflammatory metabolites (Supplementary Fig. 12A–C). The strong concordance between differential abundance analysis and functional analysis suggested that pathologic species may produce pro-inflammatory metabolites in MC, or pro-inflammatory metabolites produced by MC gut may inhibit the growth of protective species. Although not observed in our differential abundance analysis, a number of notable species that have previously been linked to MC or IBD were also functionally correlated with the altered metabolites in our study. For instance, Ruminococcus gnavus, which has been reported to be strongly enriched in IBD26, was positively correlated with sphingolipids, lysophospholipids, and lysoplasmalogens, while Akkermansia spp., which have been shown to be depleted in IBD and MC4,10, were negatively correlated with these metabolites.
For correlations between all microbial metabolic pathways and altered metabolites, we presented the pathways highly correlated with sphingolipids, lysophospholipids, and lysoplasmalogens (Supplementary Fig. 13A–C). Several important pathways were correlated with elevated levels of pro-inflammatory metabolites. Enriched sphingolipids were correlated with altered carbohydrate metabolism (increased degradation and decreased synthesis of sugar) in bacteria, increased pentose phosphate pathway and purine/pyrimidine synthesis and salvage (DNA/RNA synthesis), upregulated peptidoglycan maturation (bacterial cell wall) and phospholipid synthesis (bacterial cell membrane), suggesting some bacteria may proliferate faster in inflamed gut (Supplementary Fig. 14A). Elevated levels of lysoplasmalogens were correlated with downregulated glycogen synthesis (Supplementary Fig. 14B), likely suggesting increased environmental stress during active MC phase27.
Discussion
In this large cohort study, we comprehensively characterized the composition of the stool microbiome and metabolome in patients with MC compared to chronic diarrhea and controls without diarrhea. Consistent with prior work from our group and others4,5, we broadly saw a decrease in microbiome richness in active MC as compared to controls without diarrhea and the remission phase of the disease. In per-feature analysis, we found enrichment in pathogenic oral-typical species such as H. parainfluenzae and Veillonella spp. in patients with active MC as compared to controls, while species with anti-inflammatory properties such as B. glucerasea and B. stercoris were reduced in relative abundance. Our analysis also revealed a marked increase in pro-inflammatory metabolites such as LCERs, ceramides, lysophospholipids, and lysoplasmalogens. Lastly, multi-omics analysis revealed a strong functional association across the microbiome and metabolome suggesting that compositional changes in the gut microbiome closely correlate with structural alterations in gut metabolome in MC.
Our findings are in line with prior studies of the gut microbiome in MC and IBD. Specifically, consistent with our findings, pro-inflammatory species such as Intestinibacter bartlettii, Veillonella spp., and H. parainfluenzae have been widely reported to be enriched in patients with MC or IBD5,8,13,14,15,28,29,30, while species such as Clostridiales spp., C. bacterium, Collinsella spp., M. butyricigenes, B. glucerasea, and B. stercoris have been shown to be depleted in the gut microbiome of patients with MC or IBD6,8,9,17,20. Interestingly and in contrast to prior studies in MC, we did not observe an association with Ruminococcaceae6, Coriobacteriaceae7, Akkermansia4,10, Desulfovibrionales7, Prevotella8, or Alistipes5. This may in large be explained by our adoption of multiple control groups, eliminating confounding effects by stool consistency, which has previously been shown may lead to false association with species such as Ruminococcaceae, Akkermansia, and Prevotella11. No prior study has examined stool metabolites in MC; however, our results were highly consistent with those previously reported in other forms of IBD12,31,32.
Our study significantly extends our current knowledge of gut microbiome in MC. First, through comparisons to multiple control groups and longitudinal sampling according to disease activity, we identified stool microbiome compositional and functional changes that appear to be unique to MC. Notably, some taxonomic alterations observed in our study, such as the enrichment of C. spiroforme and Clostridium_sp_AF20_17LB and the depletion of Methylobacterium_SGB15164, have not been previously associated with UC or CD. C. spiroforme is known to produce a clostridial toxin that can induce diarrhea and colitis in animal models33. In contrast, Clostridium_sp_AF20_17LB has been linked to a lower risk of immune checkpoint inhibitor-related colitis in a recent study, a finding that differs from our observations in MC and suggests that the microbiome alterations in MC may be different than those of other colitides34. Second, we identified key metabolites that are disturbed in MC and compared them to those reported in UC and CD12,22,23,24. For instance, lactosyl-N-palmitoyl-sphingosine (d18:1/16:0) (an LCER), N-palmitoyl-sphingosine (d18:1/16:0) (a ceramide), 1-stearoyl-GPC (18:0) (a lysophospholipid), and 1-stearyl-GPE (O-18:0)* (a lysoplasmalogen) are highly enriched in active MC relative to controls and those in remission. These findings have also been reported in other types of IBD12,22,23,24. We also identified several metabolites that are uniquely enriched in MC such as 1-dihomo-linolenylglycerol (20:3) (a monoacylglycerol) and alanylhistidine (a dipeptide), that have previously not been associated with CD or UC, suggesting distinct metabolic pathways that are disturbed in MC. These microbiome and metabolome alterations provide valuable insights into disease mechanisms and can help identify potential biomarkers specific to MC. Third, through correlation analysis, we demonstrated that many of the perturbations in the gut microbiome composition are closely linked to gut metabolome, suggesting that the shift in the gut microbiome likely has biological significance. Lastly, we explored differences in the gut microbiome and metabolome according to histologic subtypes of MC. Although the gut metabolomic landscapes were similar across CC and LC, we found that more species in the CC microbiome were significantly disturbed than those in LC, indicating the gut microbiome compositions may be distinct between MC subtypes.
Our findings are biologically plausible. In MC, an inflammatory milieu is expected to increase oxidative stress which in turn may lead to reduction in obligate anaerobic bacteria such as Collinsella spp. and C. bacterium with anti-inflammatory properties. Both Veillonella spp. and H. parainfluenzae are oral-typical bacteria enriched in MC, and their ectopic transplantation to the intestine has been shown to induce T helper 1 (TH1) cells and gut inflammation, implicated in IBD pathogenesis and disease exacerbation30. In our metabolomics analysis, sphingolipids were highly enriched in active MC, most of which were human-derived and pro-inflammatory25,35. For example, LCERs, also known as CD17 on leukocytes, promote inflammation through releasing arachidonic acids, facilitating neutrophil adhesion, and generating reactive oxygen species36,37. Similarly, ceramides can induce inflammation by activating NF-κB and COX-2 pathway38 or by being degraded to sphingosine and sphingosine-1-phosphate (S1P), which can modulate lymphocyte migration from lymph nodes12,39. Lastly, we demonstrated that biosynthetic enzymes in V. parvula and H. parainfluenzae contribute to the enrichment of lysophospholipids, which increase intestinal epithelial barrier permeability, facilitate bacterial translocation, and promote pathological Th1-mediated intestinal inflammation23,40,41.
SCFAs, produced by bacterial fermentation of lactate, play a crucial anti-inflammatory role by activating regulatory T cells (Tregs) and promoting intestinal barrier integrity42. While our metabolomic workflow only identified two SCFAs (butyrate/isobutyrate and valerate) via LC-MS, which is below our inclusion threshold of ≥ 3 metabolites per class, we compared their relative abundance and found no significant differences between MC and controls or between active and remission MC. This aligns with previous studies reporting no significant changes in SCFA levels when comparing active CC to quiescent phase or controls43. While some SCFA-producing bacteria, such as Veillonella spp., are enriched in MC, others, including B. stercoris and B. glucerasea, are depleted. Moreover, SCFA levels are influenced by additional factors such as gut transit time and luminal conditions, both of which are altered in conditions associated with chronic diarrhea such as MC and irritable bowel syndrome (IBS)43. In IBS, increased acetate and propionate have been linked to worsened symptoms44, underscoring the complexity of SCFA metabolism in gut disorders. Our finding of unchanged SCFA levels in MC likely reflects these dynamic interactions.
Lastly, we note that in addition to providing an opportunity for identifying novel biomarkers for diagnosing and monitoring MC12,45, our metabolomic analysis revealed that modulating the S1P receptor could potentially be a promising therapeutic target46,47,48. Although we did not directly identify S1P in our metabolomics profiling, its upstream metabolite sphingosine and downstream metabolite palmitate were successfully found and quantified. Patients with MC had elevated levels of sphingosine compared to controls without diarrhea, but the levels were similar to those of chronic diarrhea. In the longitudinal cohort, sphingosine levels were significantly higher in active MC compared to remission (Supplementary Fig. 15A). Similar trends were also found in palmitate analysis (Supplementary Fig. 15B).
Our study has several strengths. First, our study represents the largest study with longitudinal follow-up for MC patients which enabled us to compare the microbial and metabolic compositions according to disease type and activity and explore the difference between LC and CC. Second, by adopting multiple control groups, we decreased the possibility of confounding by stool consistency11. Third, the whole-genome shotgun metagenomic sequencing method provided higher resolution data on microbiome, generating species-level taxonomic profiles as compared to 16S rRNA sequencing, which generally only provides genus-level data4,6,7,8,9, but more importantly, cannot be mapped against curated databases for metabolic pathway reconstruction49. Fourth, we simultaneously analyzed the microbiome and metabolome, allowing us to examine both the abundance and functional changes in the gut microbiome to cross-validate multi-omic signatures of inflammation. Moreover, through additional analyses, we were able to assess the functional significance of microbiome changes as it relates to metabolic activity. We acknowledge several limitations. First, we did not systematically collect data on potential confounders such as dietary intake, history of prior gastrointestinal infection and medication use50,51,52, and therefore could not adjust for these factors in our analysis. Second, our participants were mostly White, which could limit the generalizability of our findings to other populations. Third, our sample size across histologic subtypes of MC was limited and therefore we could not make any definitive conclusion about differences in the gut microbiome between LC and CC. Lastly, we acknowledge that the observational nature of our study may limit our ability to make any conclusion about the biological roles of the observed changes in the gut microbiome and metabolome in the pathophysiology of MC.
In conclusion, patients with MC have dysbiotic gut microbiome and pro-inflammatory metabolome. In addition to deepening our understanding of MC pathogenesis, our findings have potential clinical implications including helping to identify non-invasive biomarkers for diagnosing and monitoring MC and offering potential clues for novel therapeutic targets for MC.
Methods
Study population
The study was approved by Partners Human Research Committee and the Institutional Review Board of Mass General Brigham (Protocol # 2015P001333 and # 2015P000275). All participants have provided written informed consent before enrollment. Starting January 2016, we invited all adults (≥ 18 years old) with chronic diarrhea who were seen for consultation or were scheduled for a diagnostic colonoscopy at the Massachusetts General Hospital (MGH) Gastroenterology unit to submit a stool sample and provide detailed information on their demographics and lifestyle factors (Fig. 6). Inclusion criteria for the active MC group included: (1) presence of active symptoms (≥ 3 bowel movements/day and Bristol stool score [BSS] > 5) and (2) a confirmed histologic diagnosis of MC. The chronic diarrhea cohort consisted of individuals with active symptoms (≥ 3 bowel movements/day and BSS > 5) but a normal colonoscopy with biopsies. Patients with active MC were invited to provide a second stool sample during remission phase (< 3 bowel movements/day and BSS ≤ 5) at least 8 weeks after their initial stool sample submission. At baseline, we excluded individuals with diagnoses of UC or CD, colorectal cancer, prior colorectal surgery, and recent antibiotics use (within 3 months).

Created in BioRender. Chen, A. (2025) https://BioRender.com/v34u718.
We also included individuals in our ongoing colonoscopy-based longitudinal cohort of GIDER (GastroIntestinal Disease and Endoscopy Registry)53,54,55,56, which included adult participants who were scheduled for a screening or surveillance colonoscopy at MGH (Fig. 6). Before their colonoscopy, consented individuals were asked to complete a baseline medical history questionnaire and provide a stool sample. Participants with a history of UC or CD, hereditary non-polyposis colorectal cancer (HNPCC), familial adenomatous polyposis (FAP), recent antibiotics use (within 3 months), or known bleeding disorders were not eligible for this study. We used this cohort to identify non-diarrhea controls that were matched in a 3:1 ratio to active MC participants according to age and sex.
Stool collection and processing
After consent, individuals were provided with a stool specimen collection kit prior to their scheduled colonoscopy. All participants, including MC, chronic diarrhea controls, and controls without diarrhea, were instructed to collect the sample prior to their bowel preparation and any dietary change for colonoscopy. Stool samples were processed and frozen within 48 h of collection. Participants provided stool specimens in storage tubes containing 95% ethanol. Samples were handled at room temperature and stored at −80 °C until nucleic acid extraction. Stool genomic DNA purification was performed using QIAGEN AllPrep MiniKit (Valencia, CA, USA) per the manufacturer’s instructions and consistent with the Human Microbiome Project protocols57,58.
Metagenomic sequencing, taxonomic composition, and metabolic pathway profiling
Purified DNA from stool samples were processed using the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA) following manufacturer’s protocol to generate metagenomic sequencing libraries. Then, whole-genome shotgun metagenomic sequencing was performed at the Broad Institute (Cambridge, MA, USA) using Illumina NovaSeq 6000 Sequencing System with 150-base-pair paired-end reads. The raw metagenomic reads were processed using KneadData (v0.12.0) to remove Illumina adapters and short reads (< 50 base pairs). We used Bowtie2 (v2.5.1) to identify and remove reads that are of human origin based on the Genome Reference Consortium human genome template (GRCh37/hg19). After this, quality control of the metagenomes was conducted using FastQC (v0.12.0). During quality control (QC), two samples with a read depth ≤ 7 million reads were removed. Samples below this threshold consistently exhibited a markedly lower number of identified species than expected, strongly suggesting poor sequencing quality. Four samples with unknown BSS were excluded, while seven chronic diarrhea samples were removed since initial collection was done after resolution of symptoms. After exclusion, 820 samples (out of 835 samples, 98.2%) were included in our study, with a post-quality control average read depth of 26.6 ± 9.8 million (~ 3.7 Gbp per sample).
Taxonomic composition and functional pathways were profiled from metagenomes using the bioBakery meta’omics workflow59. The Metagenomic Phylogenetic Analysis tool (MetaPhlAn v4.0.6) was used to compare our samples’ shotgun sequences with the tool’s database to characterize taxonomies and species relative abundance in each sample60. We adopted the HMP Unified Metabolic Analysis Network (HUMAnN3 v3.8) to profile the relative abundance of functional pathways61. HUMAnN3 aligns the metagenomic sequences to two databases, ChocoPhlAn pangenome database62 and UniRef90 protein database63, to reconstruct the microbial metabolic pathway abundances based on species identified from MetaPhlAn. Finally, before downstream diversity and enrichment analyses, we applied a filter to retain taxonomic species with relative abundance ≥ 0.01% and microbial metabolic pathways with relative abundance ≥ 0.1% that were present in ≥ 10% of samples54.
Metabolite profiling
Metabolomic profiling was performed using ultra-high performance liquid chromatography–mass spectrometry (UPLC-MS/MS) by Metabolon Inc. (Morrisville, NC, USA) in a subset of participants. A total of 335 samples were analyzed, including 92 patients with MC, 138 chronic diarrhea controls, and 105 controls without diarrhea. Stool samples were collected using standardized protocols, stored at −80 °C, and aliquoted after a single thaw to minimize degradation. Sample preparation and run order were randomized to reduce batch effects. System suitability and analytical reproducibility were monitored using Metabolon’s pooled human plasma matrix (MTRX), recovery and internal standards, and process blanks (ultra-pure water processed alongside study samples). Instrument and process variability were evaluated calculating the median relative standard deviation (RSD) for internal standards and endogenous metabolites in pooled QC samples. Each biological sample was analyzed once (n = 1), and pooled QC samples, process blanks, and internal standards were systematically interspersed throughout the analytical sequence to ensure performance consistency and reproducibility. Metabolites were extracted using the automated MicroLab STAR® system (Hamilton Company), where proteins were precipitated with methanol for removal, followed by centrifugation. The resulting supernatant was divided into four fractions for analysis: two for reverse phase (RP)/UPLC-MS/MS with positive ion mode electrospray ionization (ESI+), one for RP/UPLC-MS/MS with negative ion mode ESI (ESI−), and one for hydrophilic interaction chromatography (HILIC)/UPLC-MS/MS with negative ion mode ESI (ESI−).
Chromatographic separation was performed using Waters ACQUITY UPLC BEH C18 (2.1 × 100 mm, 1.7 µm) and BEH Amide HILIC (2.1 × 150 mm, 1.7 µm) columns. The UPLC-MS/MS analysis utilized four complementary methods tailored for different compound characteristics. The PosEarly method (ESI+, C18 column) was optimized for hydrophilic compounds, using a mobile phase of water and methanol containing 0.05% perfluoropentanoic acid (PFPA) and 0.1% formic acid (FA). The PosLate method (ESI+, C18 column) was optimized for hydrophobic compounds, using higher organic content with methanol, acetonitrile, water, 0.05% PFPA, and 0.01% FA. The Neg method (ESI−, C18 column) was optimized for basic conditions, using methanol and water with 6.5 mM ammonium bicarbonate (pH 8). Finally, the HILIC method (ESI−, BEH Amide column) was designed for hydrophilic compounds, using a mobile phase of water and acetonitrile containing 10 mM ammonium formate (pH 10.8). All analyses were performed using a Waters ACQUITY UPLC system coupled to a Thermo Scientific Q-Exactive Orbitrap mass spectrometer operating at 35,000 mass resolution64. The instrument used heated electrospray ionization (HESI-II) in both positive and negative ion modes. Mass spectra were acquired in full scan mode with data-dependent MS/MS (MSn) using dynamic exclusion, across a scan range of 70–1000 m/z.
Metabolite identification was based on Metabolon’s proprietary library, which includes retention time/index (RI), accurate monoisotopic mass (± 10 ppm), observed m/z, and MS/MS fragmentation spectra. Metabolites were annotated with standard chemical identifiers such as HMDB, KEGG, and PubChem when available. Peak areas were extracted and processed through established Metabolon untargeted metabolomics workflows. Raw peak area values were log-transformed, normalized to account for batch variation (median scaling to 1), and imputed by the minimum observed value per metabolite (Supplementary Data 2). For analysis, we used the log-transformed, normalized imputed dataset. Out of 1775 identified metabolite features, 1423 (80.2%) metabolites were annotated. For subsequent analyses, we limited metabolites to those annotated with relative abundance ≥ 0.1% and prevalence ≥ 10%.
Metagenomic and metabolomic analysis
From the MetaPhlAn taxonomic outputs, we calculated the alpha diversity using Chao 1 index and Shannon index for each sample at species level. The average alpha diversity was then compared between the three groups using non-parametric Wilcoxon rank-sum tests. To compare the difference in microbiome composition across samples, we calculated the Bray–Curtis dissimilarity and Aitchinson distances65. We performed classical multidimensional scaling with Bray–Curtis dissimilarity matrix or Aitchinson distances and constructed principal coordinates analysis (PCoA) plots. Subsequently, permutational multivariate analysis of variance (PERMANOVA) was conducted with 999 permutations to quantify the proportion of observed variation in microbiome composition explained by patient factors including disease type, stool consistency as defined by BSS, and demographics (age, sex, and BMI). The analyses were performed using the R package Vegan (v2.6–4)66.
In the cross-sectional analysis, to identify unique taxonomic features in MC, we compared relative abundance of species and pathways between MC versus non-MC controls using the Microbiome Multivariable Association with Linear Models (MaAsLin2 v3.18, http://huttenhower.sph.harvard.edu/maaslin2)67, while adjusting for BSS (> 5 vs ≤ 5), age, sex, and BMI. Age and BMI were standardized into z-scores in the regression model by MaAsLin2 (Supplementary Method 1). As an exploratory analysis, we also assessed the relative abundance of species in LC and CC, the histologic subtypes of MC. The linear model for cross-sectional analysis could be expressed as:
Metabolites were categorized into different classes based on Metabolon classification system, which assigns a metabolite class to each identified metabolite based on the relevant biochemical pathway. Using the same MaAsLin2 model, we compared the relative abundance of each metabolite in MC to controls. Metabolites altered in the same direction comparing MC to both controls were selected, and we then calculated and ranked the medians of t-statistics of metabolites within each metabolite class using box plots. These box plots provided a qualitative overview to help prioritize key metabolites and metabolite classes for subsequent quantitative analyses. Metabolite classes with a median > 0 were considered to be enriched in MC, while those with a median < 0 were considered depleted25. After identifying metabolite classes that were enriched or depleted in MC compared to both controls, we also compared the abundance of individual metabolites within each class cross-sectionally.
For the longitudinal comparison, we limited our analyses to species and metabolites that were significantly different between MC and both controls and used Wilcoxon signed-rank tests to compare the relative abundances between active and remission samples. We also explored whether our observed differences in relative abundances of metabolites were consistent across LC and CC histologic subtypes. For all analyses, multiple testing corrections were implemented using the Benjamini–Hochberg method when appropriate, and a false discovery rate (FDR) q < 0.25 was considered statistically significant, consistent with prior discovery-based analysis of microbiome and metabolomics data15,54,68. All p-values were two-sided.
Microbe-metabolite-microbial metabolic pathway correlation analysis
To understand the extent of metabolome alterations attributable to dysbiosis in MC, we performed hierarchical correlation analyses between microbiome compositions, microbial metabolic pathways, and metabolomic profiles using Hierarchical All-against-All association testing (HAllA v0.8.20)69. HAllA reports significant Spearman’s correlation coefficient (rho) between paired large multi-omics datasets, with statistical significance determined by permutations. Two correlation analyses were conducted. First, to determine if our observed metabolite alterations were linked to perturbations in species composition, we calculated the correlations of altered metabolites against all microbes and reported metabolite-microbe correlations. Second, to evaluate whether the metabolite alterations may be attributable to microbial metabolism, we estimated the correlations between altered metabolites and microbial metabolic pathways and reported metabolite-pathway correlations. For all the correlation analyses, we only reported those with Spearman’s rho > 0.15 or < −0.15, consistent with prior metabolite-microbe correlation analysis70. All correlation coefficients were calculated using the MaAsLin2 residuals to account for potential confounding effects of disease type, BSS, age, sex, and BMI.
Statistics & reproducibility
This is a prospective cohort study with longitudinal follow-up of a subset of participants. Statistical analyses are reproducible with our reported methods, datasets, and codes. No statistical method was used to predetermine sample size. Less than 2% of data were excluded after quality control. Our study is observational; therefore, no randomization or blinding were performed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All sequencing data that support the findings of this study have been deposited in Sequence Read Archive (BioProject: PRJNA1197998). All the metabolomic data are available within the paper and its supplementary information files (Supplementary Data 2). Source data are provided with this paper.
Code availability
All codes have been deposited in GitHub repository (https://github.com/albertsyc/Association-of-Distinct-Microbial-and-Metabolic-Signatures-with-Microscopic-Colitis) (https://doi.org/10.5281/zenodo.15170779)71.
Change history
02 July 2025
In this article the affiliation 'Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden' for Hamed Khalili was missing. The original article has been corrected.
References
Nyhlin, N., Wickbom, A., Montgomery, S. M., Tysk, C. & Bohr, J. Long-term prognosis of clinical symptoms and health-related quality of life in microscopic colitis: a case–control study. Aliment. Pharmacol. Ther. 39, 963–972 (2014).
Burke, K. E. et al. Microscopic colitis. Nat. Rev. Dis. Prim. 7, 39 (2021).
Daferera, N. et al. Fecal stream diversion and mucosal cytokine levels in collagenous colitis: a case report. World J. Gastroenterol. 21, 6065–6071 (2015).
Rindom Krogsgaard, L., Kristian Munck, L., Bytzer, P. & Wildt, S. An altered composition of the microbiome in microscopic colitis is driven towards the composition in healthy controls by treatment with budesonide. Scand. J. Gastroenterol. 54, 446–452 (2019).
Morgan, D. M. et al. Microscopic colitis is characterized by intestinal dysbiosis. Clin. Gastroenterol. Hepatol. 18, 984–986 (2020).
Carstens, A. et al. The gut microbiota in collagenous colitis shares characteristics with inflammatory bowel disease-associated dysbiosis. Clin. Transl. Gastroenterol. 10, e00065 (2019).
Millien, V., Rosen, D., Hou, J. & Shah, R. Proinflammatory sulfur-reducing bacteria are more abundant in colonic biopsies of patients with microscopic colitis compared to healthy controls. Dig. Dis. Sci. 64, 432–438 (2019).
Hertz, S. et al. Microscopic colitis patients possess a perturbed and inflammatory gut microbiota. Dig. Dis. Sci. 67, 2433–2443 (2022).
Sun, S. et al. Microbial associations with microscopic colitis. Clin. Transl. Gastroenterol. 13, e00528 (2022).
Fischer, H. et al. Altered microbiota in microscopic colitis. Gut 64, 1185–1186 (2015).
Vandeputte, D. et al. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 65, 57–62 (2016).
Vich Vila, A. et al. Faecal metabolome and its determinants in inflammatory bowel disease. Gut 72, 1472–1485 (2023).
Schirmer, M. et al. Compositional and temporal changes in the gut microbiome of pediatric ulcerative colitis patients are linked to disease course. Cell Host Microbe 24, 600–610.e604 (2018).
Vieira-Silva, S. et al. Quantitative microbiome profiling disentangles inflammation- and bile duct obstruction-associated microbiota alterations across PSC/IBD diagnoses. Nat. Microbiol 4, 1826–1831 (2019).
Lloyd-Price, J. et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019).
Atarashi, K. et al. Ectopic colonization of oral bacteria in the intestine drives T(H)1 cell induction and inflammation. Science 358, 359–365 (2017).
Inciuraite, R. et al. Constituents of stable commensal microbiota imply diverse colonic epithelial cell reactivity in patients with ulcerative colitis. Gut Pathog. 16, 16 (2024).
Morgan, X. C. et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13, R79 (2012).
Sun, M., Wu, W., Liu, Z. & Cong, Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J. Gastroenterol. 52, 1–8 (2017).
Ishikawa, D. et al. Anti-inflammatory effects of bacteroidota strains derived from outstanding donors of fecal microbiota transplantation for the treatment of ulcerative colitis. Inflamm. Bowel Dis. https://doi.org/10.1093/ibd/izae080 (2024).
Ni, J., Wu, G. D., Albenberg, L. & Tomov, V. T. Gut microbiota and IBD: causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 14, 573–584 (2017).
Daniluk, U. et al. Untargeted metabolomics and inflammatory markers profiling in children with Crohn’s disease and ulcerative colitis—A preliminary study. Inflamm. Bowel Dis. 25, 1120–1128 (2019).
Otake-Kasamoto, Y. et al. Lysophosphatidylserines derived from microbiota in Crohn’s disease elicit pathological Th1 response. J. Exp. Med. 219 https://doi.org/10.1084/jem.20211291 (2022).
Hakoi, H. et al. Lysophospholipase D from Thermocrispum limits psoriatic inflammation by hydrolyzing epidermal lysoplasmalogen produced by group IIF secreted phospholipase A(2). Biochimie 215, 75–87 (2023).
Franzosa, E. A. et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 4, 293–305 (2019).
Hall, A. B. et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 9, 103 (2017).
Wilson, W. A. et al. Regulation of glycogen metabolism in yeast and bacteria. FEMS Microbiol. Rev. 34, 952–985 (2010).
Read, E., Curtis, M. A. & Neves, J. F. The role of oral bacteria in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 18, 731–742 (2021).
Dahal, R. H., Kim, S., Kim, Y. K., Kim, E. S. & Kim, J. Insight into gut dysbiosis of patients with inflammatory bowel disease and ischemic colitis. Front. Microbiol 14, 1174832 (2023).
Forbes, J. D. et al. A comparative study of the gut microbiota in immune-mediated inflammatory diseases—does a common dysbiosis exist? Microbiome 6, 221 (2018).
Ström, A., Stenlund, H. & Ohlsson, B. The metabolomic profile of microscopic colitis is affected by smoking but not histopathological diagnosis, clinical course, symptoms, or treatment. Metabolites 14, 303 (2024).
Di’Narzo, A. F. et al. Integrative analysis of the inflammatory bowel disease serum metabolome improves our understanding of genetic etiology and points to novel putative therapeutic targets. Gastroenterology 162, 828–843.e811 (2022).
Borriello, S. P. & Carman, R. J. Association of iota-like toxin and Clostridium spiroforme with both spontaneous and antibiotic-associated diarrhea and colitis in rabbits. J. Clin. Microbiol. 17, 414–418 (1983).
Björk, J. R. et al. Longitudinal gut microbiome changes in immune checkpoint blockade-treated advanced melanoma. Nat. Med. 30, 785–796 (2024).
Le, H. H., Lee, M.-T., Besler, K. R. & Johnson, E. L. Host hepatic metabolism is modulated by gut microbiota-derived sphingolipids. Cell Host Microbe 30, 798–808.e797 (2022).
Nakamura, H. et al. Lactosylceramide-induced phosphorylation signaling to group IVA phospholipase A(2) via reactive oxygen species in tumor necrosis factor-α-treated cells. J. Cell Biochem. 118, 4370–4382 (2017).
Arai, T., Bhunia, A. K., Chatterjee, S. & Bulkley, G. B. Lactosylceramide stimulates human neutrophils to upregulate Mac-1, adhere to endothelium, and generate reactive oxygen metabolites in vitro. Circ. Res. 82, 540–547 (1998).
Nixon, G. F. Sphingolipids in inflammation: pathological implications and potential therapeutic targets. Br. J. Pharmacol. 158, 982–993 (2009).
Maceyka, M. & Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 510, 58–67 (2014).
Tan, S. T., Ramesh, T., Toh, X. R. & Nguyen, L. N. Emerging roles of lysophospholipids in health and disease. Prog. Lipid Res. 80, 101068 (2020).
Sawai, T., Drongowski, R. A., Lampman, R. W., Coran, A. G. & Harmon, C. M. The effect of phospholipids and fatty acids on tight-junction permeability and bacterial translocation. Pediatr. Surg. Int. 17, 269–274 (2001).
Deleu, S., Machiels, K., Raes, J., Verbeke, K. & Vermeire, S. Short chain fatty acids and its producing organisms: an overlooked therapy for IBD? eBioMedicine 66 https://doi.org/10.1016/j.ebiom.2021.103293 (2021).
Wildt, S., Nordgaard-Lassen, I., Bendtsen, F. & Rumessen, J. J. Metabolic and inflammatory faecal markers in collagenous colitis. Eur. J. Gastroenterol. Hepatol. 19, 567–574 (2007).
Tana, C. et al. Altered profiles of intestinal microbiota and organic acids may be the origin of symptoms in irritable bowel syndrome. Neurogastroenterol. Motil. 22, 512–e115 (2010).
Chen, P. et al. Serum biomarkers for inflammatory bowel disease. Front. Med. 7 https://doi.org/10.3389/fmed.2020.00123 (2020).
Danese, S., Furfaro, F. & Vetrano, S. Targeting S1P in inflammatory bowel disease: new avenues for modulating intestinal leukocyte migration. J. Crohn’s. Colitis 12, S678–S686 (2017).
Verstockt, B. et al. Sphingosine 1-phosphate modulation and immune cell trafficking in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 19, 351–366 (2022).
Sandborn, W. J. et al. Etrasimod as induction and maintenance therapy for ulcerative colitis (ELEVATE): two randomised, double-blind, placebo-controlled, phase 3 studies. Lancet 401, 1159–1171 (2023).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 42, D459–D471 (2013).
Nielsen, H. L., Dalager-Pedersen, M. & Nielsen, H. High risk of microscopic colitis after <em>Campylobacter concisus</em> infection: population-based cohort study. Gut 69, 1952 (2020).
Vich Vila, A. et al. Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat. Commun. 11, 362 (2020).
Nagata, N. et al. Metagenomic identification of microbial signatures predicting pancreatic cancer from a multinational study. Gastroenterology 163, 222–238 (2022).
Maxner, B. et al. Fruit and vegetable consumption is associated with lower prevalence of asymptomatic diverticulosis: a cross-sectional colonoscopy-based study. BMC Gastroenterol. 20, 221 (2020).
Hua, X. et al. Gut microbiome structure and function in asymptomatic diverticulosis. Genome Med. 16, 105 (2024).
Mashayekhi, R. et al. Obesity, but not physical activity, is associated with higher prevalence of asymptomatic diverticulosis. Clin. Gastroenterol. Hepatol. 16, 586–587 (2018).
Lee, J. W. J. et al. Association of distinct microbial signatures with premalignant colorectal adenomas. Cell Host Microbe 31, 827–838.e823 (2023).
Integrative HMP (iHMP) Research Network Consortium. The integrative human microbiome project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe 16, 276–289 (2014).
Peterson, J. et al. The NIH human microbiome project. Genome Res. 19, 2317–2323 (2009).
McIver, L. J. et al. bioBakery: a meta’omic analysis environment. Bioinformatics 34, 1235–1237 (2018).
Blanco-Míguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 41, 1633–1644 (2023).
Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 10, e65088 (2021).
Franzosa, E. A. et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 15, 962–968 (2018).
Suzek, B. E., Huang, H., McGarvey, P., Mazumder, R. & Wu, C. H. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23, 1282–1288 (2007).
Ford, L. et al. Precision of a clinical metabolomics profiling platform for use in the identification of inborn errors of metabolism. J. Appl Lab Med 5, 342–356 (2020).
Bray, J. R. & Curtis, J. T. An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 27, 325–349 (1957).
Oksanen, J. et al. vegan: Community Ecology Package (Version 2.6-4) [R package] (2022).
Mallick, H. et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 17, e1009442 (2021).
Wang, D. D. et al. The gut microbiome modulates the protective association between a Mediterranean diet and cardiometabolic disease risk. Nat. Med. 27, 333–343 (2021).
Ghazi, A. R. et al. High-sensitivity pattern discovery in large, paired multiomic datasets. Bioinformatics 38, i378–i385 (2022).
Dekkers, K. F. et al. An online atlas of human plasma metabolite signatures of gut microbiome composition. Nat. Commun. 13, 5370 (2022).
Chen, A. S.-Y. et al. Association of distinct microbial and metabolic signatures with microscopic colitis. https://doi.org/10.5281/zenodo.15170779 (2025).
Acknowledgements
This work is supported by the National Institute on Aging grant R01AG068390 (H.Kh.) and the National Institute of Diabetes and Digestive and Kidney Diseases grant P30DK043351 (R.J.X.). Funding sources did not participate in study design, analysis, interpretation, drafting of manuscript, or submission process.
Author information
Authors and Affiliations
Contributions
Study design and concept: H.Kh. Study funding: D.C.C., H.Kh., and R.J.X. Acquisition of data: D.C.C., H.Kh., J.G., J.M., K.W., and R.J.X.Acquisition of data: D.C.C., H.K.h., J.G., J.M., K.W., and R.J.X. Statistical analysis: A.S.Y.C., E.N., H.K.i., J.S., L.N. Drafting of the manuscript: A.S.Y.C. and H.K.h. Critical revision of the manuscript: A.S.Y.C., H.K.i., E.N., J.S., K.W., J.G., J.M., K.E.B., J.C.Y., L.H.N., K.S., D.C.C., R.J.X., H.K.h.
Corresponding author
Ethics declarations
Competing interests
H.Kh. has served on clinical advisory board for Cylinder and has received consulting fees from Aditium Bio for work unrelated to the topic. K.S. has served as a consultant to Ardelyx, Gemelli Biotech, Laborie, Mahana, Salix, and Takeda and has received research support from Ardelyx and ReStalsis for work unrelated to the topic. Other authors have no competing interests to disclose.
Ethical approval
The study was approved by Partners Human Research Committee and the Institutional Review Board of Mass General Brigham (Protocol # 2015P001333 and # 2015P000275).
Peer review
Peer review information
Nature Communications thanks Suguru Nishijima, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, A.SY., Kim, H., Nzabarushimana, E. et al. Association of distinct microbial and metabolic signatures with microscopic colitis. Nat Commun 16, 4644 (2025). https://doi.org/10.1038/s41467-025-59566-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59566-9
