
Abstract
Metal-catalyzed hydrosilylation of alkynes is recognized as a straightforward and atom economic method for synthesizing alkenylsilanes. While substantial advancements have been made with terminal alkynes, achieving precise regio- and stereocontrol with unsymmetrical internal alkynes remains a significant challenge. In this study, we report the utilization of an intriguing β-boron effect in metal catalysis, enabling an exclusively regioselective Ru-catalyzed hydrosilylation of propargylic N-methyliminodiacetic acid boronates (B(MIDA)) to synthesize alkenylsilanes. Variations in the Ru catalyst can lead to stereo-divergency without compromising regioselectivity. Density functional theory (DFT) calculations indicate that the hyperconjugative effect of the σ(C–B) bond, which stabilizes the electrophilic metallacyclopropene intermediate with Fischer carbene character, is crucial for achieving high regioselectivity. The observed switch in stereoselectivity is attributed to the different steric effects of 1,2,3,4,5-pentamethylcyclopenta-1,3-diene (Cp*) and cyclopenta-1,3-diene (Cp) ligands in the catalyst. This method produces a diverse array of regio- and stereodefined products incorporating boryl, silyl, and alkene functionalities, each of which serves as a valuable handle for further functionalization.
Similar content being viewed by others
Introduction
Organosilicon compounds hold significant utility across diverse fields including synthetic chemistry1,2,3,4,5,6,7, medicinal chemistry8, and materials science9. Among these, alkenylsilanes stand out as crucial building blocks in organic synthesis. Their possession of a polarized alkene moiety and a silicon center renders them environmentally friendly and user-friendly carbon-centered nucleophiles, facilitating numerous nucleophilic addition10,11, oxidation12,13,14, and cross-coupling reactions2,3.
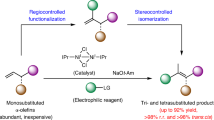
While numerous methods exist for synthesizing alkenylsilanes, metal-catalyzed hydrosilylation of alkynes offer the most direct and atom-economic approach15,16. However, the primary focus has been on the hydrosilylation of terminal alkynes17,18,19,20,21,22,23,24,25,26. In contrast, the utilization of unsymmetrical internal alkynes has faced challenges in achieving precise regio- and stereocontrol (Fig. 1A). Notably, employing unsymmetrical (internal) alkynes can yield up to four regio- and/or stereoisomers. To tackle this issue, attaching electronically (e.g., aryl24,27,28, carbonyl29,30,31,32, CF333, or heteroatom34,35,36,37) or sterically diverse substituents38,39,40,41 directly to the acetylenic carbon has proven effective (Fig. 1B). In addition, for internal alkynes with similar substituents, utilizing pendent directing groups has emerged as another efficient protocol14,42,43,44,45,46. Specifically, directing groups based on electron-negative heteroatoms capable of coordinating14,42,43,46 with the metal center or hydrogen-bonding44,45,46,47 to the metal ligand have been elegantly developed (Fig. 1B). Despite these advancements, there remains a pressing demand for alternative strategies to achieve precise regio- and stereocontrol for practical applications.

A metal-catalyzed hydrosilylation of internal alkynes. B traditional strategies for solving regioselectivity problems. C β-boron effect-enabled regioselective functionalizations of alkynes.
Recently, we have unveiled the capability of the sp3-B hybridized B(MIDA) (N-methyliminodiacetic acid) moiety48,49 to induce the high-lying σ(C–B) orbital interaction with the low-lying p*(C), effectively stabilizing adjacent carbocations. The β-boron effect has enabled us to achieve regioselective functionalization reactions involving internal alkenes50,51, alkynes52, and allenes53 (left, Fig. 1C). Motivated by this discovery, we sought to explore whether a similar effect could be harnessed in the realm of regio-controlled metal catalysis.
Our attention was drawn to the intriguing Ru-catalyzed hydrosilylation of alkynes, a transformation pioneered by Trost19,54 and further developed by Fürstner44,47, among others32,35,38,45,55. Mechanistically, an unusual oxidative hydrometallation of alkyne leads to the formation of a metallacyclopropene intermediate, wherein the hydrogen atom and the silyl group of the silane are arranged on opposite sides of the ruthenacyclopropene plane. Following a silyl migration, an unusual anti-addition product is formed54,55,56. We hypothesized that the electrophilic nature of the metallacyclopropene intermediate, akin to a Fischer carbene57, could potentially benefit from stabilization by an adjacent σ(C–B) bond, thereby facilitating high regioselectivity (right, Fig. 1C). This hypothesis aligns with the proton-like property of the hydrogen atom in Ru–H35,54 and the nucleophilic nature of the γ position of propargylic B(MIDA)s52. Consequently, the β-silylated products should be preferentially formed.
In this report, we present a β-boron effect-guided regioselective Ru-catalyzed hydrosilylation of propargylic B(MIDA)s. Intriguingly, subtle variations in the Ru catalyst led to a switch in stereoselectivity without altering the regioselectivity. Such a pronounced alteration in stereo-selectivity is a rare occurrence in metal-catalyzed hydrosilylation of internal alkynes. This reaction enables the synthesis of diverse regio- and stereo-defined building blocks that amalgamate the synthetic potential of boryl, silyl, and alkene functionalities (right, Fig. 1C)58,59,60,61,62,63,64,65,66. The involvement of the β-boron effect in stabilizing the electrophilic Ru-carbene is theoretically supported, accounting for the observed high level of regioselectivity. Furthermore, our findings regarding stereo-divergence have been rationalized through DFT studies.
Results
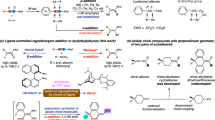
In our initial study, we employed propargylic B(MIDA) S-1 and triethoxysilane as model starting materials (Table 1). The cationic catalyst [Cp*Ru(MeCN)3]PF6 was chosen for initial evaluation due to its proven catalytic reactivity in a range of alkyne functionalization reactions. When conducted in THF, the reaction provided the exclusive β-selective (β/γ > 20:1) product 1, validating our working hypothesis. Additionally, exceptional anti-selectivity (Z/E > 20:1) and yield (95%) were achieved (entry 1). The reaction performed equally well in 1,4-dioxane (entry 2), but MeCN proved unsuitable as a solvent, likely due to its strong coordinating nature (entry 3)67. An intriguing observation arose when we substituted [Cp*Ru(MeCN)3]PF6 with its analogue [CpRu(MeCN)3]PF6, featuring a smaller Cp ligand. This change resulted in a switch of stereochemistry, with the syn-addition product increasing (E/Z = 1:1), while maintaining high regioselectivity (entry 4). This outcome underscores the significant impact of ligand steric effects on stereoselectivity28,38,68,69 and emphasizes the potential for achieving stereo-divergence through judicious catalyst selection. Indeed, employing CpRu(PPh3)2Cl as the catalyst in THF led to excellent syn-selectivity and regioselectivity, yielding compound 2 with a 65% yield, E/Z = 16:1, and β/γ > 20:1 (entry 5). Conversely, the use of the bulkier Cp*Ru(PPh3)2Cl catalyst resulted in increased formation of the anti-addition product (entry 6). Conducting the reaction in 1,4-dioxane improved both yield and stereoselectivity (entry 7). Exploration of other ruthenium-based catalysts such as [Cp*RuCl2]n, Ru(PPh3)3Cl2, and [Ru(pcymene)Cl2]2 proved ineffective (entries 8–10). In addition, attempts with other hydrosilylation catalysts, including [Rh]70, [Co]41, and [Pt]37 complexes following either the classic Chalk-Harrod or modified Chalk-Harrod mechanism, yielded either poorer regioselectivity or no reactivity (entries 11–13).
The scope for the synthesis of both stereoisomers was then explored (Fig. 2). A diverse array of silanes, including trialkyl- (3–5), aryldialkyl- (6), mono-alkoxyl- (7), di-alkoxyl- (8, 9), and trialkoxyl-substituted (10, 11) ones, were successfully applied to the anti-addition reactions, yielding the corresponding products with excellent regio- and stereoselectivities. The lower Z/E ratio (3:1) for HSiMe(OTMS)2 is likely due to steric hindrance (9). Next, a wide variety of internal alkynes were subjected to the reaction. It was found that valuable functional groups such as halogens (14-16), aryl groups (17–22), cyano (26), sulfonamide (27, 29), amide (28, 30), ester (31), hydroxy (32-34), OTBDPS (35, 36), OBz (37), OTs (38), and OBn (39) were all well tolerated, demonstrating the robustness and mildness of the protocol. Terminal alkynes were applicable as well, but in lower yields, with about 50% starting materials remained (41, 42). The installation of a secondary alkyl substituent on the distal side of the triple bond had minimal impact on the selectivity (40). However, the introduction of a substituent α- to the boryl moiety did reduce the Z/E ratio to 4.5:1, but the regioselectivity remained unaffected (43). Enyne (44) was also well tolerated with good yield and stereoselectivity. However, aromatic alkyne (45) was not applicable even when heated to 100 °C.

Reaction conditions: propargylic B(MIDA) (0.1 mmol, 1.0 equiv.), silane (0.2 mmol, 2.0 equiv.), [Cp*Ru(MeCN)3]PF6 (5 mol%), in 1,4-dioxane or THF (1.0 mL), room temperature.
The scope for the CpRu(PPh3)2Cl-catalyzed syn-hydrosilylation was also impressive (Fig. 3). While all the silanes tested exhibited excellent regioselectivity, trialkyl- (46, 47) and aryldialkylsilanes (48) showed slightly diminished stereoselectivity. Notably, HSi(OTMS)2Me performed well in this reaction (49), and the resulting vinyl-Si(OTMS)2Me is a suitable Hiyama-Denmark cross-coupling partner22. For various propargylic B(MIDA)s, consistently high levels of regio-control were observed. It is noteworthy that, in previous studies, alkynes containing tethered hydrogen bond donors (e.g., OH, NHTf) that can induce inter-ligand interactions with the [Ru–Cl] bond and the silanes tend to undergo proximal silylation to the directing group44,45,47. However, in our case, the presence of the B(MIDA) moiety reversed this regioselectivity, yielding exclusively the distal functionalization product (61, 63, 64). These results highlight the profound directing effect of the B(MIDA) moiety. Our DFT calculations revealed that the activation energies for compounds 59, 61, and 62, influenced by the directing effect of the B(MIDA) moiety, are 2.7–4.8 kcal/mol lower than those governed by hydrogen bonding (For further details, see the Supplementary Information). Propargylic B(MIDA)s with cyclic (58, 69) or branched substituent (59, 68) were also competent substrates. Commonly encountered functional groups such as chloro (53), aryl (54–57), cyano (60), amide (61), ester (62), hydroxy (63, 64), OTBDPS (65), OBz (66), and OTs (67) were all well tolerated. Interestingly, this protocol is sensitive to steric hindrance at the B(MIDA) side. When a methyl group was installed at its α position, increasing the reaction temperature to 60 °C was necessary to ensure high conversion, which unfortunately compromised both regio- and stereoselectivity. In this circumstance, we found that replacing CpRu(PPh3)2Cl with its cationic analog [CpRu(MeCN)3]PF6 was beneficial (70). However, enyne (71), aromatic alkyne (72), and terminal alkyne (41) was not applicable.

Reaction conditions: propargylic B(MIDA) (0.1 mmol, 1.0 equiv.), silane (0.2 mmol, 2.0 equiv.), CpRu(PPh3)2Cl (5 mol%), in 1,4-dioxane (1.0 mL) at room temperature. aReaction temperature: 60 oC, b[CpRu(MeCN)3]PF6 (5 mol%) as the catalyst.
Synthetic utilities
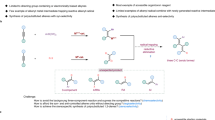
The presence of chemically distinct boryl, silyl, and alkene functionalities in the products offers ample opportunities for follow-up decoration. As shown in Fig. 4, the gram-scale synthesis of 6 could be achieved in 87% yield with simple crystallization. The silyl group in 6 could be easily converted to iodide 73 upon treatment with NIS and 2,6-lutidine, which would allow further cross-coupling reactions such as Suzuki-Miyaura cross-coupling (74), Sonogashira cross-coupling (75), and Mizoroki-Heck cross-coupling (76). The B(MIDA) moiety can be transferred to Bpin, and the resulting allyl Bpin can be chemoselectively oxidized to a silylated allyl alcohol (77). The silyl group can undergo Hiyama-Denmark cross-coupling without difficulty (78). In addition, the stereochemistry of the double bond in the syn- or anti-product can be effectively translated into the high diastereoselectivity when reacting with benzaldehyde (79–81)71,72.

A gram-scale synthesis. B synthetic applications. Reaction conditions: a 6 (0.1 mmol, 1.0 equiv.), NIS (0.2 mmol, 2.0 equiv.), 2,6-lutidine (0.7 equiv.), HFIP (0.2 M), 0 oC; b 73 (0.1 mmol, 1.0 equiv.), PhB(OH)2 (0.3 mmol, 3.0 equiv.), Pd(PPh3)2Cl2 (10 mol%), SPhos (15 mol%), K2CO3, (0.15 mmol, 1.5 equiv.), Ag2O (0.25 mmol, 2.5 equiv.), 4 Å MS, THF (0.5 M), 60 oC; c 73 (0.1 mmol, 1.0 equiv.), phenylacetylene (0.15 mmol, 1.5 equiv.), Pd(PPh3)2Cl2 (5 mol%), CuI (10 mol%), Et3N (0.3 mmol, 3.0 equiv.), DMF, (0.5 M), rt; d 73 (0.1 mmol, 1.0 equiv.), ethyl acrylate (0.2 mmol, 2.0 equiv.), Pd(PPh3)4 (5 mol%), Et3N (2.0 equiv.), MeCN (0.5 M), 70 oC; e 6 or 3 or 49 (0.1 mmol, 1.0 equiv.), pinnacol (0.5 mmol, 5.0 equiv.), NaHCO3 (0.5 mmol, 5.0 equiv.), CH3OH, 50 oC, then PhCHO (0.11 mmol, 1.1 equiv.), Et2O (0.2 M), rt.; f 49 (0.1 mmol, 1.0 equiv.), pinnacol (0.5 mmol, 5.0 equiv.), NaHCO3 (0.5 mmol, 5.0 equiv.), CH3OH, 50 oC, then NaBO3·4H2O (0.3 mmol, 3.0 equiv.), THF/H2O (1:1, 0.2 M), rt; g 77 (0.1 mmol, 1.0 equiv.), methyl 4-iodobenzoate (0.15 mmol, 1.5 equiv.), Pd2dba3 (5 mol%), TBAF (2.0 equiv.), THF (0.2 M), 60 oC.
Experimental mechanistic studies
To highlight the role of the B(MIDA) moiety in this regio- and stereoselective reaction, several control experiments were conducted. The use of sp²-B propargylic Bpin S-40 in this reaction also led to an anti-addition product, but with a diminished β/γ ratio of 4:1 (Fig. 5A). Our calculations indicated that Bpin exhibits a lower tendency to engage in σ(C–B) hyperconjugation (For further details, see the Supplementary Information). In contrast, employing propargylic TIDA (tetramethyl N-methyliminodiacetic acid) boronate S-41, a bulkier sp³-B analogue of B(MIDA), resulted in the corresponding product with excellent regioselectivity as well. When using propargylic BF3K S-42, the reaction did not occur, even when the reaction was performed in various solvents such as THF, dioxane, MeCN, or acetone to rule out solubility issues. Bdan S-43 also demonstrated excellent β-selectivity, though with a relatively low yield. Taken together, these results suggest that the β-boron effect may be present in multiple boron species, but B(MIDA) delivers the most favorable outcomes. The reaction of homopropargylic MIDA-boronate S-44 with one additional carbon linkage showed a regioselectivity of 4:1 (Fig. 5B). This result indicates that the directing ability of the B(MIDA) moiety decreases with increased distance. The hydrosilylation of hex-4-yn-1-ylbenzene, an internal alkyne without boron groups, showed a low regioselectivity of 2:1 (87, 88). It should also be mentioned that, when using CpRu(PPh3)2Cl as the catalyst, the hydrosilylation of hex-4-yn-1-ylbenzene showed both low regio- and stereoselectivity, forming including four isomers (Z-α, Z-β, E-α, E-β) (Fig. 5C). We were also curious about the origin of the unusual syn-selectivity observed with CpRu(PPh₃)₂Cl as the catalyst. When 5 mol% of AgPF₆ was added to precipitate the chloride from the catalyst, we observed a complete loss of stereoselectivity. When 10 mol% of PPh3 was added to the reaction with [CpRu(MeCN)3]PF6 as the catalyst, the reaction did not occur. When both PPh3 and LiCl were added to this catalyst system, good regio- and stereoselectivity were observed. All these results above indicated the crucial role of the Cl ligand in determining the stereochemical outcome of the reaction instead of PPh3 (Fig. 5D). Deuteration experiments with DSiEt3 (99% D) yielded 3-d and 46-d smoothly with 99% D-incorporation at the γ-position (Fig. 5E). The kinetic isotope effect measured via intermolecular competition between HSiEt3 and DSiEt3 gave an inverse KIE (kH/kD = 0.67 for 3 & 3-d, kH/kD = 0.47 for 46 & 46-d) with both Ru catalytic (Fig. 5F). Similar to this result, a parallel kinetic isotope experiment with CpRu(PPh3)2Cl as the catalyst also gave an inverse KIE of 0.32 (Fig. 5G). We proposed that during the concerted oxidative-addition and hydride-insertion, the weaker Si–H/D bond are partially weakened and C–H/D bond are partially formed. Due to the higher bond energy of C–D bond than C–H bond, the driving force of forming C–D bond should be stronger73,74,75.

A selectivity of other [B] groups. B selectivity of homopropargylic B(MIDA). C selectivity of hex-4-yn-1-ylbenzene. D selectivity of hex-4-yn-1-ylbenzene. E deuteration experiments. F intermolecular KIE experiment. G parallel KIE experiment.
Computational mechanistic studies
To gain a clearer understanding of the mechanisms underlying the regioselectivity and stereoselectivity in this reaction, DFT calculations were conducted (Fig. 6, Supplementary Data 1 & Supplementary Data 2). In the [Cp*Ru(MeCN)₃]PF₆-catalyzed system (Fig. 6A), the propargyl MIDA boronate and triethoxysilane reagent initially exchange with the MeCN ligands in the [Cp*Ru(MeCN)₃]PF₆ catalyst, forming the intermediates INT-1β and INT-1γ. For β-selectivity, a concerted hydrometallation occurs through transition state TS-1β (ΔG≠ = 12.3 kcal/mol), leading to the formation of the ruthenium three-membered ring intermediate INT-2β-anti (ΔG = 2.5 kcal/mol). In comparison, achieving γ-selectivity requires a higher activation barrier (via TS-1γ, ΔG≠ = 14.1 kcal/mol), and the corresponding intermediate INT-2γ-anti is also higher in energy (ΔG = 10.7 kcal/mol). This indicates that the concerted hydrometallation process is the regioselectivity-determining step, with TS-1γ and TS-1β differing in energy by ΔΔG≠ = 1.8 kcal/mol.

A the [Cp*Ru(MeCN)3]PF6-catalyzed system. B the CpRu(PPh3)2Cl-catalyzed system.
NBO orbital interaction analysis reveals that INT-2β-anti exhibits a significant orbital interaction between σ(C–B) and π*(C = Ru), with a second-order perturbation energy (E(2)) of 7.6 kcal/mol. This suggests that the hyperconjugative effect of the C–B bond significantly stabilizes the C = Ru bond. In contrast, no such orbital interaction is present in INT-2γ-anti. Therefore, the hyperconjugative interaction between σ(C–B) and π*(C = Ru) likely plays a crucial role in driving the β-regioselectivity during the concerted hydrometallation process.
Next, INT-2β-anti undergoes reductive elimination (via TS-2β-anti, ΔG≠ = 9.9 kcal/mol) to yield the anti-product β-PC-anti. However, to obtain the syn-product from INT-2β-anti, an additional Cβ–Cγ bond rotation is required. Nevertheless, the scanning results reveal that the Cβ–Cγ bond directly rotation from INT-2β-anti is more likely to lead to retro-hydrosilylation. Since the MeCN can facilely dissociate from the Ru center in INT-2β-anti and re-coordinate to the metal center to form INT-2β’-anti (see Supplementary Information for details), the INT-2β’-syn can be formed by the Cβ–Cγ bond rotation from INT-2β’-anti (via TSβ-rot, ΔG≠ = 13.7 kcal/mol). Although INT-2β’-syn is thermodynamically more stable than INT-2β’-anti due to steric effects, the energetically demanding TSβ-rot prevents the facile conversion of INT-2β’-anti to INT-2β’-syn. Once INT-2β’-syn is formed, it can undergo reductive elimination (via TS-2β’-syn, ΔG≠ = 7.8 kcal/mol) to produce the syn-product β-PC-syn. The energy difference between TSβ-rot and TS-2β-anti is 3.8 kcal/mol, which aligns with the experimentally observed high stereoselectivity favoring the anti-configuration over the syn-configuration for the [Cp*Ru(MeCN)₃]PF₆-catalyzed reaction.
In the CpRu(PPh₃)₂Cl-catalyzed system (Fig. 6B), initial ligand exchange leads to the formation of intermediates CpINT-1β and CpINT-1γ. For β-selectivity, a concerted hydrometallation occurs via CpTS-1β (ΔG≠ = 22.9 kcal/mol), forming the three-membered ring intermediate CpINT-2β-syn. Similar to the [Cp*Ru(MeCN)₃]PF₆-catalyzed system, the γ-site selective concerted hydrometallation CpTS-1γ requires a higher energy barrier (ΔG≠ = 27.3 kcal/mol) to form the intermediate CpINT-2γ-anti, which is also higher in energy (ΔG = 19.9 kcal/mol). NBO orbital interaction analysis indicates that CpINT-2β-syn exhibits a significant orbital interaction between σ(C–B) and π*(C = Ru) (E(2) = 10.1 kcal/mol), whereas CpINT-2γ-anti does not show such interaction. Thus, the β-regioselectivity is attributed to the hyperconjugative effect between σ(C–B) and π*(C=Ru). Following this, CpINT-2β-syn undergoes reductive elimination via CpTS-2β-syn (ΔG≠ = 21.3 kcal/mol), yielding the syn-product β-PC-syn. The high activation barrier of CpTSβ-rot (ΔG≠ = 27.3 kcal/mol) prevents the rotation of the Cβ–Cγ bond, despite the subsequent reductive elimination via CpTS-2β-anti being relatively low in energy (ΔG≠ = 20.9 kcal/mol). The significant energy difference of 6.0 kcal/mol between CpTSβ-rot and CpTS-2β-syn aligns with the experimentally observed high stereoselectivity between the anti and syn configurations for CpRu(PPh₃)₂Cl. It was found that Cl⁻ plays a crucial role in stereoselectivity by imposing steric hindrance (see the Supplementary Information for further details).
The key distinction between the [Cp*Ru(MeCN)₃]PF₆-catalyzed system and the CpRu(PPh₃)₂Cl-catalyzed system lies in their respective concerted hydrometallation processes. Specifically, in the [Cp*Ru(MeCN)₃]PF₆-catalyzed system, the transition state TS-1β results in the generation of INT-2β-anti, the anti-product precursor. Conversely, in the CpRu(PPh₃)₂Cl-catalyzed system, the transition state CpTS-1β leads to the formation of CpINT-2β-syn, the syn-product precursor. To investigate the underlying reasons behind the anti/syn selectivity in these different catalytic systems, we performed Born-Oppenheimer molecular dynamics (BOMD) calculations76,77 on the transition states TS-1β and CpTS-1β (Fig. 7A). For the [Cp*Ru(MeCN)₃]PF₆-catalyzed system, among 100 dynamic trajectories, 58 led to INT-1β preceding TS-1β, 41 resulted in the anti-product precursor INT-2β-anti, and only one yielded the syn-product precursor. In contrast, for the CpRu(PPh₃)₂Cl-catalyzed system, 32 out of 100 trajectories produced the syn-product precursor CpINT-2β-syn, while only 8 led to the anti-product precursor. These BOMD results align with the reaction potential energy surfaces described above, indicating that the anti/syn selectivity in these catalytic processes is kinetically controlled.

A Sum of the BOMD Results. B Conformational Analysis of the Dynamic Trajectories.
The detailed conformational analysis of dynamic trajectories based on TS-1β and CpTS-1β is depicted in Fig. 7B. For the [Cp*Ru(MeCN)₃]PF₆ catalyst, when the H atom moves downward, it induces significant steric hindrance between the CH₂–B(MIDA) group and the bulky Cp ring (the distance between the nearest two H atoms is 2.17 Å, which is shorter than the sum of the van der Waals radii of two H atoms, 2.40 Å). This hindrance arises because, prior to the formation of the three-membered ring intermediate, both the H atom and the CH₂-B(MIDA) group are connected to the same plane of the double bond. Consequently, as the H atom moves downward, the CH₂–B(MIDA) group shifts upward, drawing closer to the Cp* ring. Conversely, in dynamic trajectories where the H atom moves upward, it does not cause associated steric hindrance, making this pathway more favorable and leading to the formation of the anti-product precursor INT-2β-anti. In contrast, with the CpRu(PPh₃)₂Cl catalyst, minimal or no steric hindrance from the Cp ligand is observed, regardless of whether the H atom moves upward or downward to form the three-membered ring. Consequently, the dynamic trajectory leading to the formation of the thermodynamically more stable syn-product precursor CpINT-2β-syn is favored.
To sum up, the DFT calculation results indicate that concerted hydrometallation is the determining step for regioselectivity and stereoselectivity. The hyperconjugative effect of the C–B bond50,52 significantly stabilizes the C = Ru bond at the β position of the boron atom, a crucial factor influencing β-regioselectivity. Meanwhile, the differing steric effects of Cp* and Cp in different catalytic systems contribute to the stereoselectivity of this reaction.
Discussion
In summary, we have demonstrated the significant impact of the β-boron effect in achieving regioselective Ru-catalyzed hydrosilylation of propargylic B(MIDA)s. This approach leverages the interaction between the σ(C–B) orbital and the electrophilic metallacyclopropene intermediate, akin to a Fisher carbene, to achieve high regioselectivity. Additionally, subtle variations in the Ru catalyst have led to a switch in stereoselectivity without affecting regioselectivity, showcasing a rare instance of stereo-divergence in metal-catalyzed hydrosilylation. Our work introduces a versatile synthesis route for regio- and stereo-defined building blocks that incorporate boryl, silyl, and alkene functionalities. This study paves the way for future exploration of β-boryl effect-guided strategies in metal-catalyzed transformations, offering avenues for the precise and efficient synthesis of complex organic molecules.
Methods
General procedure A for the synthesis of products in Z isomers
In the glove box, to an oven-dried 15 mL vial were added the propargylic MIDA boronate (0.10 mmol), the silane (0.20 mmol), [Cp*Ru(MeCN)3]PF6 (2.5 mg, 5.0 μmol), and THF or 1,4-dioxane (1.0 mL). The vial was capped and removed from the glove box. The reaction mixture was stirred at room temperature for 30 min, and then concentrated under reduced pressure. The residue was purified by silica gel flash column chromatography (ethyl acetate–petroleum as the eluent) to give the desired product.
General procedure B for the synthesis of products in E isomers
In a glove box, to an oven-dried 15 mL vial were added the propargylic MIDA boronate (0.10 mmol), the silane (0.20 mmol), CpRu(PPh3)2Cl (3.6 mg, 5.0 μmol), and 1,4-dioxane (1.0 mL). The vial was capped and removed from the glove box. The reaction mixture was stirred at room temperature for 3 h, and then concentrated under reduced pressure. The residue was purified by silica gel flash column chromatography (ethyl acetate–petroleum as the eluent) to give the desired product.
Data availability
The authors declare that the main data supporting the findings of this study, including experimental procedures, characterization of materials and products, general methods, and NMR spectra, are available within the article and its Supplementary Information. All data are available from the corresponding author upon request.
References
Fleming, I., Barbero, A. & Walter, D. Stereochemical control in organic synthesis using silicon-containing compounds. Chem. Rev. 97, 2063–2192 (1997).
Denmark, S. E. & Liu, J. H. C. Silicon-based cross-coupling reactions in the total synthesis of natural products. Angew. Chem. Int. Ed. 49, 2978–2986 (2010).
Nakao, Y. & Hiyama, T. Silicon-based cross-coupling reaction: an environmentally benign version. Chem. Soc. Rev. 40, 4893–4901 (2011).
Cheng, C. & Hartwig, J. F. Catalytic silylation of unactivated C–H bonds. Chem. Rev. 115, 8946–8975 (2015).
Siegel, J. S. Silylium ions: from controversial beginnings to useful catalysts. Nat. Rev. Chem. 4, 4–5 (2020).
Klare, H. F. T. et al. Silylium ions: From elusive reactive intermediates to potent catalysts. Chem. Rev. 121, 5889–5985 (2021).
Fan, X. et al. Stepwise on-demand functionalization of multihydrosilanes enabled by a hydrogen-atom-transfer photocatalyst based on eosin Y. Nat. Chem. 15, 666–676 (2023).
Ramesh, R. & Reddy, D. S. Quest for novel chemical entities through incorporation of silicon in drug scaffolds. J. Med. Chem. 61, 3779–3798 (2018).
Park, S. S., Santha Moorthy, M. & Ha, C.-S. Periodic mesoporous organosilicas for advanced applications. NPG Asia Mater. 6, e96–e96 (2014).
Overman, L. E., Castaneda, A. & Blumenkopf, T. A. Acetal-initiated cyclizations of vinylsilanes. A general synthesis of allylically unsaturated oxacyclics. J. Am. Chem. Soc. 108, 1303–1304 (1986).
Kishi, N., Mikami, K. & Nakai, T. Vinylsilane-terminated cycloacylation: A general synthetic approach to four- to six-membered cyclic ketones and its regiochemical features. Tetrahedron 47, 8111–8118 (1991).
Johns, G. R. & Landais, Y. The oxidation of the carbon-silicon bond. Tetrahedron 52, 7599–7662 (1996).
Barrett, A. G. M. et al. Fleming-Tamao oxidation and masked hydroxyl functionality: Total synthesis of (+)-pramanicin and structural elucidation of the antifungal natural product (-)-pramanicin. J. Org. Chem. 64, 6005–6018 (1999).
Trost, B. M., Ball, Z. T. & Jöge, T. Regioselective hydrosilylation of propargylic alcohols: An aldol surrogate. Angew. Chem. Int. Ed. 42, 3415–3418 (2003).
Chen, J., Guo, J. & Lu, Z. Recent advances in hydrometallation of alkenes and alkynes via the first row transition metal catalysis. Chin. J. Chem. 36, 1075–1109 (2018).
He, P., Hu, M.-Y., Zhang, X.-Y. & Zhu, S.-F. Transition-metal-catalyzed stereo- and regioselective hydrosilylation of unsymmetrical alkynes. Synthesis 54, 49–66 (2022).
Na, Y. G. & Chang, S. B. Highly stereoselective and efficient hydrosilylation of terminal alkynes catalyzed by RuCl2(p-cymene)2. Org. Lett. 2, 1887–1889 (2000).
Denmark, S. E. & Wang, Z. G. Highly stereoselective hydrocarbation of terminal alkynes via Pt-catalyzed hydrosilylation/Pd-catalyzed cross-coupling reactions. Org. Lett. 3, 1073–1076 (2001).
Trost, B. M. & Ball, Z. T. Markovnikov alkyne hydrosilylation catalyzed by ruthenium complexes. J. Am. Chem. Soc. 123, 12726–12727 (2001).
Sridevi, V. S., Fan, W. Y. & Leong, W. K. Stereoselective hydrosilylation of terminal alkynes catalyzed by [Cp*IrCl2]2: A computational and experimental study. Organometallics 26, 1157–1160 (2007).
Gao, R., Pahls, D. R., Cundari, T. R. & Yi, C. S. Experimental and computational studies of the ruthenium-catalyzed hydrosilylation of alkynes: mechanistic insights into the regio- and stereoselective formation of vinylsilanes. Organometallics 33, 6937–6944 (2014).
Mutoh, Y., Mohara, Y. & Saito, S. (Z)-Selective Hydrosilylation of Terminal Alkynes with HSiMe(OSiMe3)2 Catalyzed by a Ruthenium Complex Containing an N-Heterocyclic Carbene. Org Lett. 19, 5204–5207 (2017).
Teo, W. J., Wang, C., Tan, Y. W. & Ge, S. Cobalt-catalyzed z-selective hydrosilylation of terminal alkynes. Angew. Chem. Int. Ed. 56, 4328–4332 (2017).
Wen, H. A., Wan, X. L. & Huang, Z. Asymmetric synthesis of silicon-stereogenic vinylhydrosilanes by cobalt-catalyzed regio- and enantioselective alkyne hydrosilylation with dihydrosilanes. Angew. Chem. Int. Ed. 57, 6319–6323 (2018).
Wu, C. Z., Teo, W. J. & Ge, S. Cobalt-catalyzed (E)-selective anti-markovnikov hydrosilylation of terminal alkynes. ACS Catal. 8, 5896–5900 (2018).
Yang, L. et al. Regio- and stereoselective hydrosilylation of alkynes with alkoxysilanes for the synthesis of β-(Z) vinylsilanes catalyzed by a dirhodium(ii)/XantPhos complex. Org. Chem. Front. 11, 448–457 (2024).
Wu, G., Chakraborty, U. & Jacobi von Wangelin, A. Regiocontrol in the cobalt- catalyzed hydrosilylation of alkynes. Chem. Commun. 54, 12322–12325 (2018).
Yang, X. X. & Wang, C. Y. Dichotomy of manganese catalysis via organometallic or radical mechanism: stereodivergent hydrosilylation of alkynes. Angew. Chem. Int. Ed. 57, 923–928 (2018).
Trost, B. M. & Ball, Z. T. Synthetic stitching with silicon: Geminal alkylation-hydroxylation of alkynyl carbonyl compounds. J. Am. Chem. Soc. 126, 13942–13944 (2004).
Rooke, D. A. & Ferreira, E. M. Platinum-catalyzed hydrosilylations of internal alkynes: Harnessing substituent effects to achieve high regioselectivity. Angew. Chem. Int. Ed. 51, 3225–3230 (2012).
Sumida, Y., Kato, T., Yoshida, S. & Hosoya, T. Palladium-catalyzed regio- and stereoselective hydrosilylation of electron-deficient alkynes. Org. Lett. 14, 1552–1555 (2012).
Wang, Z.-L., Wang, Y., Sun, Y.-C., Zhao, J.-B. & Xu, Y.-H. Regiodivergent hydrosilylation of polar enynes to synthesize site-specific silyl-substituted dienes. Angew. Chem. Int. Ed. 63, e202405791 (2024).
Konno, T., Taku, K., Yamada, S., Moriyasu, K. & Ishihara, T. Remarkable access to fluoroalkylated trisubstituted alkenes via highly stereoselective cobalt-catalyzed hydrosilylation reaction of fluoroalkylated alkynes. Org. Biomol. Chem. 7, 1167–1170 (2009).
Huang, K.-H. & Isobe, M. Highly regioselective hydrosilylation of unsymmetric alkynes using a phenylthio directing group. Eur. J. Org. Chem. 2014, 4733–4740 (2014).
Ding, S. et al. Highly regio- and stereoselective hydrosilylation of internal thioalkynes under mild conditions. Angew. Chem. Int. Ed. 54, 5632–5635 (2015).
Song, L. J. et al. Ir-Catalyzed regio- and stereoselective hydrosilylation of internal thioalkynes: A combined experimental and computational study. J. Org. Chem. 81, 6157–6164 (2016).
Stefanowska, K., Sokolnicki, T., Walkowiak, J., Czapik, A. & Franczyk, A. Directed cis-hydrosilylation of borylalkynes to borylsilylalkenes. Chem. Commun. 58, 12046–12049 (2022).
Ding, S. T. et al. Ligand-controlled remarkable regio- and stereodivergence in intermolecular hydrosilylation of internal alkynes: Experimental and theoretical studies. J. Am. Chem. Soc. 135, 13835–13842 (2013).
Rivera-Hernández, A. et al. Regio- and stereoselective hydrosilylation of unsymmetrical alkynes catalyzed by a well-defined, low-valent cobalt catalyst. Org. Lett. 18, 4242–4245 (2016).
Hu, M. Y. et al. Iron-catalyzed regiodivergent alkyne hydrosilylation. J. Am. Chem. Soc. 142, 16894–16902 (2020).
Li, H., Yang, C., Wang, D. & Deng, L. Cobalt-catalyzed regio- and stereoselective hydrosilylation of alk-2-ynes with tertiary silanes. Organometallics 42, 1693–1698 (2023).
Kawasaki, Y., Ishikawa, Y., Igawa, K. & Tomooka, K. Directing group-controlled hydrosilylation: regioselective functionalization of alkyne. J. Am. Chem. Soc. 133, 20712–20715 (2011).
Garcia-Rubia, A., Romero-Revilla, J. A., Mauleon, P., Gomez Arrayas, R. & Carretero, J. C. Cu-catalyzed silylation of alkynes: A traceless 2-pyridylsulfonyl controller allows access to either regioisomer on demand. J. Am. Chem. Soc. 137, 6857–6865 (2015).
Rosca, D.-A. et al. Ruthenium-catalyzed alkyne trans-hydrometalation: Mechanistic insights and preparative implications. J. Am. Chem. Soc. 139, 2443–2455 (2017).
Dai, W. et al. Regio-selective and stereo-selective hydrosilylation of internal alkynes catalyzed by ruthenium complexes. RSC Adv. 8, 28261–28265 (2018).
Gao, W., Ding, H., Yu, T., Wang, Z. & Ding, S. Iridium-catalyzed regioselective hydrosilylation of internal alkynes facilitated by directing and steric effects. Org. Biomol. Chem. 19, 6216–6220 (2021).
Rummelt, S. M., Radkowski, K., Rosca, D. A. & Fürstner, A. Hydrogen bonding to a chloride ligand as a steering principle in catalysis. J. Am. Chem. Soc. 137, 5506–5519 (2015).
He, Z. & Yudin, A. K. Amphoteric alpha-boryl aldehydes. J. Am. Chem. Soc. 133, 13770–13773 (2011).
Li, J. & Burke, M. D. Pinene-derived iminodiacetic acid (PIDA): A powerful ligand for stereoselective synthesis and iterative cross-coupling of C(sp3) boronate building blocks. J. Am. Chem. Soc. 133, 13774–13777 (2011).
Li, Y. et al. β-Boron effect enables regioselective and stereospecific electrophilic addition to alkenes. J. Am. Chem. Soc. 145, 7548–7558 (2023).
Liu, Y. et al. Boryl-dictated site-selective intermolecular allylic and propargylic C–H amination. J. Am. Chem. Soc. 144, 14380–14387 (2022).
Li, Y. et al. Regioselective electrophilic addition to propargylic B(MIDA)s enabled by β-boron effect. Adv. Sci. 10, 2304282 (2023).
Qian, J. et al. Synthesis of allenyl-B(MIDA) via hydrazination/fragmentation reaction of B(MIDA)-propargylic alcohol. Chin. Chem. Lett. 34, 107479 (2023).
Chung, L. W., Wu, Y. D., Trost, B. M. & Ball, Z. T. A theoretical study on the mechanism, regiochemistry, and stereochemistry of hydrosilylation catalyzed by cationic ruthenium complexes. J. Am. Chem. Soc. 125, 11578–11582 (2003).
Yang, Y.-F., Chung, L. W., Zhang, X., Houk, K. N. & Wu, Y.-D. Ligand-controlled reactivity, selectivity, and mechanism of cationic ruthenium-catalyzed hydrosilylations of alkynes, ketones, and nitriles: A theoretical study. J. Org. Chem. 79, 8856–8864 (2014).
Rummelt, S. M. & Fürstner, A. Ruthenium-catalyzed trans-selective hydrostannation of alkynes. Angew. Chem. Int. Ed. 53, 3626–3630 (2014).
Fischer, E. O., Reitmeier, R. & Ackermann, K. Synthesis and Structure of (CO)4W[C(NEt2)PMePh], a novel cyclic carbene complex. Angew. Chem. Int. Ed. 22, 411–412 (1983).
Ito, H., Kosaka, Y., Nonoyama, K., Sasaki, Y. & Sawamura, M. Synthesis of optically active boron–silicon bifunctional cyclopropane derivatives through enantioselective copper(I)-catalyzed reaction of allylic carbonates with a diboron derivative. Angew. Chem. Int. Ed. 47, 7424–7427 (2008).
Hall, D. G. in Boronic Acids (2011).
Tellis, J. C. et al. Single-electron transmetalation via photoredox/nickel dual catalysis: unlocking a new paradigm for sp3–sp2 cross-coupling. Acc. Chem. Res. 49, 1429–1439 (2016).
Bootwicha, T., Feilner, J. M., Myers, E. L. & Aggarwal, V. K. Iterative assembly line synthesis of polypropionates with full stereocontrol. Nat. Chem. 9, 896–902 (2017).
Diner, C. & Szabó, K. J. Recent advances in the preparation and application of allylboron species in organic synthesis. J. Am. Chem. Soc. 139, 2–14 (2017).
Jin, S., Liu, K., Wang, S. & Song, Q. Enantioselective cobalt-catalyzed cascade hydrosilylation and hydroboration of alkynes to access enantioenriched 1,1-silylboryl alkanes. J. Am. Chem. Soc. 143, 13124–13134 (2021).
Tian, Y.-M., Guo, X.-N., Braunschweig, H., Radius, U. & Marder, T. B. Photoinduced borylation for the synthesis of organoboron compounds. Chem. Rev. 121, 3561–3597 (2021).
Li, Y. & Yin, G. Nickel chain-walking catalysis: A journey to migratory carboboration of alkenes. Acc. Chem. Res. 56, 3246–3259 (2023).
Chen, M., Tugwell, T. H., Liu, P. & Dong, G. Synthesis of alkenyl boronates through stereoselective vinylene homologation of organoboronates. Nat. Synth. 3, 337–346 (2024).
Feng, Q. et al. Ru-catalyzed geminal hydroboration of silyl alkynes via a new gem-addition mechanism. J. Am. Chem. Soc. 142, 13867–13877 (2020).
Yong, L., Kirleis, K. & Butenschoen, H. Stereodivergent formation of alkenylsilanes: syn or anti hydrosilylation of alkynes catalyzed by a cyclopentadienyl cobalt(I) chelate bearing a pendant phosphane tether. Adv. Synth. Catal. 348, 833–836 (2006).
Katayama, H. et al. Ruthenium-catalyzed hydrosilylation of terminal alkynes: stereodivergent synthesis of (E)- and (Z)-alkenylsilanes. J. Organomet. Chem. 645, 192–200 (2002).
Takeuchi, R., Nitta, S. & Watanabe, D. A selective synthesis of (E)-vinylsilanes by cationic rhodium complex-catalyzed hydrosilylation of 1-alkynes and tandem hydrosilylation/isomerization reaction of propargylic alcohols to.beta.-silyl ketones. J. Org. Chem. 60, 3045–3051 (1995).
Chang, K. J., Rayabarapu, D. K., Yang, F. Y. & Cheng, C. H. Unusual palladium-catalyzed silaboration of allenes using organic iodides as initiators: Mechanism and application. J. Am. Chem. Soc. 127, 126–131 (2005).
Yamamoto, Y. & Asao, N. Selective reactions using allylic metals. Chem. Rev. 93, 2207–2293 (1993).
Bustos, I., Mendicute-Fierro, C. & Huertos, M. A. Inverse isotope effect for aldehyde hydrosilylation catalyzed by a hydrido-silyl-iridium(III) complex. Organometallics 43, 3236–3241 (2024).
Gomez-Gallego, M. & Sierra, M. A. Kinetic isotope effects in the study of organometallic reaction mechanisms. Chem. Rev. 111, 4857–4963 (2011).
Schneider, N. et al. Multiple reaction pathways in rhodium-catalyzed hydrosilylations of ketones. Chem. Eur. J. 15, 11515–11529 (2009).
McFadden, T. R. et al. Synthesis of boriranes by double hydroboration reactions of N-heterocyclic carbene boranes and dimethyl acetylenedicarboxylate. J. Am. Chem. Soc. 139, 1726–1729 (2017).
Zheng, C., Xia, Z.-L. & You, S.-L. Unified mechanistic understandings of Pictet-Spengler reactions. Chem 4, 1952–1966 (2018).
Acknowledgements
This work was supported by Noncommunicable Chronic Diseases-National Science and Technology Major Project (2023ZD0507600 to H. W.), the National Natural Science Foundation of China (22371311 to H.W., 22403067 to T.-Y.S. and 22171293 to Q.L.), Guangdong Provincial Key Laboratory of Construction Foundation (2023B1212060022 to Q.L.), Guangzhou Municipal Science and Technology Project (2025A04J7101 to H.W.), Shenzhen Bay Laboratory High Performance Computing and Informatics Facility, Open Research Fund of State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, and the GBRCE for Functional Molecular Engineering.
Author information
Authors and Affiliations
Contributions
J.Q. and S.L. contributed equally. J. Q. conducted reaction optimization and contributed to the substrate scope. J.Q. and Q.L. contributed to the analysis of the data. S. L. and T.-Y.S. performed the DFT calculations. J.Q., S.L., T.-Y.S., and H.W. wrote the paper. Z.-H.C., J.H., and W.Z. contributed to the synthesis of substrates. All authors contributed to discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Qiuling Song, and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qian, J., Lin, S., Chen, ZH. et al. Harnessing the β-boron effect for regioselective Ru-catalyzed hydrosilylation of internal alkynes. Nat Commun 16, 4469 (2025). https://doi.org/10.1038/s41467-025-59823-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59823-x
