
Abstract
Fluorine-containing compounds hold pivotal importance in life sciences. Recent decades have witnessed significant research efforts toward developing practical fluorination methods. Radical-mediated decarboxylative fluorination has proven to be a robust approach for incorporating diverse monofluoroalkyl groups. Here we show a radical-mediated modular synthesis of alkyl fluorides through a decarboxylative-desulfonylative gem-difunctionalization under mild photochemical conditions. The multi-component reaction proceeds in a controlled sequence of radical decarboxylation and heteroaryl migration, governed by radical polarity and kinetic effects, resulting in a wide range of valuable alkyl fluorides. Two C-C bonds and one C-F bond are concurrently formed throughout the process. Both styrenes and aliphatic alkenes serve as suitable substrates for this transformation. Furthermore, this method can be applied to the incorporation of a monofluoroalkyl moiety into complex alkene molecules at a late stage.
Similar content being viewed by others

Introduction
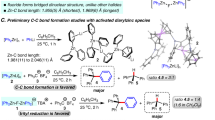
The integration of a fluorine atom or fluorinated groups into the molecular frameworks of pharmaceuticals, agrochemicals, and organic materials frequently elicits substantial enhancements in their physical, chemical, and biological functionalities, including improvements in lipophilicity, permeability, polarity, and metabolic resilience1,2,3,4. Owing to such profound impacts, fluorine-containing compounds are pivotal in fields related to life sciences, with estimates suggesting that over 20% of pharmaceuticals on the market today are fluoro-pharmaceuticals5,6,7,8. Consequently, the past decades have witnessed an intense focus on the development of versatile and efficient synthetic methods for fluorination3,9,10,11,12,13,14,15. In this vein, radical-mediated decarboxylative fluorination emerges as a distinguished approach, offering a gateway to a variety of monofluoroalkyl moieties prevalent in top-tier medications and an array of preclinical entities (Fig. 1a). Despite the remarkable evolution in reaction modalities that encompass traditional transition-metal catalysis inspired by Hunsdiecker-type reactions to modern photo-/electro-catalytic processes16,17,18,19,20,21,22,23,24,25, the shortcomings of these protocols (e.g., the limited reaction mode that constricts the diversity of products, and the poor availability of precursors required for complex product constructions) remain inadequately addressed (Fig.1b). In pursuit of expanding the chemical landscape, there is a high demand for preparing intricate alkyl fluoride compounds via multi-component decarboxylative fluorination, employing readily available alkenes as substrates.

a The importance of monofluoroalkyl moiety in bioactive molecules. b Classic decarboxylative fluorination. c Fluorination via decarboxylation-desulfonylation.
We envision a modular pathway to synthesize alkyl fluorides via a multi-component reaction that involves α-sulfonyl carboxylic acid 1, alkene, and Selectfluor (Fig. 1c). This fluorination process unfolds through an orchestrated radical decarboxylation-desulfonylation cascade26. Presumably, the decarboxylation of 1 through single-electron transfer (SET) yields the intermediate a27, which subsequently adds across the alkene to generate intermediate b. The following process involves the R3 group migrating from the sulfone to the carbon-centered radical with a concurrent elimination of SO2, leading to intermediate c28,29,30,31,32. The reaction with Selectfluor converts intermediate c into the desired product 3. Theoretically, both radical intermediates a and b could potentially precede radical c in reacting with the fluorinating agent, thereby disrupting the formation of product 3. Several considerations, however, offer insight into achieving the desired chemoselectivity: 1) The sulfone-associated alkyl radical a is electrophilic, making it less inclined to abstract a fluorine atom from Selectfluor due to mismatched polarity; 2) Alkyl radical b favors intramolecular migration of the R3 group through a kinetically preferred five-membered cyclic transition state over intermolecular fluorine abstraction; and 3) The nucleophilic alkyl radical c is likely to engage in fluorine abstraction rather than addition to the electron-rich alkene, forestalling the formation of intermediate d. These rationales contribute to an anticipation of selectivity governed by radical polarities and kinetic influences. Nevertheless, the presence of competitive pathways still renders the overall transformation somewhat unpredictable.
Herein we report preliminary evidence in support of the hypothesis. The desired multi-component reaction proceeds under photochemical conditions, exhibiting remarkable chemoselectivity. Utilization of α-sulfonyl carboxylic acids provides double reacting sites compared to conventional fatty acids, facilitating the synthesis of an extensive spectrum of intricate alkyl fluorides. This process forges two C-C bonds and a C-F bond within the radical cascade of decarboxylation and desulfonylation.
Results
Reaction parameters survey
Optimization of the reaction parameters was pursued using styrene 1a and α-sulfonyl carboxylic acid 2a under photochemical conditions (Table 1). A thorough investigation revealed that employing Selectfluor as the fluorine donor, KHCO3 as the base, and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 as the photoredox catalyst in dichloromethane (DCM) yielded the targeted alkyl fluoride 3a in a synthetically useful yield (Table 1, entry 1). Substitution of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 with other frequently used photoredox catalysts, including Fukuzumi’s salt (Mes-Acr+ClO4-) and 4CzIPN, resulted in diminished yields of 3a (Table 1, entries 2 and 3). Although possible, achieving 3a via silver-catalyzed decarboxylative fluorination19,33,34,35 proved less efficacious (Table 1, entry 4). Evaluation of various bases and solvents indicated that the replacement of KHCO3 and DCM negatively affected the overall effectiveness of the reaction (Table 1, entries 5-10). Control experiments underscored the indispensability of light, catalyst, and base for this transformation, with exposure to air entirely inhibiting the conversion (Table 1, entries 11-14).
Substrate scope
With the optimized reaction conditions in hand, we started to evaluate the generality of the protocol (Fig. 2). Various styrenes were initially tested, showing that the corresponding products were consistently produced (3a-3n), independent of the electronic characteristics and steric hindrance of the aryl substituents. Naphthyl and hetaryl such as thienyl ethylene are also suitable substrates (3o and 3p). Both enamide and enol ether served as effective substrates, yielding the anticipated adducts (3q and 3r). Notably, the protocol demonstrated robustness through its application to a range of unactivated aliphatic alkenes (3s-3ag). 1,1-Disubstituted alkenes, including low-boiling-point isobutene, efficiently formed all-carbon quaternary centers in the products (3w and 3x). Many sensitive functional groups remained intact during the reaction, including alkyl bromide (3aa), epoxide (3ab), unprotected alcohol (3ac), and nitro group (3ae). A set of α-sulfonyl carboxylic acids was conveniently synthesized and then subjected to standard conditions. The sulfonyl part could be substituted with various heteroaryl groups. Beyond the parent benzothiazolyl group, functionalized benzothiazolyl, thiazolyl, quinolyl, pyridyl, and thienyl groups were readily integrated into the products (3ah-3am). Moreover, the aliphatic backbone of the carboxylic acids was diversified, accommodating acyclic, cyclic, and even heterocyclic alkyl groups for decarboxylative coupling to alkenes (3an-3ar). However, secondary and primary carboxylic acids are so far unsuitable for the transformation, probably attributed to the low efficiency to generate relatively high-energy primary and secondary alkyl radicals via radical decarboxylation, as well as the lower stability of these radicals. Remarkably, this method was also applicable to the monofluoroalkylation of complex alkenes derived from natural products and drug molecules (3as-3aw).

Reaction conditions: 1 (0.2 mmol), 2 (0.4 mmol), [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (2 mol %), Selectfluor (0.6 mmol), and KHCO3 (0.6 mmol) in DCM (2 mL), irradiated with 30 W blue LEDs at 20 oC under N2 for 48 h.
Synthetic applications
The products were proven to be versatile precursors to several valuable compounds, further underscoring the practicality of the synthetic approach (Fig. 3). The benzothiazolyl moiety in compound 3a was efficiently transformed into a formyl group, yielding the fluorinated aldehyde 4. Upon the treatment with diethylaminosulfur trifluoride (DAST), compound 4 was converted into the 1,1,4-trifluoroalkane 5. A different sequence involving initial reduction of 4 with NaBH4 and subsequent exposure to DAST generated the 1,4-difluoroalkane 6. The oxidation of 4 via the Bayer-Villiger reaction with meta-chloroperoxybenzoic acid (m-CPBA) led to the formation of the formate 7. Additionally, the reaction of 4 with a carbene generated from ethyl diazoacetate produced the fluorinated β-keto ester 8.

a The synthesis of 4. Reaction conditions: (1) 3a (0.1 mmol), 4 Å molecular sieves powders (150 mg), Me3OBF4 (0.5 mmol), CH2Cl2, 10 min; (2) MeOH (1 mL), 0 °C, NaBH4 (0.25 mmol); (3) AgNO3 (0.3 mmol), CH3CN (1.5 mL)/H2O(0.18 mL). Further transformations of 4. Reaction conditions: 4 (0.1 mmol). b DAST (1.2 equiv), rt, 4 h; c Na2HPO4 (1.2 equiv), m-CPBA (3 equiv), 0 oC, 12 h; d (1) NaBH4 (1.2 equiv), MeOH, rt, 4 h; (2) DAST (1.2 equiv), CH2Cl2, rt, 8 h; e MoO2Cl2 (5 mol %), N2CHCOOEt (1.2 equiv), CH2Cl2, 30 °C. Isolated yields are shown.
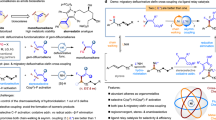
To gain further insight into the reaction pathways, a set of mechanistic studies was carried out. There was no change in the yield under dark conditions and no change in the yield after the reaction time was extended to 48 hours, confirming the light-dependent nature of the reaction (Fig. 4a). Quantum yield measurements (Φ = 0.55) further support the involvement of a photocatalytic pathway in the reaction (refer to ‘Quantum Yield Measurements’ in the Supplementary Information for detailed information). However, radical chain pathway could not be entirely ruled out from the transformation. Cyclic voltammetry revealed that the oxidation potential of the corresponding potassium salt of 2a (Ep/2 = +1.49 V vs SCE in MeCN) is substantially higher than that of IrIV (E1/2III*/II = +1.21 V vs SCE in MeCN)36,37, ruling out the possibility of single-electron oxidative decarboxylation initiated by the excited IrIII species (Fig. 4b). This conclusion is bolstered by Stern-Volmer studies, where the [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 was not reductively quenched by 2a, but was quenched by Selectflour (E1/2red = +0.33 V vs SCE in MeCN; E1/2III*/IV = −0.89 V vs SCE in MeCN) (Fig. 4c)38,39,40. The notable decrease in the Stern-Volmer plot slope for 2a was ascribed to the overlapping light absorption by 2a, which caused an increase in fluorescence intensity.

a Light on/off experiment. b Cyclic voltammogram of the potassium salt of 2a in MeCN. c Stern-Volmer Studies. d Proposed reaction mechanism.
Proposed mechanism
The proposed mechanism is illustrated in Fig. 4d. Initially, exposing [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 to visible light excites it to the IrIII* state, which then engages in the oxidative quenching with N-radical cation (III) to generate IrIV species. A single-electron transfer (SET) from IrIV to the carboxylate anion (I)-formed by deprotonating carboxylic acid (2)-yields the carboxy radical (V). This radical rapidly undergoes decarboxylation, producing the alkyl radical (VI). The alkyl radical (VI) then adds to the alkene, initiating an intramolecular heteroaryl migration followed by SO2 expulsion. The resultant alkyl radical (VIII) abstracts a fluorine atom from Selectfluor (II) to create the targeted product (3), simultaneously regenerating the radical cation (III).
Discussion
We present a modular synthesis of alkylfluorides through a sequence of radical decarboxylation-desulfonylation reactions. Departing from conventional radical decarboxylative fluorination approaches, this protocol constructs multiple chemical bonds simultaneously and introduces a versatile means to assemble alkylfluorides with remarkable structural intricacy and extensive diversity. The multi-component reaction proceeds orderly under mild photochemical conditions, steered by the interplay of radical polarity and kinetic influences. Both styrene derivatives and the more challenging non-activated alkenes, particularly aliphatic alkenes, are suitable substrates for this transformation. Furthermore, this method allows for the incorporation of a monofluoroalkyl moiety into complex alkene frameworks.
Methods
General procedure for the synthesis of alkyl fluorides
1 (0.2 mmol), 2 (0.4 mmol), [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (2 mol %), Selectfluor (0.6 mmol), and KHCO3 (0.6 mmol) in DCM (2 mL) was loaded in a 4 mL reaction vial. The mixture was stirred with 30 W blue LEDs at 20 °C under N2 for 48 h. After evaporation of the solvent under reduced pressure, the residue was purified by flash column chromatography on silica gel to give the desired product.
Data availability
The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author on request.
References
Dolbier, W. R. Structure, reactivity, and chemistry of fluoroalkyl radicals. Chem. Rev. 96, 1557–1584 (1996).
Müller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881–1886 (2007).
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320–330 (2008).
Meanwell, N. A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 54, 2529–2591 (2011).
Wang, J. et al. Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade. Chem. Rev. 114, 2432–2506 (2014).
Campbell, M. G. et al. Bridging the gaps in 18F PET tracer development. Nat. Chem. 9, 1–3 (2017).
Meanwell, N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 61, 5822–5880 (2018).
Tantawy, A. H. et al. Design, synthesis, biological evaluation, and computational studies of novel fluorinated candidates as PI3K inhibitors: targeting fluorophilic binding sites. J. Med. Chem. 64, 17468–17485 (2021).
Singh, R. P. & Shreeve, J. M. Recent highlights in electrophilic fluorination with 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate). Acc. Chem. Res. 37, 31–44 (2004).
Grushin, V. V. The organometallic fluorine chemistry of palladium and rhodium: studies toward aromatic fluorination. Acc. Chem. Res. 43, 160–171 (2010).
Hollingworth, C. & Gouverneur, V. Transition metal catalysis and nucleophilic fluorination. Chem. Commun. 48, 2929–2942 (2012).
Petrone, D. A., Ye, J. & Lautens, M. Modern transition-metal-catalyzed carbon-halogen bond formation. Chem. Rev. 116, 8003–8104 (2016).
Yan, H. & Zhu, C. Recent advances in radical-mediated fluorination through C-H and C-C bond cleavage. Sci. China Chem. 60, 214–222 (2017).
Szpera, R., Moseley, D. F., Smith, L. B., Sterling, A. J. & Gouverneur, V. The fluorination of C-H bonds: developments and perspectives. Angew. Chem. Int. Ed. 58, 14824–14848 (2019).
Zhang, Y. et al. Ketones as ideal photocatalysts for decarboxylative fluorination and a competition with C(sp3)-H fluorination. Chem. Catal. 4, 101162 (2024).
Xiao, P., Pannecoucke, X., Bouillon, J.-P. & Couve-Bonnaire, S. Wonderful fusion of organofluorine chemistry and decarboxylation strategy. Chem. Soc. Rev. 50, 6094–6151 (2021).
Patrick, T. B., Johri, K. K. & White, D. H. Fluoro-decarboxylation with xenon difluoride. J. Org. Chem. 48, 4158–4159 (1983).
Wang, C. M. & Mallouk, T. E. New photochemical method for selective fluorination of organic molecules. J. Am. Chem. Soc. 112, 2016–2018 (1990).
Yin, F., Wang, Z., Li, Z. & Li, C. Silver-catalyzed decarboxylative fluorination of aliphatic carboxylic acids in aqueous solution. J. Am. Chem. Soc. 134, 10401–10404 (2012).
Rueda-Becerril, M. et al. Direct C-F bond formation using photoredox catalysis. J. Am. Chem. Soc. 136, 2637–2641 (2014).
Ventre, S., Petronijevic, F. R. & MacMillan, D. W. C. Decarboxylative fluorination of aliphatic carboxylic acids via photoredox catalysis. J. Am. Chem. Soc. 137, 5654–5657 (2015).
Huang, X., Liu, W., Hooker, J. M. & Groves, J. T. Targeted fluorination with the fluoride ion by manganese-catalyzed decarboxylation. Angew. Chem. Int. Ed. 54, 5241–5245 (2015).
Zhang, R., Ni, C., He, Z. & Hu, J. Decarboxylative fluorination of β-Ketoacids with N-fluorobenzenesulfonimide (NFSI) for the synthesis of α-fluoroketones: Substrate scope and mechanistic investigation. J. Fluor. Chem. 203, 166–172 (2017).
Wang, Z., Guo, C.-Y., Yang, C. & Chen, J.-P. Ag-catalyzed chemoselective decarboxylative mono-and gem-difluorination of malonic acid derivatives. J. Am. Chem. Soc. 141, 5617–5622 (2019).
Zhang, Y., Qian, J., Wang, M., Huang, Y. & Hu, P. Visible-light-induced decarboxylative fluorination of aliphatic carboxylic acids catalyzed by iron. Org. Lett. 24, 5972–5976 (2022).
Wnuk, S. F., Rios, J. M., Khan, J. & Hsu, Y. L. Stannyl Radical-mediated cleavage of π-deficient heterocyclic sulfones. Synthesis of α-fluoro esters. J. Org. Chem. 65, 4169–4174 (2000).
Zhang, L., Huang, Y. & Hu, P. Iron-catalyzed SO2-retaining smiles rearrangement through decarboxylation. Org. Lett. 26, 10940–10945 (2024).
Wu, X. & Zhu, C. Radical-mediated remote functional group migration. Acc. Chem. Res. 53, 1620–1636 (2020).
Wu, X., Ma, Z., Feng, T. & Zhu, C. Radical-mediated rearrangements: past, present, and future. Chem. Soc. Rev. 50, 11577–11613 (2021).
Yu, J., Wu, Z. & Zhu, C. Efficient docking-migration strategy for selective radical difluoromethylation of alkenes. Angew. Chem. Int. Ed. 57, 17156–17160 (2018).
Liu, J. et al. Polarity umpolung strategy for the radical alkylation of alkenes. Angew. Chem. Int. Ed. 59, 8195–8202 (2020).
Yu, J. et al. Metal-free radical difunctionalization of ethylene. Chem 9, 472–482 (2023).
Schwarz, J. & König, B. Decarboxylative reactions with and without light-a comparison. Green. Chem. 20, 323–361 (2018).
Liu, C., Wang, X., Li, Z., Cui, L. & Li, C. Silver-catalyzed decarboxylative radical azidation of aliphatic carboxylic acids in aqueous solution. J. Am. Chem. Soc. 137, 9820–9823 (2015).
Tan, X. et al. Silver-catalyzed decarboxylative trifluoromethylation of aliphatic carboxylic acids. J. Am. Chem. Soc. 139, 12430–12433 (2017).
Prier, C. K., Rankic, D. A. & MacMillan, D. W. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322–5363 (2013).
Lowry, M. S. et al. Single-layer electroluminescent devices and photoinduced hydrogen production from an ionic iridium (III) complex. Chem. Mater. 17, 5712 (2005).
Girina, G. P., Fainzil’berg, A. A. & Feoktistov, L. G. Reduction potential as a measure of activity of the NF fluorinating agents. Russ. J. Electrochem. 36, 162–163 (2000).
Stavber, S. & Zupan, M. Selectfluor F-TEDA-BF4 as a versatile mediator or catalyst in organic chemistry. Acta Chim. Slov. 52, 13–26 (2005).
González-Esguevillas, M., Miró, J., Jeffrey, J. L. & MacMillan, D. W. Photoredox-catalyzed deoxyfluorination of activated alcohols with Selectfluor®. Tetrahedron 75, 4222–4227 (2019).
Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (grant numbers 22171201 and 22371185, C.Z.), the Fundamental Research Funds for the Central Universities (23×010301599 and 24×010301678, C.Z.), and the Program of Shanghai Academic/ Technology Research Leader (23XD1421900, C.Z.).
Author information
Authors and Affiliations
Contributions
C.Z. conceived the concept and directed the project. X.-J.W., H.L., Y.C., and Z.W. performed experiments. X.-J.W., H.L., and X.-X.W. prepared the Supplementary Information. C.Z. and X.-X.W. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Li, H., Chen, Y. et al. Modular access to alkylfluorides via radical decarboxylative-desulfonylative gem-difunctionalization. Nat Commun 16, 4702 (2025). https://doi.org/10.1038/s41467-025-60011-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60011-0
