
Abstract
The SARS-CoV-2 papain-like protease (PLpro) is a cysteine protease that cleaves viral polyproteins and antagonizes the host immune response during viral replication. Jun12682 and PF-07957472 are the first-in-class PLpro inhibitors showing potent in vivo antiviral efficacy in mouse models. In this study, we characterize naturally occurring mutations at residues located at the drug-binding site of Jun12682. The results reveal several PLpro mutants showing significant drug resistance while maintaining comparable enzymatic activity as the wild-type PLpro. The physiological relevance of the identified drug-resistant mutants, including E167G and Q269H, is validated through independent serial viral passage experiments. Molecular dynamics simulations and perturbative free energy calculations show that drug-resistant PLpro mutants weaken hydrogen bonding and π-π stacking interactions. Collectively, this study identifies E167, Y268, and Q269 as drug-resistant hotspots for PLpro inhibitors that bind to the BL2 loop and groove region, which are valuable in informing the design of the next-generation PLpro inhibitors.
Similar content being viewed by others


Introduction
SARS-CoV-2 has caused catastrophic damage to the global economy and public health. Although the COVID-19 pandemic is winding down, SARS-CoV-2 viruses are likely to continue to circulate among humans and animals in the foreseeable future1,2. SARS-CoV-2 is an RNA virus with a high mutation rate, and multiple variants have emerged since the beginning of the pandemic. The RNA-dependent RNA polymerase (RdRp) inhibitors remdesivir and molnupiravir, and the main protease (Mpro) inhibitors nirmatrelvir and ensitrelvir, have been approved for human use. However, the virus’s ability to mutate and adapt poses challenges to maintaining their efficacy3,4,5. Clinical and laboratory studies have identified mutations in RdRp and Mpro conferring drug resistance to these approved antivirals6,7,8,9,10,11,12,13,14. As such, additional antivirals with novel mechanisms of action are needed to complement existing RdRp and Mpro inhibitors.
Papain-like protease is one of the two viral-encoded cysteine proteases. It plays a pivotal role in processing the viral polyproteins to produce functional nonstructural proteins that assemble into the replication-transcription complex15. In addition, PLpro exhibits deubiquitinating and deISGylating functions, enabling the virus to evade host immune responses16,17. The dual role of PLpro as a protease and immunomodulator renders it a promising therapeutic target18. Compared to Mpro, PLpro is a more challenging antiviral drug target18. PLpro specifically recognizes two consecutive glycines and cleaves the peptide bond following the second glycine15. The lack of S1 and S2 substrate binding pockets near the catalytic cysteine C111 poses a grand challenge in designing PLpro inhibitors. The naphthalene analog GRL0617 and its derivatives are among the most extensively studied non-covalent PLpro inhibitors18,19. However, their moderate antiviral potency and suboptimal pharmacokinetic properties pose significant hurdles to advancing to animal models. The COVID-19 pandemic reignites the interest in developing PLpro inhibitors20,21,22,23,24. Our group recently reported Jun12682 as the first-in-class PLpro inhibitor to demonstrate in vivo antiviral efficacy in a SARS-CoV-2-infected mouse model22. Jun12682 targets a novel drug binding site, Val70Ub, and the previously reported the blocking loop 2 (BL2) groove and BL2 loop21. In parallel, Pfizer disclosed a quinoline inhibitor PF-07957472 with robust efficacy in a mouse-adapted model of SARS-CoV-2 infection23. Given the promise of PLpro inhibitors as oral antiviral drug candidates, we are interested in elucidating their drug resistance mechanism.
In this study, we choose Jun12682 as a chemical probe and systematically analyze the naturally occurring mutations near its drug-binding site. Our study reveals several PLpro mutants showing comparable enzymatic activity as the wild type (WT) but with significant drug resistance against Jun12682. Cross-resistance is also observed for Jun12682 and PF-07957472. The physiological relevance of the drug-resistant mutants predicted by structural analysis is further validated by independent serial viral passage experiments in cell culture, which reveal similar mutations, including E167G and Q269H. Molecular dynamics (MD) simulations and perturbative free energy calculations provide a molecular basis for drug resistance. Collectively, this study identifies E167, Y268, and Q269 as drug resistance hot spots for SARS-CoV-2 PLpro inhibitors that bind to the BL2 loop and groove region, such as Jun12682 and PF-07957472.
Results
Identification of naturally occurring mutations at drug-binding site residues of SARS-CoV-2 PLpro
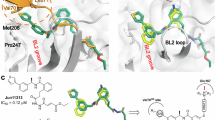
For the prediction of drug-resistant mutants, we first focus on residues that form direct interactions with Jun12682, including M208, P247, P248, Y264, Y268, Q269, D164, and E167 (PDB: 8UOB) (Fig. 1a–c). We hypothesize that mutations at these key residues may lead to drug resistance. The N-ethyl pyrazole substituent in Jun12682 binds to the Val70Ub site, forming CH–π and S–π interactions with M208 S-methyl moiety. In addition, the N-ethyl pyrazole also makes van der Waals contact with the P247 ring. The N-methyl pyrazole substituent binds to the BL2 groove and forms hydrophobic interactions with P248, Y264, and Y268. The phenyl group engages in π–π interactions with the side chain of Y268. The central amide linker is stabilized through hydrogen bonds with the D164 side chain carboxylate and the Y268 mainchain amide NH. The terminal dimethylamino moiety makes electrostatic interactions with E167 side chain carboxylate. The oxygen atom in the linker forms a hydrogen bond with the Q269 side chain amide NH2.

a X-ray crystal structure of PLpro with Jun12682 (PDB: 8UOB). b Active site view of Jun12682 with side chains from residues that form direct contact with the inhibitor. c 2D scheme showing the interactions between Jun12682 and PLpro. d X-ray crystal structures of PLpro with PF-07957472 (PDB: 9CSY). e Active site view of PF-07957472. f 2D scheme showing the interactions between PF-07957472 and PLpro. Jun12682 is colored in orange, PF-07954742 is colored in magenta, and BL2 loop is colored in cyan. c, f were generated using Molecular Operating Environment (MOE) software.
PF-07957472 binds with a similar pose as Jun12682 except in the Val70Ub site (Fig. 1d–f). Key interactions, including hydrophobic interactions in the BL2 groove site, BL2 loop, hydrogen bonds in the central amide linker, and electrostatic interactions with the terminal ammonium, were conserved between these two inhibitors.
Next, we aimed to identify naturally occurring mutations at these critical residues. For this, we analyzed the SARS-CoV-2 sequences deposited in the GISAID database and focused on high-frequency mutations at each residue (Fig. 2)4. We expressed and purified the identified high-frequency mutants at each residue. The enzymatic activity (kcat/Km) of the mutant proteins was determined in the enzymatic assay using a FRET substrate containing the viral polyprotein nsp2/3 cleavage sequence22. The drug sensitivity was characterized by determining the half-maximal inhibitory concentration (IC50) and inhibitory constant (Ki). In addition to our rationally designed PLpro inhibitor Jun12682, we included Pfizer’s PLpro inhibitor PF-07957472 and GRL0671 for assessing cross-resistance. The drug resistance of selected PLpro mutants was further validated in the differential scanning fluorimetry (DSF) binding assay. The complete dataset is listed in the Supplementary Table 1. In total, we attempted the expression of 34 PLpro mutants at eight residues. The expression and purification failed for the three mutants, D164A, E167Q, and P248F, possibly due to protein instability or misfolding. Six mutants, D164V, P248L/T/S, and Y264L/H, were enzymatically inactive. The remaining 25 mutants were characterized for enzymatic activity (kcat, Vmax, Km, kcat/Km), and drug binding (IC50, Ki, and DSF assay).

The results were obtained from CoVsurver of the GISAID, developed by A*STAR Bioinformatics Institute (BII), Singapore. In total, 12,777,211 NSP3 sequences containing the PLpro domain were retrieved from the database as of December 16, 2024. The total mutational occurrence of each amino acid residue is labeled in the bracket at the center of each pie chart, and specific mutations are on the right of their respective pie charts.
The 25 mutants, including D164E/G, E167K/A/S/V/G/D, M208I/V/T, P247T/Q/L/S, Y268H/C/N/F/I, and Q269R/L/K/H/S, showed similar stability as the WT (Fig. 3a), indicating that these mutations did not perturb the structural integrity of PLpro.

a DSF assay to characterize the thermal stability of PLpro mutants. Error bars denote mean ± SD of four biological replicates. b Enzymatic activity of PLpro mutants. The dashed line at the bottom indicates a 10-fold reduction compared to WT, The values are the mean of two biological replicates. Source data are provided as a source data file.
One crucial factor to consider in studying antiviral drug resistance is the fitness of replication of the mutant virus. As mutations that significantly impact the enzymatic activity might attenuate viral replication4,25, we, therefore, define physiologically relevant drug-resistant mutants as those showing comparable enzymatic activity as the WT (kcat/Km < 10-fold change compared to WT). Among the 25 mutants, D164E/G and P247T had significantly reduced enzymatic activities compared to WT (kcat/Km < 10-fold change compared to WT) (Fig. 3b) and did not proceed to the drug sensitivity characterization. Interestingly, M208V displayed enhanced enzymatic activity with kcat/Km of 1.4-fold higher than WT.
In addition, we define significant drug resistance as the inhibitory consistent Ki increase by more than 10-fold compared to WT. Collectively, PLpro mutants with kcat/Km < 10-fold change and Ki > 10-fold change compared to WT are classified as potential physiologically relevant drug-resistant mutants.
Naturally occurring mutations at PLpro residues D164, P248, and Y264 significantly reduced the enzymatic activity of PLpro
D164 is located at the back of the BL2 loop region. It forms a hydrogen bond with the central amide NH in Jun12682 (Fig. 1). The D164G and D164E mutants had significantly reduced enzymatic activities with kcat/Km values being 9% and 10% of WT, respectively (Fig. 3b). D164G mutant also showed significant resistance against GRL0617, Jun12682, and PF-07957472 with inhibitory constant Ki values increased by more than 100-fold (Supplementary Table 1). The purification of D164A failed, and the D164V mutant was inactive.
P248 is located at the BL2 groove binding pocket and forms hydrophobic interactions with the N-methyl pyrazole substituent from Jun12682 (Fig. 1). Three mutants, P248L/T/S, were inactive (Supplementary Table 1). The expression of P248F failed, suggesting that P248 is a critical residue for the enzymatic activity of PLpro.
Y264 is located at the bottom of the BL2 loop and forms a T-shaped π–π stacking with the phenyl ring from Jun12682 (Fig. 1). Both Y264L/H mutants were enzymatically inactive and were not further characterized (Supplementary Table 1).
Naturally occurring mutations at PLpro residues M208 and P247 do not confer drug resistance
M208 is located at the Val70Ub site, and its S-methyl moiety forms CH–π and S–π interactions with the N-ethyl pyrazole substituent in Jun12682 (Fig. 1). The three mutants at M208 residue had similar or enhanced enzymatic activities (kcat/Km) compared to WT: M208T (85%), M208I (51%), and M208V (136%) (Fig. 3b). All three mutants remain sensitive to Jun12682, PF-07957472, and GRL0617 with the increase in Ki values less than 2.5-fold (Fig. 4a).

a Plot of inhibitory constant Ki values. Dash lines indicate the Ki values of GRL0617, Jun12682, and PF-07957472 against PLpro WT. The values are the mean of three biological replicates. b DSF assay of Jun12682 against PLpro M208 mutants. c DSF assay of PF-07957472 against PLpro M208 mutants. d DSF assay of Jun12682 against PLpro P247 mutants. e DSF assay of PF-07957472 against PLpro P247 mutants. Error bars denote mean ± SD of two biological replicates. Source data are provided as a source data file.
P247 is located between the Val70Ub site and the BL2 groove, making van der Waals contact with the N-ethyl pyrazole (Fig. 1). Except for P247T, which had only 8% of the enzymatic activity as the WT, the remaining three mutants had comparable enzymatic activity (kcat/Km) as WT: P247Q (46%), P247S (40%), and P247L (51%) (Fig. 3b). No significant resistance was observed for these three mutants against Jun12682, PF-07957472, and GRL0617 (Ki increase ≤ 4.3-fold) (Fig. 4a).
DSF binding assay showed that Jun12682 and PF-07957472 led to dose-dependent stabilization of PLpro mutants M208T/I/V and P247Q/S/L, with reduced sensitivity compared to WT (Fig. 4b–e), corroborating with the enzymatic assay Ki results (Fig. 4b–e).
In summary, the high-frequency naturally occurring mutations at residues M208 (M208T/I/V) and P247 (P247Q/S/L) do not confer significant drug resistance (Ki increase ≤ 10-fold), and have comparable enzymatic activities as the WT.
E167, Y268, and Q269 are PLpro drug resistance hotspots
E167 side chain carboxylate forms an electrostatic interaction with the terminal positively charged ammonium from Jun12682 (Fig. 1). This electrostatic interaction is conserved in the co-crystal structures of XR8-23, Jun12682, and PF-07957472 (Fig. 1b). It contributes to their improved binding affinity compared to GRL0617. It is expected that E167 mutations will abolish this key interaction, leading to drug resistance. To test this hypothesis, we chose the top 7 high-frequency mutations according to the GISAID gene sequence analysis, E167G/K/A/S/V/D/Q. Except for E167Q, the remaining six mutants were successfully expressed and showed similar thermal stability to the WT according to the DSF assay results (Fig. 3a). It is noted that the occurrence of natural mutations at E167 remains low, with the second frequent mutation, E167G, appearing 23 times as of 12/16/2024 (Supplementary Table 1). It was found that mutations at E167 did not significantly affect the enzymatic activity of PLpro, with all six mutants displaying similar enzymatic activities (kcat/Km) as the WT: E167G (30%), E167K (29%), E167A (42%), E167S (17%), E167V (61%), and E167D (41%) (Fig. 3b). E167G, E167K, E167A, E167S, and E167V showed significant resistance against Jun12682 and PF-07957472 with inhibitory constant Ki values increased by more than 24-fold compared to WT, among which E167G, E167K, and E167S are the most resistant mutants with Ki values increased by more than 130-fold (Fig. 5a). In comparison, the E167D mutant remains sensitive to Jun12682 and PF-07957472 with Ki values increased by 2- and 6-fold, respectively, suggesting both compounds may still retain electrostatic interactions with the E167D side chain carboxylate. Similarly, all six mutants remain sensitive to GRL0617 with Ki increase less than 7-fold, which corroborates with its binding mode: GRL0617 does not engage in electrostatic interaction with E167 side chain carboxylate, and E167 mutations are not expected to impact drug binding.

a Plot of inhibitory constant Ki values against E167 mutants. Dash lines indicate a 10-fold increase in Ki values of Jun12682 and PF-07957472 against the PLpro mutants compared to WT. b DSF assay of Jun12682 against PLpro E167 mutants. c DSF assay of PF-07957472 against PLpro E167 mutants. d Plot of inhibitory constant Ki values against Y268 mutants. Dash lines indicate a 10-fold increase in Ki values of Jun12682 and PF-07957472 against the PLpro mutants compared to WT. e DSF assay of Jun12682 against PLpro Y268 mutants. f DSF assay of PF-07957472 against PLpro Y268 mutants. g Plot of inhibitory constant Ki values against Q269 mutants. Dash lines indicate a 10-fold increase in Ki values of Jun12682 and PF-07957472 against the PLpro mutants compared to WT. h DSF assay of Jun12682 against PLpro Q269 mutants. i DSF assay of PF-07957472 against PLpro Q269 mutants. Error bars of DSF assays denote mean ± SD of two biological replicates. And the values of Ki are the mean of three biological replicates. Source data are provided as a source data file.
The drug resistance was further validated in the DSF assay (Fig. 5b, c). Jun12682 and PF-07957472 showed reduced binding to E167D, E167V, E167A, E167G, E167S, and E167K mutants compared to WT. The degree of drug resistance from the DSF assay is consistent with the Ki values from the enzymatic assay, with E167K, E167S, and E167G being the most resistant mutants.
Y268 is located at the BL2 loop and is a critical residue for drug binding. Y268 forms a T-shaped π–π stacking with the phenyl ring from Jun12682 (Fig. 1a). Y268H is a high-frequency mutation with an occurrence 285 (Supplementary Table 1). Y268H mutant had similar enzymatic activity as the WT (kcat/Km 51%) and had cross-resistance against all three inhibitors: Jun12682 (Ki increase 41-fold), PF-07957472 (Ki increase 75-fold), and GRL0617 (Ki increase 22-fold) (Fig. 5d). The other three mutants had lower occurrence, Y268N (3), Y268F (14), and Y268I (5), and had comparable enzymatic activities of WT (kcat/Km 39%, 92%, 104%) (Fig. 3b). Y268N similarly showed a high degree of cross-resistance against Jun12682 (Ki increase > 385-fold), PF-07957472 (Ki increase 969-fold), and GRL0617 (Ki increase > 100-fold) (Fig. 5d). Y268F remained sensitive to all three inhibitors, suggesting the Y268F might retain the π–π stacking interaction with Jun12682. Y268I had a significant but lower degree of cross-resistance against Jun12682 (Ki increase 11-fold), PF-07957472 (Ki increase 28-fold), and GRL0617 (Ki increase 14-fold) (Fig. 5d).
The dose-dependent DSF assay results further validated the drug resistance with Y268F/I/H/N displaying decreased stabilization by Jun12682 and PF-0795742 compared to WT (Fig. 5e, f). Y268N had the highest resistance, corroborating with the enzymatic assay results.
Q269 is also located at the BL2 loop. The carbonyl from the central amide linker in Jun12682 forms a hydrogen bond from the mainchain amide NH of Q269 (Fig. 1a). In addition, the Q269 side chain amide NH2 forms a hydrogen bond with the oxygen in the linker of Jun12682. Among the five Q269 mutants, Q269H showed comparable enzymatic activity as the WT (kcat/Km 69%) and was resistant to Jun12682 (Ki increase 13.2-fold) and PF-07957472 (Ki increase 13.3-fold) (Fig. 5g). The Q269L/K/S/R did not show significant drug resistance against these three inhibitors (Ki increase < -fold) and had comparable enzymatic activities as the WT (kcat/Km 47-113%) (Fig. 5g). The DSF assay results validated that Q269H is the most drug-resistant mutant (Fig. 5h, i) against Jun12682 and PF-07957472.
In summary, structure-based predictions revealed eight naturally occurring PLpro mutants, including E167G/K/A/S/V, Y268H/N, and Q269H, showing significant resistance against Jun12682 and PF-07957472 (Ki increase > 10-fold), while maintaining comparable enzymatic activities as the WT PLpro (kcat/Km < 10-fold change).
Validation of the physiological relevance of PLpro drug-resistant mutants through viral passage experiments
To validate the physiological relevance of the drug-resistant PLpro mutants predicted by the structure-based approach, we performed serial viral passage experiments to see whether the same PLpro mutant(s) would emerge. For this, SARS-CoV-2 (USA-WA1/2020 strain) was passaged in the presence of increasing concentrations of Jun12682, starting with 1 µM (2 × EC50 in Vero cells) and increasing by two folds every two passages (Fig. 6a). We conducted the passage in triplicate in Vero-E6 cells expressing human angiotensin-converting enzyme 2 (hACE2) and human transmembrane protease, serine 2 (hTMPRSS2) (Vero-AT) as they have been used broadly to propagate SARS-CoV-2 and readily present cytopathic effect (CPE) during infection, allowing rapid observation of drug resistance selection. The passaged virus was given sufficient time to propagate until 30–50% of the cells showed CPE before the next passage. The passaging was conducted 12 times, where the Jun12682 concentration reached 32 µM (32-fold relative to the starting concentration), and the next concentration (64 µM) would be cytotoxic. To identify what mutations might confer resistance, we sequenced the PLpro coding sequence of the three lineages collected every two passages. We found all three lineages started acquiring a mutation of Q269H as early as passage 4 (P4) and became dominant at P6 and maintained through the rest of the passage (Fig. 6b). A L152F mutation appeared in lineage 1 and 2 starting at P10, but not in lineage 3, which acquired different V98L and E167G mutations. The V98L mutation was detected starting at P6, and its frequency peaked around P8-P10 before declining at P11, where the E167G mutation started to arise, suggesting a stepwise manner of increasing drug resistance. In addition, despite no E167G substitution being detected in lineage 2, a T168I mutation adjacent to the E167 site was detected, implying these residues may be involved in drug resistance (Fig. 6b). To confirm these sequencing results and determine if additional mutations occur in the PLpro cleavage sites (Nsp1/2, Nsp2/3, and Nsp3/4 junctions), we performed next-generation sequencing (NGS) to sequence the whole genome of the passage 12 (P12) viruses from each lineage. We found that the PLpro mutations were identical to those revealed by Sanger sequencing, and the frequencies of these mutations were similar between the two assays (Supplementary Table 2). Two non-synonymous Nsp3 mutations (E121G, A1803V) outside the PLpro domain were found in only the lineage 2 P12 virus and some minor Nsp3 mutations (frequency < 15%) were also found. Notably, no mutations in the PLpro cleavage sites were detected. Additional non-synonymous mutations were detected in other genes, such as the spike and the nucleocapsid genes, which was expected for SARS-CoV-2 cell culture adaptation26,27.

a Schematic diagram of virus passaging experiments (See details in “Methods”). The graph was created with Biorender.com. Deng, X. (2025) https://BioRender.com/15xra8k. b PLpro mutations were found in the indicated passages from each lineage. Dots indicate WT at that residue. Mutations are shaded according to frequency. c Virus competition experiments were performed in the absence or presence of Jun12682. Frequencies of PLpro mutations at each passage are shown. d The co-crystal structure of the PLpro/Jun12682 complex with mutations identified during virus passaging. e Plaques formed by recombinant PLpro mutants and size comparison. Isolated plaques (>30 plaques of each virus) were selected and size-measured using ImageJ. The relative size was calculated based on the median plaque size of rWT-nLuc and analyzed with ordinary one-way ANOVA test with multiple comparisons. The p values of each mutant virus compared to rWT-nLuc are p < 0.0001 for rE167G, rY268H, rQ269H, rE167G/Q269H, and p = 0.0004 for rL152F/Q269H. Groups with significant differences (P < 0.05) are indicated with different letters. f Growth kinetic assay with recombinant PLpro mutants carrying single (left) and double mutations (right). Vero-AT cells were infected with 0.001 multiplicity of infection (MOI) of a virus, and luciferase reporter activity was quantified at the indicated time points. The luciferase reporter activity of rE167G and rE167G/Q269H at 30 h post-infection (HPI) are significantly lower than rWT-Luc (two-way ANOVA with the Geisser–Greenhouse correction followed by Sidak’s multiple comparisons test; P = 0.0029, P = 0.0002, respectively). Representative curves from a single experiment from two independent experiments are shown. Error bars denote the mean ± SD of four biological replicates. RLU, relative luminescence units. g Plaque assay EC50 plots of Jun12682. h Plaque assay EC50 plots of PF-07957472. i Plaque assay EC50 plots of nirmatrelvir. EC50 values are mean ± SD of two biological repeats. Source data are provided as a source data file.
To assess the drug resistance of the selected viral lineages rapidly, we performed a virus competition experiment in the absence or presence of 32 µM Jun12682. We chose lineage 3 P11 (L3P11) to compete with the wild-type parental WA1 because L3P11 harbors three mutations with different frequency trends. By passaging the mixture of two viruses, we could monitor the frequency changes of these mutations and determine the fitness of the selected virus relative to the parental strain. As shown in Fig. 6c, the frequencies of E167G and Q269H mutations maintained or slightly increased in the absence of Jun12682, while the V98L mutation became undetectable after four passages. When Jun12682 was present, as expected, the E167G and Q269H mutations rapidly became dominant after passaging. The mutation frequencies were confirmed by the NGS results of the passage 4 virus (Supplementary Table 2). These results suggest that L3P11 acquired resistance to Jun12682 without significantly compromising fitness.
Among the mutations identified in the serial viral passage experiments, E167G and Q269H are located at the drug binding site of Jun12682, while V98L, L152F, and T168I are more than 8 Å far away from the inhibitor and are not directly involved in drug binding (Fig. 6d).
To determine if the PLpro mutations identified through structure prediction and serial passaging attenuate the fitness of SARS-CoV-2 viral replication, we generated a panel of recombinant PLpro mutant viruses by introducing nucleotide substitutions into the infectious cDNA clone of a recombinant SARS-CoV-2 expressing a NanoLuc luciferase reporter (rWT-nLuc)28. These PLpro mutant viruses carry either single mutation E167G (termed rE167G), Y268H (rY268H), Q269H (rQ269H) or double mutations L152F/Q269H (rL152F/Q269H) and E167G/Q269H (rE167G/Q269H). Among these mutations, E167G and Q269H were identified through structure-based prediction and passaging experiments, while a structure-based approach revealed Y268H. Double mutations L152F/Q269H and E167G/Q269H were only identified through virus passaging. After successful recovery, these viruses were subjected to full-genome sequencing followed by plaque formation and viral growth analyses. Plaque size comparison revealed that all single and double mutants formed relatively smaller plaques than those of rWT-nLuc, with rE167G forming the smallest plaques (Fig. 6e). We further determined luciferase reporter activities in infected cells at different hours post-infection (HPI) as a proxy of viral growth kinetics. We found these PLpro mutants expressed moderately reduced luciferase activities, with a statistical significance at 30 HPI for rE167G and rE167G/Q269H (Fig. 6f). These results together suggest that these PLpro mutations mildly attenuate viral replication compared to WT (Fig. 6f), consistent with the slightly reduced enzymatic activities of these PLpro mutants (Fig. 3b).
Next, we assessed the drug resistance of these PLpro mutants against Jun12682 and PF-0795742 using antiviral plaque assays. As expected, rWT-nLuc, which has a wild-type PLpro, had an EC50 of 0.5 µM against Jun12682, similar to WA1 as we reported previously22. In contrast, rE167G and rY268H had EC50 values of 3.0 and 4.8 µM, respectively (Fig. 6g). The rQ269H had an EC50 of 5.6 µM against Jun12682, 11.2-fold relative to that of rWT-nLuc. Importantly, double mutant rL152F/Q269H and rE167G/Q269H exhibited the strongest resistance to Jun12682, with EC50 values greater than 30 μM, suggesting additive effects of the single mutants. Similarly, we determined the resistance of these PLpro mutants to PF-0795742. The rWT-nLuc had an EC50 of 0.1 µM in Vero cells, while rE167G and rY268H had EC50 values of 3.5 and 4.0 µM, respectively (Fig. 6h). The rQ269H had an EC50 of 9.5 µM, 95-fold relative to that of rWT-nLuc. Interestingly, double mutant rL152F/Q269H had similar EC50 as the single mutant rQ269H (11.9 µM vs 9.5 µM), whereas rE167G/Q269H exhibited strong resistance to PF-0795742, with EC50 values greater than 30 μM. Taken together, these results indicate these PLpro mutations identified through structure-based prediction or virus passaging render resistance to Jun12682 and PF-0795742. The single Q269H mutation that appeared rapidly and became dominant during virus passaging confers a stronger resistance effect than the other two single mutations E167G and Y268H. Consistent with viral passages, a secondary mutation such as L152F led to a higher degree of drug resistance. Nirmatrelvir remained sensitive to all five recombinant PLpro mutant viruses and had no cross-resistance with PLpro inhibitors Jun12682 and PF-07957472 (Fig. 6i).
The drug sensitivity of these recombinant SARS-CoV-2 viruses was further characterized in Caco-2 cells (Supplementary Fig. 1). Caco-2 is a human intestinal epithelial cell line shown to be immunocompetent and has been used in SARS-CoV-2 studies29,30,31,32. The results showed that rQ269H, rL152F/Q269H, and rE167G/Q269H exhibited strong resistance to Jun12682 and PF-07957472, with EC50 values increased by more than 25-fold compared to WT. The rE167G and rY268H mutant viruses displayed moderate resistance against Jun12682 and PF-07957472, with EC50 values increased by 6-13-fold compared to WT. Overall, the phenotypic drug resistance is consistent between Caco-2 and Vero-AT cells.
To characterize the mutants selected in the viral passage experiments in enzymatic assay and DSF binding assay, we expressed and purified eight single, double, and triple PLpro mutants including V98L, L152F, T168I, V98L/Q269H, L152F/Q269H, E167G/Q269H, V98L/E167F/Q269H, and L152F/T168I/Q269H. The assay results of E167G and Q269H obtained earlier were included for comparison (Fig. 5). DSF assay showed that L152F, T168I, L152F/Q269H, and the L152F/T168I/Q269H mutants had reduced thermal stability, while V98L, E167G, Q269H, V98L/Q269H, E167G/Q269H, V98L/E167G/Q269H had increased thermal stability (Fig. 7a). Nevertheless, the results indicated that all PLpro mutants folded properly.

a Thermal stability of PLpro mutants. b Enzymatic activity of PLpro mutants. c Plot of inhibitory constant Ki values against PLpro mutants. Dash lines indicate a 10-fold increase in Ki values of Jun12682 and PF-07957472 against the PLpro mutants compared to WT. d DSF assay of Jun12682 against PLpro mutants V98L, L152F, and T168I. e DSF assay of PF-07957472 against PLpro mutants V98L, L152F, and T168I. f DSF assay of Jun12682 against PLpro mutants E167G, Q269H, V98L/Q269H, L152F/Q269H, E167G/G269H, V98:/E167G/Q269H, and L152F/T168I/Q269H. g DSF assay of PF-07957472 against PLpro mutants E167G, Q269H, V98L/Q269H, L152F/Q269H, E167G/G269H, V98:/E167G/Q269H, and L152F/T168I/Q269H. Error bars of all DSF assays denote mean ± SD of two biological replicates. The values of Ki are the mean of three biological replicates. And the value of Kcat/Km are the mean of two biological replicates. Source data are provided as a source data file.
The enzymatic assay showed that the V98L mutant is more active than the WT (kcat/Km 148% of WT) (Fig. 7b). The remaining double and triple mutants had comparable enzymatic activity as the WT: L152F (30%), E167G (30%), T168I (27%), Q269H (69%), V98L/Q269H (70%), L152F/Q269H (60%), E167G/Q269H (16%), V98L/E167G/A269H (22%), and L152F/T168I/Q269H (31%) (Fig. 7b).
The allosteric V98L, L152F, and T168I mutants (Fig. 6d) do not confer drug resistance against Jun12682, PF-07957472, and GRL0617 (Fig. 7c). In contrast, the E167G or Q269H-containing double and triple mutants, including V98L/Q269H, L152F/Q269H, E167G/Q269H, V98L/E167G/Q269H, and L152F/T168I/Q269H displayed significant resistance against Jun12682, PF-07957472, and GRL0617 (Ki increase > 10-fold) (Fig. 7c). The E167G/Q269H and V98L/E167G/Q269H showed the highest drug resistance against Jun12682 and PF-07957472 (Ki increase > 385-fold).
DSF assay results further confirmed that the V98L, L152F, and T168I mutants remained sensitive to Jun12682 and PF-07957472 as the WT (Fig. 7d, e), while the V98L/Q269H, L152F/Q269H, E167G/Q269H, V98L/E167G/Q269H, and L152F/T168I/Q269H mutants were resistant to Jun12682 and PF-07957472 (Fig. 7f, g).
In summary, the antiviral assay results (Fig. 6g, h), coupled with the enzymatic and binding assay results (Fig. 7c–g), confirmed that the E167G and Q269H mutants are the main drug-resistant mutants. The allosteric mutations L152F and T168I do not confer directly to drug resistance, but when combined with E167G or Q269H, they further increase the degree of drug resistance.
To further characterize the identified PLpro drug-resistant mutants in the cleavage of ubiquitin and ISG15 modifications, we determined the enzymatic activity (kcat/Km) of PLpro mutants using the Ub-AMC and ISG15-AMC substrates (Supplementary Fig. 2 and Supplementary Table 3). It was found that E167G and E167G/Q269H had significantly reduced enzymatic activity in digesting Ub-AMC and ISG15-AMC substrates compared to WT (kcat/Km > 10-fold reduction), while Y268H and L152F/Q269H showed enhanced enzymatic activities (Supplementary Fig. 2a, c). All five PLpro mutants, including E167G, Y268H, Q269H, L152F/Q269H, and E167G/Q269H, displayed significant drug resistance against Jun12682 and PF-07957472 (Ki increase > 10-fold) (Supplementary Fig. 2b, d), corroborating the drug-resistant results obtained using the nsp2/3 FRET substrate (Fig. 7c).
MD simulations of PLpro mutants with Jun12682 provide the molecular basis of drug resistance
To provide a molecular basis for the drug resistance, we performed molecular docking calculations and MD simulations to gain insight into the binding mode of Jun12682 inside the SARS-CoV-2 PLpro WT, and the E167G, Y268H, Q269L, Q269H, and E167G/Q269H mutants (Fig. 8).

a, d, g, j, m, p Representative frame from the MD simulations showing the complex between Jun12682 and PLpro WT and its mutants. b, e, h, k, n, q Stabilizing interactions inside the binding site of PLpro with Jun12682; hydrogen bonding interactions bar is depicted in yellow, hydrophobic interactions in magenta and π-stacking interaction in green, and water bridges in light blue. c, f, i, l, o, r RMSD of Cα carbons plots of PLpro (magenta diagram) and RMSD of the ligand’s heavy atoms (orange diagram). For the MD simulations, the X-ray structure of Jun12682 with PLpro WT was used (PDB ID 8UOB22).
The binding interactions between Jun12682 and the viral PLpro proteins were explored using 500 ns-MD simulations with the amber19sb force field (ff19sb33) and the Amber22 software34. For the simulations, the X-ray structure of Jun12682 with SARS-CoV-2 PLpro was used (PDB ID 8UOB22) to generate homology models of the Jun12682-PLpro mutant complexes.
The MD simulations showed that the Jun12682-WT, Jun12682-E167G, Jun12682-Y268H, Jun12682-Q269L, Jun12682-Q269H, and Jun12682-E167G/Q268H all formed stable complexes: the RMSD of Cα carbons and ligand’s heavy atoms were <3 Å for Jun12682-WT (Fig. 8c); the RMSD of Cα carbons and ligand’s heavy atoms were <2 Å for Jun12682-E167G (Fig. 8f); the RMSD of Cα carbons and ligand’s heavy atoms were <~3 Å for Jun12682-Y268H (Fig. 8i); the RMSD of Cα carbons was <3 Å and RMSD of ligand’s heavy atoms was <2.5 Å for Jun12682-Q269L (Fig. 8l); the RMSD of Cα carbons and ligand’s heavy atoms were <2.5 Å for Jun12682-Q269H (Fig. 8o); and the RMSD of Cα carbons and ligand’s heavy atoms were <~2.3 Å for Jun12682-E167G/Q269H (Fig. 8r).
However, compared to the Jun12682-WT complex, altered binding interactions were observed in the Jun12682-PLpro mutant complexes, which account for the phenotypical drug resistance. In the Jun12682-E167G complex, the hydrogen bonding interactions between Jun12682 and residue at positions 167 and R166 are missing, while the hydrogen bonding interactions with D164 are reduced (Fig. 8d, e). In the Jun12682-Y268H complex, the hydrogen bonding of Jun12682 with R166 is missing and the phenyl moiety connecting to the pyrazole rings does not form π-stacking with p-hydroxy phenyl of H268 (Fig. 8g, h). In the Jun12682-Q269L complex, the only difference observed was that the hydrogen bonding of Jun12682 with R166 is missing, while the hydrogen bond with E167 has been considerably reduced (Fig. 8j, k). In the Jun12682-Q269H complex, the hydrogen bonding and hydrophobic interactions of Jun12682 with glutamine acid at position 269 have been replaced by similar interactions with H269 (Fig. 8m, n). However, the hydrogen bonding interaction with R166 is missing while the hydrogen bond with E167 has been considerably reduced. In the Jun12682-E167G/Q269H complex, the direct hydrogen bonding interactions of Jun12682 with D164, R166 and E167 are missing and only water-mediated hydrogen bonding interactions with D164 are observed (Fig. 8p, q).
The differences in stabilizing interactions between Jun12682 and PLpro WT or E167G, Y268H, Q269L, Q269H, and E167G/Q269H mutant proteins are summarized in Table 1. As is shown generally in the mutations that reduce the affinity of Jun12682, the number of important stabilizing hydrogen bonding or π-π stacking interactions were reduced compared to the PLpro WT, with the most pronounced case of the double mutation E167G/Q269H where all the direct hydrogen bonds were disrupted. Hydrophobic interactions with L162, M208, P247, P248, Y264, Y268, and Y273 remain similar in the complexes with mutants and WT proteins.
Accurate assessment of the drug resistance interactions in the complexes of Jun12682-WT and PLpro mutants by perturbative free energy calculations
To compare the calculated and experimental thermodynamic effect of mutations E167G, Y268H, Q269L, Q269H, and E167G/Q269H in the PLpro protein that cause resistance to Jun12682, we calculated the relative binding free energies between Jun12682-WT and each of the Jun12682-PLpro mutant complexes. Toward this aim, we performed perturbative free energy calculations with the thermodynamic integration coupled with the MD simulations (TI/MD) method implemented in Amber22 software34 using the ff14sb35. The calculated values were ΔΔGb,TI/MD (E167→G) = 2.09 kcal mol−1, ΔΔGb,TI/MD (Y268→H) = 2.07 kcal mol−1, ΔΔGb,TI/MD (Q269→) = 1.41 kcal mol−1, ΔΔGb,TI/MD (Q269→H) = 1.70 kcal mol−1, and ΔΔGb,TI/MD (E167→G/Q269→H) = 9.90 kcal mol−1 (Table 2). Considering that the method error is 1 kcal/mol, the calculated values agreed with the experimental ones, as shown in Table 2.
Discussion
The recent design of PLpro inhibitors with in vivo antiviral efficacy in the SARS-CoV-2 infection mouse models offers hope for the translational development of oral bioavailable PLpro inhibitors22,23,24. As new coronavirus variants continue to emerge, developing PLpro inhibitors with broad-spectrum activity could enhance preparedness for future outbreaks. PLpro inhibitors act on a distinct mechanism of action compared to existing Mpro and RdRp inhibitors, thus holding the promise of tackling drug resistance. Nevertheless, antiviral drug resistance is ultimately inevitable. Understanding the drug resistance mechanism guides monitoring drug resistance in clinics and informs the design of next-generation antivirals. To profile the genetic barrier of drug resistance of PLpro inhibitors, we chose Jun12682 as a chemical probe. We adopted two independent approaches to identify PLpro drug-resistant mutations: structure-based prediction and serial viral passage. We recently disclosed Jun12682 as the first-in-class PLpro inhibitor showing in vivo antiviral efficacy in the SARS-CoV-2 infection mouse model22. Jun12682 exploits a previously unknown drug-binding site, Val70Ub, and shows broad-spectrum antiviral activity against SARS-CoV-2 variants and nirmatrelvir-resistant strains22.
In the structure-based prediction approach, we expressed and purified high-frequency naturally occurring PLpro mutations at residues within 4 Å in the drug binding site. Mutations at D164, P248, and Y264 significantly impaired the enzymatic activity of PLpro, suggesting that they are unlikely to become predominant in circulating viruses. The high-frequency naturally occurring mutations at residues M208 (M208T/I/V) and P247 (P247Q/S/L) did not confer significant drug resistance (Ki increase ≤ 10-fold) and have comparable enzymatic activities as the WT. Therefore, mutations at M208 and P247 are also unlikely to lead to drug resistance in circulating viruses. On the contrary, eight naturally occurring PLpro mutants, including E167G/K/A/S/V, Y268H/N, and Q269H, displayed significant resistance against Jun12682 and PF-07957472 (Ki increase > 10-fold) while maintaining similar enzymatic activities as the WT PLpro (kcat/Km < 10-fold change). These results suggest that SARS-CoV-2 variants encoding these PLpro mutations are likely resistant to Jun12682 and PF-07957472. In parallel, we conducted the serial viral passage experiment with infectious SARS-CoV-2 (USA-WA1/2020 strain) under escalating drug selection pressure of Jun12682 in cell culture, a traditional approach to predicting antiviral drug resistance. Interestingly, the serial viral passage approached similarly identified E167G and Q269H mutants. In addition, allosteric mutations V98L, L152F, and T168I were also identified. Although these allosteric mutations do not confer direct drug resistance, they enhance it when combined with E167G or Q269H. MD simulations and perturbative free energy calculations provided a molecular-level understanding of the drug resistance caused by PLpro mutations, showing reduced hydrogen bonding and π-π stacking interactions. Collectively, this study suggests that the PLpro E167, Y268, and Q269 are drug-resistance hot spots for PLpro inhibitors that bind to the BL2 loop and BL2 groove region. SARS-CoV-2 circulating strains with mutations at these three residues should be closely monitored.
E167 side chain carboxylate forms electrostatic interactions with the terminal ammonium in Jun12682 and PF-07957472 (Fig. 1). To circumvent drug resistance caused by E167 mutations, including E167G, analogs that do not form electrostatic interactions with E167 could be designed. Similarly, Q269 side chain amide NH2 forms a hydrogen bond with the oxygen linker in Jun12682. To bypass the drug resistance of Q269 mutations, analogs that do not contain para-substituents can be designed. However, these modifications might lead to reduced binding affinity and antiviral activity.
The FRET substrate we used in characterizing PLpro drug resistance is Dabcyl-FTLRGG/APTKV(Edans), which contains the viral polyprotein nsp2/3 cleavage site. PLpro also cleaves at the nsp1/2 (LNGG/A) and nsp3/4 (LKGG/K) junctions15. In addition, PLpro has deubiquitinase and deISGlyase activities16. The phenotypic drug resistance in the antiviral assay is a combined result of digesting nsp1/2, nsp2/3, nsp3/4, and ubiquitin and ISG15-conjugated substrates. To justify that the FRET substrate containing the nsp2/3 junction is a valid substrate for studying PLpro drug resistance, we further characterized the drug resistance of PLpro mutants E167G, Y268H, Q269H, L152F/Q269H, and E167G/Q269H using Ub-AMC and ISG15-AMC substrates. The results showed that all five PLpro mutants displayed significant drug resistance against Jun12682 and PF-07957472 (Ki increase > 10-fold), corroborating the results using the FRET substrate. Overall, the FRET substrate containing the nsp2/3 cleavage site is valid for studying drug resistance.
The physiological relevance of these PLpro mutations, as predicted by our in vitro findings, warrants further investigation in animal models should they arise in clinical contexts. Moreover, due to the limited sequence conservation of PLpro drug-binding residues across the coronavirus family36,37, Jun12682, PF-07957472, and GRL0617 and their derivatives generally lack broad-spectrum antiviral activity against other coronaviruses, such as MERS-CoV23. Consequently, our conclusions regarding drug-resistant mutations are specific to SARS-CoV-2 and its variants. Furthermore, this study focuses primarily on the in vivo lead candidates Jun12682 and PF-07957472, which exhibit similar binding poses within the BL2 loop and groove region. Therefore, the predicted resistance mutations might not apply to PLpro inhibitors targeting alternative sites, such as allosteric inhibitors38,39. Similarly, our structure-based prediction approach centers on residues within the drug-binding pocket, potentially overlooking allosteric mutations that could contribute to drug resistance.
Methods
Ethical statement
All procedures with live SARS-CoV-2 were performed in certified biosafety level 3 (BSL3) facilities at Oklahoma State University using biosafety protocols approved by the Institutional Biosafety Committee (IBC), which comprises scientists, biosafety and compliance experts, and members of the local community. All research personnel received rigorous biosafety, biosecurity, and BSL3 training before participating in experiments. Personal protective equipment, including scrubs, disposable overalls, shoe covers, double-layered gloves, and powered air-purifying respirators, were used. Biosecurity measures are built in the environment through building and security systems and are reinforced through required training programs, standing meetings, and emergency exercises. The researchers working with live viruses received the SARS-CoV-2 vaccines voluntarily before the study started. Finally, all researchers were medically cleared by the Oklahoma State University Occupational Health Program.
SARS-CoV-2 PLpro expression and purification
The SARS-CoV-2 PLpro gene fragment (ORF 1564–1876) form BetaCoV/Wuhan/WIV04/2019 was codon optimized for Escherichia expression and inserted in the pET28a (+) vector with N-terminus 6X-Histidine and SUMO tag by GenScript22. Mutants were generated by QuikChange II site-directed mutagenesis kit (Agilent) following the manufacturer’s directions. Sequencing primers for mutagenesis of recombinant SARS-CoV-2 PLpro include the forward primer (T7) TAATACGACTCACTATAGGG and the reverse primer (T7 terminator) GCTAGTTATTGCTCAGCGG. The expression vector was transformed to BL21 competent cells and was induced by 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) at 18 °C overnight for the expression of PLpro. The competent cell was then lysed by sonication on ice with 1 sec on and 1 s off in the lysis buffer (25 mM Tris, 750 mM NaCl, 0.5 mg/ml lysozyme, 0.02 mg/ml DNAse I, 0.5 mM PMSF, 10% glycerol, pH 7.6). The supernatant containing PLpro was separated by centrifuge and incubated with nickel chelating resin (Biosciences). PLpro was eluted by escalating imidazole in wash buffer (50 mM Tris, 150 mM NaCl, pH 7.6), and fractions with more than 95% homogeneity were collected for dialysis in dialysis buffer (50 mM Tris, 150 mM NaCl, 2 mM DTT, 10% glycerol, pH 7.6). The PLpro was incubated with 0.02 mg/ml SUMO protease I at 30 °C for 1 h, followed by incubation with nickel chelating resin at 4 °C to remove 6X-Histidine-SUMO tag and SUMO protease I. The purity of PLpro was confirmed by SDS-PAGE. The PLpro was fast-frozen by liquid nitrogen and stored at -80 °C.
FRET-based enzymatic assay
The FRET enzymatic assays were performed in the reaction buffer (50 mM HEPES, 5 mM DTT, 0.01% TritonX-100, pH 7.5). The FRET substrate we used in the enzymatic assay is Dabcyl-FTLRGG/APTKV(Edans), which contains the viral polyprotein nsp2/3 cleavage site. The Km and Vmax measurements were performed using optimized PLpro concentrations. PLpro WT and mutant proteins were titrated against various FRET substrates ranging from 0.78-100 µM. The signal of digested FRET substrate at excitation of 360/40 nm and emission of 460/40 nm was detected using BioTek Cytation 5 imaging reader (Agilent). The initial velocity was determined by linear regression of first 600 s of the reaction. The Km and Vmax were calculated by fitting the initial velocity against substrate concentrations with the classic Michaeli-Menten curve fitting (Y = Vmax*X/(Km + X), X = substrate concentration; Y = enzyme velocity) by GraphPad Prism 8. To determine drug sensitivity and resistance, Ki was measured by titrating various compound concentrations using optimized PLpro WT and mutant protein concentrations (The concentration gives at least 1 h of linear initial velocity) at the FRET substrate concentration of 20 µM. The initial velocity was determined by linear regression of the first 1 h of the reaction. The Ki was fitted by plotting the initial velocity against inhibitor concentrations using the Morrison equation for tight binding (Y = V0*(1 − ((((Et + X + (Ki*(1 + (S/Km)))) − (((Et + X + (Ki*(1 + (S/Km))))2̂) − 4*Et*X)0.5̂))/(2*Et))), X = inhibitor concentration; Y = enzyme velocity; Et = enzyme concentration; V0 = enzyme velocity in the absence of inhibitor). Jun12682 and PF-07957472 were synthesized according to the reported procedures22,23.
Ub-AMC and ISG15-AMC enzymatic assay
The enzymatic assays of SARS-CoV-2 PLpro mutants digesting rhUbiquitin-AMC (U-550, R&D Systems) and rhISG15-AMC (UL-553, R&D Systems) substrates were carried out in the reaction buffer containing 50 mM HEPES, 5 mM DTT, and 0.01% TritonX-100 with pH of 7.5. For the Km and Vmax measurement with Ubiquitin AMC substrate, PLpro mutant proteins were diluted in reaction buffer to the optimized concentrations (WT 0.05 µM, E167G 1 µM, Y268H 0.01 µM, Q269H 0.01 µM), and the reaction was initiated by addition of Ub-AMC substrate at series of concentrations (5.46, 2.73, 1.36, 0.68, 0.34, 0.17, 0.08, and 0.04 µM). For the Km and Vmax measurement with ISG15-AMC substrate, PLpro mutant proteins were diluted to the optimized concentrations (WT 0.2 nM, E167G 5 nM, Y268H 0.1 nM, Q269H 0.05 nM), and the reaction was initiated by addition of ISG15-AMC substrate at series of concentrations (0.64, 0.32, 0.16, 0.08, 0.04, 0.02, 0.01, and 0.005 µM). The reaction was monitored every 71 s in the Bio Tek Cytation 5 plate reader with an excitation of 360 nm and an emission of 460 nm. The initial velocity was calculated by linear regression of the first 300 s of kinetic progress curves. The Km and Vmax were determined by plotting the initial velocity against substrate concentrations by classic Michaelis- Menten curve fitting in GraphPad Prism 8.
For the Ki measurement with Ub-AMC substrate, PLpro mutant proteins were diluted in reaction buffer to the optimized concentrations (WT 0.05 µM, E167G 1.5 µM, Y268H 0.05 µM, Q269H 0.01 µM). The reaction was initiated by addition of Ub-AMC substrate to 0.1 µM with various concentrations of testing compounds. For the ISG15-AMC Ki measurements, PLpro mutant proteins were diluted to the optimized concentrations (WT 0.8 nM, E167G 20 nM, Y268H 0.3 nM, Q269H 0.8 nM). The reaction was initiated by addition of ISG15-AMC substrate to 0.05 µM with various concentrations of testing compounds. The reaction was monitored for 2 h, and the first 60 min of curve was plotted in linear regression to determine the initial velocity. The Ki was determined using the Morrison equation for tight binding by plotting initial velocity versus compound concentrations in the GraphPad Prism 8.
Differential scanning fluorimetry (DSF) assay
The thermal shift assay was performed in the reaction buffer (50 mM HEPES, 5 mM DTT, 0.01% TritonX-100, pH 7.5) using ThermoFisher QuantStudio 5 Real-Time PCR system. 4 µM of PLpro WT and mutant proteins were incubated with various concentrations of compounds at 30 °C for 30 min, followed by addition of 1× SUPRO orange dye (Thermo Scientific). The fluorescence was monitored under an escalating temperature of 0.05 °C/s from 25 °C to 95 °C. The melting temperature (Tm) was determined by Boltzmann model using Protein Thermal Shift Software v1.3. The Δ Tm was calculated by subtracting the Tm of DMSO incubated PLpro WT and mutant proteins. The reported Δ Tm values were the average of four replicates.
Cells and viruses
A Vero-E6 line expressing human angiotensin-converting enzyme 2 (hACE2) and human transmembrane protease, serine 2 (hTMPRSS2) (Vero-AT), obtained through NIH-BEI Resources (NR-54970) was grown in DMEM containing 10% FBS, 1% Pen/Strep, 1× non-essential aminal acids, and 10 μg/mL puromycin (InVivogen, ant-pr-1) to maintain the expression of hTMPRSS2 and hACE2. A Caco-2 line expressing hACE2 and hTMPRSS2 (Caco2-AT), a gift from Dr. Mohsan Saeed (Boston University), was propagated in DMEM containing 20% FBS, 1% Pen/Strep, 1× NEAA, 1.5 μg/mL puromycin, and 1 μg/mL blasticidin (InVivogen, ant-bl-05). These cell lines were tested negative for mycoplasma contamination.
The following SARS-CoV-2 strains were obtained through BEI Resources, NIAID, NIH: Washington strain 1 (WA1) (NR-52281), recombinant SARS-CoV-2 expressing nano-luciferase reporter (rSARS-CoV-2-nLuc) (NR-54003). These viruses were propagated once with Vero-AT cells to obtain large viral stocks and titrated with Vero-AT cells. These viruses were full genome sequenced using the ARTIC method40.
Generation of PLpro mutant viruses
To generate rSARS-CoV-2-nLuc reporter viruses carrying PLpro mutants (PLproE167G, PLproY268H, PLproQ269H, PLproE167G+Q269H, PLproL152F+Q269H), mutagenesis primers (Supplementary Table 4) designed using an online primer design tool NEBaseChanger (https://nebasechanger.neb.com/) were used to introduce corresponding nucleotide substitutions into the SARS-CoV-2 infectious cDNA plasmid subclones using the Q5 site-directed mutagenesis kit (NEB, E0554S). In addition, a plasmid subclone expressing the NanoLuc luciferase (nLuc) was constructed by replacing the orf7 gene with the coding sequence of NanoLuc luciferase amplified from the genome of rSARS-CoV-2-nLuc as previously described28. These recombinant plasmid clones were Sanger-sequenced to verify the mutations. Virus recovery was conducted as previously described4. Briefly, viral genomic cDNA fragments, including those encoding PLpro mutations and nLuc, were ligated in an equal molar ratio to assemble a full-length genomic cDNA with a T4 DNA ligase (NEB, M0202L). The ligated full-length cDNA and a plasmid encoding the SARS-CoV-2 N gene were used as templates for in vitro transcription using the T7 mMESSAGE mMACHINE T7 transcription kit (ThermoFisher, AM1344). The transcribed viral genomic and N gene RNAs were subsequently electroporated into Vero-AT cells. These cells were maintained in DMEM containing 2% FBS at 37 °C. Culture supernatants containing infectious viral particles were collected when over 70% of cells showed cytopathic effects. All harvested viral stocks were titrated in Vero-AT cells and subjected to full-genome sequencing using the ARTIC method40. These recombinant SARS-CoV-2 PLpro mutant viruses are available upon request through material transfer agreements.
Virus titration
A plaque assay was used to titrate viral stocks. Vero-AT cells were seeded in 6-well plates at 6 × 105 cells per well and incubated overnight. Virus aliquots stored at −80 °C were thawed, and 100 µL of each virus sample was used to prepare a 10-fold serial dilution. Before virus inoculation, the cells were washed three times with phosphate-buffered saline (PBS), and 300 µL of each viral dilution was added per well in duplicates. The plates were incubated at 37 °C for virus adsorption. After 1 h of incubation, the viral inoculum was removed, and a 2 mL overlay (a 1:1 mixture of 2 × DMEM containing 4% FBS and 1.2% agarose [Fisher, M26944]) was added to each well. After 48 h of incubation at 37 °C, cells were fixed with 4% formaldehyde for 30 min and stained with 0.25% crystal violet solution. Plaques were counted, and average titers were calculated for each virus.
Viral growth kinetics assay
Vero-AT cells at a density of 4 × 104 cells per well were mixed with wild-type rSARS-CoV-2-nLuc or recombinant PLpro mutant viruses at an MOI of 0.001 and seeded into 96-well plates with 100 µL of DMEM containing 2% FBS per well. At 2, 12, 24, 30, and 36 h post-infection (HPI), infected cells were subjected to a Nano-Glo Luciferase assay (Promega, G9243) on a Promega GloMax Discover microplate reader (Promega, GM3000), following the manufacturer’s instructions.
Antiviral assays with infectious SARS-CoV-2
An antiviral plaque assay and a SARS-CoV-2 reporter virus assay were employed for antiviral effect evaluation22. Vero-AT cells were seeded into 12-well plates at a density of 3 × 105 cells per well and incubated at 37 °C overnight. PLpro inhibitors were prepared in 3-fold serial dilutions in 2×DMEM. Cells were infected with rSARS-CoV-2-nLuc or PLpro mutants at 50 plaque-forming units (PFU) per well. After 1-h incubation, the inoculum was removed and replaced with 1 mL 2× DMEM-1.2% Avicel (FMC polymers) mixture containing 2 µM P-glycoprotein inhibitor (CP-100356) and serially diluted PLpro inhibitors (10 µM starting concentration for rSARS-CoV-2-nLuc, 30 µM for PLpro mutants). The plates were incubated at 37 °C for 48 h, after which the overlay was removed. Cells were fixed with 4% formaldehyde for 30 min and then stained with 0.25% crystal violet solution for 15 min. After staining, the plates were rinsed with tap water and photographed. The area of virus-infected cells was then quantified using ImageJ.
For the reporter virus assay, Caco2-AT cells at a density of 2.5 × 104 cells/well were batch inoculated with icSARS-CoV-2-nLuc at a MOI of 0.2 and then added to 96-well plates with 50 μL per well of DMEM containing 4% FBS and 4 μM CP-100356. The test compounds were 3-fold serially diluted in DMEM to prepare compound solutions. After dilution, 50 μL diluted compound solution was transferred and mixed (1:1 volume ratio) with the cell-virus mixture pre-seeded in 96-well plates. Cells were incubated for 24 h at 37 °C with 5% CO2 and then subjected to a Nano-Glo Luciferase assay (Promega, G9243) on a Promega GloMax Discover microplate reader (Promega, GM3000) following the manufacturer’s instructions. The raw data were analyzed with Prism 10 using the non-linear regression curve fit method.
In vitro selection for SARS-CoV-2 resistance to Jun12682
We designed the virus passage experiment by propagating SARS-CoV-2 (USA-WA1/2020 strain) in the presence of increasing concentrations of Jun12682. Starting with 1 µM (2× EC50 of Jun12682 in Vero cells), the virus was passaged 12 times, and the inhibitor concentration was doubled every two passages until reaching 32 µM, where minimal cytotoxicity was observed. Between passages, the infected cells were incubated for sufficient time (typically within 48 h) until evident cytopathic effects were observed. Then the culture supernatant was harvested and used to infect fresh cells for the next passage. This passaging scheme allows the virus to acquire genomic mutations rapidly that confer resistance6,13. To initiate passaging, Vero-AT cells were seeded into 12-well plates at a density of 3 × 105 cells per well a day before infection. Viral inoculum prepared with DMEM containing SARS-CoV-2 (0.01 PFU per cell), 2% FBS, Jun12682, and 2 µM P-glycoprotein inhibitor (CP-100356) were added to wells in triplicates and passaged independently (designated as lineage1, 2 and 3 for replicates). When 30–50% of the cells exhibited viral-specific CPE, the culture supernatant was harvested (designated P1) and used as the viral seed to prepare the next passage of the virus.
To identify PLpro mutations that arose during the virus passaging, 200 µL culture supernatants of passages 2, 4, 6, 8, 10, and 12 of each lineage were used for RNA extraction using the Trizol Reagent (Thermo Fisher, 15-596-026) following the manufacturer’s instructions. Reverse transcription was performed using LunaScript RT SuperMix (New England Biolabs, E3010L) following the manufacturing instructions. The PLpro coding sequence was amplified by Q5® Hot Start High-Fidelity 2X Master Mix (New England Biolabs, M0494L) using primers (PLP-Fwd: 5′-ATCCTACCACATTCCACC-3′, and PLP-Rev: 5′-ACAAACAACACCATCCAA-3′). The PCR reaction steps of cycling follow: 98 °C for 30 s, 35 cycles of 98 °C for 10 s, 49 °C for 20 s, 72 °C for 30 s, and final extension at 72 °C for 2 min. PCR products were purified by Wizard SV Gel and PCR clean kit (Promega, A9282) and then sequenced by the DNA Protein Core Facility of Oklahoma State University. Sequencing results were analyzed using Snapgene software. The proportions of each nucleotide substitution of PLpro were calculated based on the peak value of the fluorescent signal on the chromatogram. To confirm the PLpro mutations revealed by Sanger sequencing and determine if additional mutations occur in the PLpro cleavage sites (Nsp1/2, Nsp2/3, and Nsp3/4 junctions), we performed the ARTIC-amplicon NGS to sequence the whole genome of the passage 12 (P12) viruses from each lineage. Bioinformatic tool iVar was used to reveal the frequencies of genomic mutations41.
Virus competition experiment
Vero-AT cells were seeded into 12-well plates at a density of 3 × 105 cells per well. The wild-type WA1 and the Lineage 3 P11 virus were mixed at a 1:1 PFU ratio and added to the cells at 0.01 MOI. Infected cells were cultured in DMEM containing 2% FBS, with or without 32 µM Jun12682 and 2 µM P-glycoprotein inhibitor. The mixed viruses were passed four times. For each passage, RNA was extracted from the supernatant and subjected to RT-PCR and sequencing of the PLpro region. The proportions of each nucleotide substitution of PLpro were calculated based on the peak value of the fluorescent signal on the chromatogram. We also performed the ARTIC-amplicon NGS to sequence the whole genome and obtain PLpro mutation frequencies of the passage 4 viruses from each competition experiment as described above.
MD simulations
Protein preparation
The protein structure PDB ID 8UOB22 was prepared using the Protein Preparation Wizard in Maestro. (Schrödinger Release 2021-2: Maestro, Schrödinger, LLC, New York, NY, 2021). The N- and C-termini were capped with acetyl and methylamino groups, respectively, and the pH was adjusted to 7 ± 0.5. The protein-ligand complex was minimized utilizing the OPLS4 force field42. Mutations were introduced by substituting the relevant amino acids using the Builder panel in Maestro, followed by a new energy minimization using OPLS442 force field to ensure optimal spacing. The specific studied mutations include E167G, Y268Y, Q269L, Q269H, and E167G/Q269H.
System setup
To perform MD simulations on the five complexes (including the wild type), we generated a simulation box with water molecules as the solvent and added Na+ and Cl- ions. The water box was created using the System Builder panel in Maestro (Schrödinger Release 2021-2: Maestro, Schrödinger, LLC, New York, NY, 2021), employing the TIP3P water model43. Na+ and Cl- ions were added to neutralize the system and achieve a concentration of 0.150 M. The orthorhombic periodic box was set 12 Å away from the protein. The total number of atoms of the complex was ~45,000, and the simulation box dimensions were (67 × 10 × 100.5 Å3). These systems were exported as PDB files for further processing with Amber 22 program34. Using antechamber44 and prmchk2, we created parameters for the ligand molecule with the GAFF245. We then initialized tleap and, using the ff19sb33 along with the TIP3P water and ions force field43 and parameters, generated the necessary starting structure files for MD simulations with Amber22 software34.
MD simulation protocol
Prior to the production run, a brief equilibration phase was conducted. First, energy minimization was performed using 1000 steps with the steepest descent method46 followed by 500 steps with the conjugate gradient method, applying restraints on protein atoms at 100 kcal/mol · Ų.
Next, the system underwent a gradual heating process, starting from 100 K and reaching 298 K over 1 ns at constant volume (NVT), using the Langevin thermostat (dynamics) for temperature control, as implemented in Amber22 software34 employing a Langevin collision frequency of 2.0 ps and a friction coefficient constant at 1 ps−1, with the same restraints as the minimization step.
The second step involved maintaining the system at 298 K for 1 ns at constant pressure in a NPTγ (with γ = 10 dyn cm1) simulation with Berendsen barostat47 to allow the box density to stabilize, again using the same restraints.
In the third NPTγ equilibration step, the restraints were reduced to 10 kcal/mol · Ų. This was followed by two additional relaxation steps, each lasting 1 ns, with restraints further reduced to 1 kcal/mol · Ų and 0.1 kcal/mol · Ų, respectively. In each NPTγ equilibration step for the pressure control the Berendsen barostat was used to adjust the density over the simulation time, with a target pressure of 1 bar and a 2 ps pressure relaxation time47.
Bonds involving hydrogen atoms were constrained by the SHAKE algorithm and a time step of 1 fs was used for the integration of the equations of motion for the first 2 NVT and the first NPT equilibration steps and for the rest of the NPT steps the time step was set at 2 fs with the leapfrog Verlet integrator48,49. Long-range electrostatics were calculated using Particle-mesh Ewald summation (PME) with a 1 Å grid, and short-range non-bonding interactions were truncated at 12 Å with a continuum model long-range correction applied for energy and pressure.
The equilibration phase was followed by production MD simulation for 500 ns Snapshots were recorded every 100 ps during the production phase. Within this simulation time, the RMSD plots reached a plateau, and the systems were considered equilibrated and suitable for statistical analysis. Two MD simulation repeats were performed for each complex using the same starting structure and applying randomized velocities.
Particle Mesh Ewald Molecular Dynamics (pmemd) is the primary engine for running MD simulations with AMBER22 software. The energy minimization step was performed using the Central Processing Unit (CPU) of the workstations by implementing pmemd. The rest of the equilibration steps including the unrestraint production were run with Amber22 software34 on RTX 4090 GPUs in lab workstations using pmemd.CUDA algorithm34,50,51. The pmemd.CUDA executable provides the ability to use NVIDIA GPUs to run the MD simulations.
MD analysis
The visualization of the MD simulation trajectories was performed using VMD52. The analysis of all the MD simulations trajectories was performed by ptraj and cpptraj of AmberTools2234,53. To analyze the output trajectory files, we employed a series of custom Python scripts utilizing various libraries including MDAnalysis54, RDKit, Pandas, Matplotlib, and Threading. The images were generated using PyMOL (The PyMOL Molecular Graphics System, Version 3.0 Schrödinger, LLC.) from Schrödinger.
For hydrogen bond interactions distance = 2.5 Å between donor and acceptor heavy atoms, and an angle ≥120° between donor-hydrogen-acceptor atoms and ≥90° between hydrogen-acceptor-bonded atoms were considered. Non-specific hydrophobic contacts were measured if the residue fell within 3.8 Å from a ligand’s aromatic or aliphatic carbon, while π-π interactions were measured if two aromatic groups are stacked face-to-face or face-to-edge. Water-mediated interactions were measured if the distance between donor and acceptor atoms is 2.7 Å, the angle between donor-hydrogen-acceptor atoms is ≥110° and the angle between hydrogen-acceptor-bonded atoms is ≥80°.
Alchemical binding free energies calculated with MBAR method
TI/MD method’s principle
The TI/MD method has been described55. Free energy is a state function, and thus the free energy difference between states is independent of the path that connects them. To compare ligand’s binding for two protein mutants 0 and 1 the calculation of \({\Delta A}_{{{\rm{b}}},1}\) and \({\Delta A}_{{{\rm{b}}},0}\), respectively, is needed and then the difference \({\Delta \Delta A}_{{{\rm{b}}},0\to 1}\) (b) or \({\Delta \Delta A}_{{{\rm{b}}},{\mathrm{0,1}}}\) (b). The calculation of \({\Delta A}_{1}\left({{\rm{b}}}\right)\) and \({\Delta A}_{0}\left({{\rm{b}}}\right)\) is demanded because it includes large changes between the two states. Thus, the calculation of the relative binding free energies for Jun12682 bound to proteins PLpro WT and PLpro mutant (Table 2) can be performed instead using the MBAR method56 and applying a thermodynamic cycle (Supplementary Fig. 3), i.e., using the ΔG values obtained for the transformations of the proteins in the bound (b) and the solvent (s; water) state ΔGb,0,1 and ΔGs,0,1(s), respectively, according to Eq. (1) (Supplementary Fig. 3).
Using this method, we can calculate the difference between \({\Delta A}_{{{\rm{b}}},{\mathrm{0,1}}}\) \({{\rm{and}}}\) \({\Delta A}_{{{\rm{s}}},{\mathrm{0,1}}}\) which corresponds to the unphysical alchemical transformation 0 → 1 in the bound and in the water state known as alchemical transformation which may be chosen to include small change or perturbation of ligand structure to lower the error for the free energy perturbation calculation \({\Delta A}_{{{\rm{b}}},{\mathrm{0,1}}}{{\rm{or}}}\) \({\Delta A}_{{{\rm{s}}},{\mathrm{0,1}}}\).
Because the phase space overlap between two states 0, 1 of interest can be near zero, doing free energy calculations for the two states alone will often have large errors. Free energy is a state function, we can construct a thermodynamic path that takes us through a set of states that improves phase space overlap between states that can be unphysical. By this, we mean that our intermediate states do not have to be observable experimentally. To put this mathematically, we can improve our results by constructing high-phase space overlap intermediates and calculating our free energy difference \({\Delta \Delta A}_{{{\rm{b}}},0\to 1}\) by the sum of the binding free energy differences between the intermediate states.
Briefly, a thermodynamic parameter λ that smoothly connects states 0 and 1 through a λ-dependent potential U(rN; λ), such that U(rN; 0) = U0(rN) and U(rN; 1) = U1(rN). The transformation is broken down into a series of M steps corresponding to a set of λ values λ1, λ2,…, λM ranging from 0 to 1, such that there is sufficient phase space overlap between neighboring intermediate λ states. The TI estimator computes the free energy change of transformation 0 → 1 by integrating the Boltzmann averaged dU(λ)/dλ as is described in Eq. (1)
where the second sum indicates numerical integration over M quadrature points (λk, for k = 1,…, M) with associated weights wk. A linear extrapolation between states can be applied for the construction of U1(rN; λ) while with Amber18 softcore potentials57,58,59 the Lennard-Jones and Coulomb term potentials are described according to Eq. (2)
MBAR56 calculates the free energy difference between neighboring intermediate states using Eq. (3)
where w is a function of Α(λ) and Α(λ + 1). The equation is solved iteratively to give the free energy change of neighboring states ΔΑ(λ → λ + 1), which via combination yield the overall free energy change. MBAR method has been shown to minimize the variance in the calculated free energies, by making more efficient use of the simulation data56,60,61,62.
TI/MD calculations protocol
For the TI/MD calculations, the experimental structure of the protein – ligand with the solvent around, was used as starting structure for the alchemical transformations between amino acids. For the ligand their protonated form assumed at pH 8 was used.
Setups were performed by using the Amber22 software34. To start the process, we extracted the protein and ligand from the solvent box into a new PDB file. Then, by initializing pdb4amber module we mutated the necessary amino acid to create a new PDB file for each protein mutation. Afterwards we processed the mutant, and non-mutant protein—ligand PDB files to extract the protein only into a new PDB file. By using the tleap module of Amber we merged the protein – ligand and protein only PDB files (mutants and non-mutant) into a prmtop file (now the prmtop file has both protein and protein-mutant or complex and complex mutant). Then by using parmed module and the tiMerge command we were able to merge these 2 different proteins (mutant and non-mutant) into one copy of the duplicated protein and the mutate residue correctly linked to the protein. For the protein force field, we used the amber14sb force field (ff14sb)35 for the ligand we used GAFF263, for the water and ions we used the TIP3P model43.
The initial geometries were minimized using 20,000 steps of steepest descent minimization at λ = 0.5. These minimized geometries were used for simulations at all λ values. Eleven λ values were applied, equally spaced between 0.0 and 1.0. In each MD simulation the system was heated to 298 K for 500 ps using the Langevin64 thermostat (dynamics) for temperature control, as implemented in Amber22 software, employing a Langevin collision frequency of 2.0 ps−1 in the presence of harmonic restraint with force constant 10 kcal mol−1 Å−2 on all membrane, protein, and ligand atoms. The Berendsen barostat47 was used to adjust the density over the 500 ps of equilibration at constant pressure (NPTγ) simulation using γ = 10 dyn cm−1 with a target pressure of 1 bar and a 2 ps coupling time. Then, a 500 ps of NVT equilibration MD simulation was applied following by 2 ns NVT production MD simulation without restraints. Energies were recorded every 1 ps and coordinates were saved every 10 ps. Production simulations recalculated the potential energy at each λ value every 1 ps for later analysis with MBAR56,60.
The bond constraint SHAKE algorithm48 was disabled for the TI calculations performed with AMBER GPU-TI module pmemdGTI65, and therefore a time step of 1 fs was used for all MD simulations. Long range electrostatics were calculated using the PME method66,67 with a 1 Å grid, and short-range non-bonding interactions were truncated at 12 Å with a continuum model long range correction applied for energy and pressure.
For each alchemical calculation, dual topology was applied, that does not convert one species to another, but only converts between dummy atoms and an interacting species, but usually uses softcore potentials35,59 The 1-step protocol was applied where in the softcore potential29,35,38,59 the electrostatic and van der Waals interactions are scaled simultaneously.
The final states 0 and 1 of the alchemical calculations 0 → 1 or 1 → 0, complexes as resulted from the alchemical transformations were compared with these complexes structure resulted from the converged 500-ns MD simulations. This was performed to certify that the 2 ns-MD simulation for each λ-state during the alchemical calculations was enough for the complexes to converge to same structure with 500 ns-MD simulations. Two repeats of the TI/MD calculation for each alchemical transformation were performed and the mean values are shown in Table 2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw NGS data of the passaged viruses has been deposited in NCBI SRA under accession code PRJNA1244330. All other data are available in the main text or the supplementary materials. The publicly available dataset used in this study is the crystal structures of SARS-CoV-2 in complex with Jun12682 and PF-07957472 with accession codes PDB ID 8UOB and 9CSY, respectively. The following link is provided to access output frames of the complexes between Jun12682 and Mpro proteins from the MD simulations: GitHub - georgioukyriakos/SARS-CoV-2-PLpro. Source data are provided with this paper.
References
Townsend, J. P. et al. Seasonality of endemic COVID-19. mBio 14, e0142623 (2023).
Telenti, A. et al. After the pandemic: perspectives on the future trajectory of COVID-19. Nature 596, 495–504 (2021).
Li, G., Hilgenfeld, R., Whitley, R. & De Clercq, E. Therapeutic strategies for COVID-19: progress and lessons learned. Nat. Rev. Drug Discov. 22, 449–475 (2023).
Hu, Y. et al. Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. ACS Cent. Sci. 9, 1658–1669 (2023).
Tan, B., Joyce, R., Tan, H., Hu, Y. & Wang, J. SARS-CoV-2 main protease drug design, assay development, and drug resistance studies. Acc. Chem. Res. 56, 157–168 (2023).
Iketani, S. et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 613, 558–564 (2022).
Zhou, Y. et al. Nirmatrelvir-resistant SARS-CoV-2 variants with high fitness in an infectious cell culture system. Sci. Adv. 8, eadd7197 (2022).
Jochmans, D. et al. The substitutions L50F, E166A, and L167F in SARS-CoV-2 3CLpro are selected by a protease inhibitor in vitro and confer resistance to nirmatrelvir. mBio 14, e0281522 (2023).
Kiso, M. et al. In vitro and in vivo characterization of SARS-CoV-2 strains resistant to nirmatrelvir. Nat. Commun. 14, 3952 (2023).
Abdelnabi, R. et al. Nirmatrelvir-resistant SARS-CoV-2 is efficiently transmitted in female Syrian hamsters and retains partial susceptibility to treatment. Nat. Commun. 14, 2124 (2023).
Zuckerman, N. S., Bucris, E., Keidar-Friedman, D., Amsalem, M. & Brosh-Nissimov, T. Nirmatrelvir resistance—de novo E166V/L50V mutations in an immunocompromised patient treated with prolonged nirmatrelvir/ritonavir monotherapy leading to clinical and virological treatment failure - a case report. Clin. Infect. Dis. 78, 352–355 (2024).
Focosi, D., Maggi, F., McConnell, S. & Casadevall, A. Very low levels of remdesivir resistance in SARS-COV-2 genomes after 18 months of massive usage during the COVID19 pandemic: a GISAID exploratory analysis. Antivir. Res. 198, 105247 (2022).
Stevens, L. J. et al. Mutations in the SARS-CoV-2 RNA-dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 14, eabo0718 (2022).
Chen, S. A. et al. SARS-CoV-2 M(pro) protease variants of concern display altered viral substrate and cell host target galectin-8 processing but retain sensitivity toward antivirals. ACS Cent. Sci. 9, 696–708 (2023).
Rut, W. et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: a framework for anti-COVID-19 drug design. Sci. Adv. 6, eabd4596 (2020).
Shin, D. et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587, 657–662 (2020).
Wydorski, P. M. et al. Dual domain recognition determines SARS-CoV-2 PLpro selectivity for human ISG15 and K48-linked di-ubiquitin. Nat. Commun. 14, 2366 (2023).
Tan, H., Hu, Y., Jadhav, P., Tan, B. & Wang, J. Progress and challenges in targeting the SARS-CoV-2 papain-like protease. J. Med. Chem. 65, 7561–7580 (2022).
Ghosh, A. K., Mishevich, J. L., Mesecar, A. & Mitsuya, H. Recent drug development and medicinal chemistry approaches for the treatment of SARS-CoV-2 infection and COVID-19. ChemMedChem 17, e202200440 (2022).
Sanders, B. C. et al. Potent and selective covalent inhibition of the papain-like protease from SARS-CoV-2. Nat. Commun. 14, 1733 (2023).
Shen, Z. et al. Design of SARS-CoV-2 PLpro inhibitors for COVID-19 antiviral therapy leveraging binding cooperativity. J. Med. Chem. 65, 2940–2955 (2022).
Tan, B. et al. Design of a SARS-CoV-2 papain-like protease inhibitor with antiviral efficacy in a mouse model. Science 383, 1434–1440 (2024).
Garnsey, M. R. et al. Discovery of SARS-CoV-2 papain-like protease (PL(pro)) inhibitors with efficacy in a murine infection model. Sci. Adv. 10, eado4288 (2024).
Lu, Y. et al. Discovery of orally bioavailable SARS-CoV-2 papain-like protease inhibitor as a potential treatment for COVID-19. Nat. Commun. 15, 10169 (2024).
Matthew, A. N. et al. Drug design strategies to avoid resistance in direct-acting antivirals and beyond. Chem. Rev. 121, 3238–3270 (2021).
Aiewsakun, P. et al. Systematic exploration of SARS-CoV-2 adaptation to vero E6, Vero E6/TMPRSS2, and Calu-3 cells. Genome Biol. Evol. 15, evad035 (2023).
Carrascosa-Sàez, M. et al. Cell type-specific adaptation of the SARS-CoV-2 spike. Virus Evol. 10, veae032 (2024).
Hou, Y. J. et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 182, 429–446 e414 (2020).
Hui, K. P. Y. et al. Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: an analysis in ex-vivo and in-vitro cultures. Lancet Respir. Med. 8, 687–695 (2020).
Chu, H. et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: an observational study. Lancet Microbe 1, e14–e23 (2020).
Sacco, M. D. et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci. Adv. 6, eabe0751 (2020).
Ma, C. et al. Discovery of Di- and trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. 143, 20697–20709 (2021).
Tian, C. et al. ff19SB: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 16, 528–552 (2020).
Case, D. A. et al. The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 (2005).
Maier, J. A. et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Baez-Santos, Y. M., St John, S. E. & Mesecar, A. D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir. Res. 115, 21–38 (2015).
Lee, H. et al. Inhibitor recognition specificity of MERS-CoV papain-like protease may differ from that of SARS-CoV. ACS Chem. Biol. 10, 1456–1465 (2015).
Taylor, A. J. et al. Fragment-based screen of SARS-CoV-2 papain-like protease (PL(pro)). ACS Med. Chem. Lett. 15, 1351–1357 (2024).
Srinivasan, V. et al. Antiviral activity of natural phenolic compounds in complex at an allosteric site of SARS-CoV-2 papain-like protease. Commun. Biol. 5, 805 (2022).
Deng, X. et al. Breakthrough infections with multiple lineages of SARS-CoV-2 variants reveals continued risk of severe disease in immunosuppressed patients. Viruses 13, 1743 (2021).
Grubaugh, N. D. et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 20, 8 (2019).
Lu, C. et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 17, 4291–4300 (2021).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 25, 247–260 (2006).
Mobley, D. L. et al. Escaping atom types in force fields using direct chemical perception. J. Chem. Theory Comput. 14, 6076–6092 (2018).
Haug, E. J., Arora, J. S. & Matsui, K. A steepest-descent method for optimization of mechanical systems. J. Optim. Theory Appl. 19, 401–424 (1976).
Berendsen, H. J. C., Postma, J. P. M., Vangunsteren, W. F., Dinola, A. & Haak, J. R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Ryckaert, J.-P., Ciccotti, G. & Berendsen, H. J. C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Verlet, L. Computer “experiments” on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 159, 98–103 (1967).
Götz, A. W. et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J. Chem. Theory Comput. 8, 1542–1555 (2012).
Salomon-Ferrer, R. et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888 (2013).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33-38–27-38 (1996).
Roe, D. R. & Cheatham, T. E. 3rd. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013).
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B. & Beckstein, O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327 (2011).
Kirkwood, J. G. Statistical mechanics of fluid mixtures. J. Chem. Phys. 3, 300–313 (1935).
Procacci, P. Multiple Bennett acceptance ratio made easy for replica exchange simulations. J. Chem. Phys. 139, 124105 (2013).
Song, L. F., Lee, T. S., Zhu, C., York, D. M. & Merz, K. M. Using AMBER18 for relative free energy calculations. J. Chem. Inf. Model 59, 3128–3135 (2019).
Lee, T. S. et al. Alchemical binding free energy calculations in AMBER20: advances and best practices for drug discovery. J. Chem. Inf. Model 60, 5595–5623 (2020).
Steinbrecher, T., Joung, I. & Case, D. A. Soft-core potentials in thermodynamic integration: comparing one- and two-step transformations. J. Comput. Chem. 32, 3253–3263 (2011).
Shirts, M. R. & Pande, V. S. Comparison of efficiency and bias of free energies computed by exponential averaging, the Bennett acceptance ratio, and thermodynamic integration. J. Chem. Phys. 122, 144107 (2005).
Paliwal, H. & Shirts, M. R. A benchmark test set for alchemical free energy transformations and its use to quantify error in common free energy methods. J. Chem. Theory Comput. 7, 4115–4134 (2011).
Tan, Z., Gallicchio, E., Lapelosa, M. & Levy, R. M. Theory of binless multi-state free energy estimation with applications to protein-ligand binding. J. Chem. Phys. 136, 144102 (2012).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
Izaguirre, J. A., Catarello, D. P., Wozniak, J. M. & Skeel, R. D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 114, 2090–2098 (2001).
Lee, T. S., Hu, Y., Sherborne, B., Guo, Z. & York, D. M. Toward fast and accurate binding affinity prediction with pmemdGTI: an efficient implementation of GPU-accelerated thermodynamic integration. J. Chem. Theory Comput. 13, 3077–3084 (2017).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Acknowledgements
J.W. is funded by National Institutes of Health grants R01AI158775 and Rutgers HealthAdvance funding. Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number U01HL150852. X.D. is funded by National Institutes of Health grant R01AI158775 and Rutgers HealthAdvance fund. A.K. acknowledges the support of Chiesi Hellas (SERG No 10354). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
J.W., X.D., and H.T. conceived and supervised the research and designed the experiments; H.T. and K.L. performed gene expression and protein purification; H.T. performed enzymatic inhibition and DSF assays; Q.Z. and S.Z. performed the serial viral passage and antiviral assay with recombinant viruses; K.G., G.L., and A.K. performed the MD simulations; J.W., Q.Z., K.G., S.Z., K.L., G.L., A.K., and X.D. analyzed and discussed the data; and J.W., X.D., A.K., and H.T. wrote the manuscript with the assistance of Q.Z., K.G., S.Z., K.L., and G.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tan, H., Zhang, Q., Georgiou, K. et al. Identification of naturally occurring drug-resistant mutations of SARS-CoV-2 papain-like protease. Nat Commun 16, 4548 (2025). https://doi.org/10.1038/s41467-025-59922-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59922-9
