
Abstract
Targeting abnormal dysregulation of adenosine-to-inosine deamination by ADAR enzymes offers a promising therapeutic strategy in cancer research. However, the development of effective inhibitors is impeded by the incomplete structural information on ADAR1 complexes. In this study, we employ a combination of computational 3D modeling and high-speed atomic force microscopy to elucidate the atomic and molecular dynamics of ADAR1. Two distinct interface regions (IFx and IFy) on the surface of the deaminase domain and oligomerization structural models are identified. Single-molecule-level insights into the structural dynamics of ADAR1 reveal the oligomerization of ADAR1 monomers through the self-assembly of deaminase domains. In the presence of the substrate dsRNA, the N-terminal region, including RNA-binding domains, of ADAR1 dimer exhibits a controlled flexible conformation and promotes a stable dimeric interaction with dsRNA for RNA editing. These findings provide the basis for the development of targeted inhibitors to regulate ADAR1 activity in therapeutic applications.
Similar content being viewed by others


Introduction
Adenosine deaminases acting on RNA (ADARs) convert adenosines (A) into inosines (I) in double-stranded RNA (dsRNA)1,2,3. Inosines are perceived as guanosines by cellular machinery; hence, editing can result in changes in protein-coding sequences, alternative splicing, alterations in miRNA processing, and modulation of dsRNA stability4,5,6,7. In mammals, A-to-I RNA editing by two different enzymes, ADAR1 (p150:1226 aa; p110:931 aa) and ADAR2 (741 aa), plays distinct roles in cancer progression. ADAR1 is frequently overexpressed with elevated activity in many cancer types, including hepatocellular carcinoma, breast cancer, esophageal cancer, prostate cancer, and multiple myeloma7,8,9,10,11,12. In addition, recent studies have shown that the loss of ADAR1 function can inhibit cancer cell proliferation, induce apoptosis, and sensitize certain types of tumors to immunotherapy, suggesting ADAR1 as a potential cancer therapeutic target13,14,15.
The structural and RNA-binding properties of ADAR2 have been extensively studied16,17,18, whereas little information is available on the structural features of ADAR119,20,21. This disparity is primarily due to ADAR1’s large size and complexity, which includes multiple domains and regions that interact with RNA or other proteins, making it technically challenging to determine its complete structure. However, indirect observation methods, including co-immunoprecipitation (co-IP), size fractionation, and techniques such as fluorescence or bioluminescence resonance energy transfer (FRET), have demonstrated ADAR1 interactions with RNA and other RNA-processing enzymes such as Dicer22,23,24. In addition, a recent study has described the structural features of the ADAR1 deaminase domain in its specific selectivity of dsRNA over ADAR2 through high-throughput mutagenesis studies of purified mutant ADAR1 proteins and Rosetta-based molecular modeling25, However, the comparative RNA substrate binding selectivity of ADAR1 and ADAR2 for editing is influenced by structural perturbations in the substrate RNA26. Another study highlighted the structural roles of the ADAR1-third double-stranded RNA binding domain (ADAR1-dsRBD3) in dimerization and RNA editing27. Despite these studies, our understanding of ADAR1 structural dynamics in the presence and absence of substrate RNA remains limited. ADAR1 adopts multiple conformations and undergoes conformational changes during its function, posing challenges for conventional structural biology techniques to capture these dynamics. Thus, a comprehensive approach is essential to advance our understanding of the dynamic features of ADAR1, both alone and in complex with its substrate RNA. In this context, we propose a combined approach utilizing 3D modeling and high-speed atomic force microscopy (HS-AFM) to elucidate the dynamic features of the ADAR1 dimer and its interaction with substrate RNA.
3D modeling offers a suitable alternative for facilitating protein structure prediction in the absence of experimental 3D structures28,29. AlphaFold has also emerged as a tool for predicting protein structure30. Methods such as template-based modeling (TBM), which uses known structures of related proteins, provides reliable predictions31. Despite their utility, traditional 3D models often overlook the flexibility of protein structures, which is a vital aspect affecting their functionality32,33. Although the experimental structures determined by Nuclear magnetic resonance, X-ray crystallography, and cryo-electron microscopy represent an average conformation, average conformations are not accurate enough to justify their use in refinement approaches34,35. This limitation suggests the need for complementary techniques, such as HS-AFM, which have the potential to capture dynamic molecular conformations and interactions in detail36,37,38,39,40. HS-AFM, an advanced imaging technology, enables real-time observation of macromolecules in dynamic states and interaction modes. Its applications span diverse biological systems, from elucidating protein dynamics to observing molecular machinery in action36,37. Notably, HS-AFM has unraveled the mechanisms of transcription factor dimerization at the single-molecule level and provided insights into the behavior of CRISPR-Cas9 during DNA target search38,39,40.
In this study, structural models of ADAR1 oligomerization were constructed via interface regions on deaminase domains using 3D modeling and constrained by the results of real-time HS-AFM observations. We also suggest that two ADAR1 monomers initiate interaction via dsRBDs-dsRBDs, leading to dimer stabilization via interface regions on deaminase domains. Our study demonstrates the potential of integrating computational and HS-AFM approaches to address the challenges posed by conformational dynamics, particularly in contexts with limited structural information. These insights provide future directions for the development of strategies to inhibit ADAR1 in cancer treatment.
Results
Structural characterization of ADAR1 oligomerization using computational 3D modeling, simulated AFM, and HS-AFM
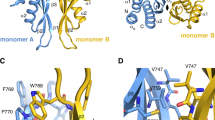
The ADAR1 short-length isoform (p110) contains a C-terminal catalytic center deaminase domain and three dsRNA binding domains (dsRBDs) with a Zβ domain in the N-terminal portion. ADAR1 full-length isoform (p150) also includes an N-terminal Zα domain in the N-terminal region (depicted in Fig. 1a). We investigated the protein-protein interface (PPI) regions within the ADAR1 deaminase domains and analyzed the oligomerization of ADAR1. The analysis was performed by constructing ADAR1 deaminase domain model structures from the AlphaFold2-predicted 3D structure of the ADAR1 monomer and superimposing the monomer onto the X-ray crystal structure of the ADAR2 dimer (Supplementary Fig. 1a, b). We predicted two distinct interface regions, designated as interface x (IFx: Pro826-Pro834; Asp877-Asp880; Ser932-Val955) and interface y (IFy: Gly1183-Asp1205), located on each deaminase domain, which were complementary and in contact with each other in a dimeric configuration (Fig. 1b). The following residues form intermolecular hydrogen bonds between the ADAR1 monomers (M1 and M2) in the dimer model: (M2: LYS1200: NZ – M1: ASP877: OD1; M1: LYS936: NZ – M2: SER1181: OG; M2: LYS1187: NZ – M1: LYS936: O; M2: ARG1190: NH1 – M1: PRO942: O) (Fig. 1b, maximized in inset). Based on these findings, we proposed oligomerization of the ADAR1 protein into trimer and tetramer 3D models (Fig. 1c). These models account for the interactions of multiple ADAR1 monomers across the identified dimerization interface regions, suggesting a higher-order organization of ADAR1 beyond the dimeric state. The results were almost identical to those of ADAR1 models constructed using another method, that is, homology modeling of the ADAR1 monomer using the experimental structure of ADAR2 as the template (Supplementary Fig. 2). Notably, the dimerization helix region of ADAR2, which is essential for its dimerization, is located near the dimerization interface regions in our 3D models (indicated in yellow in Supplementary Fig. 3a)17. This region directly interacts with the residues Pro826-Pro834, which are part of the IFx region, suggesting that this interaction may influence the dimer structure of ADAR1.

a Illustration of the domain arrangement of ADAR1 isoforms (p150:1226 aa and p110: 931 aa). b The ADAR1 dimer model predicted two distinct interfaces, named interface x (IFx) and interface y (IFy) on each deaminase domain. The IFx (Pro826-Pro834; Asp877-Asp880; Ser932-Val955) and IFy (Gly1183-Asp1205) shown in red and green colors, respectively, are complementary and contact each other. A total of 4 hydrogen bonds formed between four residue pairs in the IFx and IFy regions contribute to monomer-monomer interactions within the dimer model (inset, magnified). c Computational models of ADAR1 oligomerization highlight the complementary interactions between IFx and IFy across monomers, suggesting a mechanism for the higher-order organization of ADAR1.
In our analysis, we next focused on elucidating the binding dynamics of RNA to the ADAR1 dimer using docking simulations. The aryl hydrocarbon receptor (AhR) 3’UTR mRNA selected for docking contains two Alu elements (AluSx/Sc), which are known to be preferred target sites for ADAR141. Given the complex nature of the RNA backbone intertwining, we specifically modeled and optimized the 3D structure of the Alu element (AluSc) to facilitate accurate docking analysis. After modeling and optimization, we used the 3D structures of a 49-nucleotide double-stranded RNA (dsRNA) corresponding to the AluSc and ADAR1 dimer to predict RNA-ligand complex docking models. This prediction was based on docking energy calculations using the ZRANK functions. We selected the top ten based on cluster size and high ZDOCK score, from the generated docking models for further analysis. We illustrated the 3D model of the docked conformations of dsRNA, which exhibited a duplex conformation (Supplementary Fig. 4a–c). Docking results showed that the deaminase domain of the dimer bound RNA on one side of the duplex with several amino acids, establishing interactions with RNA nucleotides (Supplementary Fig. 4d). The magnified view shows that serine and arginine form hydrogen bonds with the targeted adenosine (A40) (Supplementary Fig. 4e).
To elucidate the structural dynamics of ADAR1 in the presence and absence of dsRNA at the single-molecule level, we employed HS-AFM imaging. A sample of ADAR1-p110 was deposited on a bare mica surface and imaged with HS-AFM. Intriguingly, the HS-AFM movie revealed the presence of ADAR1 molecules in various forms, including monomer, dimer, trimer, and tetramer structures, in an aqueous solution on the mica surface (Supplementary Fig. 5a, b). To illustrate the dynamics of ADAR1 deaminase domain-mediated oligomerization, we present selected frames captured at different times (Supplementary Fig. 5b). Additionally, we provided a movie (Supplementary Movie 1) showing the temporal evolution of ADAR1 oligomerization and dissociation events. These visual representations offer valuable insights into the dynamic behavior of ADAR1 monomers at the molecular level and highlight the intricate processes underlying their oligomeric assembly.
The analysis of our 3D model indicated that each deaminase domain possessed complementary x and y interfaces, potentially facilitating the formation of an oligomer chain-like structure (Fig. 1b). To assess the correlation between our generated 3D models of monomers, dimers, trimers, and tetramers with experimentally observed HS-AFM images of ADAR1, we conducted simulated AFM scanning (Fig. 2) using the BioAFMviewer software42. In this simulation, 3D model structures of the ADAR1 monomer, dimer, and trimer were converted into simulated AFM images and correlated with real HS-AFM images. As shown in Fig. 2, the simulated AFM images were in good agreement with the experimental HS-AFM images. This alignment between the simulated and observed HS-AFM images indicates that the experimentally observed ADAR1 deaminase domain dimerization, trimerization, and tetramerization corroborate the computationally predicted 3D model structures. This agreement supports the accuracy and reliability of the proposed 3D model structures and their representation of ADAR1 oligomeric assemblies.

Similarity comparison between the structure obtained by 3D modeling, pseudo-HS-AFM graphics, and real HS-AFM images of ADAR1 deaminase domain (monomer)-mediated dimerization, trimerization, and tetramerization in buffer solution on a mica surface.
Structural insights into ADAR1 flexibility in the absence of substrate dsRNA
We then focused on the single-molecule dynamics of the ADAR1 monomer. HS-AFM observations revealed that most ADAR1 monomers in the buffer exhibited a visible deaminase domain characterized by a globular conformation. Additionally, some monomers displayed a tail-like structure protruding from the main globular segment, potentially corresponding to the N-terminal domain region (three dsRBDs and Zβ domain) (Fig. 3a). However, a direct comparison of domain architectures and their assignment is hindered by the lack of a crystal structure for full-length ADAR1. To address this limitation, we depicted the domain arrangement of the ADAR1 monomer (Fig. 1a)43 to represent its proposed domain organization, facilitating the interpretation and analysis of the HS-AFM observations. Observations of the monomers are shown in supplementary movie 2 in the supplemental material, with selected frames from the movie shown in Fig. 3b. These frames depict the structural characteristics of the ADAR1 multifaceted domains represented in the illustrations. To further analyze the flexibility of the N-terminal domains relative to the deaminase domain, a cross-sectional analysis was conducted to illustrate the peak-to-peak distance fluctuation between the deaminase and the N-terminal domains (Fig. 3c). The analysis revealed that the protrusion of the N-terminal domain structure exhibited a high degree of flexibility and dynamic conformations, moving around the globular deaminase domain structure. This suggests that the N-terminal domains possess significant flexibility and dynamics that may be crucial for RNA recognition and capture. The height distribution of ADAR1 monomers acquired in the deaminase domain (N = 174) is presented in Fig. 3d, lower panel. The average height of the deaminase domain was measured at 6 ± 0.6 nm, consistent with the 5.98 nm height observed in the PDB structure using PYMOL software (https://pymol.org/) (Fig. 3d, upper panel). Based on size distributions, particles in the 5–6 nm range were identified as ADAR1 monomers by HS-AFM. These observations suggest that ADAR1 primarily exists as monomers in the samples analyzed. However, oligomeric forms of ADAR1 were also detected. The size distributions and structural features of these larger oligomeric forms suggest the presence of dimers, trimers, and tetramers. ADAR1 monomers and dimers have an average height of ~6 nm, with diameters ranging from 20 to 40 nm, corresponding to monomers ( ~ 20 nm) and dimers ( ~ 40 nm), respectively, in close conformation. ADAR1 trimers, which maintain a height of ~5 nm, exhibit diameters greater than 40 nm (Supplementary Fig. 5c). Dimers were characterized by fluctuating interparticle distances and variable shapes, whereas trimers and tetramers exhibited topographies consistent with their predicted 3D models. We quantified the ratios of monomers, dimers, trimers, tetramers, and larger oligomers (Supplementary Fig. 5d) within ADAR1 molecules in the absence of substrate dsRNA. In addition, to further understand the dynamics of these structures, we performed a detailed frame-by-frame analysis of HS-AFM movies, tracking the trajectories of individual particles. These HS-AFM images of the ADAR1 monomer offer direct visualization of the architectures of both the deaminase and the N-terminal domains. These observations contribute to our understanding of the structural dynamics of ADAR1 and its potential roles in RNA recognition and editing. A limitation of our study is the absence of explicit control experiments to directly test for non-specific collision interactions. However, we minimized the risk of misidentification by ensuring consistent particle size and shape across multiple frames. We acknowledge that additional experiments or controls, such as cross-validation with complementary techniques, would offer further validation of the specificity of these interactions.

a The domain structures of ADAR1-p110. b Selected frames show the structural insight of the ADAR1 monomer over time. The monomer shows the dsRBDs with the Zβ domain (N-terminal domains) and a deaminase domain in one-tailed and globular structures, respectively. The tail protruding from the globular structure hides behind it. c Cross-sectional analysis of the monomer clearly shows fluctuations in the peak-to-peak distance between dsRBDs with the Zβ and the deaminase domains. d The height of the PDB structure of the deaminase domain using PYMOL online software and the distribution of the height peaks in the HS-AFM images of the deaminase domain were observed. Imaging parameters: scanning area = 100 × 100 nm2 (150 × 100 pixels); frame rate = 3.3 frames/s. The displayed area is 43 × 40 nm2. Source data of the graphs are provided as a Source Data file.
We then closely observed the dynamics of ADAR1 dimers in the absence of the substrate dsRNA, which was detected in the same sample as the monomers. As shown in Fig. 4, ADAR1 monomers formed dimers, and this dimerization occurred independently of dsRNA binding. The dimer appeared as two globular structures, and their contact exhibited fluctuations accompanied by flexible N-terminal domains. The full range of dimer dynamics is shown in supplementary movie 3. Throughout the observation window, the two monomers of ADAR1 start interacting via dsRBDs. This interaction increases the deaminase domain-deaminase domain association to form a stable dimerization between two monomers (Supplementary Fig. 6, left panel). Notably, the dimer exhibited active dynamics of the N-terminal domains in the central region (Fig. 4a). This flexibility of the N-terminal domains leads to fluctuations in the distance between the two monomers, highlighting the dynamic nature of ADAR1 dimerization. Cross-sections taken along the primary axis of the ADAR1 dimer provided insights into fluctuations in the peak-to-peak distance between the deaminase domains of the two monomers. The distance profiles of ten frames (i–x), as depicted in Fig. 4b, and the schematic drawing presented in Fig. 4c, illustrate that the deaminase domains of the two monomers in the dimer were in close contact. However, the interaction distance between the peaks of the two deaminase domains fluctuated within a range from 11.8 to 29.9 nm. This fluctuation occurred in the presence of activated dynamic behavior of the N-terminal domains. This observation suggests that the flexible interactions of the dsRBDs increase the association of the interfaces (IFx and IFy) on the deaminase domain of two monomers. The dynamic behavior and interaction of the dsRBDs-dsRBDs play a crucial role in strengthening the interaction between the deaminase domains. We demonstrated the dynamic characteristics of ADAR1 monomers involved in dimerization through cartoon modeling (Supplementary Fig. 6, right panel). Further studies involving mutations in the contact residues will help clarify the role of these predicted interface regions in the oligomerization and function of the ADAR1 dimer.

a HS-AFM observations indicated that ADAR1 monomers interact with each other, and the distance between two monomers in the dimer fluctuates with flexible and dynamic N-terminal domain regions. b Peak-to-peak distance between monomers fluctuates, as shown by the cross-sectional analysis of the dimers. c The schematic drawing represents the distance range of fluctuations between two monomers in a dimer. Imaging parameters: scanning area = 100 × 100 nm2 (100 × 80 pixels); frame rate = 3.3 frames/s. The displayed area is 57 × 59 nm2. Representative data from more than 3 independent experiments with similar results are shown. Source data of the graphs are provided as a Source Data file.
A stable ADAR1 dimer acts in complex with dsRNA
We next studied the dynamics of ADAR1 dimerization in the presence of substrate dsRNA. We selected the aryl hydrocarbon receptor (AhR) 3’UTR mRNA as a target of ADAR1. AhR is a transcription factor that regulates the expression of enzymes involved in the metabolism of foreign substances44. Previously, our group discovered that ADAR1 extensively edits adenosines at 38 editing sites on the 3’UTR of AhR mRNA, which contains Alu repeats forming a perfect duplex RNA in human liver cells41. To visualize the structural features of AhR dsRNA, we examined the conformation of non-edited AhR 3’UTR dsRNA on a mica surface functionalized with 3-aminopropyl-triethoxysilan (APTES), hereafter referred to as AP (APTES)-treated mica surface. The HS-AFM results (Supplementary Fig. 7a and Supplementary Movie 4) revealed that Alu elements at the 3’UTR region of AhR mRNA perfectly formed a rod-Y-shaped helical structure similar to that predicted by RNAfold Webserver (Supplementary Fig. 8b). The cross-sectional analysis was performed at three different locations along the length of the dsRNA to measure the height of the dsRNA on the AP-mica. The measured height of the dsRNA ranged between 2 and 2.5 nm (Supplementary Fig. 7b). Notably, the length of the Alu region, approximately 95 nm, is closely aligned with the theoretical length (300 bp x 0.34 nm/nt = 102 nm). Furthermore, the HS-AFM dsRNA analysis revealed minor and major grooves (Supplementary Fig. 7c). These observations provide valuable insights into the structural properties of AhR 3’UTR dsRNA, offering a basis for understanding its interactions with ADAR1 and subsequent RNA editing processes.
We then attempted to gain insight into the dynamics of the ADAR1 dimer and its dimerization in the presence of the substrate dsRNA. We successfully captured images depicting the formation of a stable dimer complex with dsRNA with sufficient temporal and spatial resolutions (Figs. 5 and 6), which enabled us to visualize various dynamic conformations of the dimer, spanning from the initial search to subsequent scanning of the dsRNA substrate. In supplementary movie 5 and the selected images in Fig. 5, we show sequential images that illustrate the process of searching, recognizing, capturing, and scanning the substrate dsRNA by ADAR1 dimers. The images were captured on a weakly functionalized AP-mica surface. We observed that the ADAR1 dimer dissociated from the upper segment of the dsRNA and moved towards the middle segment, covering a distance of ~30 nm. Surprisingly, the dimer complex shifted towards a different site on the lower segment of the dsRNA at 383.0 s (Fig. 5). This movement could have been induced by the force applied by the HS-AFM tip moving from left to right or could have resulted from selective molecular attraction. At these points, we observed a long-range interaction that led to the following events: (i) A dimer initiates the search, recognition, and capture of substrate dsRNA at the backbone side. During the recognizing event ( ~ 26.4 to 88.8 s), the ADAR1 dimer approaches the dsRNA in a flexible conformation, with the dsRBDs in an elongated state. This configuration allows the dimer to transition into a more stable, anchor-like structure along the dsRNA backbone, preparing the enzyme for RNA capture. Following the capturing event ( ~ 105.5 to 171.4 s), ADAR1 engages with the dsRNA by changing anchor-like conformation to capture the dsRNA at the backbone. At this stage, the dsRBDs stabilize the interaction by securing the RNA in place, ensuring a stable binding conformation before scanning. (ii) A single ADAR1 dimer interacts with a single dsRNA substrate. (iii) A dimer loops out on the substrate dsRNA with a visibly large interfacial interaction between the deaminase domains, forming a dimer. The dimer then scans ( ~ 59.9 to 305.5 s) along the RNA, assessing potential editing sites. This scanning involves the upward motion of the monomers in the dimer, while the RNA remains stably bound. (iv) The dimer dissociated from the dsRNA and searched for a new editing site on the dsRNA. Illustrations of selected sequential HS-AFM images of ADAR1 dimers were provided to indicate the various stages of dimer movement and conformation in the presence of the substrate dsRNA (Fig. 6a). These selected images indicate the structural flexibility of the N-terminal domains for dsRNA search and capture, suggesting their significant role in ADAR1 editing activity and site selectivity. We calculated the scan rate of the ADAR1 dimer to be approximately 0.17 nm/s, covering a distance of ~ 37.8 nm during scanning of the dsRNA substrate (Fig. 6b), as measured from the lower images in Fig. 5. The distance between the two deaminase domains was measured from the maximum peak heights to determine the fluctuations between the two monomers in the dimer in the presence of the dsRNA substrate. Interestingly, the fluctuation in the monomer-monomer interaction ranged from 0 to 13 nm, which was lower than that in the absence of substrate dsRNA (Fig. 6c), indicating increased dimer stability in the complex with substrate dsRNA. Furthermore, the N-terminal domains showed less flexibility when the dsRNA substrate was present, probably because they were involved in binding to dsRNA, which made the deaminase domain more stable in its dimerized form. This stability allowed the deaminase domain to effectively scan the dsRNA substrates. Figure 6d shows the intricate conformational dynamics and fluctuations of the dimer during search, recognition, capture, and scanning of the dsRNA substrate. These real-time HS-AFM observations provide compelling evidence that ADAR1 operates within a dimeric complex structure on the dsRNA substrate, adopting a range of conformations facilitated by the coordination between its two monomers. This dynamic behavior enables the dimer to effectively locate, interact with, and ultimately form a ternary complex with the dsRNA substrate. A quantitative analysis of the separation distribution reveals a statistically significant difference between the two states (p < 0.001) (Supplementary Fig. 9). The results indicate that RNA binding reduces the separation distance between monomers, leading to a more stable dimeric conformation. This finding supports the hypothesis that dimerization is stabilized upon dsRNA binding. The difference in separation between the bound and unbound states suggests that the cooperative flexibility of the dsRBDs is key to the enzyme’s ability to capture and stabilize dsRNA. Upon RNA binding, this flexibility induces conformational shifts in the deaminase domain, promoting a more stable and functional dimer. Additionally, the inactive ADAR1 protein in our in vitro RNA editing assay was not observed in a dimer conformation by HS-AFM, indicating that ADAR1 functions exclusively as an active dimer (Supplementary Fig. 10). These findings are consistent with previous biochemical studies that show dimerization of ADAR1 is essential for RNA editing. Furthermore, the stability of the dimer on dsRNA may be facilitated by interactions involving the N-terminal domain region.

HS-AFM direct evidence of searching, recognizing, capturing, and scanning dsRNA by ADAR1 dimers. An ADAR1 dimer was observed at different sites and formed the ternary complex. The images show that ADAR1 utilizes N-terminal domains for searching, recognizing, and capturing dsRNA, and then the deaminase domain dimer loops out on the dsRNA for scanning. Imaging parameters: scanning area = 150 × 150 nm2 (100 × 100 pixels); frame rate = 5 frames/s.

a The illustration of the ADAR1 dimer interacting with dsRNA from selected sequential HS-AFM images. The images indicate that the N-terminal domains in the dimer recognize and capture dsRNA, and then the two monomers loop on dsRNA to initiate the scanning in the process of editing. b The scan rate of the ADAR1 dimer for scanning the substrate dsRNA. c The distance measured between the maximum height of both deaminase domains in the dimer shows the structural flexibility of monomers to form a stable molecular complex. d Illustration showing fluctuations in monomer-monomer interaction ranging from 0 to 13 nm in the presence of substrate dsRNA. These images represent snapshots from the highest-resolution movie obtained across numerous experiments. Source data of the graphs are provided as a Source Data file.

a RNA without ADAR1 (0 ng). b RNA with ADAR1 (25 ng) at 25 °C. c RNA with ADAR1 (25 ng) at 37 °C. Images in (a–c) RNA samples were captured before RNA purification. d RNA with ADAR1 (100 ng) at 37 °C (images captured after RNA purification). Representative sequencing electropherograms of selected RNA-edited sites on AhR 3’UTR dsRNA are shown on the right. A-to-I RNA editing was detected as A-to-G changes. Imaging parameters: scanning area = 250 × 250 nm2 (120 × 120 pixels); frame rate = 2.5 frames/s. The displayed area is 110 × 107 nm2. Representative data from more than 2 independent experiments with similar results are shown.
HS-AFM revealed the structural morphology of dsRNA with and without editing
The early studies in the RNA-editing field proposed that ADAR1 primarily unwinds dsRNAs45. Thus, we next sought to characterize the structure of dsRNA by observing the AhR RNA after the in vitro adenosine (A)-to-inosine (I) RNA editing assay with and without ADAR1 by HS-AFM (see Fig. 7a–d). We performed the in vitro editing assay with and without ADAR1 at 25 °C and 37 °C for 2 h. Subsequently, samples were deposited on AP-mica for HS-AFM observations. In parallel, the A-to-I RNA editing level at AhR RNA editing sites was detected as A-to-guanosine (G) using the direct sequencing method (shown in the right panel of Fig. 7). The analysis of dsRNA without ADAR1 did not show any conversion of A peaks to G peaks at the editing sites of AhR dsRNA (see Fig. 7a). In contrast, RNA samples containing ADAR1 (25 ng) exhibited some editing activity at 25 °C (low) and at 37 °C (moderate) (Fig. 7b, c). Increasing the ADAR1 concentration to 100 ng further enhanced the A-to-G conversion (Fig. 7d). The theoretical analysis of the 2D structures of non-edited and fully edited AhR indicates that RNA editing may enhance intramolecular base pairing by eliminating loop structures (Supplementary Fig. 8a, b). HS-AFM images of the non-edited AhR mRNA sample alone (Supplementary Fig. 7) revealed the Y-shaped rod structure of dsRNAs. While edited dsRNA showed a linear and relaxed structure on the AP-modified mica surface.
Discussion
The previously reported structures of the ADAR2 deaminase domain in the absence of dsRNA18, ADAR2 deaminase domain-dsRBD2 complexed with dsRNA17, and ADAR1-dsRBD327, along with various biochemical and computational studies, have provided foundational insights into ADAR1 dimerization, catalytic mechanisms, and target preferences. Despite these advances, the comprehensive structural characterization of ADAR1 alone and in complex with dsRNA remains limited, particularly with regard to the impact of structural stability on RNA editing efficiency.
In this study, we employed an integrative approach combining 3D modeling and HS-AFM to directly observe the structural dynamics of the ADAR1 complex during oligomerization and the interaction of the ADAR1 dimer with the substrate dsRNA. Here, we proposed a mechanism for ADAR1 oligomerization using 3D modeling. The crystal structure of the asymmetric dimer previously reported by Beal et al.17, which is composed of two monomers of the 2nd dsRBD and the deaminase domain of ADAR2, suggested that dimerization occurs through the helical region at the interface of the deaminase domain. Furthermore, the mutagenesis of key residues displayed on the helix has been shown to disrupt dimerization. They also suggested that ADAR2 dimerization occurred in an RNA-independent manner. We also analyzed the alignment of the dimerization helix of ADAR2 (PDB ID 1zy7 [https://www.rcsb.org/structure/1ZY7] and PDB ID 6vff [https://www.rcsb.org/structure/6VFF]) with the predicted dimerization interfaces in the ADAR1 model (Supplementary Fig. 3a and b). The dimer structures of ADAR2 differ between 1zy7 and 6vff; both are asymmetric, but the interfaces are distinct. In the 6vff structure, the dimerization helix is part of the interface. In contrast, our ADAR1 model (ADAR1d_AB_1zy7), derived from 1zy7, positions the predicted interfaces (IFx and IFy) near the helix (Supplementary Fig. 3a). However, the contribution of the helix to dimerization differs. When we used 6vff as a template to model ADAR1, the dimer structure of ADAR1d_AB_6vff showed a misalignment between the dimerization helix and the predicted interfaces. Specifically, this model suggests that when dsRNA binds, the predicted interfaces (IFx and IFy) no longer form the dimerization interface between monomers M1 and M2, leading to altered trimer and tetramer conformations (Supplementary Fig. 3b). However, these structural predictions are inconsistent with the conformations observed in our HS-AFM images (Fig. 2 and Supplementary Fig. 3), which show a similar arrangement using the 1zy7 template.
A more recent study on ADAR1 dsRBD3 by Barraud et al.27 showed that the deaminase domains of ADAR1 mediate self-association. Our results are in agreement with the previous findings17,27. In our 3D modeling study, we identified distinct complementary amino acid residues at the x- and y-interface regions of each deaminase domain and proposed 3D models for ADAR1 dimerization and oligomerization. The heterodimerization of ADAR1 and ADAR2 has been previously reported in in vivo FRET experiments and has been proposed as an interesting mechanism to regulate the efficiency and specificity of editing24. Based on our findings, we can assume that the association between ADAR1 and ADAR2 likely occurs via the heterodimerization of their deaminase domains, which have similar non-identical amino acid residues on the interface surfaces. We further validated these ADAR1 complex models with and without the dsRNA substrate using HS-AFM. Interestingly, our 3D models of deaminase domain alignment in dimer, trimer, and tetramer formation through the x- and y-axis interface surfaces showed good agreement with the HS-AFM images (Fig. 2).
In this study, ADAR1 showed the capability to deaminate the dsRNA substrate, suggesting that monomeric ADAR1 was reconstituted into stable homodimers upon binding to dsRNA during RNA editing. This has been suggested in previous studies using recombinant ADAR1 in enzymatic activity assays46,47.
We further described the ADAR1 deaminase domain dimer in complex with the substrate dsRNA using molecular docking analysis. The top-docked model of the ADAR1 dimer in complex with RNA suggested that one deaminase domain uses a large portion of the surface to bind dsRNA, whereas another deaminase domain interacts with dsRNA in a small area. We also proposed that the top-docked model of the ADAR1 deaminase domain dimer is bound to dsRNA as an asymmetric homodimer. This suggestion is supported by the X-ray crystal structure of a fragment of human ADAR2 comprising its deaminase domain and dsRBD2 bound to an RNA duplex17,48. Furthermore, with close observation of the ADAR1 dimer in the absence of substrate dsRNA, we identified the critical role of dynamic flexibility and the interaction of N-terminal domains between the two monomers. Specifically, the activated dynamic flexible N-terminal domain interactions strengthen the dimeric association, reinforcing the interface between the two deaminase domains. This ADAR1 deaminase domain-mediated dimerization is further stabilized upon binding to substrate dsRNA. In line with previous studies, ADARs form a stable dimer, with dimerization becoming more pronounced in the presence of substrate dsRNA22. Previously, Nishikura et al. suggested that cooperative interactions of functional dsRBDs of both ADAR dimer subunits are required for dsRNA binding, and having one monomer in the dimer complex that is unable to bind the dsRNA prevents it from binding to its substrate49,50. The ADAR1 protein, which was unable to edit dsRNA in vitro, did not exhibit a dimeric conformation when analyzed by HS-AFM. Based on this observation, we hypothesized that ADAR1 is only functional as a dimer (Supplementary Fig. 10).
HS-AFM observations also revealed that the single dimeric form of ADAR1 acted on one substrate, dsRNA. We did not observe monomeric or oligomeric forms of ADAR1 in complexes with RNA. Our data further showed that the formation of the ADAR1 dimer initiates with the interaction of the monomers through their dsRBDs. This interaction subsequently facilitates the association of the deaminase domains, resulting in the formation of a stable dimeric conformation. The stabilized ADAR1 dimer forms a complex with the dsRNA substrate. During this process, we observed a reduction in fluctuations between the two monomers, indicating enhanced stability of the complex with dsRNA. These observations suggest that the dsRBDs are critical for initiating the interaction between the deaminase domains, thereby promoting the formation of a stable, functional dimeric complex capable of efficiently binding and catalyzing the editing of dsRNA substrates.
While this study is mainly focused on the dynamics of ADAR1, we recognize the potential relevance of ADAR2 in this context. Future studies will aim to compare the dynamic behavior of ADAR1 with ADAR2, particularly their dimerization mechanisms and interactions with dsRNA, to gain a deeper understanding of the structural and functional similarities and differences between the two enzymes. In conclusion, we used an integrated approach involving 3D modeling and HS-AFM to reveal the structural dynamics of ADAR1 complexes. Initially, we constructed 3D models to elucidate the assembly of ADAR1 dimers, trimers, and tetramers, which revealed the presence of distinct interfacial surfaces within each monomer. These surfaces play a crucial role in initiating self-dimerization or oligomerization. Subsequently, using HS-AFM, we captured the dynamic conformations of the 3D models and obtained real-time images of their characteristics. Our observations highlighted the influence of N-terminal domain dynamic interactions on ADAR1 dimerization, exhibiting fluctuations in monomer-monomer interactions that lead to a stable interface association between the deaminase domains. Notably, in the presence of substrate dsRNA, the ADAR1 dimer showed a controlled flexible conformation for searching, recognizing, and capturing substrate dsRNA and recruiting the deaminase domain dimer on the RNA for scanning. Furthermore, our study revealed distinct dimer conformations in the presence of substrate dsRNA, suggesting their importance in RNA editing mechanisms. Overall, our study provided a detailed visualization of the features of ADAR1 complexes in their dynamic conformations at the atomic and molecular levels, emphasizing the critical role of N-terminal domains in the stability of ADAR1 dimerization in complex with dsRNA. Based on our HS-AFM observations, we propose that targeting interface regions on the deaminase domain of ADAR1 dimerization could offer a promising strategy for regulating ADAR1 function. Future studies on identifying inhibitors, including aptamers51, that specifically modulate ADAR1 dimerization may, therefore, be a valuable addition to therapeutic approaches for diseases associated with aberrant RNA editing.
Methods
Materials
Human Recombinant ADAR Protein NM_001025107 was purchased from OriGene (Rockville, MD, USA). StriptMAX® Thermo T7 Transcription Kit and ReverTra Ace were purchased from Toyobo (Japan). NucleoSpin® Gel and PCR Clean-up Kit, Gflex DNA polymerase, DNase I, and RNase inhibitor were purchased from Takara Bio (Japan). The RNeasy Mini Spin Column was purchased from Qiagen (Germany. The 3-aminopropyl-triethoxysilane (APTES) was purchased from Shin-Etsu Chemicals (Tokyo).
In vitro transcription
ADAR1 edits adenosine into double-stranded RNA (dsRNA) structures. Previously, our group identified the 3’UTR of aryl hydrocarbon receptor (AhR) mRNA as an RNA target of ADAR141, and therefore, it was used for HS-AFM analysis and RNA editing studies. The transcript was synthesized from the pT7/AhR 3’UTR linear plasmid as a template DNA using a T7 Transcription Kit for 30 min at 37 °C, yielding a 1138 nt long AhR 3’UTR RNA transcript with an optical density of ~2.0. at 260 nm. To eliminate residual plasmid DNA, mRNA transcripts were treated with DNase I for 30 min at 37 °C, followed by purification on an RNeasy Mini Spin Column.
In vitro RNA editing assay
In vitro editing reactions were performed in 1 x editing buffer composed of 20 mM HEPES-NaOH pH 7.8, 100 mM NaCl, 5% (w/v) glycerol, 0.01% NP-40, 1 mM DTT, RNase inhibitor (1 U/μL). To promote inter-molecular over intra-molecular binding, the AhR RNA substrate (6.6 ng/μL) was diluted in water, heated at 95 °C for 2 min, immediately cooled on ice, and then added to the reaction mixture at a concentration of 35 fmol in a total volume of 50 μL. The RNA substrate was then incubated with human recombinant adenosine deaminase ADAR1 protein (2 ng/μL) at 25 °C and 37 °C for 2 h. Subsequently, cDNA was synthesized from the RNA using ReverTra Ace. Direct sequencing of the PCR products was performed for RNA editing. The AhR 3’UTR contains an antisense-oriented AluSx and a sense-oriented AluSc. The AluSc element was amplified by PCR using cDNA as a template with the following primers: sense S4 (5′-GGA ATC CCA AAC TGA GAT GTA T −3′) and antisense AS5 (5′-CCT CAT GCT GGA AAC AAA TT −3′). The PCR mixture included cDNA, 2×Gflex PCR buffer, 0.3 μM concentrations of each primer, and 0.5 units of Gflex DNA polymerase in a final volume of 25 μL. Amplification conditions included an initial denaturation at 94 °C for 1 min, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 55 °C for 15 s, and extension at 68 °C for 20 s, with a final extension step at 72 °C for 5 min. The PCR products were analyzed by electrophoresis on a 2% agarose gel. To ensure that the amplified product originated from the reverse-transcribed cDNA rather than from contaminating DNA traces, a control experiment without reverse transcriptase was performed. The PCR product was purified using a Gel and PCR Clean-up Kit and subjected to direct sequencing. The extent of editing was quantified as a percentage and calculated from the ratio of the peak height of G over the sum of the peak heights of G and A in the sequencing electropherograms. The details of the AhR 3’UTR mRNA sequence and editing sites are mentioned in a previous study41.
In-silico 3D modeling and molecular docking
The full-length 3D structure of human ADAR1(hADAR1), consisting of 1226 amino acids, was predicted using AlphaFold2 (https://alphafold.ebi.ac.uk). However, the N-terminal domain, spanning residues from 1 to 822 amino acids, displayed structural disorder and was thus omitted from the final model. Consequently, the refined ADAR1 monomer, encompassing residues 823–1226 (the deaminase domain), was used for subsequent in silico investigations. This monomeric model structure served as the basis for the construction of higher-order structures (Fig. 1c). The dimer, trimer, tetramer, and polymer models of the ADAR1 deaminase domain were generated by superposition of the monomer or dimer model on the X-ray crystallographic structure of the ADAR2 dimer (PDB ID 1zy7 [https://www.rcsb.org/structure/1ZY7]) by sequence alignment. The protein surface of the ADAR1 monomer was drawn as a Connolly solvent surface with a probe radius of 1.4 Å52. The secondary structure of the 3’UTR AhR mRNA (1138 nt) was constructed by the RNAFold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) (Supplementary Fig. 8a). The ADAR1 dimer complex with the full-length RNA exceeded the ZDOCK limitations, which require the sum of receptor and ligand radii to be within 200 Å. As a result, the RNA was truncated while retaining its essential elements, specifically the three adenosines targeted by ADAR1, which are highlighted in bold. The resulting truncated AhR (AluSc) – 49 nt sequence was: 5’-GGAUCACGAGGUCAAGAGAUGGAGACCAUCCUGACCAACAUGGUGAAAC- ‘3. The corresponding 3D structure of the short AhR RNA model was generated and visualized using RNAcomposer (http://rnacomposer.cs.put.poznan.pl) (Supplementary Fig. 4a). Energy minimization using the CHARMM force field was then employed to refine the RNA structure53. Subsequently, protein-RNA docking simulations were conducted using ZDOCK with the ADAR1 dimer and RNA model54. The resulting 2000 docking poses from each simulation were ranked using the ZRANK score and clustered according to their docking locations. The validity of docking poses was assessed using the ZDOCK score and cluster size criteria.
HS-AFM observation
A laboratory-built HS-AFM apparatus was used to scan the samples in liquid using the tapping mode36. A glass stage (Japan Cell Co., Ltd., Japan; 1.5 mm in diameter and 2 mm in height) with a mica disc (Furuuchi Chemical Corp., Japan; 1.5 mm in diameter and 0.05 mm in thickness) was attached to the top of the Z-scanner using nail polish. Cantilevers (BL-AC10DS-A2, Olympus, Japan) with a spring constant of ∼0.15 N/m, a resonance frequency of ∼500 kHz in water, and a quality factor of ∼1.5 in water were used.
Commercially available ADAR1 protein (50 µg/mL) was supplied in a stock solution containing 25 mM Tris-HCl, 100 mM glycine, pH 7.3, with 10% glycerol. For HS-AFM observation, ADAR1 was diluted to a final concentration of 20 nM in diluting buffer A (20 mM HEPES-KOH, 5% glycerol, 10 mM KCl, 2 mM MgCl2, 0.1 mM DTT, pH 7.5).
The synthesized AhR RNA (1138 nt) was dissolved at a 6.65 ng/µL concentration in diluting buffer B (20 mM HEPES-KOH, 10 mM KCl, 2 mM MgCl2, pH 7.5) for HS-AFM observation (Supplementary Fig. 7).
For the HS-AFM observations of ADAR1 alone, ADAR1 was added to a freshly cleaved mica surface in buffer A at a concentration of 20 nM. HS-AFM observations were performed in the same manner as for buffer A. For HS-AFM observations of dsRNA, the mica surface was treated for 3 min with 0.01% AP dissolved in Milli-Q water. Subsequently, 2 µL dsRNA (13.3 nM) was applied to the AP-treated mica surface. After a 5 min incubation, HS-AFM observation was performed in buffer B. For HS-AFM observations of ADAR1 dimer complex to substrate dsRNA, 2 µL of dsRNA (3.4 nM) was applied onto the weak AP-treated mica surface (0.001%) and incubated with 20 nM of ADAR1. All HS-AFM experiments were performed at 25 °C, and the captured HS-AFM images were processed using Kodec4 and UMEX viewer software55. We applied pre-interpolation (Bilinear) and post-interpolation (Nearest Neighbor) settings to the HS-AFM images to reduce spike noise. A flattening filter was also applied to correct for substrate tilt effects.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data needed to evaluate the conclusions in the paper are provided in the main text, including the main Figures and the Supplementary information. PDB structures used in this article are available in the protein data bank (PDB) with the following accession codes: 1zy7 [https://www.rcsb.org/structure/1ZY7] and 6vff [https://www.rcsb.org/structure/6VFF]. Source data are provided with this paper.
References
Bass, B. L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 71, 817–846 (2002).
Bass, B. L. & Weintraub, H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55, 1089–1098 (1988).
Goodman, R. A. et al. ADAR proteins: structure and catalytic mechanism. Curr. Top. Microbiol. Immunol. 353, 1–33 (2012).
Wang, Q. et al. ADAR1 regulates ARHGAP26 gene expression through RNA editing by disrupting miR-30b-3p and miR-573 binding. RNA 19, 1525–1536 (2013).
Rueter, S. M. et al. Regulation of alternative splicing by RNA editing. Nature 399, 75–80 (1999).
Yeo, J. et al. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc. Natl Acad. Sci. 107, 20715–20719 (2010).
Bass, B. L. et al. A standardized nomenclature for adenosine deaminases that act on RNA. RNA 3, 947–949 (1997).
Han, L. et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 28, 515–528 (2015).
Chen, L. et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat. Med 19, 209–216 (2013).
Paz-Yaacov, N. et al. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep. 13, 267–276 (2015).
Chen, Y., Wang, H., Lin, W. & Shuai, P. ADAR1 overexpression is associated with cervical cancer progression and angiogenesis. Diagn. Pathol. 12, 12 (2017).
Ishizuka, J. J. et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48 (2019).
Bhate, A., Sun, T. & Li, J. B. ADAR1: a new target for immuno-oncology therapy. Mol. Cell 73, 866–868 (2019).
Gannon, H. S. et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun. 9, 5450 (2018).
Kung, C. P. et al. Evaluating the therapeutic potential of ADAR1 inhibition for triple-negative breast cancer. Oncogene 40, 2147 (2021).
Matthews, M. M. et al. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 23, 426–433 (2016).
Alexander, S. T. et al. Asymmetric dimerization of adenosine deaminase acting on RNA facilitates substrate recognition. Nucleic Acids Res. 48, 7958–7972 (2020).
Macbeth, M. R. et al. Inositol Hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 309, 1534–1539 (2005).
Eggington, J. M., Greene, T. & Bass, B. L. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2, 1–9 (2011).
Wang, Y., Park, S. & Beal, P. A. Selective recognition of RNA substrates by ADAR deaminase domains. Biochemistry 57, 1640–1651 (2018).
Schneider, M. F. et al. Optimal guideRNAs for re-directing deaminase activity of hADAR1 and hADAR2 in trans. Nucleic Acids Res 42, e87 (2014).
Gallo, A., Keegan, L. P., Ring, G. M. & O’Connell, M. A. An ADAR that edits transcripts encoding ion channel subunits functions as a dimer. EMBO J. 22, 3421–3430 (2003).
Cho, D. S. et al. Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. J. Biol. Chem. 278, 17093–17102 (2003).
Chilibeck, K. A. et al. FRET analysis of in vivo dimerization by RNA-editing enzymes. J. Biol. Chem. 16, 16530–16535 (2006).
Park, S. et al. High-throughput mutagenesis reveals unique structural features of human ADAR1. Nat. Commun. 11, 5130 (2020).
Zambrano-Mila, M. S. et al. Dissecting the basis for differential substrate specificity of ADAR1 and ADAR2. Nat. Commun. 14, 8212 (2023).
Allegra, M. et al. Dimerization of ADAR1 modulates site-specificity of RNA editing. Nat. Commun. 15, 10051 (2024).
Zhang, C., Liu, S., Zhu, Q. & Zhou, Y. A Knowledge-Based Energy Function for Protein–Ligand, Protein–Protein, and Protein–DNA Complexes. J. Med. Chem. 48, 2325–2335 (2005).
Schmidt, T., Bergner, A. & Schwede, T. Modelling three-dimensional protein structures for applications in drug design. Drug Discov. Today 19, 890–897 (2014).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Chothia, C. & Lesk, A. M. The relation between the divergence of sequence and structure in proteins. EMBO J.5, 823–826 (1986).
Schwede, T. Protein modeling: what happened to the “protein structure gap”? Structure. 21,1531–1540 (2013).
Cozzini, P. et al. Target flexibility: an emerging consideration in drug discovery and design. J. Med Chem. 51, 6237–6255 (2008).
MacCallum, J. L. et al. Assessment of protein structure refinement in CASP9. Proteins 79, 74–90 (2011).
Feig, M. Computational protein structure refinement: Almost there, yet still so far to go. Wiley Interdiscip. Rev. Comput. Mol. Sci. 7, e1307 (2017).
Ando, T. et al. A high-speed atomic force microscope for studying biological macromolecules. Proc. Natl Acad. Sci. USA 98, 12468–12472 (2001).
Kodera, N. et al. Video imaging of walking myosin V by high-speed atomic force microscopy. Nature 468, 72–76 (2010).
Shibata, M. et al. Real-space and real-time dynamics of CRISPR-Cas9 visualized by high-speed atomic force microscopy. Nat. Commun. 8, 1430 (2010).
Tsuji, A., Yamashita, H., Hisatomi, O. & Abe, M. Dimerization processes for light-regulated transcription factor Photozipper visualized by high-speed atomic force microscopy. Sci. Rep. 12, 12903 (2022).
Puppulin, L. et al. Dynamics of Target DNA Binding and Cleavage by Staphylococcus aureus Cas9 as Revealed by High-Speed Atomic Force Microscopy. ACS Nano 17, 4629–4641 (2023).
Nakano, M. et al. RNA Editing Modulates Human Hepatic Aryl Hydrocarbon Receptor Expression by Creating MicroRNA Recognition Sequence. J. Biol. Chem. 291, 894–903 (2016).
Amyot, R. et al. BioAFMviewer software for simulation atomic force microscopy of molecular structures and conformational dynamics. J. Struct. Biol. X 7, 100086 (2023).
Yuan, J. et al. Biological roles of A-to-I editing: implications in innate immunity, cell death, and cancer immunotherapy. J. Exp. Clin. Cancer Res 42, 149 (2023).
Ramadoss, P. et al. Role of the aryl hydrocarbon receptor in drug metabolism. Expert Opin. Drug Metab. Toxicol. 1, 9–21 (2005).
Wagner, R. W. et al. A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc. Natl Acad. Sci. USA 86, 2647–2651 (1989).
Kim, U. et al. Purification and characterization of double-stranded RNA adenosine deaminase from bovine nuclear extracts. J. Biol. Chem. 269, 13480–13489 (1994).
O’Connell, M. A. & Keller, W. Purification and properties of double-stranded RNA-specific adenosine deaminase from calf thymus. Proc. Natl Acad. Sci. USA 91, 10596–10600 (1994).
Fisher, A. J. & Beal, P. A. Structural perspectives on adenosine to inosine RNA editing by ADARs. Mol. Ther. Nucleic Acids 35, 102284 (2024).
Valente, L. & Nishikura, K. RNA binding-independent dimerization of adenosine deaminases acting on RNA and dominant negative effects of nonfunctional subunits on dimer functions. J. Biol. Chem. 282, 16054–16061 (2007).
Dan-Sung, C. C. et al. Requirement of Dimerization for RNA Editing Activity of Adenosine Deaminases Acting on RNA. J. Biol. Chem. 278, 17093–17102 (2003).
Biyani, M. et al. A novel aptamer-antibody sandwich electrochemical sensor for detecting ADAR1 in complex biological samples. Biosens. Bioelectron.: X 19, 100491 (2024).
Connolly, M. L. Solvent-accessible surfaces of proteins and nucleic acids. Science 221, 709–713 (1983).
MacKerell, A. D. Jr, Banavali, N. & Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 56, 257–265 (2000).
Iwakiri, J. et al. Improved Accuracy in RNA-Protein Rigid Body Docking by Incorporating Force Field for Molecular Dynamics Simulation into the Scoring Function. J. Chem. Theory Comput 12, 4688–4697 (2016).
Hayakawa, Y. et al. Actin-binding domain of Rng2 sparsely bound on F-actin strongly inhibits actin movement on myosin II. Life Sci. Alliance 6, e202201469 (2022).
Acknowledgements
Financial support from the Grants-in-Aid for Scientific Research (C), KAKENHI, Japan Society for the Promotion of Science (JSPS) (23K06067 to Madhu B.), and the World Premier International Research Center Initiative (WPI), MEXT, Japan, are gratefully acknowledged. The authors thank Prof. Toshio Ando, Dr. Kenichi Umeda, Ms. Wei Weilin, Ms. Aimi Makino, and Ms. Kayo Nagatani for their technical support of HS-AFM experiments.
Author information
Authors and Affiliations
Contributions
Madhu B., Miki N., and Manish B. conceived and designed the study; Madhu B. and K.S. performed HS-AFM experiments through discussion with N.K.; S.M., H.A., Y.S., and Masataka N. performed in vitro editing assay experiments; Y.I. performed in silico 3D modeling and molecular docking experiments; Madhu B. performed the simulation of HS-AFM images; Madhu B., Y.I., and Manish B. analyzed the data, discussed its validity, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks George Heath and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Biyani, M., Isogai, Y., Sharma, K. et al. High-speed atomic force microscopy and 3D modeling reveal the structural dynamics of ADAR1 complexes. Nat Commun 16, 4757 (2025). https://doi.org/10.1038/s41467-025-59987-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59987-6
