
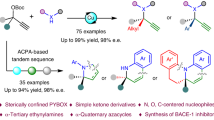
Abstract
We report the highly enantioselective Cu-catalyzed asymmetric propargylic substitution (APS) of α-tertiary propargylic electrophiles using hydrazines and hydroxylamines as a fruitful strategy to access multifunctional α-tertiary hydrazines or hydroxylamines. Using trifluoroethanol (TFE) as the solvent play a key role to decrease the nucleophilicity of hydrazines to suppress side reactions such as elimination, thus improve the yield and the enantioselectivity. NMR analysis and theoretical calculations suggest the formation of an H-bond adduct of TFE with hydrazide, stabilized by multiple H-bonding interactions, including C–F···H–N interaction. The sterically confined pyridinebisoxzolines (PYBOX), featuring a bulky benzylthio shielding group also contribute to the excellent enantioselectivity. Aryl- and aliphatic-ketone-derived α-ethynylalcohol carbonates, α-tertiary α-ethynyl epoxides, cyclic carbonates and and α-hydroxycarboxylates all are competent substrates to afford α-tertiary α-ethynylhydrazines with high structural diversity. The obtained products can be readily converted into various α-tertiary hydrazines and azacycles featuring an aza-quaternary stereocenter.
Similar content being viewed by others

Introduction
Chiral amines are ubiquitous in drugs, biological probes, and agrochemicals1,2,3,4,5,6,7. Their facile synthesis holds the key to future drug development. Hydrazine is a notable subclass of the amine family, and its derivatives constitute versatile synthetic intermediates for the synthesis of azacycles and prominent pharmacophores for drug design (Fig. 1a)8,9,10,11. For example, replacing the NH2 group of methyldopa (used to treat high blood pressure) with a hydrazine group led to a different drug, carbidopa (used for Parkinson’s disease), with a completely different pharmacology12. In addition, the N–N bond enables hydrazino acids to be used as aza-analogs of β-amino acids, allowing their application as backbone-extended peptidomimetics with a unique hydrazino turn structure10,13. With the chemical space associated with quaternary carbons emerging as an important element in drug discovery14,15,16,17,18,19, chiral α-tertiary hydrazines have found increasing numbers of applications, as shown by the representative drugs and bioactive compounds in Fig. 1a20,21,22. Accordingly, it is important to develop efficient methods for the synthesis of diverse chiral α-tertiary hydrazines for medicinal research. Several strategies are known, including enzymatic resolution23, the addition of nucleophiles to ketone hydrazones24,25,26, electrophilic amination using azodicarboxylates27,28 and C–H amination of methines29 (Fig. 1b). These methods have been developed to varying degrees, but significant gaps remain in the scope of these methodologies. Thus, strategies to develop multifunctional chiral α-tertiary hydrazines as platform molecules are still highly desirable.

a Selected bioactive molecules containing chiral α-tertiary hydrazine. b Known strategies for catalytic asymmetric synthesis of α-tertiary hydrazine. c Our previous work on Cu-catalyzed asymmetric propargylic amination enabled by sterically confined PYBOX ligands. d This work, enantioselective propargylic hydrazination to generate α-tertiary ethynylhydrazines.
Since the seminal work of Nishibayashi, Hidai, Uemura, and coworkers30, asymmetric propargylic substitution (APS) of propargylic alcohol derivatives has emerged as a powerful tool to construct multifunctional chiral synthons featuring an α-ethynyl group as a synthetic handle because its sp C–H bond and triple bond can undergo many diversifying reactions31,32. The APS of ketone-derived α-ethynyl alcohol derivatives using hydrazine derivatives would afford chiral α-tertiary α-ethynylhydrazines as a versatile synthetic platform from which to construct structurally diverse chiral hydrazines. However, this approach remains unexplored, although elegant APS of secondary α-ethynyl alcohol derivatives to give chiral 2-pyrazolines was achieved using N-phenylhydrazine33, or to give chiral secondary α-ethynylhydrazines using hydrozones34. Two difficulties face the development of APS as a route to α-tertiary α-ethynylhydrazines. First, APS of simple ketone-derived α-ethynylalcohol derivatives to give fully substituted stereocenters remains undeveloped because the prochiral center in the key metal–allenylidene intermediate is separated from the metal by three bonds35,36,37; thus, remote enantiofacial control is required to achieve excellent enantioselectivity19,38,39. Furthermore, a diminished chiral bias arises because of the similarities of the two substituents on the prochiral carbon. Second, because of the α-effect, the hydrazine derivatives are generally more nucleophilic than amines40, which might lead to both side reactions such as elimination and poisoning of the metal catalyst by occupying the empty binding. Furthermore, the linear shape and dual reactive sites of hydrazine derivatives related to the N–N bond impose further requirements on the chiral catalysts to achieve excellent regio- and enantioselectivity via remote control. As a result, even racemic versions of APS of α-tertiary α-ethynylalcohol derivatives using hydrazines remain unexplored41,42 despite the remarkable progress in the corresponding asymmetric propargylic amination reactions43,44,45,46,47,48,49,50,51,52,53,54.
Recently, we developed a variety of sterically confined pyridinebisoxazoline (PYBOX) ligands featuring a bulky C4-shielding group on the pyridine to modify the electronic properties of the ligand and relay chiral information from the oxazoline ring. These ligands proved to be effective for asymmetric Cu-catalyzed alkyne-azide cycloaddition55,56,57. Particularly, those with both a C4-shielding and extra relaying groups on the 5-position of the oxazolines enabled a highly enantioselective Cu-catalyzed asymmetric propargylic amination (ACPA) of propargylic carbonates derived from simple aryl or aliphatic ketones (Fig. 1c)52. Theoretical studies suggest that the electron-donating benzyloxy group renders the ligands more Lewis basic and forms shorter N–Cu bonds, thereby increasing the contact regions of the chiral pocket and enhancing remote enantiofacial control (Fig. 1c). These results suggest the possibility of tuning the structure of such sterically confined PYBOX to address the aforementioned challenges in synthesizing α-tertiary α-ethynylhydrazines via APS. Herein, we report the PYBOX featuring a bulky C4 benzylthio shielding group that enables highly enantioselective APS of propargylic carbonates derived from both aliphatic and aryl ketones using hydrazides and hydroxylamines, with the assistance of trifluoroethanol (Fig. 1d).
Results
Reaction development
Given that the nature of the substituents greatly influences the nucleophilicity of hydrazines40, we first evaluated the performance of various hydrazine derivatives in the APS of α-phenyl propargylic carbonate (1a) under previously developed conditions for the corresponding APS of anilines by using CuCl2 · 2H2O/L6 as the catalyst and MeOH as the solvent, with 1.0 equiv diisopropylethylamine (DIPEA)52. As expected, the structure of the hydrazine derivatives influenced the desired reaction significantly (Fig. 2a).

a Influences of hydrazine derivatives. b Effects of the ligands. c Effects of the solvent. Reagents and conditions: 1a (0.10 mmol), 2 (0.12 mmol), CuCl2 · 2H2O (10 mol%), PYBOX ligand (12 mol%), DIPEA (1.0 equiv), solvent (1.0 mL), 0 °C, 48 h. NMR yield with 1,3,5-trimethoxybenzene as internal standard. The e.e. was determined by chiral HPLC analysis. d Left:13C NMR analysis for the interaction of TFE or EtOH with 2a. Right: 19F NMR study on the interaction of TFE with 2a. e DFT calculations to examine the influence of the fluoro-atom on the interaction of alcohol with 2a. The bond distances are given in angstroms and the natural bond oribital (NBO) charges are in parentheses. aDetected by GC analysis. bDIPEA (2.0 equiv). cCuCl2 · 2H2O (2.0 mol%), DABCO (1.0 equiv) in TFE at –20 °C. dIsolated yield. RSM: recovery of starting material. DIPEA diisopropylethylamine, TFE trifluoroethanol, BP byproduct, HFIP hexafluoroisopropanol.
No desired product was obtained when hydrazine hydrochloride and acetyl hydrazine were used, with a large amount of carbonate 1a recovered, accompanied by the elimination product enyne 4 and other unidentified byproducts. On the other hand, when N-benzyl or phenyl hydrazine, p-toluenesulfonyl hydrazine, or hydrazone was used, the carbonate 1a was fully consumed, but no target product was detected; only enyne 4, substitution byproduct 5, and other unidentified products were observed. The fact that α-tertiary α-ethynyl alcohol derivatives can undergo an elimination reaction in the presence of a base is expected, but this may be aggravated by the use of hydrazine derivatives with strong nucleophilicity. It was found that without a chiral copper catalyst, no elimination occurred when stirring 1a and DIPEA in MeOH at 0 °C for 48 h, but the presence of any tested hydrazine derivative shown in Fig. 2a led to obvious side reactions to yield enyne 4 and other unclarified byproducts, with 56–72% of carbonate 1a being remained by GC analysis (see Table S5 of the Supplementary Information, SI). Fortunately, when benzoyl hydrazine 2a was used, the desired α-tertiary α-ethynylhydrazine 3a was obtained in 71% yield and 64% e.e., accompanied by a 29% yield of side products. It should be noted that, under the same conditions, the corresponding APS of 1a using anilines generally gave the desired amines in over 80% yield and 90% e.e52. This difference clearly demonstrates that the efficient synthesis of α-tertiary α-ethynylhydrazines via APS is not as straightforward as it first appeared.
Subsequently, we examined other sterically confined PYBOX ligands to identify a suitable C4-shielding group to tune the electronic and steric properties of the catalyst to suppress side reactions while improving enantioselectivity (Fig. 2b). Indeed, the electronic properties of the C4 substituent affected the reaction significantly. As compared with unmodified ligand L1, which gave chiral hydrazine 3a in 66% yield and 59% e.e., L2, with an electron-withdrawing chloro group, led to diminished e.e., whereas L3, with an electron-donating benzyloxy group, gave 3a with an improved 64% e.e. Because sulfur has a lower electronegativity than oxygen and the C–S bond is longer than the C–O bond, we then designed ligand L4 with a benzylthio group for fine-tuning the catalyst properties and found that it afforded a higher e.e. of 73%. Further studies showed that with either the electron-rich 3,5-dimethoxylbenzylthio or electron-deficient 3,5-di(trifluoromethyl)benzylthio group, the corresponding ligands L5 and L7 both gave inferior results, but still afforded better e.e values than analogous L6 and L8 bearing a benzyloxy-type C4-shielding group. Nevertheless, L9, bearing a bulky C4 3,5-di-tert-butyl benzylthio group, afforded 3a in slightly higher e.e. (77%) with similar yield.
These results justified developing sterically confined PYBOX ligands bearing a benzylthio-type group at C4 position. Notably, these C4 benzylthio-PYBOX ligands were readily prepared by a modified two-step procedure from easily available dicyanopyridine. For example, L9 can be obtained in an overall yield of 60% on a gram scale (for details, see Section 2 of SI). Next, to suppress E2 elimination, improve the yield, and enhance enantiofacial control, we examined the use of fluoroalkyl alcohols as solvents. Hexafluoroisopropanol (HFIP) and trifluoroethyl alcohol (TFE) are known to have unique properties that facilitate the synthesis of amines and azacycles58,59,60, owing to their high polarity, enhanced Brøsted acidity, and capacity to stabilize carbocations better than the analogous nonfluorinated alcohols61,62. We considered that fluoroalkyl alcohols can form multiple H-bonding interactions with benzoyl hydrazine 2a to modulate its basicity and/or nucleophilicity to suppress side elimination; furthermore, the resulting hydrazine-based H-bond adduct might have beneficial steric effects that would enhance enantiofacial discrimination. As anticipated, the use of fluoroalkyl alcohols as the solvent improved both the yield and the enantioselectivity substantially when ligand L9 was employed (Fig. 2c). The reaction in 2-fluoroethanol gave 3a in obviously improved yield (87%) and e.e. (82%).
Better results were obtained when the solvent contained more fluorine atoms, with up to 93% yield and 90% e.e. achieved when TFE was used as the solvent. For comparison, the use of EtOH, nPrOH, or nBuOH as the solvent did not improve the outcome of the reaction, and the desired APS reaction proceeded poorly in common solvents as well (for details, see Section 3 of the SI). However, no reaction occurred when using HFIP as the solvent, possibly because its higher acidity led to irreversible protonation of 2a; a similar finding that the epoxide ring-opening reaction with piperidine was inhibited by HFIP solvent was reported previously63. Further evaluating copper salts, bases, and temperature revealed that the reaction in TFE could be catalyzed by only 2.0 mol% of the CuCl2 · 2H2O/L9 complex at –20 °C, in the presence of DABCO, to furnish α-ethynylhydrazine 3a in 94% e.e. and 94% isolated yield.
Subsequently, NMR analysis and DFT calculations were conducted to establish the role of TFE. 13C NMR analysis showed a distinct interaction between hydrazide 2a and TFE. Upon adding TFE to a solution of 2a in CDCl3, the characteristic peak of the carbonyl group shifted downfield from 168.67 to 170.06 ppm. However, almost no change was observed in the same operation when EtOH was used as the solvent (Fig. 2d). 19F NMR analysis also revealed a significant shift of the characteristic peak from –77.437 to –77.333 ppm upon titrating a solution of 2a in CD2Cl2 against a solution of TFE in CD2Cl2. These results suggest that TFE forms stronger H-bonding interactions with hydrazine 2a than EtOH. In addition to TFE being a better H-bond donor than EtOH, we wondered whether its C–F bond could serve as H-bond acceptor to form C–F···H–N interactions between TFE and 2a to reinforce their binding, based on our interest in probing the influence of C–F···H–X interactions in organic reactions64,65,66,67,68, as well as reports from other groups69,70,71,72,73,74,75. This hypothesis was supported by DFT calculations (Fig. 2e). It was found that by changing from EtOH to mono-, di-, and trifluoroethanol, the binding energy (ΔE) between 2a and the alcohol increased gradually from –5.0 to –7.1 kcal/mol, indicating the formation of a more stable complex (for details, see Section 9 of the SI). In the case of TFE, the calculations revealed that O–H···O = C interactions between the hydroxyl group of TFE and the carbonyl group of 2a (bond length 1.77 Å; bond angle of O–H···O = C, 162.9°), along with the H-bonding interactions between the NH2 group of 2a and the oxygen of TFE (bond length 2.66 Å; bond angle of N–H···O–C, 126.1°), formed a seven-membered ring. Meanwhile, effective C–F···H–N interactions between TFE and the NH2 group of 2a (bond length 2.36 Å; bond angle of N–H···F–C, 128.0°) were observed, given that the sum of the van der Waals radii of hydrogen and fluorine atoms is approximately 2.55 Å. Such nonclassical H-bonding interactions between hydrazide 2a and mono- and difluoroethyl alcohol were also suggested by the DFT calculations (for details, see Section 9 of the SI). These studies cast light on the beneficial influence of TFE on the reaction. The effective H-bonding interactions between TFE and 2a both regulate the basicity and/or nucleophilicity of 2a to suppress competitive side reactions and confine the rotation of the N–N bond. This adjusts the conformation of 2a to a more favorable orientation to attack Cu–allenylidene intermediate for better enantiofacial discrimination.
Although HFIP and TFE are among the most common oxo-halogenated unconventional solvents to facilitate organic reactions61,62, to our knowledge, the correlation between their extraordinary reactivity-enabling character and the potential of their C–F bonds as H-bond acceptors has not been previously proposed. We believe our studies will bring insights into these nonclassical interactions, which can be leveraged to facilitate selective organic reactions by using fluoroalkyl alcohols as the reaction media.
Substrate scope
With the optimized conditions in hand, we examined the generality of the APS of α-tertiary α-ethynylalcohol derivatives using hydrazines (Fig. 3). Gratifyingly, propargylic carbonates derived from both aryl and aliphatic ketones worked well to give the desired chiral α-tertiary ethynylhydrazines in good to excellent yields and e.e. values. First, alkyl aryl ketone-derived α-tertiary α-ethynylalcohol carbonates were evaluated. Various α-phenyl propargylic carbonates reacted smoothly with benzoyl hydrazine 2a to give the desired chiral ethynylhydrazines 3a–m in 85–96% yield with 84–94% e.e., irrespective of the nature and position of the substituents on the phenyl group. Acetonaphthone-derived carbonate also worked well to give 3n in 93% yield with 90% e.e. Carbonates bearing ethyl, n-propyl, or n-butyl were also viable substrates, delivering the corresponding chiral α-ethynylhydrazines 3o–q in high yield and e.e. values. Subsequently, the performance of challenging aliphatic-ketone-derived α-ethynyl carbonates was investigated. A range of benzylacetone-derived propargylic carbonates reacted smoothly with hydrazide 2a to afford the corresponding α-alkyl α-ethynylhydrazines 3r–u in 80%–92% yield with 84–91% e.e., irrespective of the electron-donating or -withdrawing nature of the substituent on the phenyl group. 2-Naphthyl, 3-indolyl, 3-benzothienyl, and 2-furanyl substituted carbonates were also viable substrates, yielding the corresponding hydrazines 3v–y in 84–92% yield with 86–97% e.e. Notably, linear aliphatic carbonates bearing an alkene moiety also worked well to furnish chiral hydrazines 3z–ab in 87–90% yield with 90% e.e. Heptanone-derived carbonate gave the corresponding product 3ac in 78% yield with moderate e.e. The use of a range of substituted benzoyl hydrazines was then examined. The electronic nature of phenyl substituents had little impact on the hydrazination using carbonate 1a, giving the desired hydrazines 3ad–ai in 78–99% yield with 92–94% e.e. Hydrizines with 1-naphthyl, 2-naphthyl, 3-indolyl, or piperonyl reacted smoothly, giving 3aj–am in 88–95% yield with 91–95% e.e.

Conditions A: α-aryl propargylic ester 1 (0.2 mmol), hydrazide 2 (0.24 mmol), CuCl2·2H2O (2.0 mol%), L9 (2.4 mol%), DABCO (0.2 mmol), CF3CH2OH (2.0 mL), –20 °C, 2 days. Conditions B: α-alkyl propargylic ester 1 (0.2 mmol), hydrazide 2 (0.24 mmol), CuBr2 (10 mol%), L8 (12 mol%), DABCO (0.1 mmol), CF3CH2OH (2.0 mL), –20 °C, 2 days. Conditions C: propargylic ester 1 (0.2 mmol), hydroxylamine 6 (0.24 mmol), CuBr2 (10 mol%), L8 (12 mol%), N,N-dimethylpiperazine (0.8 mmol), nPrOH (2.0 mL), –20 °C, 4 days. Isolated yield. The e.e values were determined by chiral HPLC analysis. The absolute configuration of (S)−3e and (R)−3r was determined by X-ray analysis, for detail see Section 10 of the SI.
Encouraged by the excellent enantioselectivity achieved in the APS of α-tertiary α-ethynyl carbonates and hydrazides, we attempted to extend the reaction using hydroxylamines as the nucleophile for the synthesis of optically active α-tertiary ethynylhydroxylamines; these are multifunctional building blocks that are used to access chiral α-tertiary hydroxylamines, which are prominent structural motifs in pharmaceutically active compounds that are difficult to synthesize76,77,78. Gratifyingly, the APS of O-benzylhydroxylamine 6a with various benzylacetone-derived propargylic carbonates proceeded well to yield the chiral hydroxylamines 7a–d in 73–80% yield with 90–92% e.e. Carbonates bearing 2-naphthyl, 2-furanyl, 3-benzothienyl, and 3-indolyl readily afforded the desired products 7e–h in 78–85% yield with 75–94% e.e. Hydroxylamines bearing benzyl or allyl substituents were also tolerated, allowing access to hydroxylamines 7i–k in 61–75% yield with 91–94% e.e. However, α-aryl carbonates afforded the desired products in moderate yield and e.e., as shown by the synthesis of 7 l (65% yield, 52% e.e.).
To demonstrate further the capacity of the C4 benzylthio-PYBOX ligands, we then examined the Cu-catalyzed propargylic hydrazination using α-tertiary α-ethynyl epoxides36, cyclic carbonates37,38, and α-hydroxycarboxylates40. These propargylic electrophiles have been widely used in amination reactions, but their propargylic hydrazination remains undeveloped. We envisioned that this approach could provide pathways to access chiral β-hydrazino alcohols or α-hydrazino acids, which are valuable targets for medicinal research. As shown in Fig. 4, α-aryl epoxides reacted well with benzoyl hydrazines to afford the desired β-hydrazino alcohols 8a–f in 70–80% yield with 86–93% e.e. A variety of α-aryl cyclic carbonates also readily reacted with benzoyl hydrazine 2a to give the corresponding α-aryl β-hydrazino alcohols 8a–i in 77–90% yield with 92–98% e.e. Furthermore, both α-aryl and α-alkyl α-ketoester derived α-ethynyl carbonates were also suitable, delivering chiral α-tertiary α-hydrazino acid esters 8j–n in up to 85% yield and 90% e.e.

Reaction conditions: 1 (0.2 mmol), hydrazide 2 (0.24 mmol), CuCl2·2H2O (10 mol%), L9 (12 mol%), DABCO (0.2 mmol), CF3CH2OH (2.0 mL), –20 °C, 2 days. aL13 was used; for details see Section 6 of the SI. Isolated yield. The e.e. values were determined by chiral HPLC analysis. The absolute configuration of (S)−8i was determined by X-ray analysis. The absolute configuration of (S)−8j was determined by converting the ester group into a hydroxymethyl group and comparing the optical rotation value with that of the (S)−8a.
Application explorations
Having established the highly enantioselective propargylic hydrazination, its synthetic utility was further explored (Fig. 5). First, the scalability of this protocol was shown by the facile access to 1.2 g of chiral α-tertiary α-ethynylhydrazine (S)−3a by using 2.0 mol% chiral catalyst under the optimized conditions, without erosion of enantioselectivity. Leveraging the ethynyl or hydrazine groups as synthetic handles enabled diversity-oriented synthesis based on 3a to be achieved; this allowed the construction of structurally diverse α-tertiary hydrazines and azacycles that are difficult to access by previous protocols. For example, the Au-catalyzed intramolecular hydroamination led to chiral cyclic hydrazine 9 in an 82% yield. Starting from 3a and isocyanate, a sequential nucleophilic addition and base-mediated cyclization process afforded imidazolidinone 10 in 79% yield, and a tandem nucleophilic addition and Au-catalyzed cyclization sequence unexpectedly delivered chiral oxadiazole 11 in 75% yield. Furthermore, taking advantage of the ethynyl group, it was possible to merge the resulting α-tertiary α-ethynylhydrazines for late-stage modification of drugs and bioactive compounds, as shown by a CuAAC, which enabled the effective fusion of a zidovudine fragment (a drug commonly used in the treatment of AIDS-related syndromes and HIV infection), leading to compound 12, bearing a triazole moiety, in a remarkable 99% yield and >20:1 dr. Gratifyingly, the selective hydrogenation of the alkyne moiety mediated by Pd/C or Lindlar catalyst afforded α-ethyl hydrazine 13 or α-vinyl hydrazine 14 in 90% and 89% yield, respectively. The Sonogashira coupling gave chiral hydrazine 15 in 83% yield with 94% e.e.

a Product elaboration: Reaction conditions: (a) Ph3PAuCl (20 mol%), MeOH, 50 °C, 8 h; (b) 4-fluorophenyl isocyanate (1.0 equiv), Et2O, 4 h, then NaOMe (2.0 equiv), MeOH, 50 °C, 2 h, Ar = 4-FC6H4; (c) 4-fluorophenyl isocyanate (1.0 equiv), Et2O, 4 h, then Ph3PAuCl (5.0 mol%), MeOH, 50 °C, 24 h, Ar = 4-FC6H4; (d) zidovudine (1.0 equiv), CuSO4 ∙ 5H2O (10 mol%), sodium ascorbate (20 mol%), tBuOH/H2O (1:1, v/v), rt, 4 h; (e) Pd/C (10 mol% Pd), H2 (1.0 atm), MeOH (1.0 mL), 50 °C, 6 h; (f) Lindlar catalyst (10 mol% Pd), quinoline (2.0 equiv), H2 (1.0 atm), EtOAc, rt, 10 min; (g) Pd(PPh3)4 (10 mol%), CuI (10 mol%), PhI (1.5 equiv), Et3N (10 equiv), DMF, 55 °C, 3 h. Isolated yield. The e.e. values were determined by chiral HPLC analysis. b Tandem reaction: The propargylic hydrazination/carboxylative cyclization sequence. For hydrazination step: according to standard condition; For carboxylative cyclization step: AgOTs (50 mol%), 1,3-diphenylguanidine (1.2 equiv), CO2 (3.0 MPa) at 25 oC for 72 h. Isolated yield. The e.e. values were determined by chiral HPLC analysis.
Interestingly, the Cu-catalyzed propargylic hydrazination can be further coupled with an Ag-catalyzed carboxylative cyclization (CC) for tandem synthesis of chiral 2-oxazolidinones using CO2 as a C1 synthon. Based on our recently reported tandem ACPA/CC reactions79, we further developed a similar tandem propargylic hydrazination/CC sequence using a modified condition, paving way to chiral 5-methylidene-2-oxazolidinones 16 bearing αza-quaternary stereocenters in 83%-87% e.e., slightly lower than that of the corresponding chiral α-tertiary ethynylhydrazines. It is worth mentioning that optically active 2-oxazolidinones are privileged scaffolds in drugs and bioactive compounds, but methods to those bearing an aza-quaternary stereocenter are rare80, despite incorporating a fully substituted carbon might significantly enhance the pharmaceutical properties of bioactive compounds14.
Mechanistic studies
Mechanistic studies were conducted to probe the beneficial role of the C4 benzylthio group of the ligands in the highly enantioselective Cu-catalyzed propargylic hydrazination (Fig. 6). First, single crystals of complexes formed from CuCl2 ∙ 2H2O with unmodified ligand L1 or benzylthio-PYBOX L9 were obtained. Comparative analysis revealed that the Cu–N distances in complex Cu(II)/L9 were substantially shorter than those in the complex Cu(II)/L1 (1.992, 1.922, and 1.969 Å vs. 2.033, 1.995, and 2.025 Å, respectively), as shown in Fig. 6a. These data showed that the electron-donating C4 benzylthio group enhanced the Lewis basicity of the PYBOX ligand, thereby strengthening the binding of L9 to Cu(II). This analysis is consistent with our previous theoretical calculations that in the key Cu(I)-allenylidene intermediates, PYBOX ligand L3 (featuring a C4 benzyloxy group) formed shorter Cu–N bonds than unmodified ligand L1. Therefore, we believe the role of the electron-donating C4 benzylthio group was to enhance the Lewis basicity of the ligands to form shorter Cu–N bonds, which, in turn, led to the formation of a more congested chiral pocket. In addition, the sterically hindered C4 benzylthio group could interact with the two relaying phenyls at the 5-position of the oxazolines, leading to a more confined chiral environment. Consequently, the electronic and steric effects of the C4 benzylthio group have a concerted effect that reinforces the remote enantiofacial control during the nucleophilic attack of hydrazide 2 to the Cu–allenylidene intermediate.

a X-ray structure of CuCl2/L1 and CuCl2/L9. b Nonlinear effects of the e.e. values of ligand L9 and product 3a. c Variations of CuCl2/L9 ratios of propargylic hydrazination of 1a and 2a. NMR yield. d UV–vis spectroscopy experiments with Cu complexes. e DFT calculations of the optimized structures for TS modes to chiral 3a catalyzed by benzylthio–PYBOX L4 in the absence of TFE. f DFT calculations of the optimized structures in the presence of TFE.
To obtain more information on the reactive chiral copper species involved in the reaction, the e.e. value of PYBOX L9 versus that of α-ethynylhydrazine 3a was examined. The data exhibited a pronounced positive nonlinear effect (NLE), which implied that more than one chiral ligand might be involved in the stereoselective amination step (Fig. 6b). Investigations into the Cu/L9 stoichiometric ratio further demonstrated that the optimal catalytic performance occurred at a ratio near 1:1.2 (Fig. 6c), supporting the hypothesis of a dicopper complex stabilized by two chiral ligands as the active species. Additionally, UV-vis spectroscopy of the reaction mixture under standard conditions with CuCl2·2H2O/L9 displayed nearly identical absorption profiles to that with CuCl/L9 (Fig. 6d), suggesting the dominance of a Cu(I)-ligand complex rather than a Cu(II) species in the catalytic cycle, which was further supported by EPR spectroscopy experiments (for details, see Section 9 of the SI).
Based on these results and our previously established working model for the benzyloxy-PYBOX-enabled Cu-catalyzed propargylic amination, DFT calculations were performed to study the possible transition states (TSs) for the formation of chiral 3a catalyzed by benzylthio-PYBOX L4. Initially, the possible TSs in the absence of TFE were examined. As shown in Fig. 6e, in the optimized TS-L4-S for the major (S)−3a, two key stabilizing interactions could be identified: the π–π stacking interaction between the phenyl group of 1a and the 4-phenyl group of the oxazoline ring in ligand L4, as well as the C–H/π interaction involving the phenyl group of 1a and the phenyl group of the benzylthio moiety. In contrast, the optimized TS-L4-R corresponding to the minor (R)−3a enantiomer lacked these stabilizing interactions and exhibited destabilizing steric repulsion between the methyl group of 1a and the phenyl group of L4, resulting in a 2.3 kcal/mol higher free energy.
Next, the optimized TSs in the presence of TFE, incorporating H-bonding interactions, were studied based on the established 2a-TFE interaction model. As shown in Fig. 6f, the H-bonding-induced spatial constraint between 2a and TFE enhances the favorable π–π stacking interactions in TS- L4/TFE-S-1 while exacerbating unfavorable steric repulsions in TS-L4/TFE-R-1. This is evidenced by the shorter Ph···Ph distances in TS-L4/TFE-S-1 (3.83 Å) compared to TS-L4-S (3.91 Å), and more pronounced repulsive interactions between the methyl group of 1a and the phenyl group of L4 in TS-L4/TFE-R-1 (2.23 Å) versus TS-L4-R (2.26 Å). Consequently, DFT calculations revealed a substantial free-energy gap (3.7 kcal/mol) between transition states TS-L4/TFE-S-1 and TS-L4/TFE-R-1, matched well with experimental results. These theoretical results indicate that the H-bonding interactions between TFE and 2a plays an important role in achieving the excellent enantioselectivity through two complementary mechanisms: stabilizing π–π interactions in the S-type transition state and exacerbating steric repulsion between the methyl group of 1a and the phenyl group of L4 in the R-type transition state.
Discussion
We have developed a highly enantioselective Cu-catalyzed APS of α-tertiary propargylic electrophiles using hydrazides and hydroxylamines, enabled by the sterically confined PYBOX ligands featuring a C4 benzylthio group. The scope of the propargylic electrophiles was broad, with suitable substrates including not only aryl- and aliphatic-ketone-derived α-ethynylalcohol carbonates, but also α-tertiary α-ethynyl substituted epoxides, cyclic carbonates, and α-hydroxycarboxylates. To our knowledge, no previous report can achieve highly enantioselective APS reaction with such a broad scope of α-tertiary propargylic electrophiles. The use of TFE as the solvent played an important role in suppressing side reactions, leading to increased yield and improved enantioselectivity. As suggested by NMR analysis and theoretical calculations, this beneficial effect is believed to originate from the formation of a H-bond adduct between the TFE and the hydrazide, which is stabilized by multiple H-bonding interactions, including C–F···H–N interactions, which is a reactivity-enabling characteristic of fluoroalkyl alcohols that has previously been overlooked. This research also highlights the potential of our sterically confined PYBOX ligands in developing other APS reactions. X-ray crystallographic analysis of the CuCl2/L1 and CuCl2/L9 complexes as well as theoretical calculations revealed that the C4 benzylthio group increased the congestion of the chiral pocket, leading to increased remote enantiofacial control. This approach facilitates the synthesis of α-tertiary α-ethynylhydrazines and α-ethynylhydroxylamines with high structural diversity. The multifunctional chiral α-tertiary α-ethynylhydrazines that are generated provide versatile platform molecules that can undergo various diversifying transformations. We believe that our method should attract the interest of researchers in medicinal, agrochemical, and materials science.
Methods
General procedure for the synthesis of chiral α-tertiary propargylhydrazines 3 and 8. To a 5 mL vial equipped with a screw-cap were added CuCl2 · 2H2O or CuBr2 (0.004 or 0.02 mmol, 2.0 or 10 mol%) and PYBOX ligand L8 or L9 (0.0048 or 0.024 mmol, 2.4 or 12 mol%), followed by 2.0 mL of anhydrous CF3CH2OH. The solution was stirred at 40 oC for 2.0 h, and then propargylic esters 1 (0.2 mmol, 1.0 equiv) was added. After cooling the mixture to −20 oC for 0.5 h, DABCO (0.1 or 0.2 mmol, 0.5 or 1.0 equiv) was added, and the mixture was continued to stir at −20 oC for another 0.5 h, following by the addition of hydrazides 2 (0.24 mmol, 1.2 equiv). The resulting mixture was stirred at −20 oC for 2 days till almost full conversion of 1 by TLC analysis, and then quickly passed through a short column to remove copper catalyst using EtOAc as the eluent. The thus obtained solution was concentrated under reduced pressure, and the residue was directly subjected to column chromatography purification using petroleum ether/EtOAc (10:1 to 3:1, v/v) as the eluent, to afford the desired chiral α-tertiary α-ethynylhydrazines 3 and 8.
Data availability
Materials and methods, detailed optimization studies, experimental procedures, mechanistic studies, NMR spectra and computational data are available in the Supplementary Information. Source data are present. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition numbers 3e (CCDC 2326713), 3r (CCDC 2389394), 8i (CCDC 2362807), 11 (CCDC 2327368), CuCl2/L1 (CCDC 2323626) and CuCl2/L9 (CCDC 2324124). These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. The Cartesian coordinates of the optimized transition states are recorded in Supplementary Data 1. All source data in support of the findings of this study are available within the Article and its Supplementary Information or from the corresponding author upon request.
References
Royer, J. Chiral Amine Synthesis: Methods, Developments and Applications (Wiley-VCH: Weinheim, 2010).
Li, M.-L., Yu, J.-H., Li, Y.-H., Zhu, S.-F. & Zhou, Q.-L. Highly enantioselective carbene insertion into N–H bonds of aliphatic amines. Science 366, 990–994 (2019).
Xi, Y.-M., Ma, S.-J. & Hartwig, J. F. Catalytic asymmetric addition of an amine N–H bond across internal alkenes. Nature 588, 254–260 (2020).
Chen, C.-Y., Peters, J. C. & Fu, G. C. Photoinduced copper-catalysed asymmetric amidation via ligand cooperativity. Nature 596, 250–256 (2021).
Chen, J.-J. et al. Enantioconvergent Cu-catalyzed N-alkylation of aliphatic amines. Nature 618, 294–300 (2023).
Wang, M. et al. Asymmetric hydrogenation of ketimines with minimally different alkyl groups. Nature 631, 556–562 (2024).
Wu, X. et al. Modular α-tertiary amino ester synthesis through cobalt-catalysed asymmetric aza-Barbier reaction. Nat. Chem. 16, 398–407 (2024).
Rothgery, E. F. Hydrazine and its Derivatives. In Kirk−Othmer Encylopedia Chemical Technology, (Wiley: New York, 2004).
Le Goff, G. & Ouazzani, J. Natural hydrazine-containing compounds: Biosynthesis, isolation, biological activities and synthesis. Bioorg. Med. Chem. 22, 6529–6544 (2014).
Avan, I., Hallb, C. D. & Katritzky, A. R. Peptidomimetics via modifications of amino acids and peptide bonds. Chem. Soc. Rev. 43, 3575–3594 (2014).
Cabré, A., Verdaguer, X. & Riera, A. Recent advances in the enantioselective synthesis of chiral aminesvia transition metal-catalyzed asymmetric hydrogenation. Chem. Rev. 122, 269–339 (2022).
Zhu, H. et al. Carbidopa, a drug in use for management of Parkinson disease inhibits T cell activation and autoimmunity. PLOS ONE 12, e0183484 (2017).
Katoh, T. & Suga, H. Consecutive ribosomal incorporation of α‑aminoxy/α-hydrazino acids with L/D‑configurations into nascent peptide chains. J. Am. Chem. Soc. 143, 18844–18848 (2021).
Talele, T. T. Opportunities for tapping into three-dimensional chemical space through a quaternary carbon. J. Med. Chem. 63, 13291–13315 (2020).
Christoffers, J. & Baro, A. Quaternary Stereocenters. Challenges and Solutions for Organic Synthesis. (Wiley-VCH: Weinheim, 2006)
Quasdorf, K. W. & Overman, L. E. Catalytic enantioselective synthesis of quaternary carbon stereocentres. Nature 516, 181–191 (2014).
Liu, Y., Han, S.-J., Liu, W.-B. & Stoltz, B. M. Catalytic enantioselective construction of quaternary stereocenters: assembly of key building blocks for the synthesis of biologically active molecules. Acc. Chem. Res. 48, 740–751 (2015).
Cao, Z.-Y., Zhou, F. & Zhou, J. Development of synthetic methodologies via catalytic enantioselective synthesis of 3,3-disubstituted oxindoles. Acc. Chem. Res. 51, 1443–1454 (2018).
Xu, P.-W., Zhou, F., Zhu, L. & Zhou, J. Catalytic desymmetrization reactions to synthesize all-carbon quaternary stereocentres. Nat. Synth. 2, 1020–1036 (2023).
Zuhl, A. M. et al. Competitive activity-based protein profiling identi-fies Aza-β-lactams as a versatile chemotype for serine hydrolase inhibition. J. Am. Chem. Soc. 134, 5068–5071 (2012).
Oubella, A. et al. Diastereoselective synthesis and cytotoxic evaluation of new isoxazoles and pyrazoles with monoterpenic skeleton. J. Mol. Struct. 1198, 126924 (2019).
Alakurtti, S. et al. Synthesis and anti-leishmanial activity of heterocyclic betulin derivatives. Bioorg. Med. Chem. 18, 1573–1582 (2010).
Yee, C., Blythe, T. A., McNabb, T. J. & Walts, A. E. Biocatalytic resolution of tertiary alpha-substituted carboxylic acid esters: Efficient preparation of a quaternary asymmetric carbon center. J. Org. Chem. 57, 3525–3527 (1992).
Keith, J. M. & Jacobsen, E. N. Asymmetric hydrocyanation of hydrazones catalyzed by lanthanide−PYBOX complexes. Org. Lett. 6, 153–155 (2004).
Hashimoto, T., Omote, M. & Maruoka, K. Catalytic asymmetric alkynylation of C1-substituted C,N-cyclic azomethine imines by CuI/Chiral Brønsted acid co-catalyst. Angew. Chem. Int. Ed. 50, 8952–8955 (2011).
Lykke, L., Carlsen, B. D., Rambo, R. S. & Jorgensen, K. A. Catalytic asymmetric synthesis of 4-nitropyrazolidines: An access to optically active 1,2,3-triamines. J. Am. Chem. Soc. 136, 11296–11299 (2014).
Zhou, F., Liao, F.-M., Yu, J.-S. & Zhou, J. Catalytic asymmetric electrophilic amination reactions to form nitrogen-bearing tetrasubstituted carbon stereocenters. Synthesis 46, 2983–3003 (2014).
Lu, Q., Du, Y., Xu, M.-M., Xie, P.-P. & Cai, Q. Catalytic asymmetric aza-electrophilic additions of 1,1-disubstituted styrenes. J. Am. Chem. Soc. 146, 21535–21545 (2024).
Yang, C.-J. et al. Cu-catalysed intramolecular radical enantioconvergent tertiary β-C(sp3) −H amination of racemic ketones. Nat. Catal. 3, 539–546 (2020).
Nishibayashi, Y., Onodera, G., Inada, Y., Hidai, M. & Uemura, S. Synthesis of diruthenium complexes containing chiral thiolate-bridged ligands and their application to catalytic propargylic alkylation of propargylic alcohols with acetone. Organometallics 22, 873–876 (2003).
Lauder, K., Toscani, A., Scalacci, N. & Castagnolo, D. Synthesis and reactivity of propargylamines in organic chemistry. Chem. Rev. 117, 14091–14200 (2017).
Li, M.-D., Wang, X.-R. & Lin, T.-Y. Recent advances in copper-catalyzed asymmetric propargylic substitution. Tetrahedron Chem. 11, 100082 (2024).
Zhang, D.-Y., Shao, L., Xu, J. & Hu, X.-P. Copper-catalyzed asymmetric formal [3+2] cycloaddition of propargylic acetates with hydrazines: enantioselective synthesis of optically active 2-pyrazolines. ACS Catal. 5, 5026–5030 (2015).
Liu, S.-Y., Tanabe, Y., Kuriyama, S., Sakata, K. & Nishibayashi, Y. Ruthenium- and copper-catalyzed propargylic substitution reactions of propargylic alcohol derivatives with hydrazones. Chem. Eur. J. 27, 15650–15659 (2021).
Hattori, G. et al. Copper-catalyzed enantioselective propargylic amination of propargylic esters with amines: Copper-allenylidene complexes as key intermediates. J. Am. Chem. Soc. 132, 10592–10608 (2010).
Sakata, K. & Nishibayashi, Y. Mechanism and reactivity of catalytic propargylic substitution reactions via metal–allenylidene intermediates: a theoretical perspective. Catal. Sci. Technol. 8, 12–25 (2018).
Roh, S. W., Choi, K. & Lee, C. Transition metal vinylidene- and allenylidene-mediated catalysis in organic synthesis. Chem. Rev. 119, 4293–4356 (2019).
Clayden, J., Lund, A., Vallverdú, L. & Helliwell, M. Ultra-remote stereocontrol by conformational communication of information along a carbon chain. Nature 431, 966–971 (2004).
Liao, S.-H., Sun, X.-L. & Tang, Y. Side arm strategy for catalyst design: Modifying bisoxazolines for remote control of enantioselection and related. Acc. Chem. Res. 47, 2260–2272 (2014).
Nigst, T. A., Antipova, A. & Mayr, H. Nucleophilic reactivities of hydrazines and amines: The futile search for the α‑effect in hydrazine reactivities. J. Org. Chem. 77, 8142–8155 (2012).
Ohta, K., Takahashi, N. & Yoshimatsu, M. Propargyl hydrazides: Synthesis and conversion into pyrazoles through hydroamination. Chem. Eur. J. 18, 15602–15606 (2012).
Qian, J. et al. Synthesis of allenyl-B(MIDA) via hydrazination/fragmentation reaction of B(MIDA)-propargylic alcohol. Chin. Chem. Lett. 34, 107479 (2023).
Detz, R. J., Delville, M. M. E., Hiemstra, H. & van Maarseveen, J. H. Enantioselective copper-catalyzed propargylic amination. Angew. Chem. Int. Ed. 47, 3777–3780 (2008).
Hattori, G., Matsuzawa, H., Miyake, Y. & Nishibayashi, Y. Copper-catalyzed asymmetric propargylic substitution reactions of propargylic acetates with amines. Angew. Chem. Int. Ed. 47, 3781–3783 (2008).
Hattori, G., Yoshida, A., Miyake, Y. & Nishibayashi, Y. Enantioselective ring-opening reactions of racemic ethynyl epoxides via copper-allenylidene intermediates: efficient approach to chiral β-amino alcohols. J. Org. Chem. 74, 7603–7607 (2009).
Gómez, J. E., Guo, W.-S., Gaspa, S. & Kleij, A. W. Copper-catalyzed synthesis of γ-amino acids featuring quaternary stereocenters. Angew. Chem. Int. Ed. 56, 15035–15038 (2017).
Tian, L., Gong, L. & Zhang, X. Copper-catalyzed enantioselective synthesis of β-amino alcohols featuring tetrasubstituted tertiary carbons. Adv. Synth. Catal. 360, 2055–2059 (2018).
Guo, W.-S., Zuo, L.-H., Cui, M.-Y., Yan, B.-W. & Ni, S.-F. Propargylic amination enabled the access to enantioenriched acyclic α-quaternary α-amino ketones. J. Am. Chem. Soc. 143, 7629–7634 (2021).
Liu, T., Ni, S.-F. & Guo, W.-S. Practical asymmetric amine nucleophilic approach for the modular construction of protected α-quaternary amino acids. Chem. Sci. 13, 6806–6812 (2022).
Cai, Q. et al. Well-defined chiral dinuclear copper complexes in enantioselective propargylic substitution: For a long-standing supposition on binuclear mechanism. Chem 10, 265–282 (2024).
Qian, H.-D. et al. Remote copper-catalyzed enantioselective substitution of Yne-thiophene esters. Sci. China Chem. 67, 1175–1180 (2024).
Zhang, Z. et al. Enantioselective propargylic amination and related tandem sequences to α-tertiary ethynylamines and azacycles. Nat. Chem. 16, 521–532 (2024).
Chen, P., Zhang, M.-M., Xiao, W.-J. & Lu, L.-Q. Dynamic steric confinement effect expands the frontier of Cu-catalyzed asymmetric propargylic aminations. Sci. China Chem. 67, 3181–3183 (2024).
Lan, S. et al. Copper-catalyzed asymmetric nucleophilic opening of 1,1,2,2-tetrasubstituted donor−acceptor cyclopropanes for the synthesis of α-tertiary amines. J. Am. Chem. Soc. 147, 1172–1185 (2025).
Zhu, R.-Y., Chen, L., Hu, X.-S., Zhou, F. & Zhou, J. Enantioselective synthesis of P-chiral tertiary phosphine oxides with an ethynyl group via Cu(I)-catalyzed azide–alkyne cycloaddition. Chem. Sci. 11, 97–106 (2020).
Liao, K. et al. Highly enantioselective CuAAC of functional tertiary alcohols featuring an ethynyl group and their kinetic resolution. Angew. Chem. Int. Ed. 60, 8488–8493 (2020).
Gong, Y. et al. Sulfonyl–PYBOX ligands enable kinetic resolution of α-tertiary azides by CuAAC. Angew. Chem. Int. Ed. 62, e202301470 (2023).
Cox, L. et al. Alcohols as alkylating agents in the cation-induced formation of nitrogen heterocycles. Angew. Chem. Int. Ed. 61, e202206800 (2022).
Zhu, Y., Smith, M. J. S., Tu, W. & Bower, J. F. A stereospecific alkene 1,2-aminofunctionalization platform for the assembly of complex nitrogen-containing ring systems. Angew. Chem. Int. Ed. 62, e202301262 (2023).
Xie, J. et al. Hexafluoroisopropanol-assisted selective intramolecular synthesis of heterocycles by single-electron transfer. Nat. Synth. 3, 1021–1030 (2024).
Yu, C., Patureau, J. F. W. & Pérez-Temprano, M. H. Emerging unconventional organic solvents for C–H bond and related functionalization reactions. Chem. Soc. Rev. 49, 1643–1652 (2020).
Motiwala, H. F. et al. HFIP in organic synthesis. Chem. Rev. 122, 12544–12747 (2022).
Vuluga, D. et al. Influence of the structure of polyfluorinated alcohols on Brønsted acidity/hydrogen-bond donor ability and consequences on the promoter effect. J. Org. Chem. 76, 1126–1133 (2011).
Liu, Y.-L. et al. Organocatalytic asymmetric strecker reaction of Di- and trifluoromethyl ketoimines. Remarkable fluorine effect. Org. Lett. 13, 3826–3829 (2011).
Yu, J.-S., Liu, Y.-L., Tang, J., Wang, X. & Zhou, J. Highly efficient “On Water” catalyst-free nucleophilic addition reactions using difluoroenoxysilanes: Dramatic fluorine effects. Angew. Chem. Int. Ed. 53, 9512–9516 (2014).
Cao, Z.-Y. et al. Catalytic enantioselective synthesis of cyclopropanes featuring vicinal all-carbon quaternary stereocenters with a CH2F group; study of the influence of C–F⋯H–N interactions on reactivity. Org. Chem. Front. 5, 2960–2968 (2018).
Mu, B.-S. et al. The bifunctional silyl reagent Me2(CH2Cl)SiCF3 enables highly enantioselective ketone trifluoromethylation and related tandem processes. Angew. Chem. Int. Ed. 61, e202208861 (2022).
Hao, Y.-J., Yu, J.-S., Zhou, Y., Wang, X. & Zhou, J. Influence of C–F…H–X interactions on organic reaction. Acta Chim. Sin. 76, 925–939 (2018).
Yuan, H.-N. et al. Hydrogen-bond-directed enantioselective decarboxylative mannich reaction of β-ketoacids with ketimines: Application to the synthesis of anti-HIV drug DPC-083. Angew. Chem. Int. Ed. 52, 3869–3873 (2013).
Jin, M.-L. et al. Effective synthesis of chiral N-fluoroaryl aziridines through enantioselective aziridination of alkenes with fluoroaryl azides. Angew. Chem. Int. Ed. 52, 5309–5313 (2013).
Champagne, P. A., Benhassine, Y., Desroches, J. & Paquin, J.-F. Friedel–crafts reaction of benzyl fluorides: Selective activation of C-F bonds as enabled by hydrogen bonding. Angew. Chem. Int. Ed. 53, 13835–13839 (2014).
Lee, K. et al. Catalytic enantioselective addition of organoboron reagents to fluoroketones controlled by electrostatic interactions. Nat. Chem. 8, 768–777 (2016).
Johnston, C. P. et al. Anion-initiated trifluoromethylation by TMSCF3: Deconvolution of the siliconate-carbanion dichotomy by stopped-flow NMR/IR. J. Am. Chem. Soc. 140, 11112–11124 (2018).
Zhang, Q.-X. et al. B(C6F5)3/chiral phosphoric acid catalyzed ketimine-Ene reaction of 2-Aryl-3H-indol-3-ones and α-methylstyrenes. Angew. Chem. Int. Ed. 59, 4550–4556 (2020).
Zhao, X.-J. et al. Enantioselective synthesis of 3,3′-disubstituted 2-amino-2′-hydroxy-1,1′-binaphthyls by copper-catalyzed aerobic oxidative cross-coupling. Angew. Chem. Int. Ed. 60, 7061–7065 (2021).
Zhang, X., Zou, J. & Dai, J. Microbial transformation of (–)-Huperzine A. Tetrahedron Lett. 51, 3840–3842 (2010).
Gao, X. et al. Fungal indole alkaloid biosynthesis: genetic and biochemical investigation of the tryptoquialanine pathway in penicillium aethiopicum. J. Am. Chem. Soc. 133, 2729–2741 (2011).
Gao, X. et al. Photoenzymatic synthesis of α‑tertiary amines by engineered flavin-dependent “Ene”-reductases. J. Am. Chem. Soc. 143, 19643–19647 (2021).
Zhang, Z. et al. Tandem asymmetric propargylic aminationcarboxylative cyclization reaction to chiral 5-methylidene-2-Oxazolidinones using CO2 as C1 synthon. Sci. China Chem. 68, 1402–1411 (2025).
Arshadi, S. et al. Three-component coupling of CO2, propargyl alcohols, and amines: An environmentally benign access to cyclic and acyclic carbamates (A Review). J. CO2 Util. 21, 108–118 (2017).
Acknowledgements
We thank the financial support from the NSFC (21971067, 22171090), National Key Research and Development Program of China (2020YFA0710200), the Shanghai Science and Technology Innovation Action Plan (No. 20JC1416900), the Innovation Program of Shanghai Municipal Education Commission (2023ZKZD37) and the Ministry of Education (PCSIRT) and the Fundamental Research Funds for the Central Universities are highly appreciated.
Author information
Authors and Affiliations
Contributions
J.Z. and F.Z. conceived the idea; Y.G. performed the experiments, collected and analyzed the data; Z. Z., H. L., T. W., M. J., N. F., P. P. and H. W. prepared the starting materials; X.W. performed the DFT calculation studies; F.Z. and J.Z. directed the project and co-wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Zhihui Shao, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gong, Y., Zhang, Z., Liu, H. et al. Trifluoroethanol-assisted asymmetric propargylic hydrazination to α-tertiary ethynylhydrazines enabled by sterically confined pyridinebisoxazolines. Nat Commun 16, 4571 (2025). https://doi.org/10.1038/s41467-025-59845-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59845-5
