
Abstract
Lipase represents one of the most important industrial biocatalysts, with a global market value of $590.5 million by 2020. However, their catalytic efficiency is often hindered by a closed “lid” conformation. Here, we present an aggregation-induced conformation locking strategy that enables the facile synthesis of highly activated lipase hybrid biocatalysts. Lipase is self-activated into an open-lid conformation via solvent-mediated aggregation, followed by conformational locking within a two-dimensional metal-organic framework (MOF). The resulting MOF biocatalyst provides high accessibility to the locked lipase aggregates through its long-range ordered pore channels, achieving a hydrolytic efficiency 5.30 times greater than that of native lipase. To the best of our knowledge, this represents a record-high activation efficiency for ester hydrolysis among the reported lipase-based hybrid biocatalysts to date. We also demonstrate its feasibility to catalytically accelerate transesterification and esterification reactions, showing up to as 6.64 times higher yield than native lipase and impressive recyclability.
Similar content being viewed by others
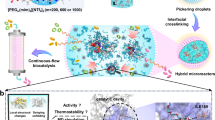
Introduction
Biocatalysis represents a green and sustainable synthetic technology that facilitates the catalytic conversion of high-value chemical products1. Lipase is a class of hydrolases enabling the catalytical cleavage of ester bonds under environment-friendly condition2, with a global market worth $590.5 million by 20203. Due to the merits of low cost and thermal stability, lipase holds enormous potential in important industrial fields including food processing, environmental remediation, organic synthesis, and the production of biofuels and pharmaceuticals, etc. 4,5,6. The thermal stability of lipase is superior to other fragile enzymes, largely owing to their unique “lid” conformation, in which the active center, composed of a Ser-His-Asp catalytic triad, is embedded within a hydrophobic pocket covered by a movable α-helical “lid”7,8,9. This lid, to a certain degree, shields the catalytic center against environmental stressors, but also restricts access to the active site, leading to reduced catalytic activity10. Addressing the trade-off between stability and activity in lipase catalysis is thus of critical importance.
Leveraging nanocarriers to immobilize lipase is an ideal means to circumvent this predicament11,12,13, when taking these into account: (1) nanocarriers can set an “armor” to stabilize the global conformation of an enzyme14,15; (2) the interfacial interactions between enzyme and nanomaterials offer the possibility to modulate the lid into a fit-for-purpose conformation16,17. For example, Ge et al. immobilized lipase on the surface of graphene and showcased the feasibility to modulate lipase’s conformation by means of the molecular interactions between lipase and graphene16. Nevertheless, surface-immobilized lipases are prone to leaching, and their catalytic stability is often compromised as large portions of the enzyme remain exposed to the external environment. To address this, the researchers, including our group, have pursued the engineering of nanopores based on reticular chemistry, allowing the entrapment, rather than, the surface immobilization of an enzyme17,18. When lipase is confined in such a nanopore, the specific host-guest interactions, also known as interfacial activation, can mediate the trapped lipase into a lid-opening conformation19,20. Despite the promise of this nanopore strategy, several challenges remain: (1) the geometry and chemistry of the nanopores must be precisely tailored to accommodate the bulky lipase, complicating the synthetic steps, and (2) the orientation of the lipase within the nanopore is unpredictable, making interfacial activation a trial-and-error process, which can result in suboptimal efficiency.
Given the structural dynamics of an enzyme, an alternative strategy involves the activation of the enzyme conformation prior to immobilization, facilitating the directional design of functional biocatalysts. For instance, Rathod et al. demonstrated that lipase conformation could be regulated by ultrasound treatment. When followed by encapsulation in a microporous metal-organic framework (zeolite imidazole framework, ZIF-8), the resulting lipase biocatalyst exhibited 155% of the activity of the native enzyme21. Similarly, ultrasound pre-activation has been used to synthesize metalloenzymes within ZIF-8, showing considerable activity enhancement compared to counterparts without ultrasound activation22. Despite these advancements, enzyme activation highly relies on the precise optimization of ultrasound parameters, including intensity, work/pause cycles, and duration. In addition, the ZIF-8 nanocarriers has narrow pore aperture (3.4 Å)23, limiting the mass transfer and therefore compromising the efficacy of this approach.
In this contribution, we describe an aggregation-induced conformation locking (ACL) strategy based on a two-dimensional (2D) metal-organic framework (MOF), enabling the facile synthesis of highly activated lipase biocatalyst (Fig. 1). We discover that the lipase can be converted into a self-activated, lid-opening conformation by aggregation in polar organic solvents. A 2D-MOF with long-range ordered pore channels (ca. 1.90 nm width) is developed to encapsulate the self-activated lipase aggregates in situ, achieving the rapid locking of the catalytically favorable conformation inside the quasi-mesoporous 2D-MOF. This ACL method well circumvents the limitations of complicated pore design and unpredictable activation behavior that existed in the previous nanopore methods. In addition, the long-range ordered pore channels in this 2D-MOF can bridge the gap between inside lipase aggregates and outside catalytic substrates, conferring the lipase hybrid biocatalyst high reactivity, which is 5.30 times higher than native lipase. This hybrid biocatalyst is utilized to catalytically accelerate the important industrial reactions involving transesterification and esterification, presenting much higher performances than native lipase in terms of catalytic rate, yield, and recyclability.

Results
The self-activation of lipase by aggregation
Lipase from Thermomyces lanuginosus (Lipase TL) is a commercially available enzyme extensively utilized in industries such as food processing, environmental remediation, organic synthesis, and the production of biofuels and pharmaceuticals, owing to its cost-effectiveness4,6,24. In addition, the unequivocable “lid”-covered catalytic center has been well resolved by single crystal X-ray diffraction25. This not only contributes to its high thermal stability but also facilitates a better understanding of the relationship between lid conformation and catalytic activity. Therefore, Lipase TL was selected as the model enzyme in this study.
In this ACL strategy, it entails the pre-activation of Lipase TL prior to the conformational locking by a 2D-MOF. It is well acceptable that polar organic solvents can disturb the hydration layer of a protein, leading to protein aggregates. This organic solvent-induced aggregation behavior, in general, will cause the unfavorable conformation and decrease an enzyme’s activity26,27. In the case of lipase of which the catalytic center is covered by a flexible hydrophobic lid chain10, we envision that the intermolecular interactions by aggregation may move the hydrophobic lid in Lipase TL, resulting in an activated, rather than, devitalized conformation. With this in mind, we monitored the aggregation behavior and activity change of Lipase TL incubated in water solution with varied polar organic solvents including dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF), methanol, n-propanol, and isopropanol. All organic solvent contents in water solution were covered from 2.44 to 80% (v/v). The theoretical dipole moments of these organic solvent molecules were calculated28 and summarized in Supplementary Table 1, with the polarity order of DMSO > DMF > methanol > isopropanol > n-propanol. The apparent phenomena of Lipase TL ultrasonically dispersed in different solutions were recorded in Supplementary Figs. 1–5, and the turbidity of the solution was synchronously monitored to examine the aggregation behavior (Fig. 2a). For highly polar DMSO and DMF, Lipase TL could be completely dissolved (Supplementary Figs. 1, 2), also well evidenced by the high transmittance in Fig. 2a. This was caused by the high solvation of surface polar amino acid of Lipase TL. However, in the low polar organic solvents of methanol, n-propanol, and isopropanol, the aggregation of Lipase TL gradually occurred when increasing the organic solvent contents, attributing to the hydration shell disturbance of Lipase TL by these organic solvents29,30. Of note, when further utilizing lower polar organic solvents such as toluene and cyclohexane (Supplementary Table 1), the stratification of the mixture solution appeared, in which Lipase TL completely dissolved in the water phase (Supplementary Fig. 6).

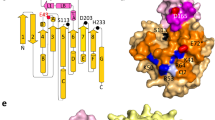
The transmittance (a) and the relatively catalytic activity (b) of Lipase TL in different content of organic solvents. p-Nitrophenyl butyrate (p-NPB) was used as the ester substrate for the activity assay. Error bars = standard deviation (SD); Data were presented as mean values ± SD (n = 3; n presents the sample size used to derive statistics). Source data are provided as a Source Data file. c Fluorescence spectra of 80% (v/v) organic solvents-induced Lipase TL aggregate dispersed in buffer solution (1 mg/mL). Excitation at 280 nm. d The simulated Lipase TL aggregate in n-propanol solution. For clarity, the n-propanol molecules were omitted. In the Lipase TL molecule, the peptide chain of the lid was highlighted in orange, while the tryptophan fluorescent was highlighted in yellow. The original Lipase TL structure is available in the Protein Data Bank under the accession number PDB 1DT3. e The snapshot of the lid domain structures of the native Lipase TL and the aggregated Lipase TL based on MD simulation. In the Ser-His-Asp catalytic triad, the red, gray, and blue represented the O, C, and N atoms, respectively.
Using p-Nitrophenyl butyrate (p-NPB) as the substrate, we found that the hydrolytic activity of Lipase TL significantly decreased in DMF- and DMSO-involved solutions, and Lipase TL completely lost its activity in the high DMF/DMSO content of 80% (v/v) (Fig. 2b). However, in high content of n-propanol and isopropanol solutions where Lipase TL aggregates arose, the self-activated biocatalysis behaviors were observed. Particularly, the hydrolytic activities of Lipase TL aggregate in 80% (v/v) n-propanol and isopropanol solutions were 8.27 times and 5.51 times higher than the native Lipase TL. Considering that the catalytic assay was conducted in a 50 mM buffer solution, dynamic light scattering (DLS) analysis was performed to confirm that the Lipase TL remained aggregated under these conditions (Supplementary Fig. 7). At the same time, we verified that the organic solvents themselves could not impact the activity test (Supplementary Fig. 8), indicating that the biocatalytic activation was a consequence of Lipase TL aggregation. Of specific note, although the Lipase TL aggregation also appeared in high content of methanol solution (Fig. 2a and Supplementary Fig. 3), no self-activation phenomenon was found (Fig. 2b). This may be caused by the fact that methanol with relatively high polarity will change the global conformation of an enzyme through destroying the intramolecular hydrogen-bond networks31.
Further insight into the molecular interaction was surveyed by Fourier transform infrared spectroscopy (FT-IR). The amide I band at 1600–1700 cm−1 of an enzyme, mainly ascribing to the C=O stretching vibration of the amido bonds, is sensitive to the conformation change32,33. It was found that, in all aggregated Lipase TL samples, the C=O stretching vibrations were shifted to high wavenumber (Supplementary Fig. 9). This could be interpreted by the structural restriction of peptide chains after molecular aggregation, which necessitated a higher radiation energy to induce the bond vibration. To ascertain that the self-activation was associated with the lid-opening conformation after aggregation, we carried out the fluorescence experiments. The fluorescent tryptophan-89 (Trp89) is located on the lid chain of lipase34, responsible for 60% of the emission fluorescence of a lipase molecule (Fig. 2d)35. We found that the emission wavelengths were red-shifted in the Lipase TL samples well dispersed in DMF and DMSO solutions (Fig. 2c), suggesting a strong solvation of lid region through polar interactions36,37. This solvation effect would decrease the accessibility of buried active center of Lipase TL, leading to an activity reduction as we observed in Fig. 2b. As a comparison, the opposite blue shifts of emission wavelengths were observed in all aggregated samples (Fig. 2c), indicating that the lid chain has undergone a different conformation change in the aggregated form38.
To get a glimpse of the conformation change in aggregated Lipase TL, we performed the molecular dynamics (MD) simulation experiment (The detailed simulation setup was provided in the Supplementary Information). In the presence of n-propanol solvent, the strong electrostatic and van der Waals intermolecular interactions of Lipase TL and between Lipase TL and n-propanol resulted in the formation of Lipase TL aggregates (Supplementary Figs. 10, 11 and Supplementary Tables 2, 3). As depicted in Fig. 2d, the intermolecular hydrogen-bond interactions in the simulated aggregates led to the structural disturbance of the lid chain (highlighted in orange, and the n-propanol molecules were omitted for clarity), rendering the buried Trp89 fluorophore exposed. This was well supported by the fluorescence shift in Fig. 2c. Figure 2e presented the structural changes of the lid chain covered on the “Ser-His-Asp” catalytic triad. It found that the lid chain was changed from a closed conformation to an open one, with the pocket enlarged from 3.62 to 7.13 Å, indicating that the accessibility of the catalytic triad was greatly enhanced in the aggregated Lipase TL. These simulations and experimental data demonstrated that the Lipase TL could be activated readily by an organic solvent-induced aggregation strategy.
Developing in situ 2D-MOF approach for enzyme encapsulation
3D microporous zeolite imidazole frameworks (ZIF-839, ZIF-9040, MAF-741, and MAF-642, etc.), to date, are mainstream MOF biocatalysts suitable for in situ enzyme encapsulation. However, the locked enzyme usually suffers from compromised activity because of the narrow micropores that inhibit mass transfer. From a structural view, the 2D-MOFs with straight and large pore channels are an ideal platform for encapsulating enzymes and consequently locking its conformation, as their structural advantages of pore channels for mass transfer. Metal-2,3,6,7,10,11-Triphenylenehexol coordination polymers (M-HHTP, M represents metal ions) are classes of 2D-MOFs family43, and their synthesis procedures mainly comprise solvothermal method44, spray layer-by-layer liquid-phase epitaxial method45, and phase transformation method46, etc. which are either enzyme-unfriendly or involved complicated steps. Herein, we developed a mild and convenient synthesis method for M-HHTP coordination polymers (M = Zn2+ and Cu2+ ions, detailed procedures seen in Supporting Information), enabling the rapid encapsulation and locking of enzymes in a mild pathway (Fig. 3a). Upon adding an HHTP-dissolved ethanol aqueous solution into a metal salts aqueous solution at pH=10, numerous precipitates were produced instantaneously. Note that the ethanol concentration used in the reaction system (12.5% (v/v)) had a minimal impact on the Lipase TL conformation and activity (Supplementary Fig. 12). Additionally, the concentrations of Zn2+/Cu2+ and HHTP ligand used exhibited limited effects on the catalytic activity of Lipase TL (Supplementary Fig. 13). The optimal pH for the reaction system was found to be 10 (Supplementary Fig. 14) for the following reasons: (1) a weak alkaline environment is required for the deprotonation of the HHTP ligand, which facilitates the formation of Zn–O coordination bonds during nucleation; (2) a strongly alkaline environment accelerates the reaction, promoting the formation of a dynamically favorable amorphous phase, which, in turn, reduces crystallinity. The powder X-ray diffraction (PXRD) (Fig. 3b and Supplementary Fig. 15) and scanning electron microscopy (SEM) (Supplementary Fig. 16) suggested that the highly crystalline and rod-like Zn-HHTP and Cu-HHTP were formed, and the PXRD patterns were well in line with the simulated models with slipped AA packing47,48 (Supplementary Fig. 17). In addition, the low-dose cryo-EM clearly identified the abundant (110) and (002) lattice planes throughout these two samples, implying that the long-range ordered pore channels (ca. 1.90 nm width) were formed by layer-by-layer packing (Supplementary Figs. 18,19).

a Schematic diagram of in situ encapsulation of Lipase TL in 2D-MOF. b PXRD characterization of Zn-HHTP and Lipase TL@Zn-HHTP. c The representative cryo-EM image of Lipase TL@Zn-HHTP, which presents the honeycomb-like pore structure. More than three times were repeated independently with similar EM results. d Schematic diagram of Enzyme@ZIF-8 and Enzyme@Zn-HHTP, in which the crystallographic pores were highlighted. In ZIF-8, the light blue, pink, and green presented C, Zn, and N atoms; while in M-HHTP, the light blue, light purple, and rose red balls presented C, O, and metal atoms. All the H atoms are omitted for clarity. e The relative activities of different Enzyme@MOFs biocatalysts in terms of catalytic rates in the initial phase. Error bars = standard deviation (SD); Data were presented as mean values ± SD (n = 3; n presents the sample size used to derive statistics). Source data are provided as a Source Data file.
The feasibility and advantage of these M-HHTP crystalline polymers for in situ Lipase TL encapsulation were then verified. The Lipase TL loading efficiency was examined by a standard Bradford assay, in which the Lipase TL concentrations in the supernatant before and after encapsulation were quantified. Almost 100% Lipase TL was encapsulated within these two M-HHTP (Supplementary Table 4), with a Lipase TL loading of 30.77 wt% and 28.57 wt% in resulting Lipase TL@Zn-HHTP and Lipase TL@Cu-HHTP (Supplementary Table 4), surpassing the reported approaches using the nanocarriers involving other MOFs and covalent organic frameworks (COFs), etc. (Supplementary Table 5). At the same time, the Lipase TL fluorescence labeling method also evidenced this high loading efficiency (Supplementary Fig. 20). In this experiment, the Lipase TL was labeled by a red fluorescence dye (rhodamine isothiocyanate B, Rhb) prior to the M-HHTP encapsulation, and the fluorescence in the supernatant disappeared after encapsulation, suggesting the complete encapsulation of lipase inside M-HHTP. Structural insights from confocal laser scanning microscope (CLSM) imaging revealed that red fluorescence overlapped with the Zn-HHTP or Cu-HHTP crystals, in contrast to the control sample, where Lipase TL was adsorbed onto the as-synthesized MOF particles (termed as Lipase TL-on-Zn-HHTP or Lipase TL-on-Cu-HHTP), thereby confirming the successful encapsulation of Lipase TL (Supplementary Figs. 21–24). Additionally, X-ray energy dispersive spectroscopy (EDS) mapping showed that the N and S elements, attributed to the Lipase TL molecules, were uniformly distributed within the M-HHTP samples, further supporting the incorporation of Lipase TL (Supplementary Figs. 25, 26). The structural examination by PXRD and SEM showed that the crystalline phase and morphology of Lipase TL@M-HHTP were not changed after Lipase TL encapsulation (Fig. 3b and Supplementary Figs. 15, 27). N2 adsorption/desorption isotherms showed that both the Brunauer–Emmett–Teller (BET) surface area and nonlocal density functional theory (NLDFT) pore size distribution of M-HHTP decreased after Lipase TL entrapment (Supplementary Fig. 28). This suggested that the Lipase TL was indeed confined inside the M-HHTP, leading to the reduction in N2 adsorption and pore volume. Further insight into the cryo-EM images certified that the straight pore channels were well retained after Lipase TL encapsulation (Fig. 3c and Supplementary Fig. 29), with a honeycomb-like pore window of 1.9 nm width (Fig. 3c), which favored the mass transfer for enzymatic reaction under confinement (vide infra).
We next demonstrated the biocatalytic advantages of this M-HHTP approach. At the same time, the Lipase TL-encapsulated ZIF-8 (Lipase TL@ZIF-8), a more widely explored lipase-MOF biocatalyst42, was also synthesized as a control (Supplementary Figs. 30,31 and Supplementary Table 4). Owing to the larger pore channels set up in M-HHTP (Fig. 3d), Lipase TL@M-HHTP presented 2.29 to 2.83-fold catalytic rates for the hydrolysis of p-NPB compared to Lipase TL@ZIF-8 under the same amount of Lipase TL (Fig. 3e and Supplementary Fig. 32). To further elucidate that the activity promotion was not an accidental phenomenon only observed in Lipase TL catalysis, we also encapsulated other enzymes including glucose oxidase (GOx), horseradish peroxidase (HRP) and Cutinase into M-HHTP and ZIF-8, respectively (Supplementary Figs. 33–35), and their catalytic performances were compared under the same conditions. The MOF materials themselves presented negligible catalytic activity (Supplementary Fig. 36). All the catalytic substrates including glucose (10.06 × 6.63 × 6.14 Å, for GOx), 2,2’-azinobis-(3-ethylbenzthiazoline-6-sulfonate (ABTS, 19.36 × 10.19 × 5.26 Å, for HRP), and p-NPB (11.53 × 7.69 × 6.68 Å, for Cutinase) could freely pass through the straight channel of M-HHTP (Supplementary Fig. 37). Yet, they were too large to pass through the crystallographic pore window of ZIF-8 (ca. 3.4 Å aperture, Fig. 3d). As a result, the catalytic activities of all Enzyme@ZIF-8 were significantly inhibited compared to the native enzymes (Fig. 3e and Supplementary Fig. 32). Of note, the Enzyme@ZIF-8 biocatalyst presented partial activity, rather than being completely inactive, because (1) the defect pores were usually formed in this ZIF-8 biohybrid material49, which offered the possibility for the entrance of large-sized substrates; and (2) partial enzymes were likely located on the surface domain of ZIF-850, which were accessible by the large-sized substrates in the solution phase. As a comparison, owing to the large and straight pore channels set up in M-HHTP, all the catalytic activities of Enzyme@M-HHTP were increased compared to those of Enzyme@ZIF-8. Particularly, Enzyme@Zn-HHTP still held the best catalytic power, all of which were close to the activity of native enzymes. This was plausibly benefited by the higher biocompatibility of Zn2+ compared to Cu2+ 51. Furthermore, the crystallinity of the Zn-HHTP biocatalysts was well preserved across a pH range of 3 to 11, indicating their excellent chemical stability (Supplementary Fig. 38). These findings demonstrated that this 2D-MOF approach rendered the encapsulated enzyme highly accessible, laying a foundation for activating lipase through locking their aggregated conformation.
Locking the self-activated lipase aggregates in 2D Zn-HHTP
Having demonstrated the structural advantages of 2D Zn-HHTP for enzyme encapsulation, we next attempted to lock the aggregated conformation of Lipase TL (Lipase TL aggregates denoted as LA thereinafter) in this 2D-MOF through in situ encapsulation (Fig. 4a). After mixing LA, formed in an 80% (v/v) organic solvent solution, with a Zn2+ aqueous solution at pH 10, numerous precipitates were generated upon the rapid addition of the HHTP solution (termed as LA-n-prop@Zn-HHTP and LA-isoprop@Zn-HHTP). Dynamic light scattering (DLS) measurements confirmed that LA remained aggregated in the Zn2+ aqueous solution (Supplementary Fig. 39). The aggregated Lipase TL conformation in both Zn2+ and ligand solutions was further confirmed by fluorescence and FT-IR spectra (Supplementary Fig. 40). The PXRD and SEM imaging attested the high crystallinity of both the LA-n-prop@Zn-HHTP and LA-isoprop@Zn-HHTP (Supplementary Fig. 41), and the appearance of amide I band in the FT-IR spectra indicated the successful encapsulation of lipase molecules (Supplementary Fig. 42).

a Schematic diagram of the conformation locking of self-activated Lipase TL aggregates using the M-HHTP encapsulation method. b Representative PDDFs for LA-n-prop in LA-n-prop@Zn-HHTP. The q-range is shown from 0.03 to 0.10 Å−1. Reg. constant is 2.4e10. Inset presents the estimated particle size of MOF-locked LA-n-prop. c The p-NBP concentration-dependent catalytic rates of Lipase TL, LA-n-prop @Zn-HHTP and LA-isoprop@Zn-HHTP, respectively. Error bars = standard deviation (SD); Data were presented as mean values ± SD (n = 3; n presents the sample size used to derive statistics). Comparison of the activation efficiency in terms of catalytic rate enhancement for ester hydrolysis between LA-n-prop@Zn-HHTP and other reported lipase hybrid biocatalysts. e The stability test. f Schematic diagram of loading discrete Lipase TL into the NU-1003 pore. In NU-1003, the light blue, light pink, and rose red presented C atom, O atom, and Zr atom, respectively. Error bars = standard deviation (SD); Data were presented as mean values ± SD (n = 3; n presents the sample size used to derive statistics). g The catalytic activity of the as-synthesized Lipase TL-NU-1003 sample and the samples after incubating in organic solvents for 1 h. Error bars = standard deviation (SD); Data were presented as mean values ± SD (n = 3; n presents the sample size used to derive statistics). Source data are provided as a Source Data file.
To validate that the locked Lipase TL was in an aggregated conformation, a small-angle X-ray scattering (SAXS) experiment was implemented (Supplementary Fig. 43a and Supplementary Table 6). The SAXS spectrum of the locked Lipase TL was acquired by a normalization subtraction approach52 (Supplementary Fig. 43b). The pair distance distribution function (PDDF) analysis described that the radius of gyration (Rg) of locked aggregated Lipase TL was approximately 69.4 Å (i.e. 13.9 nm diameter, Fig. 4b), which was slightly smaller than the hydrated particle size (21.67 nm) of Lipase TL aggregate before encapsulation, which was examined by DLS (Supplementary Fig. 39). This discrepancy can be attributed to the hydration layer, which contributes to the larger particle size observed in the DLS measurement. Considering that an individual Lipase TL molecule is ca. 38 Å × 44 Å × 53 Å with a Rg of ca. 20 Å, the much larger Rg value suggested the aggregated conformation of Lipase TL locked inside Zn-HHTP.
The biocatalytic performances of the LA@Zn-HHTP were then evaluated. LA-n-prop@Zn-HHTP and LA-isoprop@Zn-HHTP presented 4.64 times and 3.86 times higher catalytic rates than the native Lipase TL (Supplementary Fig. 44). We further investigated the catalytic rates as a function of substrate concentrations and fitted the data with Michaelis–Menten model to acquire the kinetic parameters (Fig. 4c), wherein p-NPB was selected as the ester substrate and its hydrolytic product of p-nitrophenol was quantified by a standard curve (Supplementary Fig. 45). The maximal catalytic rates (Vm) of both LA-n-prop@Zn-HHTP, and LA-isoprop@Zn-HHTP were significantly improved compared to native Lipase TL. While the Michaelis constant (Km), which reflects the substrate-binding ability of the active site, was found to be reduced in both LA-n-prop@Zn-HHTP and LA-isoprop@Zn-HHTP (Supplementary Table 7). The lower Km value suggested a higher substrate-binding capacity, and this further manifested that the Lipase TL aggregates locked inside Zn-HHTP still presented a lid-opening conformation. Additionally, the catalytic efficiencies, estimated by the kcat /Km value, were found to be 5.30 times and 3.63 times higher in LA-n-prop@Zn-HHTP and LA-isoprop@Zn-HHTP than that in native Lipase TL, with LA-n-prop@Zn-HHTP hold the highest catalytic efficiency. Note that LA-n-prop@Zn-HHTP presented the highest activation efficiency in terms of catalytic rate enhancement for ester hydrolysis (compared to native Lipase TL) among the reported lipase hybrid biocatalyst (Fig. 4d and Supplementary Table 8). Importantly, the activated catalytic ability based on this 2D-MOF-locking approach could be well maintained in water solution after 1 month (Fig. 4e). Particularly, the activity of native Lipase TL dropped to 73%, while LA-n-prop@Zn-HHTP and LA-isoprop@Zn-HHTP still retained the activities of 247%, 260% as native Lipase TL, respectively. We also demonstrated that this aggregation-induced conformation-locking approach is adaptable to activate other lipase species, including Lipase from Pseudomonas cepacia (Lipase PS) and Lipase from Candida rugosa (Lipase CRL) (details provided in Supplementary Figs. 46–48). It was worth noting that a similar aggregate locking procedure using ZIF-8 could not result in an activation of Lipase TL catalysis (Supplementary Figs. 49,50), ascribed to the narrow pore structure of ZIF-8 inhibiting the mass transfer (Fig. 3d).
To further validate that the activated biocatalysis was indeed originated from the locking of aggregated conformation by Zn-HHTP, a series of control experiments were then conducted. Firstly, we encapsulated the water-dispersive Lipase TL into Zn-HHTP in situ and incubated this Lipase TL@Zn-HHTP materials in different organic solvents, the molecule sizes of which were smaller than the pore channels of Zn-HHTP (Supplementary Fig. 51a). We found that the post treatment step by organic solvents could not induce an activity enhancement, excluding the potential interference by organic solvents (Supplementary Fig. 51b). Secondly, we prepared a NU-1003 MOF53, the pore channel (ca. 4.4 nm) of which allowed the accommodation of individual Lipase TL molecules but not of Lipase TL aggregates (Fig. 4f). Lipase TL was loaded into the pore channels of as-synthesized NU-1003 through an infiltration method, with a loading estimated to be 308.4 mg Lipase TL per 1 g NU-1003 (Supplementary Figs. 52–55 and Supplementary Table 4). The Lipase TL@NU-1003 biocatalyst only retained ca. 40% activity of pristine Lipase TL and incubating it with organic solvents such as n-propanol or isopropanol also could not lead to a distinct activity change (Fig. 4g and Supplementary Fig. 56). These control experiment excluded the possibility that the catalytic activation was originated from the post treatment of organic solvent. Collectively, it could be concluded that the biocatalytic activation in LA@Zn-HHTP was attributed to the locking of the aggregated conformation of Lipase TL.
We evaluated the structural stability of the LA-n-prop@Zn-HHTP biocatalyst under conditions relevant to industrial applications, including aqueous solutions with pH values ranging from 3 to 11, as well as various common organic solvents (methanol, acetonitrile, n-propanol, isopropanol, DMF, acetone, tetrahydrofuran (THF), and 1,4-dioxane). The results demonstrated that the MOF biocatalyst retained its structural integrity after 30 min of incubation in these aqueous solutions or organic solvents, as confirmed by PXRD analyses (Supplementary Figs. 57, 58). Furthermore, following exposure to these challenging conditions, the LA-n-prop@Zn-HHTP biocatalyst exhibited significantly higher catalytic activity compared to native Lipase TL, by a factor of 2.25- to 4.99-fold (Supplementary Fig. 59). Additionally, while aggregated Lipase TL is generally more sensitive to heat due to the exposure of its catalytic center, our MOF-locking strategy effectively protected the aggregated Lipase TL from thermal inactivation (Supplementary Fig. 60).
High-value ester synthesis by activated transesterification and esterification reaction
Aromatic esters and alkyl esters are important industrial chemicals, which are extensively used in diesel fuel, agrochemical, and pharmaceutical54,55. Transesterification represents one of the most important industrial reactions for high-value ester synthesis56. We next examined the feasibility of LA@Zn-HHTP as the heterogeneous catalyst for catalytically accelerating transesterification reactions. As a proof-of-concept, benzyl alcohol was chosen as the alcohol while vinyl acetate was selected as the acyl donor for transesterification (Fig. 5a). In this experiment, 1.6 mg of native Lipase TL or a certain amount of LA-n-prop@Zn-HHTP with 1.6 mg Lipase TL loading was dispersed in 1,4-dioxane solution involving 200 mM benzyl alcohol and 1 M vinyl acetate, respectively. The product of benzyl acetate was monitored by 1H nuclear magnetic resonance spectra (NMR). The double peak at δ = 4.69 ppm was assigned to the proton adjacent to the hydroxyl group of the reactant of benzyl alcohol (Supplementary Fig. 61), while the single peak at δ = 5.11 ppm belonged to the proton adjacent to the hydroxyl group of the product of benzyl acetate. Therefore, quantifying the signal changes of these two NMR peaks allowed us to evaluate the kinetics of the transesterification reaction. The time-dependent consumption of benzyl alcohol and generation of benzyl acetate were described in Fig. 5b, c, and the obtained transesterification kinetics were depicted in Fig. 5d. LA-n-prop@Zn-HHTP showed a higher catalytic rate for transesterification and reached over 80% yield after 8 h. While the yield was only 28% using native Lipase TL as a catalyst at the same time, suggesting a ca. 2.56 times higher yield using LA-n-prop@Zn-HHTP heterogeneous catalyst (Supplementary Fig. 62). Of note, such accelerated transesterification was also observed in other reactions using a series of benzyl alcohol derivatives including 4-ethylbenzyl alcohol, 4-isopropylbenzyl alcohol, 4-tert-butylbenzyl alcohol, 4-fluorobenzyl alcohol, and 4-chlorobenzyl alcohol as the reactants, with a 2.21 to 6.64 times enhancement on catalytic yield after 8 h (Fig. 5e and Supplementary Fig. 63).

a The principle of the transesterification reaction. The time-dependent 1H NMR (CDCl3) spectra of the transesterification conversion of benzyl alcohol using b Lipase TL and c LA-n-prop@Zn-HHTP. d Time-dependent yield of benzyl acetate and conversion of benzyl alcohol based on 1H NMR using LA-n-prop@Zn-HHTP and native Lipase TL as the catalysts, respectively. Error bars = standard deviation (SD); Data were presented as mean values ± SD (n = 3; n presents the sample size used to derive statistics). e The catalytic yield of transesterification for a series of benzyl alcohol derivatives with Lipase TL and LA-n-prop@Zn-HHTP catalyst after 8 h. Source data are provided as a Source Data file.
Enzymatic esterification was another essential strategy for the large-scale synthesis of esters in the chemical industry57. We also investigated the esterification performance using our LA-n-prop@Zn-HHTP heterogeneous catalyst, wherein oleic acid and the aliphatic alcohol with varied chain lengths (n = 5, 10, 18, n represented the C atom numbers of the aliphatic chain) were chosen as the reactants (Fig. 6a). The catalytic conversion was evaluated by monitoring ester formation via 1H NMR spectroscopy58. Taking the esterification reaction between oleic acid and n-decanol (n = 10) as a representative example, the triplet peak observed at δ = 2.37 ppm was assigned to the protons adjacent to the carboxyl group of oleic acid (Supplementary Fig. 64). Upon ester formation, this peak shifted up-field to δ = 2.31 ppm, corresponding to the generation of decyl oleate (Fig. 6b and Supplementary Fig. 65). Time-dependent integration of these two peak areas allowed us to estimate the conversion and it revealed a high ester yield of 94.15% after 24 h using the LA-n-prop@Zn-HHTP heterogeneous catalyst (Fig. 6c), surpassing most of the reported lipase biocatalyst (Supplementary Table 9). While the ester yield was only 23.42% using the native Lipase TL catalyst. Additionally, this heterogeneous biocatalysis conferred the esterification reaction with desirable recyclability, and 85.00 % yield was retained after five cycles (Fig. 6d and Supplementary Fig. 66).

a The principle of the esterification reaction. b The time-dependent 1H NMR (CDCl3) spectra of the esterification reaction between oleic acid and n-decanol by LA-n-prop@Zn-HHTP biocatalyst. The Lipase TL was normalized to 1 mg/mL in the reaction system. c Time-dependent yield of decyl oleate and conversion of oleic acid based on 1H NMR using LA-n-prop@Zn-HHTP and native Lipase TL as the catalysts, respectively. Error bars = standard deviation (SD); Data were presented as mean values ± SD (n = 3; n presents the sample size used to derive statistics). d Recyclability of LA-n-prop@Zn-HHTP catalyst for esterification reaction. Error bars = standard deviation (SD); Data were presented as mean values ± SD (n = 3; n presents the sample size used to derive statistics). e The esterification yield of Lipase TL and LA-n-prop@Zn-HHTP for pentanol and oleanol. Error bars = standard deviation (SD); Data were presented as mean values ± SD (n = 3; n presents the sample size used to derive statistics). Source data are provided as a Source Data file.
To further clarify the generalizability of our LA-n-prop@Zn-HHTP heterogeneous catalyst for promoting esterification, the aliphatic alcohol substrate was extended from n-decanol to shorter chain aliphatic alcohol of pentanol (n = 5) and to longer chain aliphatic alcohol of oleanol (n = 18). The ester yields were 90.30 and 90.02% by using LA-n-prop@Zn-HHTP heterogeneous catalyst after 24 h, while the native Lipase TL only presented 21.76 and 17.29% yield for pentanol and oleanol, respectively (Fig. 6e).
To further assess the scalability of the esterification reaction, we conducted a tenfold scale-up by proportionally adjusting the substrate and biocatalyst amounts while maintaining a reaction time of 24 h. Following purification by column chromatography (petroleum ether/ethyl acetate, 40:1, Rf = 0.6) and structural validation by 1H NMR and 13C NMR (Supplementary Figs. 67–69), the yields of pentyl oleate, decyl oleate, and oleyl oleate were determined to be 0.99, 1.15, and 1.40 g, respectively, corresponding to high conversion of 93.40, 90.68, and 87.57%. Additionally, we demonstrated the feasibility of gram-scale synthesis of the LA-n-prop@Zn-HHTP biocatalyst (see Supplementary Fig. 70 for details). These findings well demonstrated the practicability of LA-n-prop@Zn-HHTP heterogeneous catalyst for accelerating esterification.
Discussion
In summary, we report an aggregation-induced conformation locking strategy based on a 2D-MOF to synthesize highly activated and structurally stable lipase hybrid biocatalysts. This confirmation locking strategy is straightforward and essentially different from the unpredictable methods based on nanopore interfacial activation. The locking principle herein can well preserve a catalytically favorable conformation of lipase inside a robust 2D-MOF, which possesses long-range ordered pore channels that facilitate the mass transfer for confined biocatalysis. As a result, the 2D MOF-locked Lipase TL aggregate presents much higher catalytic power than the native Lipase TL, representing the highest activation efficiency for ester hydrolysis among the reported hybrid biocatalysts. The feasibility of this robust MOF biocatalyst for high-value esters synthesis based on transesterification and esterification reactions is also canvassed, and its catalytic performance significantly outperforms the native Lipase TL in terms of catalytic rate, yield, and recyclability. We anticipate that this aggregation-induced conformation locking strategy can offer a facile and straightforward technique to access highly activated lipase hybrid catalysis, holding numerous potential in lipase-associated industrial scenarios.
Methods
Synthesis of M-HHTP
The ligand solution of HHTP with a concentration of 10 mM was prepared by dissolving HHTP in a mixture solvent of water/ethanol (3/1, v/v). In a 2 mL glass vial, NaOH (8 μL, 0.008 mmol) was added in an aqueous solution of Zn(NO3)2 or CuCl2 (0.8 mL, 0.016 mmol) to adjust the pH to 10. Subsequently, 0.8 mL of prepared HHTP solution was added to generate a black suspension. The mixture was stood for 12 h at room temperature. Finally, the precipitates were collected by centrifugation, washed twice with deionized water and once with ethanol, and dried under vacuum overnight.
Synthesis of enzyme@M-HHTP
The ligand solution of HHTP with a concentration of 10 mM was prepared by dissolving HHTP in a mixture solvent of water/ethanol (3/1, v/v). In a 2 mL glass vial, NaOH (8 μL, 0.008 mmol) was added in an aqueous solution of Zn(NO3)2 or CuCl2 (0.8 mL, 0.016 mmol) to adjust the pH to 10. Then, 0.8 mg enzyme (Lipase TL, GOx, HRP, Cutinase, Lipase PS, or Lipase CRL) was added, followed by adding 0.8 mL of the prepared HHTP solution. The mixture was stood for 12 h at room temperature. Finally, the precipitates were collected by centrifugation, washed twice with deionized water and once with ethanol, and dried under vacuum overnight.
Synthesis of LA@Zn-HHTP
The ligand solution of HHTP with a concentration of 10 mM was prepared by dissolving HHTP in a mixture solvent of water/ethanol (3/1, v/v). In a 2 mL glass vial, NaOH (8 μL, 0.008 mmol) was added to an aqueous solution of Zn(NO3)2 (0.8 mL, 0.016 mmol) to adjust the pH to 10. Then, 0.08 mL of Lipase solution pre-dissolved in different contents of organic solvent at a concentration of 10 mg/ml was added, followed by adding 0.8 mL of the prepared HHTP solution immediately. The mixture was stood for 12 h at room temperature. Finally, the precipitates were collected by centrifugation, washed twice with deionized water and once with ethanol, and dried under vacuum overnight.
Catalytic transesterification reaction
In a typical transesterification reaction42, 0.5 mL of stock solution of benzyl alcohol/benzyl alcohol derivatives (200 mM) and vinyl acetate (1000 mM) in 1,4-dioxane were added to a 1,4-dioxane suspension of Lipase TL or LA-n-prop@Zn-HHTP (1.6 mg in 0.5 mL 1,4-dioxane) in a 3 mL glass bottle. The glass bottle was sealed and shaken at 250 rpm at room temperature. Subsequently, the liquid supernatant was sampled at different time intervals (10, 20, 30, 60, 120, 240, 480, and 720 min). About 20 μL of the collected supernatant was added to 500 μL of deuterated chloroform. The products at different time intervals were monitored by 1H NMR, and the conversion of the transesterification reaction was estimated via integral analysis by 1H NMR. This transesterification was repeated three times, and the catalytic yield data were represented as an average.
Catalytic esterification reaction
The esterification reaction was carried out according to a reported method59. In a flask containing the lipase or lipase@MOFs biocatalysts (Lipase TL dosage was kept at 0.1% w/v) was added, the oleic acid (100 μL) and the respective alcohol (1 equivalent) in 1.6 mL of n-Hexane. The suspension was placed at 37 °C under 200 rpm stirring. Subsequently, 100 μL of the liquid supernatant was sampled at different time intervals (2, 5, 8, 10, and 24 h). About 20 μL of the collected supernatant was added to 500 μL of deuterated chloroform. The products at different time intervals were monitored by 1H NMR, which were also qualitatively analyzed by gas chromatography-mass spectrometry (details seen in Supplementary Methods Section and Supplementary Figs. 71,72)59. The yield was calculated based on the integral analysis of the characteristic proton signals of the ester products and the original oleic acid in 1H NMR.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated in this study are provided in the Supplementary Information. Source data are provided with this paper. The Lipase TL structure used herein is available in the PDB database under accession code 1DT3. Source data are provided with this paper.
References
Bornscheuer, U. T. et al. Engineering the third wave of biocatalysis. Nature 485, 185–194 (2012).
Palomo, J. M. Synthetic complexity created by lipases. Nat. Catal. 3, 335–336 (2020).
Lipase market by source (microbial lipases, animal lipases), application (animal feed, dairy, bakery, confectionery, others), & by geography (North America, Europe, Asia-Pacific, Latin America, RoW)—global forecast to 2020. https://www.marketsandmarkets.com/Market-Reports/lipase-market-205981206.html (2020).
Gagnon, C. et al. Biocatalytic synthesis of planar chiral macrocycles. Science 367, 917–921 (2020).
Wang, L. et al. Enzyme conformation influences the performance of lipase-powered nanomotors. Angew. Chem. Int. Ed. 59, 21080 (2020).
Seddigi, Z. S., Shaheer Malik, M., Ahmed, S. A., Babalghith, A. O. & Kamal, A. Lipases in asymmetric transformations: recent advances in classical kinetic resolution and lipase-metal combinations for dynamic processes. Coord. Chem. Rev. 348, 54–70 (2017).
Brady, L. et al. A serine protease triad forms the catalytic centre of a triacylglycerol lipase. Nature 343, 767–770 (1990).
Schrag, J. D., Li, Y., Wu, S. & Cygler, M. Ser-His-Glu triad forms the catalytic site of the lipase from Geotrichum candidum. Nature 351, 761–764 (1991).
Lescic Asler, I., Stefanic, Z., Marsavelski, A., Vianello, R. & Kojic-Prodic, B. Catalytic dyad in the SGNH hydrolase superfamily: in-depth insight into structural parameters tuning the catalytic process of extracellular lipase from Streptomyces rimosus. ACS Chem. Biol. 12, 1928–1936 (2017).
Xu, W. et al. Graphene oxide enabled long-term enzymatic transesterification in an anhydrous gas flux. Nat. Commun. 10, 2684 (2019).
Weltz, J. S., Kienle, D. F., Schwartz, D. K. & Kaar, J. L. Reduced enzyme dynamics upon multipoint covalent immobilization leads to stability-activity trade-off. J. Am. Chem. Soc. 142, 3463–3471 (2020).
Ding, C. Y. et al. Photothermal enhanced enzymatic activity of lipase covalently immobilized on functionalized Ti3C2TX nanosheets. Chem. Eng. J. 378, 122205 (2019).
Arana-Peña, S., Rios, N. S., Carballares, D., Gonçalves, L. R. B. & Fernandez-Lafuente, R. Immobilization of lipases via interfacial activation on hydrophobic supports: production of biocatalysts libraries by altering the immobilization conditions. Catal. Today 362, 130–140 (2021).
Huang, S., Kou, X., Shen, J., Chen, G. & Ouyang, G. Armor-plating” enzymes with metal-organic frameworks (MOFs). Angew. Chem. Int. Ed. 59, 8786–8798 (2020).
Pan, Y. et al. A general Ca-MOM platform with enhanced acid-base stability for enzyme biocatalysis. Chem. Catal. 1, 146–161 (2021).
Zhang, J. et al. Amphiphilic nanointerface: inducing the interfacial activation for lipase. Acs. Appl. Mater. Interfaces 14, 39622–39636 (2022).
Guo, L. et al. A synergetic pore compartmentalization and hydrophobization strategy for synchronously boosting the stability and activity of enzyme. J. Am. Chem. Soc. 146, 17189–17200 (2024).
Huang, S. et al. Hydrogen-bonded supramolecular nanotrap enabling the interfacial activation of hosted enzyme. J. Am. Chem. Soc. 146, 1967–1976 (2024).
Sun, Q. et al. Pore environment control and enhanced performance of enzymes infiltrated in covalent organic frameworks. J. Am. Chem. Soc. 140, 984–992 (2018).
Zheng, Y. et al. Green and scalable fabrication of high-performance biocatalysts using covalent organic frameworks as enzyme carriers. Angew. Chem. Int. Ed. 61, e202208744 (2022).
Nadar, S. S. & Rathod, V. K. Encapsulation of lipase within metal-organic framework (MOF) with enhanced activity intensified under ultrasound. Enzym. Microb. Tech. 108, 11–20 (2018).
Liang, J. et al. Locking the ultrasound-induced active conformation of metalloenzymes in metal–organic frameworks. J. Am. Chem. Soc. 144, 17865–17875 (2022).
Fairen-Jimenez, D. et al. Opening the gate: framework flexibility in ZIF-8 explored by experiments and simulations. J. Am. Chem. Soc. 133, 8900–8902 (2011).
Villeneuve, P. et al. Chemoenzymatic synthesis of structured triacylglycerols with conjugated linoleic acids (CLA) in central position. Food Chem. 100, 1443–1452 (2007).
Brzozowski, A. M. et al. Structural origins of the interfacial activation in thermomyces (Humicola) lanuginosa lipase. Biochemistry 39, 15071–15082 (2000).
Monera, O. D. et al. Protein denaturation with guanidine hydrochloride or urea provides a different estimate of stability depending on the contributions of electrostatic interactions. Protein Sci. 3, 1984–1991 (1994).
Chen, G. et al. A convenient and versatile amino-acid-boosted biomimetic strategy for the nondestructive encapsulation of biomacromolecules within metal–organic frameworks. Angew. Chem. Int. Ed. 58, 1463–1467 (2019).
Frisch, M. J. et al. Gaussian 09, revision D.01. (Gaussian, Inc., 2013).
Ryu, K. & Dordick, J. S. How do organic solvents affect peroxidase structure and function? Biochemistry 31, 2588–2598 (1992).
Liao, F. et al. Shielding against unfolding by embedding enzymes in metal–organic frameworks via a de novo approach. J. Am. Chem. Soc. 139, 6530–6533 (2017).
Serdakowski, A. L., Munir, I. Z. & Dordick, J. S. Dramatic solvent and hydration effects on the transition state of soybean peroxidase. J. Am. Chem. Soc. 128, 14272–14273 (2006).
Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta Bioenerg. 1767, 1073–1101 (2007).
Jackson, M. & Mantsch, H. H. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 30, 95–120 (1995).
Fernandez-Lafuente, R. Lipase from Thermomyces lanuginosus: Uses and prospects as an industrial biocatalyst. J. Mol. Catal., B Enzym. 62, 197–212 (2010).
Stobiecka, A., Wysocki, S. & Brzozowski, A. M. Fluorescence study of fungal lipase from Humicola lanuginose. J. Photochem. Photobiol. 45, 95–102 (1998).
Gonçalves, K. M. et al. Conformational dissection of Thermomyces lanuginosus lipase in solution. Biophys. Chem. 185, 88–97 (2014).
Masters, B. R. Principles of Fluorescence Spectroscopy 3rd edn (Springer, 2008).
José, N. S. et al. Translocation of enzymes into a mesoporous MOF for enhanced catalytic activity under extreme conditions. Chem. Sci. 10, 4082–4088 (2019).
Park, K. S. et al. Exceptional chemical and thermal stability of zeolitic imidazolate frameworks. Proc. Natl Acad. Sci. USA 103, 10186–10191 (2006).
Ma, S. M. et al. Redirecting the cyclization steps of fungal polyketide synthase. J. Am. Chem. Soc. 130, 38–39 (2008).
Zhang, J.-P., Zhu, A.-X., Lin, R.-B., Qi, X.-L. & Chen, X.-M. Pore surface tailored SOD-type metal-organic zeolites. Adv. Mater. 23, 1268–1271 (2011).
Liang, W. et al. Enhanced bioactivity of enzyme/MOF biocomposite via host framework engineering. J. Am. Chem. Soc. 145, 20365–20374 (2023).
Wrogemann, J. et al. Overcoming diffusion limitation of Faradaic processes: property-performance relationships of 2D conductive metal-organic framework Cu3(HHTP)2 for reversible lithium-ion storage. Angew. Chem. Int. Ed. 62, e202303111 (2023).
Eagleton, A. M. et al. Fabrication of multifunctional electronic textiles using oxidative restructuring of copper into a Cu-based metal-organic framework. J. Am. Chem. Soc. 144, 23297–23312 (2022).
Yao, M.-S. et al. Layer-by-layer assembled conductive metal–organic framework nanofilms for room-temperature chemiresistive sensing. Angew. Chem. Int. Ed. 56, 16510 (2017).
Huang, C. et al. Hierarchical conductive metal-organic framework films enabling efficient interfacial mass transfer. Nat. Commun. 14, 3850 (2023).
Hmadeh, M. et al. New porous crystals of extended metal-catecholates. Chem. Mater. 24, 3511–3513 (2012).
Ko, M. et al. Employing conductive metal–organic frameworks for voltammetric detection of neurochemicals. J. Am. Chem. Soc. 142, 11717–11733 (2020).
Tong, L. et al. Atomically unveiling the structure-activity relationship of biomacromolecule-metal-organic frameworks symbiotic crystal. Nat. Commun. 13, 951 (2022).
Liu, Y. et al. Direct imaging of protein clusters in metal–organic frameworks. J. Am. Chem. Soc. 146, 12565–12576 (2024).
Meyer, J. S., Ranville, J. F., Pontasch, M., Gorsuch, J. W. & Adams, W. J. Acute toxicity of binary and ternary mixtures of Cd, Cu, and Zn to Daphnia magna. Environ. Toxicol. Chem. 34, 799–808 (2015).
Murty, R. et al. Interrogating encapsulated protein structure within metal–organic frameworks at elevated temperature. J. Am. Chem. Soc. 145, 7323–7330 (2023).
Li, P. et al. Nanosizing a metal–organic framework enzyme carrier for accelerating nerve agent hydrolysis. ACS Nano 10, 9174–9182 (2016).
Canet, A., Bonet-Ragel, K., Benaiges, M. D. & Valero, F. Lipase-catalysed transesterification: viewpoint of the mechanism and influence of free fatty acids. Biomass. Bioenerg. 85, 94–99 (2016).
Senthamarai, T. et al. A “universal” catalyst for aerobic oxidations to synthesize (hetero)aromatic aldehydes, ketones, esters, acids, nitriles, and amides. Chem 8, 508–531 (2022).
Amini, Z., Ilham, Z., Ong, H. C., Mazaheri, H. & Chen, W. H. State of the art and prospective of lipase-catalyzed transesterification reaction for biodiesel production. Energ. Convers. Manag. 141, 339–353 (2017).
Khan, Z. et al. Current developments in esterification reaction: a review on process and parameters. J. Ind. Eng. Chem. 103, 80–101 (2021).
Liu, G.-F. et al. Rapid crystallization and versatile metalation of acetylhydrazone-linked covalent organic frameworks for heterogenous catalysis. J. Am. Chem. Soc. 147, 1840–1850 (2025).
Noro, J., Cavaco-Paulo, A. & Silva, C. Chemically modified lipase from Thermomyces lanuginosus with enhanced esterification and transesterification activities. ChemCatChem 13, 4524 (2021).
Acknowledgements
We acknowledge financial support from projects of National High-Level Talents Special Support Program—Young Talents (2024WRQB006, G.C.), National Natural Science Foundation of China (22174164, G.C. and 22104159, S.H.), Guangdong Basic and Applied Basic Research Foundation (2024B1515020070, G.C.), Fundamental Research Funds for the Central Universities, Sun Yat-sen University (23lgbj005, G.C.), and Guangdong Basic Research Center of Excellence for Functional Molecular Engineering (31000-42080002, G.O.).
Author information
Authors and Affiliations
Contributions
G.C. conceived the idea, designed the experiments, and revised the draft. A.H. performed material synthesis, characterization and wrote the draft. Z.-W.L., L.G., N.Z., L.T., and Y.X. helped with the material synthesis, characterization and data analysis. X.M. performed the cryo-EM experiment and helped with the data analysis. F.Z. participated in the discussion. G.C., S.H., and G.O. supervised the experiments and provided financial support.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xiuting Li, Weibin Liang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, A., Li, ZW., Guo, L. et al. An aggregation-induced conformation locking strategy facilitates the activation of lipase biocatalyst. Nat Commun 16, 4660 (2025). https://doi.org/10.1038/s41467-025-59824-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59824-w
